

SUMMARY
Y-Family DNA polymerases (DNAPs) are often required in cells to synthesize past DNA containing lesions, such as [+ta]-B[a]P-N2-dG, which is the major adduct of the potent mutagen/carcinogen benzo[a]pyrene (B[a]P). The current model for the non-mutagenic pathway in E. coli involves DNAP IV inserting dCTP opposite [+ta]-B[a]P-N2-dG and DNAP V doing the next step(s), extension. We are investigating what structural differences in these related Y-Family DNAPs dictate their functional differences. X-ray structures of Y-Family DNAPs reveal a number of interesting features in the vicinity of the active site, including: (1) the “roof-aa,” which is the amino acid that lies above the nucleobase of the dNTP and is expected to play a role in dNTP insertion efficiency, and (2) a cluster of three amino acids, including the roof-aa, which anchors the base of a loop, whose detailed structure dictates several important mechanistic functions. Since no X-ray structures existed for UmuC (the polymerase subunit of DNAP V) or DNAP IV, we previously built molecular models. Herein, we test the accuracy of our UmuC(V) model by investigating how amino acid replacement mutants affect lesion bypass efficiency. A ssM13-vector containing a single [+ta]-B[a]P-N2-dG is transformed into E. coli carrying mutations at I38, which is the roof-aa in our UmuC(V) model, and output progeny vector yield is monitored as a measure of the relative efficiency of the non-mutagenic pathway. Findings show that: (1) the roof-aa is almost certainly I38, whose β-carbon branching R-group is key for optimal activity, and (2) I38/A39/V29 form a hydrophobic cluster that anchors an important mechanistic loop aa29-39. In addition, bypass efficiency is significantly lower for both the I38A mutation of the roof-aa and for the adjacent A39T mutation; however, the I38A/A39T double mutant is almost as active as wild type UmuC(V), which probably reflects the following. Y-Family DNAPs fall into several classes with respect to the [roof-aa/next amino acid]: one class has [isoleucine/alanine] and includes UmuC(V) and DNAP η (from many species), while the second class has [alanine (or serine)/threonine] and includes DNAP IV, DNAP κ (from many species) and Dpo4. Thus, the high activity of the I38A/A39T double mutant probably arises because UmuC(V) was converted from the V/η-class to the IV/κ-class with respect to the [roof-aa/next amino acid]. Structural and mechanistic aspects of these two classes of Y-Family DNAPs are discussed.
Keywords: mutations, cancer, Y-Family DNA polymerases, DNA polymerase V, benzo[a]pyrene
INTRODUCTION
Cells possess many DNA polymerases (DNAPs); e.g., human, yeast (S. cerevisae) and E. coli have at least fifteen, eight and five, respectively.1 - 3 The cellular role of some DNAPs can be understood by noting that DNA is constantly being damaged by radiation and chemicals, and most adducts/lesions that are not removed by DNA repair block replicative DNA polymerases. To avoid such lethal blockage, cells possess lesion-bypass DNAPs,1 - 12 which conduct translesion DNA synthesis (TLS). Many lesion-bypass DNAPs are in the Y-family,1 - 12 where humans have three (DNAPs η, ι and κ), yeast have one (DNAP η) and E. coli has two (DNAPs IV and V).
Y-Family DNAPs have a conserved ~350aa core, which includes the polymerase active site [representative references 13 - 22]. As with all DNA polymerases, Y-Family members resemble a right-hand with thumb, palm and fingers domains, although their “stubby” fingers and thumb result in more solvent accessible surface around the template/dNTP-binding pocket,7 presumably to accommodate the bypass of bulky and/or deforming DNA adducts/lesions, which protrude into these open spaces. Y-Family DNAPs grip DNA with an additional domain,7, 13, 17, 18 usually called the “little finger”. Steps in the mechanism of Y-Family DNAPs have been proposed for both protein structural changes based on a series of X-ray structures22-24 and for chemical catalysis based on theoretical studies.25
Our work has focused on benzo[a]pyrene (B[a]P), which is a well-studied DNA damaging agent that is a potent mutagen/carcinogen and an example of a polycyclic aromatic hydrocarbon (PAH), a class of ubiquitous environmental substances produced by incomplete combustion.26 - 27 PAHs in general and B[a]P in particular induce the kinds of mutations thought to be relevant to carcinogenesis and may be important in human cancer (representative reference, 28). B[a]P mutational spectra were established with the biologically relevant metabolite (+)-anti-B[a]PDE in E. coli,29 yeast30, 31 and mammalian (CHO) cells [reference 32 and references therein]. Mutagenesis has also been studied with [+ta]-B[a]P-N2-dG (+BP, Figure 1), the major adduct of (+)-anti-B[a]PDE, and G->T mutations predominate in most cases [reference 33 and references therein].
Figure 1.
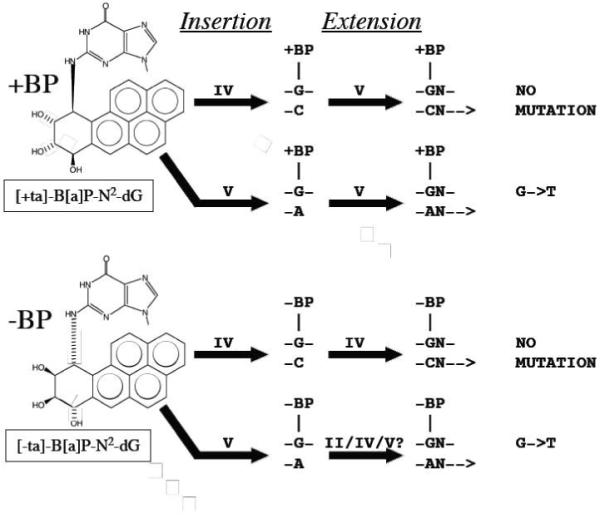
Structures of [+ta]- and [-ta]-B[a]P-N2-dG and a summary of the roles of Y-Family DNAPs IV and V in TLS in E. coli (see Introduction)
Based on genetic studies, DNAPs IV and V are both involved in the non-mutagenic translesion synthesis (TLS) pathway with +BP in E. coli.34 - 40 Why might two DNAPs required? Certain lesions need one DNAP for insertion (dCTP in this case) and a second for extension, which (following adduct-G:C formation) involves dNTP insertion opposite the next 5′-template base (and additional bases).41 - 42 Figure 1 presents the current understanding of insertion and extension for both +BP and -BP as supported by the following observations. Purified DNAP IV principally inserted dCTP (>99%) opposite +BP and -BP in a 5′-CGA sequence,34 and evidence in cells suggests DNAP IV does dCTP insertion opposite +BP35 - 39 and -BP [37], as well as other N2- dG adducts.38, 39 In contrast, purified DNAP V almost exclusively inserted dATP opposite +BP in a 5′-CGA sequence (>99%),34 and genetic findings show that DNAP V must be responsible for dATP insertion opposite +BP in the G->T pathway in E. coli.40 Collectively, these findings suggest it is DNAP IV, and not DNAP V, that does dCTP insertion opposite +BP, implying that the role of DNAP V is likely to be for extension in the non-mutagenic pathway. In the non-mutagenic pathway in E. coli with -BP, only DNAP IV is required,37 implying that DNAP IV does insertion and extension with -BP. The notion that DNAP IV does extension with -BP while DNAP V does extension with +BP is consistent with kinetic findings with purified proteins: DNAP IV is significantly worse than DNAP V at the extension step in the case of +BP compared to -BP.34 The fact that G->T mutations in a 5′-TGT sequence depend on DNAP V, but not DNAPs II or IV,40 suggest that DNAP V does both dATP insertion and extension in the mutagenic pathway. In vitro evidence also suggests that DNAP V does dATP insertion opposite -BP,34 and we have preliminary evidence that DNAP V is involved in the G->T pathway in E. coli with -BP, though involvement by DNAPs II and IV has not yet been investigated. Though random mutagenesis studies with [+anti]-B[a]PDE suggest that most G->T mutations with B[a]P-adducts require SOS-induction, implying involvement of a lesion-bypass DNAP, a minor non-SOS-inducible G->T pathway does exist [discussed in reference 37] and was studied in a 5′-GGA sequence, in which dATP insertion opposite +BP by DNAP III was proposed.35
The study of E. coli’s Y-Family DNAPs may provide insights about Y-Family DNAPs in general. Human DNAP κ was originally discovered because its sequence closely resembles E. coli DNAP IV,43 - 45 and dNTP insertion opposite a variety of adducts/lesions, including +BP, is remarkably similar for the DNAP IV/κ pair (Table 1), suggesting they are functional orthologs [discussed in reference 46]. This notion was substantiated when the identical mutation in a conserved residue in the active site of DNAP IV and DNAP κ (the “steric gate”, which excludes rNTPs) had a similar effect on lesion bypass vs. normal replication both in vitro and in cells.38 E. coli DNAP V and human DNAP η are also functional orthologs, based on their similarity of dNTP insertion opposite a variety of adducts/lesions (Table 147).
Table 1.
Dominant dNTP insertions opposite various DNA adducts/lesions by E. coli DNAPs IV and V and human DNAPs κ and η1
| Lesion | DNAP V | DNAP η | DNAP IV | DNAP κ |
|---|---|---|---|---|
| [+ta]-BP-N2-dG | A/C | A≥G | C | C |
| AAF-C8-dG | C | C | C/T | C/T |
| AF-C8-dG | - | - | C | C |
| TT-CPD | AA | AA | n | n |
| T(6-4)T | AG | nG | n | n |
| AP site | A | A | n | A* |
Dominant dNTP insertion using purified DNAPs, where “n” indicates “no” or low activity, “A*” indicates bypass by an unusual mechanism, and “-” indicates data unavailable. Data, as reviewed in reference 47.
DNAPs IV and κ have been shown to accurately bypass a variety of N2-dG adducts,34 - 39 including those formed from endogenous trioses,39 which may be the main cellular rationale for the genesis of the IV/κ-class. A case has been made that the main cellular rationale for the DNAP V/η-class is TLS of UV-induced CPDs (discussed in reference 46).
There must be structural reasons why the insertion preference opposite adducts/lesions is different for the DNAP IV/κ-class vs. the DNAP V/η-class (Table 147). The key differences, though, are not obvious given that the orthologs UmuC(V) and hDNAP η are only 20% identical by alignment, and not more identical than (e.g.) the non-ortholog pair UmuC(V) and hDNAP κ, which are 21% identical.47 Understanding the structural basis for mechanistic differences is further complicated by the fact that no X-ray structures exist for UmuC (the polymerase subunit of DNAP V), DNAP IV or hDNAP η, though X-ray structures exist for hDNAP κ with DNA.21 The need for protein structural information to guide our investigation of protein functional differences induced us to build models of UmuC(V), DNAP IV and hDNAP η taking a homology modeling approach.46 - 48 Though analysis of X-ray structures, modeled structures and sequence alignment suggest that Y-Family DNAPs lend themselves to accurate homology modeling,46 - 48 we wished to evaluate whether our models are likely to be correct, especially in the vicinity of the active site. Herein, we have investigated aspects of the structure of UmuC(V), which we believe is unlikely to have an X-ray structure in the near term given its complexity and how difficult it is to purify (e.g., see reference 49). We have taken a classic structure-activity approach: mutating the protein and inferring aspects of structure from changes in activity.
One residue of interest in Y-Family DNAPs is the “roof-amino acid”, which is a positionally conserved residue that lies above the nucleobase of the dNTP, as seen in the active site of Dpo413 - 16, yeast DNAP η,18, 19 human DNAP ι20 and hDNAP κ21 (purple residues in Figures 2A-D). The roof-aa might influence dNTP insertion. Figure 3 shows the sequence alignment in the vicinity of the roof-aa of all Y-Family DNAPs for which X-ray or modeled structures exist,13 - 21, 46 - 48 along with sequences of DNAPs κ and η from several other species. Based on the alignment in Figure 3, I38 is the roof-aa. Figure 2E shows that I38 (purple) in our UmuC model is positioned similarly to the roof-aa in other Y-Family DNAPs for which X-ray structures exist (Figures 2A-D). Figure 4A shows a top view of our UmuC model with I38 (purple) stacked on top of the nucleobase of the dNTP (white). Isoleucine is the roof-aa for the functional ortholog hDNAP η, as well as for DNAP η from other species (Figure 3).48 Serine is the roof-aa in our DNAP IV model,47 and serine is the roof-aa for its functional ortholog hDNAP κ, as well as for DNAP κ from most other species.48 (The roof-aa can also be cysteine or alanine in DNAP κ.) This correlation reinforces the notion that the roof-aa probably plays an important conserved role in protein function, and a role in dNTP insertion is a sensible possibility. Experiments described herein show that I38-mutants affect DNAP V bypass efficiency in a pattern that can be rationalized if I38 is truly the roof-aa, and that the β-branched structure of isoleucine is important to optimize DNAP V activity.
Figure 2.

Comparing the structures of Y-family DNAPs in the region corresponding to aa24-68 of Dpo4 (see Figure 3 for alignment). The figures are from X-ray coordinates for Dpo4 (1SOM, B subunit, aa24-6814), scDNAP η (1JIH,19 aa47-84), hDNAP ι (1TN3,20 aa51-88, but designated as aa471-508 in the B subunit), hDNA Ä (1T94,21 aa124-161), as well as the same regions for our models of E. coli UmuC(V) (the polymerase subunit of DNAP V) and DNAP IV.47 The template base (dT, turquoise) and dATP (red) are shown for Dpo4. The view is into the developing minor groove with the R-group of the roof-aa (purple) extending into the developing minor groove. The roof-amino acids (purple) are A44 (Dpo4), V64 (hDNAP ι), I60 (scDNAP η), S137 (hDNAP κ), I38 (UmuC(V)) and S41 (DNAP IV). The brown portions of the ribbons in Figures 2A and 2B are the corresponding non-homologous regions in Dpo4 (aa31-46) and scDNAP η (aa53-62), respectively.
Figure 3.
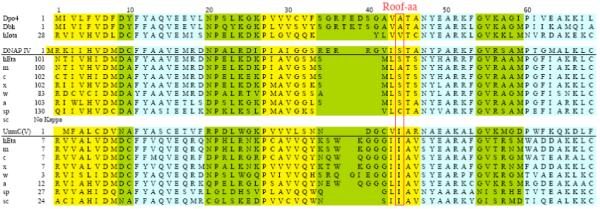
Amino acid alignment for the regions corresponding to aa1-69 in Dpo4 for eight Y-family DNAPs whose structure has been determined by X-ray crystallography (Dpo4, Dbh, scDNAP η, hDNAP ι and hDNAP κ) or by homology molecular modeling (hDNAPs η, E. coli DNAP IV and UmuC of DNAP V), along with additional sequences for DNAPs κ and η from several other species. Data are from references 47 and 48. α helices (blue), β sheets (yellow) and turns (green) are shown. Boxes indicate conserved amino acids (five of eight) or other notable amino acid similarities. Abbreviations: ss (Sulfolobus solfataricus) h, human (Homo sapien); m, mouse (Mus musculus); c, chicken (Gallus gallus); x, Xenopus laevis (frog); w, worm (Caenorhabditis elegans); a, Arabidopsis thaliana (plant); sc, Saccharomyces pombe (fission yeast); sp, Saccharomyces cerevisae (budding yeast). Colors: blue, α-helicies; yellow, β-strands; green, loops.
Figure 4.
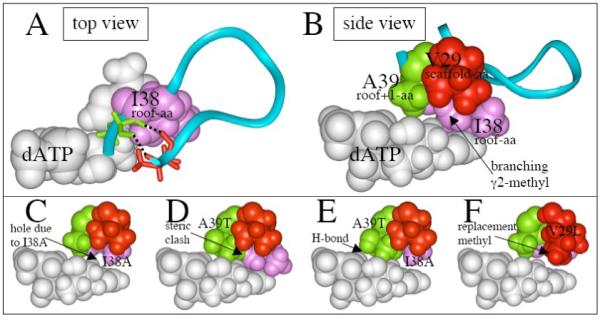
Structure of various regions of UmuC(V). (A) Top view showing how isoleucine-38 (purple) sits above dATP (white) in our UmuC(V) model, and is located in a loop (aa29-39) that includes V29 (scaffolding-aa, red), and A39 (roof+1-aa, green). The upper lip of this loop opens into the major groove, while the lower lip opens into a hole/cleft on the minor groove side, which can be analyzed based on analogy to a “chimney,”48 and which others call the “gap.”17 (B) Side view of the same structure as in panel A. (C) Model of UmuC(V) with isoleucine-38 converted to alanine-38 (I38A). (D) Model of UmuC(V) with alanine-39 converted to threonine-39 (A39T). (E) Model of UmuC(V) with both I38A and A39T. (F) Model of UmuC(V) with I38A and with valine-29 converted to isoleucine-29 (I38A/V29I).
Based on the alignment in Figure 3 and our UmuC(V) model,46-48 the roof-aa is in a loop that extends from aa29 to aa39. As depicted in Figure 4A, the top half of this loop faces into the major groove, which is extensively solvent exposed, while the bottom half faces into the minor groove, where Y-Family DNAPs have a hole/cleft in the protein surface. We call this hole/cleft the “chimney,”46-48 while others call it the “gap.”17 The size of this hole/cleft/gap/chimney is small both in our model of UmuC(V)48 and in the X-ray structure of its functional ortholog scDNAP η,18, 19 while the hole/cleft/gap/chimney is large in our model of DNAP IV48 and in the X-ray structure of its ortholog hDNAP κ.21 (Figures 2, 5A, 5B, 8B and 8D in reference 48 show these differences in opening size.) We recently discussed a likely mechanism by which the identity of several amino acids in this loop controls the size of the opening of the hole/cleft/gap/chimney, and how the large opening for the IV/κ-class might favor dCTP insertion opposite +BP, while the small opening for the V/η-class might lead to dATP insertion.48 To assess whether we have correctly aligned the aa29-39 loop in our UmuC(V) model, we investigated the following. In X-ray structures of other Y-Family DNAPs, this loop is anchored by a cluster of three amino acids, which correspond to V29/I38/A39 in our UmuC(V) model. Experiments herein support the notion that V29/I38/A39 are indeed clustered, which suggests that we have correctly aligned the amino acids in this loop and bolsters the likelihood that UmuC(V) has a small opening for its the hole/cleft/gap/chimney.
RESULTS
[+ta]-B[a]P-N2-dG was built into a 5′-TGC sequence in the single stranded genome of the phage M13mp7L2 (Materials and Methods) to give the adduct-containing vector +BP-TGC-ssM13, along with the unadducted control C-ssM13. Each vector was transformed (via electroporation) into E. coli that were proficient in DNAP V due to the presence of plasmid encoded umuD’+/C+ genes (Experimental Procedures), which were expressed from a constitutive umuD/C promoter, yielding UmuC(V) (the polymerase subunit of DNAP V), along with its regulatory subunit UmuD’ in its truncated and activated form, which precludes the need to activate DNAP V via SOS-induction. Plaque yield was used to assess progeny vector output, which is a measure of lesion bypass efficiency.
In a previous study with this identical vector,37 we showed that progeny vectors from individual plaques overwhelmingly had G at the original genome location of the adduct in +BP-TGC-ssM13 (>95%), which was confirmed in this study (data not shown). Thus, this assay provides information about the overall efficiency of the non-mutagenic pathway. Compared to progeny yield with +BP-TGC-ssM13 (taken as 100%), C-ssM13 yield was ~220% (±35%), based on eleven independent determinations, in which equal molar amounts of the two vectors were used. This finding indicates that E. coli has a reasonably efficient mechanism to bypass B[a]P-adducts as we noted previously.37 Progeny yield from +BP-TGC-ssM13 was much lower in DNAP V-deficient cells (~2.9%, Table 2) compared to DNAP V-proficient cells (100%), which is consistent with previous observations using another ΔumuD/C strain (yield ~3.4%37), and confirms a significant role for DNAP V in the non-mutagenic pathway.
Table 2.
A comparison of progeny phage output from a M13mp7L2 vector containing [+ta]-B[a]P-N2-dG as determined in E. coli carrying amino acid replacements at the roof-amino acid I38 in UmuC(V)1
| I38 | β | γ1 | γ2 | Yield | Protein Level3 |
|---|---|---|---|---|---|
| wt | -CH | -CH2CH3 | -CH3 | 100% (16) | 100% (3.9) |
| I38G | -H | -H | -H | 10% (2.9) | 48% (11) |
| I38A | -CH3 | -H | -H | 19% (4.0) | 99% (17) |
| I38C | -CHs | -SH | -H | 40% (16) | 130% (20) |
| I38V | -CH | -CH3 | -CH3 | 138% (9.9) | 95% (11) |
| I38M | -CH2 | -CH2SCH3 | -H | 12% (3.5) | 101% (20) |
| I38F | -CH2 | -Ph | -H | 20% (3.2) | 79% (22) |
| I38H | -CH2 | -Im | -H | 12% (4.5) | 140% (11) |
| I38L | -CH3 | -C3H72 | -H | 12% (3.0) | 119% (21) |
| I38S | -CH2 | -OH | -H | 16% (2.0) | 77% (13) |
| No UmuC | 2.9% (0.9) | <1% |
In parallel, a M13mp7L2 vector containing either [+ta]-B[a]P-N2-dG or no adduct was transformed into E. coli strains containing either wild type UmuC(V) (I38-wt) or various I38-mutants (e.g., I38G), or a cell with no UmuC (ΔumuC/D). The amino acid R-group is classified according to the atoms at the β-position, or the atoms attached to the β-carbon: γ1-only for straight-chain R-groups, or γ1- and γ2- if the β-carbon is branched. Progeny phage vector output (a measure of lesion bypass efficiency) was determined via plaque count (see text). Values (in percent) are the average of at least three experiments with standard deviation in parenthesis.
The amino acid leucine branches, but at the γ-position.
Protein level as determined by Western blotting. Values (in percent) are the average of at least three experiments with standard deviation in parenthesis.
+BP-TGC-ssM13 and unadducted control C-ssM13 were also transformed in parallel into E. coli strains containing I38-mutants of UmuC(V). In a representative experiment with unadducted control C-ssM13, plaque count was 1389 for I38-wt (100%), 1419 for I38L (102%), 2082 for I38C (150%) and 1494 for I38F (108%), which indicates little change in yield, as expected since DNAP V is not required to replicate undamaged templates. In contrast progeny yield with +BP-TGC-ssM13 varied for the I38-mutants as shown in Table 2 with standard deviations in parenthesis. Amino acid changes at positions other than I38, including double mutants, were also studied (Table 3). Western blotting was used to evaluate relative protein level of each mutant-UmuC(V). A typical Western blot is shown in Figure 5, and mutant-UmuC(V) protein level in comparison to wt-UmuC(V) is reported in Tables 2 and 3 with standard deviations in parenthesis.
Table 3.
A comparison of progeny phage output from a M13mp7L2 vector containing [+ta]-B[a]P-N2-dG as determined in E. coli carrying amino acid replacements at the roof-amino acid I38 and/or other sites in UmuC(V)1
| I38 | 2nd Site | Yield | Protein Level2 |
|---|---|---|---|
| wt | wt | 100% (16) | 100% (3.9) |
| I38A | wt | 19% (4.0) | 99% (17) |
| wt | A39T | 22% (2.8) | 120% (8.0) |
| I38A | A39T | 60% (16) | 104% (25) |
| wt | V29L | 32% (1.0) | 72% (11) |
| I38A | V29L | 34% (1.0) | 39% (14) |
| wt | V29I | 99% (25) | 157% (13) |
| I38A | V29I | 42% (6.0) | 126% (13) |
| wt | V29M | 244% (25) | 32% (3.0) |
| I38A | V29M | 23% (6.0) | 46% (1.0) |
Figure 5.
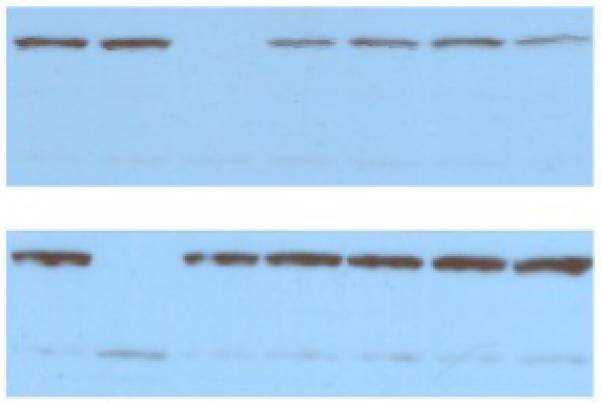
Western blotting of wt- and mutant-UmuC(V)s. Upper Panel (left-to-right): wt-UmuC(V), Δ-UmuC(V), I38A, I38A, I38V, I38V and I38C. Lower Panel (left-to-right): wt-UmuC(V), wt-UmuC(V), Δ-UmuC(V), I38G, I38G, I38F and I38F. The prominent band migrates at ~45K Daltons (compared to standards, data not shown), which is consistent with UmuC(V)’s calculated MW (47.7 Daltons).
DISCUSSION
The Effect of UmuC(V) Protein Concentration on Progeny Vector Yield
Given that each mutant-UmuC(V) was produced from the same plasmid, protein level was not expected to vary dramatically, as observed (Tables 2 and 3). If mutant-UmuC(V) protein level played a major role in determining progeny vector yield, then progeny vector yield should be proportional to protein level, which is not the case (Tables 2 and 3). For example, the protein levels for wt-UmuC(V), I38V, I38A, I38M and A39T/I38A, varied by less than 10%, even though their progeny vector yield varied by ~10-fold (12% - 138%, Tables 2 and 3).
These findings are not consistent with UmuC(V) protein level being a major determinant of progeny vector yield, which makes intuitive sense, given that our UmuC(V)/UmuD’ plasmid expression system is likely to result in more than a thousand copies of DNAP V per cell (see Materials and Methods), which is in vast excess to the one copy of vector +BP-TGC-ssM13 that a cell receives. One mutant-UmuC(V) is particularly revealing: V29M gave higher progeny yield (244%) than wt-UmuC(V), even though its protein level was relatively low (32%). In fact, if one normalizes for protein level, progeny vector yield for V29M-UmuC(V) would be 760% (= 244%/32% × 100%); this value is ostensibly impossible given that the highest possible yield from the +BP-TGC-ssM13 vector can only be 220%, which is the progeny yield for the unadducted control C-ssM13. This analysis suggests that as long as the cell has a threshold level, UmuC(V) can join the replication apparatus and conduct its bypass function. It also suggests that normalizing progeny vector yield for protein level does not make sense, so we did not do so, though none of the conclusions reached below would be significantly affected if normalization were invoked. (It will be of interest to investigate why V29M-UmuC(V) gives such high progeny yield.)
An R-group with a Branching β-Carbon Is Important for I38’s Role in UmuC(V)
The absence of an X-ray structure induced us to build a model of UmuC(V),46 - 48 which we hoped would help us begin to understand how UmuC(V) structure might affect its activity/mechanism. Herein we evaluate whether aspects of our UmuC(V) model are likely to be correct, taking a structure-activity approach, beginning with an evaluation of whether the properties of mutants at position-38 are consistent with it being the roof-aa.
The significance to TLS efficiency of the arrangement of atoms in the isoleucine R-group at I38 in UmuC(V) can be inferred from data in Table 2. The importance of R-group branching from the β-carbon is demonstrated by the fact that I38-wt (100%, R = -CH(Et)(Me)) and I38V (138%, R = -CH(Me)2) gave the highest yields. The low yield for I38L (12%, R = -CH2-CH(Me)2) shows specificity for branching from the β-carbon, since leucine branches at the γ-carbon. In general amino acids with R-groups not branching at the β-carbon gave progeny yields in the 10-20% range. For example, progeny output for the bulky I38F mutation (20%) is virtually the same as for I38A (19%), which shows that bulk pre se is not the key.
In fact, the adenine in dATP contacts the α-, β-, γ1- and γ2-carbons of the I38 R-group (Figures 4A and 4B), which is consistent with the findings that bypass efficiency is maximal for wt-I38 and I38V. I38-mutants lacking a branching γ2-methyl, such as I38A (Figure 4C), have an empty hydrophobic pocket, which undoubtedly leads to a perturbation of overall protein structure, making it suboptimal, and, thus, lowering catalytic activity.
Clustering of I38, A39 and V29 in the UmuC(V) Model
As outlined in Introduction, Y-Family DNAPs have a loop, which is aa29-39 in our UmuC model (Figures 4A and 4B) that contributes one edge to the hole/cleft/gap/chimney on the developing minor groove side of DNA near the active site (see Figures 2, 5A, 5B, 8B and 8D in reference 48). At its base, this loop is held together by two backbone hydrogen bonds (black dashed lines in Figure 4A), along with hydrophobic contacts in the V29/I38/A39 cluster (Figure 4B). We sought evidence that we have correctly identified the amino acids in this cluster in our UmuC(V) model.
Model building suggested that in a A39T-UmuC(V) mutant, threonine’s extra bulk would lead to disruptive crowding in the vicinity of the active site; e.g., as might be driven by the clash between threonine’s methyl group and the branching-γ2-methyl in I38 (Figure 4D). Indeed a decrease in bypass efficiency is observed with A39T (22%, Table 3). Model building revealed that threonine’s extra bulk might be better accommodated in the double mutant I38A/A39T (Figure 4D), or (alternatively formulated) that threonine’s extra bulk might compensate in part for the destablizing hole present in the I38A mutant. Consistent with this expectation, bypass efficiency was higher for the double mutant I38A/A39T (60%, Table 3) than for either single mutant I38A (19%) or A39T (22%). Based on a thermodynamic analysis,50 the yield from a double mutant should be the product of the yields from the two independent single mutants, which would be ~4% based on the yields for I38A and A39T, whereas the actual yield for I38A/A39T (60%) is much higher (~14-fold). (A discussion of both the thermodynamic analysis, along with assumptions and caveats, is given in Supplementary Materials.) Findings like this suggest that I38A and A39T are not behaving independently, but rather are functionally coupled, and where this kind of coupling has been observed in other proteins the two amino acids are usually in van der Waals contact (analyzed in reference 50), as observed for I38A and A39T in our model of UmuC(V).
V29 in UmuC(V) is aligned with V30 in Dpo4, and they are similarly positioned when comparing our UmuC(V) model (Figure 4A) and Dpo4 as revealed in X-ray structures.13 - 16 If V29 were positioned as shown in Figure 4A, then the hole left behind by the loss of the branching-γ2-methyl group with (e.g.) I38A (Figure 4C) could be filled by a V29L mutation, at least partially, since the R-group of leucine (Figure 4F) is longer than the R-group of valine (Figure 4C). Based on the single mutants I38A (19%) and V29L (32%), the double mutant V29L/I38A was expected to give an ~6% yield, if the mutations were operating independently. In fact, the yield for V29L/I38A (34%) is ~5.6-fold higher than expected. This suggests that V29L is compensating for something lost in the I38A mutant and suggests that they are in contact as observed in our UmuC(V) model. The expected yield for V29I/I38A is ~19% based on the single mutants V29I (99%) and I38A (19%), while the observed yield for V29I/I38A (42%) is ~2.2-fold higher than expected if V29I and I38A operated independently, which is also consistent with isoleucine being able to compensate for the hole left in I38A. Collectively, these findings suggest that I38 and V29 are in van der Waals contact.
Whatever advantage the V29M mutation alone provided to improve UmuC(V) efficiency is lost in the double mutant I38A/V29M (23%), whose yield is approximately the same as I38A itself (19%). The I38A/V29M yield is below the expected yield of 48% (= 244% × 19%) based on the yield for the individual single mutants, and I38A/V29M can be thought of as a kind of negative control, in that it shows that not all bulky R-groups at position-29 can compensate for the loss of bulk in the roof-aa position due to the I38A mutation.
Perspective on the UmuC(V) Model
As described in the previous section, the findings in Tables 2 and 3 can be rationalized by the arrangement of the three clustered amino acids I38, A39 and V29 in our UmuC(V) model (Figures 4A and 4B). In this subsection we reflect on whether X-ray structures of other Y-Family DNAPs, along with amino acid alignments, also support this line of thinking.
In Dpo4, the amino acid residues in the vicinity of the nucleobase of the dNTP are located in the aa32-68 region, which can be aligned with other Y-family DNAPs (Figure 3) because of extensive homology in the regions aa7-30 and aa47-68, as noted by Yang, Woodgate and colleagues.6 However, there is virtually no homology in the aa31-46 region, which is ostensibly problematic, given that the roof-aa (A44 in Dpo4) and other key amino acids in the active site loop are in this protein region. For example, Dpo4 and scDNAP η have no homology between aa31-46, and Dpo4 has seven more amino acids; yet their X-ray structures are virtually superimposable, including for the non-homologous stretch for aa31-46, where Dpo4 merely has a larger loop. (Compare the brown portions of ribbons in Figures 2A and 2C, which depict Dpo4 aa31-46 and the analogous region in scDNAP η aa53-62, respectively.) This is one of many examples showing structural similarities in various regions of Y-Family DNAPs (discussed more extensively in reference 47).
In fact, the entire aa24-68 region of Dpo4 is very similar for hDNAP ι, scDNAP η and hDNAP κ based on X-ray coordinates, with the only significant difference being the sizes of the right-most loops in Figures 2A-D, which correspond to the aa29-39 loop in UmuC(V) (Figures 2E, 4A and 4B). Each roof-amino acid has its R-group pointing toward the developing minor groove (purple residues in Figure 2), even though the roof-aa varies (A44 for Dpo4, S137 for hDNAP κ, V64 for hDNAP ι and I60 for scDNAP η). The roof-aa is similarly oriented in our model of UmuC(V) (Figure 2E), and it would be surprising if this orientation were not present in the UmuC(V) protein.
V29 in UmuC(V) is a conserved valine in most other Y-Family DNAPs (Figure 3), and it serves a multi-functional scaffolding role (discussed in reference 48). In addition to backbone hydrogen bonding with the roof+1-aa and forming hydrophobic contacts with the roof-aa and roof+1-aa, this scaffolding-aa (V29 in UmuC) contacts the steric-gate (Y11 in UmuC), which in turn face-stacks with another conserved amino acid (Y77 in our UmuC model) that helps orient one of the other chimney lips (aa71-74 in UmuC, blue, Figure 4G).
Thus, the clustering of I38/A39/V29 in our model of UmuC(V) is both consistent with the data in Tables 2 and 3, and reasonable based on analogy to A44/T45/V30 in Dpo413 - 16 and Dbh17, to I60/A61/C54 in scDNAP η, 18, 19 V64/T65/V57 in hDNAP ι,20 and to S137/T137/V130 in hDNAP κ.21 Collectively, these observations suggest that our alignment of UmuC(V) based on homologous regions aa7-30 and aa47-68 (Figure 3) is largely correct, and that our UmuC(V) model is likely to be reasonably accurate in the vicinity of the roof-aa and the active site.
Roof-aa and Roof+1-aa Region in Y-Family DNAPs
The I38A/A39T-UmuC(V) double mutant was chosen for study in part because Dpo4 has A44/T45 for its [roof-aa/roof+1-aa] (Figure 3). Thus, I38A/A39T-UmuC(V) is undoubtedly relatively active because it resembles Dpo4 structurally, which is apparent when comparing I38A/A39T in our UmuC(V) model (Figure 4E) with A44/T45 in various Dpo4 structures.13-16 These findings provide additional experimental support that we have correctly aligned I38/A39 in UmuC(V) with A44/T45 in Dpo4 (Figure 3), as first proposed in the alignment by Yang, Woodgate and colleagues.6 In I38A/A39T-UmuC(V) the threonine methyl sits near where the branching-γ2-methyl of I38 is in wt-UmuC(V) (compare Figures 4E and 4B), while the hydroxyl of threonine forms a hydrogen bond with a non-bonded oxygen on Pβ of the dNTP (Figure 4E), which is also observed in Dpo4 structures.13 - 16
In fact, a correlation is observed vis-a-vis the [roof-aa/roof+1-aa] in the case of Y-Family DNAPs. In the alignment in Figure 3, all of the V/η-class DNAPs have isoleucine as the roof-aa, followed by alanine, while most IV/κ-class DNAPs have serine as the roof-aa, and all are followed by threonine; the exceptions still have a non-bulky roof-aa, such alanine, which is also observed in Dpo4 and Dbh. In fact in the first ~350 amino acids, only a few positions show this kind of correlation, where one amino acid is preferred in the V/η-class and a different amino acid is preferred in the IVκ-class.
Though we do not know the mechanistic reason(s) for this difference in the [roof-aa/roof+1-aa] for the IV/κ-vs. V/η-class, we can offer the following thoughts. 46,48 Regarding the roof-aa, both a non-bulky alanine (or serine) and a bulky isoleucine are able to stack on top of the nucleobase when the dNTP adopts its “chair-like” shape, which is the canonical shape of dNTPs as observed in the active sites of non-Y-Family DNAPs in addition to the X-ray structures of many Y-Family DNAPs; however, a second non-canonical “goat-tail-like” shape15 has also been observed in Y-Family DNAP X-ray structures (discussed in references 46 and 48). (For simplicity we refer to the “chair-like” shape as S1-dNTP and the “goat-tail-like” shape as S2-dNTP.) The nucleobase in the non-canonical S2-dNTP shape lies lower down in the active site, and in our modeling studies isoleucine is able to rearrange downward in order to contact the nucleobase of the S2-dNTP, while a non-bulky roof-aa (serine or alanine) cannot, thus leaving a gap above the nucleobase in the S2-dNTP shape. This observation may mean that the V/η-class might be better able to use the S2-dNTP shape during catalysis, though this is highly speculative and the purpose/need for an alternate S2-dNTP shape (if any) is not clear.46,48
For the roof+1-aa position, threonine appears to be the better inherent choice, because its -OH can form a hydrogen bond with an oxygen on Pβ of the dNTP as seen in numerous Dpo4 structures.13-16 Threonine is compatible with a non-bulky roof-aa (serine or alanine) in the IV/κ-class, but it seems less compatible with isoleucine as the roof-aa in the V/η-class, because of overcrowding (Figure 4E), and thus alanine is the dominant roof+1-aa. Unlike threonine, alanine cannot form a hydrogen bond; however, in scDNAP η there is a compensating mechanism: an arginine (R79) on an adjacent β-strand forms a hydrogen bond with this same oxygen on Pβ. This arginine is conserved in DNAP η from most species.48 All X-ray structures of Y-family DNAPs have a highly conserved arginine (e.g., R73 in scDNAP η) that lies above the dNTP and forms a second hydrogen bond to this same oxygen on Pβ. Interestingly, most bacterial UmuC(V) orthologs appear to use a different structural strategy to interact with the oxygen on Pβ of the dNTP: they have a lysine (K45 in ecUmuC(V)) instead of arginine at the R73-equivalent position of scDNAP η, while they lack arginine at the R79-equivalent position, having a methionine instead (M51 in ecUmuC(V). It will be of interest to investigate possible reasons and consequences of these differences between bacterial UmuC(V) and DNAP η.
Finally, a meta-analysis of the sequences from a large number of Y-Family DNAPs reveals that there are many variations on the themes alluded to in the previous paragraphs (data not shown).
MATERIALS AND METHODS
A +BP-containing oligonucleotide (5′-GAAGACCTGCAGG) was built into ss-M13mp7L2 following the approach of Lawrence and LeClerc,51 as refined by Delaney and Essigmann,52, 53 and exactly as we have done previously37. The product is called +BP-TGC-ssM13, and a non-adduct-containing control C-ssM13 was constructed in parallel.
+BP-TGC-ssM13 and C-ssM13 were transformed in parallel via electroporation into E. coli exactly as done previously37, except as noted below. Strain GW8023 and plasmid pGY9738 were used (obtained from Graham Walker, M.I.T.). GW802354 has ΔumuD/C595::cat in GW2771 (AB1157 with sulA, ilvts, ΔlacU169, pro+). pGY973855 contains umuD’/C with an operator mutation, leading to constitutive expression of UmuC(V) and UmuD’, where the latter is a truncated version that mimics UmuD cleavage and activates DNAP V in the absence of SOS-induction. Thus, unlike our previous study,37 cells were not treated with UV light to induce the SOS response prior to transformation. When fully SOS induced, a cell has ~200 copies of DNAP V.56 Given this level of expression from a single chromosome, we expect over a thousand copies of DNAP V in cells based on constitutive expression from the same promoter in pGY9738, which is a pSC101-derived plasmid with ~7 copies per cell (reference 57 and references therein). Mutations were introduced into UmuC(V) in pGY9738 using the QuickChange kit (Stratagene), and were confirmed by DNA sequencing.
Following E. coli growth (to OD600 = 0.5) and electroporation, the transformed cells were immediately iced and diluted into ice-cold SOC media (<2 min), followed by plating (<20 min) using NR9050 cells as the plating bacteria.37 In-frame progeny M13 output was determined based on (blue) plaque count. Plaque count using the no-adduct control C-ssM13 was virtually similar from all E. coli strains, independent of what I38-mutant it contained (see Results), as expected, since DNAP V is not required to replicate undamaged DNA. We assume that variation in plaque count in different strains using C-ssM13 is attributable to variations in transformation efficiencies due to subtle variations in cell growth and/or preparation of cells for electroporation. Thus, we normalized results as the following example illustrates. C-ssM13 gave 1389 plaques with I38-wt (100%) and 1494 plaques (108%) with I38F, while +BP-TGC-ssM13 gave 711 plaques with I38-wt and 172 plaques with I38F. For this experiment the value of I38F is thus 23% (= 100% × [172/711]/[1494/1389]). Normalized values were determined in at least three independent experiments and average values are reported in Tables 2 and 3, along with standard deviations; e.g., I38A is 19% ±4%, which is about the typical standard deviation (average = 0.20). Statistical significance is generally taken as a difference of two standard deviations [i.e., P < 0.05], which is achieved if two values being compared do not have overlapping standard deviations; all comparisons in Discussion satisfy this criterion.
Western blotting was conducted as follows. E. coli were grown identically to the way they were grown for progeny output experiments. Cells (1.4 mL) were pelleted and resuspended in 80 μL loading buffer (10% glycerol, 5% 2-mercaptoethanopl, 2.3% SDS, 62.5 mM Tris, pH 6.8). Cells were lysed by boiling (5 min), and 10 μL aliquots were subjected to SDS PAGE (stacking gel: 5% acrylamide, 125mM Tris, 0.1% SDS, pH 6.8, for 30 min, 16mA, 75 V; resolving gel: 12% acrylamide, 375mM Tris, 0.1% SDS, pH 8.8, 1 hour, 30mA, 200V). The gel was then placed in an envelop: sponge, 3MM paper, gel, Immobillon membrane (Millipore, presoaked in absolute methanol for 15 seconds followed by de-ionized water for 1 minute), 3MM paper, sponge. Protein was transferred to the Immobillon membrane (1 hour, 220 mA, 100V in 20 mM Tris, 150 mM Glycine, 20% methanol). The membrane was washed four times with PBS-Tween (0.14M NaCl, 2mM KCl, 6 mM, Na2PO4, 1.7 mM KH2PO4, 0.1% Tween-20) for seven minutes and then blocked in 5% milk (Carnation) in PBS-Tween for 12 hours. Anti-UmuC(V) (obtained from Roger Woodgate,58 diluted 1:25,000 in SuperBlock Blocking Buffer, Thermo Scientific) was added to 50 mL PBS-Tween and 2.5% milk (3 hours) and then washed with PBS-Tween three times for seven minutes each. (Each Anti-UmuC(V) blotting solution was reusable ~15 times.) The membrane was probed with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (Pierce, diluted 1:15,000 in SuperBlock Blocking Buffer). Bound secondary antibodies were detected with Supersignal antibody and substrate (Pierce), where horseradish peroxidase catalyzed the reaction between H2O2 and luminol. Luminescence was detected using various time exposures on CL-X Posure Film (Pierce). Four exposure times were performed for each blot. Size markers were Broad Range Prestained Markers (New England BioLabs).
Films from Western blots were scanned with a CanoScan LiDE Color Image Scanner and converted to a .jpg file. Band intensity was quantitated using the ImageJ freeware program. Scanning of bands gave peaks, whose areas were determined above baseline. Signal intensity (peak area) was linearly related to the volume of cells loaded in the gel (data not shown). The [mutant-UmuC(V)/wt-UmuC(V)] peak area ratio did not vary significantly as a function of the volume of cells loaded in the gel or film exposure time for the blot (data not show). The area ratio [mutant-UmuC(V)/wt-UmuC(V)] was computed for three exposure times from each blot, and the average ratio determined. Each mutant-UmuC(V) was blotted at least three times and the average of these three values (and standard deviation) is reported in Tables 2 and 3.
Many X-ray structures were viewed in the course of this work, but for simplicity, we refer only to examples, which include Dpo4 (1SOM, subunit B14), scDNAP η (1JIH19), hDNAP ι (1TN3, subunit B20) and hDNA κ (1T9421), whose coordinates were accessed from the RCSB Protein Data Bank.59 Structures for UmuC(V) and DNAP IV are from our modeling work.46 - 48 Simple amino acid replacement was done to generate the UmuC(V)-mutant structures in Figures 4C-4F, which has the advantage of emphasizing the defect that the mutant protein must overcome, and the disadvantage that it does not show an accurate view of how the protein might accommodate the mutation(s), which would only be revealed following additional computational studies.
Supplementary Material
ACKNOWLEDGEMENT
We are grateful to Nicholas E. Geacintov and colleagues for the synthesis of the B[a]P-containing oligoncleotides, to James C. Delaney and John M. Essigmann for assistance in the use of the M13mp7L2 vector system, to Graham Walker and Penny Beuning for strains and plasmids, and to Roger Woodgate for a UmuC antibody. Supported by United States Public Health Services Grant R01ES03775.
Abbreviations used
- B[a]P
benzo[a]pyrene
- (+)-anti-B[a]PDE
7R,8S-dihydroxy-9S,10R-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene
- [+ta]-B[a]P-N2-dG
the major adduct of (+)-anti-B[a]PDE, formed by trans addition of N2-dG to (+)-anti-B[a]PDE (Figure 1)
- +BP-TGC-ssM13
a ss-M13 vector containing a single +BP adduct in a 5′-TGC sequence context
- C-ssM13
a ss-M13 vector containing a dG at the position equivalent to +BP in +BP-TGC-ssM13
- PAH
polycyclic aromatic hydrocarbon
- DNAP
DNA polymerase
- UmuC(V)
polymerase subunit of DNAP V
- MF
mutation frequency
- TLS
translesion synthesis
- aa
amino acid
- roof-aa
the amino acid lying above the nucleobase of the dNTP in a Y-family DNAP
- roof+1-aa
the amino acid that follows the roof-aa
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bebenek K, Kunkel TA. Functions of DNA Polymerases. Adv. Protein Chem. 2004;69:137–165. doi: 10.1016/S0065-3233(04)69005-X. [DOI] [PubMed] [Google Scholar]
- 3.Rothwell PJ, Waksman G. Structure and mechanism of DNA polymerases. Adv. Protein Chem. 2005;71:401–440. doi: 10.1016/S0065-3233(04)71011-6. [DOI] [PubMed] [Google Scholar]
- 4.Ohmori H, Friedberg EC, Fuchs RP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T, Prakash L, Prakash S, Todo T, Walker GC, Wang Z, Woodgate R. The Y-(2001) Family of DNA polymerases. Mol. Cell. :7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- 5.Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- 6.Boudsocq F, Ling H, Yang W, Woodgate R. Structure-based interpretation of missense mutations in Y-family DNA polymerases and their implications for polymerase function and lesion bypass. DNA Repair. 2002;1:343–358. doi: 10.1016/s1568-7864(02)00019-8. [DOI] [PubMed] [Google Scholar]
- 7.Yang W. Damage Repair DNA polymerases. Curr. Opin. Struct. Biol. 2003;13:23–30. doi: 10.1016/s0959-440x(02)00003-9. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs RP, Fujii S, Wagner J. Properties and Functions of Escherchia coli: Pol IV and Pol V. Adv. Protein Chem. 2004;69:229–264. doi: 10.1016/S0065-3233(04)69008-5. [DOI] [PubMed] [Google Scholar]
- 9.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 2005;7:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 10.Nohmi T. Environmental stress and lesion-bypass DNA polymerases. Annu. Rev. Microbiol. 2006;60:231–253. doi: 10.1146/annurev.micro.60.080805.142238. [DOI] [PubMed] [Google Scholar]
- 11.Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc. Natl. Acad. Sci. USA. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jarosz DF, Beuning PJ, Cohen SE, Walker GC. Y-Family DNA polymerases in Escherichia coli. Trends Microbiol. 2007:70–77. doi: 10.1016/j.tim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-Family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 14.Ling H, Sayer JM, Plosky BS, Boudsocq F, Woodgate R, Jerina DM, Yang W. Crystal structure of a benzo[a]pyrene diol epoxide adduct in a ternary complex with a DNA polymerase. Proc. Natl. Acad. Sci. USA. 2004;101:2265–2269. doi: 10.1073/pnas.0308332100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaisman A, Ling H, Woodgate R, Yang W. Fidelity of Dpo4: effect of metal ions, nucleotide selection and pyrophosphorolysis. EMBO J. 2005;25:2957–2967. doi: 10.1038/sj.emboj.7600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bauer J, Xing G, Yagi H, Sayer JM, Jerina DM, Ling H. A structural gap in Dpo4 supports mutagenic bypass of a major benzo[a]pyrene dG adduct in DNA through template misalignment. Proc. Natl. Acad. Sci. USA. 2007;104:14905–14910. doi: 10.1073/pnas.0700717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou BL, Pata JD, Steitz TA. Crystal structure of a DinB lesion bypass DNA polymerase catalytic fragment reveals a classic polymerase catalytic domain. Mol Cell. 2001;8:427–437. doi: 10.1016/s1097-2765(01)00310-0. [DOI] [PubMed] [Google Scholar]
- 18.Trincao J, Johnson RE, Escalante CR, Prakash S, Prakash L, Aggarwal AK. Structure of the catalytic core of S. cerevisiae DNA polymerase eta: implications for translesion DNA synthesis. Mol Cell. 2001;8:417–426. doi: 10.1016/s1097-2765(01)00306-9. [DOI] [PubMed] [Google Scholar]
- 19.Alt A, Lammens K, Chiocchini C, Lammens A, Pieck JC, Kuch D, Hopfner KP, Carell T. Bypass of DNA lesions generated during anticancer treatment with cisplatin by DNA polymerase eta. Science. 2007;318:967–970. doi: 10.1126/science.1148242. [DOI] [PubMed] [Google Scholar]
- 20.Nair DT, Johnson RE, Prakash S, Prakash L, Aggarwal AK. Replication by human DNA polymerase-iota occurs by Hoogsteen base-pairing. Nature. 2004;430:377–380. doi: 10.1038/nature02692. [DOI] [PubMed] [Google Scholar]
- 21.Lone S, Townson SA, Uljon SN, Johnson RE, Brahma A, Nair DT, Prakash S, Prakash L, Aggarwal AK. Human DNA polymerase kappa encircles DNA: implications for mismatch extension and lesion bypass. Mol. Cell. 2007;23:601–614. doi: 10.1016/j.molcel.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 22.Rechkoblit O, Malinina L, Cheng Y, Kuryavyi V, Broyde S, Geacintov E, Patel DJ. Stepwise translocation of Dpo4 polymerase during error-free bypass of an oxoG lesion. PLoS Biol. 2006;4:25–42. doi: 10.1371/journal.pbio.0040011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong JH, Fiala KA, Suo Z, Ling H. Snapshots of a Y-family DNA polymerase in replication: substrate-induced conformational transitions and implications for fidelity of Dpo4. J. Mol. Biol. 2008;379:317–330. doi: 10.1016/j.jmb.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 24.Beckman JW, Wang Q, Guengerich FP. Kinetic analysis of correct nucleotide insertion by a Y-family DNA polymerase reveals conformational changes both prior to and following phosphodiester bond formation as detected by tryptophan fluorescence. J. Biol. Chem. 2000;283:36711–36723. doi: 10.1074/jbc.M806785200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Yu X, Hu P, Broyde S, Zhang Y. A water-mediated and substrate-assisted catalytic mechanism for Sulfolobus solfataricus DNA polymerase IV. J Am Chem Soc. 2007;129:4731–4737. doi: 10.1021/ja068821c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harvey RG. Polycyclic Aromatic Hydrocarbons: Chemistry and Cancer. Wiley-VCH, Inc.; New York: 1997. [Google Scholar]
- 27.Dipple A. Polycyclic aromatic hydrocarbon carcinogens. In: Harvey RG, editor. Polycyclic Aromatic Hydrocarbons and Carcinogenesis. American Chemical Society Press; Washington, D.C.: 1985. pp. 1–17. [Google Scholar]
- 28.Pfeifer GP, Hainaut P. On the origin of G-to-T transversions in lung cancer. Mutat. Res. 2003;526:39–43. doi: 10.1016/s0027-5107(03)00013-7. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez H, Loechler EL. Mutagenesis by the (+)-anti-diol epoxide of benzo[a]pyrene: what controls mutagenic specificity? Biochemistry. 1993;32:373–383. doi: 10.1021/bi00058a009. [DOI] [PubMed] [Google Scholar]
- 30.Xie Z, Braithwaite E, Guo D, Zhao B, Geacintov NE, Wang Z. Mutagenesis of benzo[a]pyrene diol epoxide in yeast: requirement for DNA polymerase zeta and involvement of DNA polymerase eta. Biochemistry. 2003;42:11253–62. doi: 10.1021/bi0346704. [DOI] [PubMed] [Google Scholar]
- 31.Yoon JH, Lee CS, Pfeifer GP. Simulated sunlight and benzo[a]pyrene diol epoxide induced mutagenesis in the human p53 gene evaluated by the yeast functional assay: lack of correspondence to tumor mutation spectra. Carcinogenesis. 2003;24:113–119. doi: 10.1093/carcin/24.1.113. [DOI] [PubMed] [Google Scholar]
- 32.Schiltz M, Cui XX, Lu YP, Yagi H, Jerina DM, Zdzienicka MZ, Chang RL, Conney AH, Wei SJ. Characterization of the mutational profile of (+)-7R,8S-dihydroxy-9S, 10R-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene at the hypoxanthine (guanine) phosphoribosyltransferase gene in repair-deficient Chinese hamster V-H1 cells. Carcinogenesis. 1999;20:2279–2286. doi: 10.1093/carcin/20.12.2279. [DOI] [PubMed] [Google Scholar]
- 33.Seo K-Y, Nagalingam A, Loechler EL. Mutagenesis studies on four stereoisomeric N2-dG benzo[a]pyrene adducts in the identical 5′-CGC sequence used in NMR studies: Although adduct conformation differs, mutagenesis outcome does not as G->T mutations dominate in each case. Mutagenesis. 2005;20:441–448. doi: 10.1093/mutage/gei061. [DOI] [PubMed] [Google Scholar]
- 34.Shen X, Sayer JM, Kroth H, Ponten I, O’Donnell M, Woodgate R, Jerina DM, Goodman MF. Efficiency and accuracy of SOS-induced DNA polymerases replicating benzo[a]pyrene-7,8-diol 9,10-epoxide A and G adducts. J. Biol. Chem. 2002;277:5265–5674. doi: 10.1074/jbc.M109575200. [DOI] [PubMed] [Google Scholar]
- 35.Lenne-Samuel N, Janel-Bintz R, Kolbanovskiy A, Geacintov NE, Fuchs RP. The processing of a benzo(a)pyrene adduct into a frameshift or a base substitution mutation requires a different set of genes in Escherichia coli. Mol. Microbiol. 2000;38:299–307. doi: 10.1046/j.1365-2958.2000.02116.x. [DOI] [PubMed] [Google Scholar]
- 36.Napolitano R, Janel-Bintz R, Wagner J, Fuchs RP. All three SOS-inducible DNA polymerases (Pol II, Pol IV and Pol V) are involved in induced mutagenesis. EMBO J. 2000;19:6259–6265. doi: 10.1093/emboj/19.22.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seo K-Y, Nagalingam A, Miri S, Yin J, Kolbanovskiy A, Shastry A, Loechler EL. Mirror Image Stereoisomers of the Major Benzo[a]pyrene N2-dG Adduct Are Bypassed by Different Lesion-Bypass DNA Polymerases in E. coli. DNA Repair. 2006;5:515–522. doi: 10.1016/j.dnarep.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 38.Jarosz DF, Godoy VG, Delaney JC, Essigmann JM, Walker GC. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature. 2006;439:225–228. doi: 10.1038/nature04318. [DOI] [PubMed] [Google Scholar]
- 39.Yuan B, Cao H, Jiang Y, Hong H, Wang Y. Efficient and accurate bypass of N2-(1-carboxyethyl)-2′-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 2008;105:8679–8684. doi: 10.1073/pnas.0711546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin J, Seo K-Y, Loechler EL. A role for DNA polymerase V in G-to-T mutagenesis from the major benzo[a]pyrene N2-dG adduct when studied in a 5′-TGT sequence in Escherichia coli. DNA Repair. 2004;3:323–334. doi: 10.1016/j.dnarep.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 41.Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L. Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature. 2000;406:1015–1019. doi: 10.1038/35023030. [DOI] [PubMed] [Google Scholar]
- 42.Yuan F, Zhang Y, Rajpal DK, Wu X, Guo D, Wang M, Taylor JS, Wang Z. Specificity of DNA lesion bypass by the yeast DNA polymerase eta. J Biol Chem. 2000;275:8233–8239. doi: 10.1074/jbc.275.11.8233. [DOI] [PubMed] [Google Scholar]
- 43.Johnson RE, Prakash S, Prakash L. The human DINB1 gene encodes the DNA polymerase Poltheta. Proc Natl Acad Sci U S A. 2000;97:3838–3843. doi: 10.1073/pnas.97.8.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohashi E, Ogi T, Kusumoto R, Iwai S, Masutani C, Hanaoka F, Ohmori H. Error-prone bypass of certain DNA lesions by the human DNA polymerase kappa. Genes Dev. 2000;14:1589–1594. [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Yuan F, Wu X, Wang M, Rechkoblit O, Taylor JS, Geacintov NE, Wang Z. Error-free and error-prone lesion bypass by human DNA polymerase kappa in vitro. Nucleic Acids Res. 2000;28:4138–4146. doi: 10.1093/nar/28.21.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chandani S, Lee CH, Loechler EL. Molecular Modeling Benzo[a]pyrene N2-dG Adducts in Two Partially Overlapping Active Sites of the Y-Family DNA Polymerase Dpo4. Journal of Molecular Graphics and Modelling. 2007;25:658–670. doi: 10.1016/j.jmgm.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 47.Lee CH, Chandani S, Loechler EL. Homology modeling of four lesion-bypass DNA polymerases: structure and lesion bypass findings suggest that E. coli pol IV and human Pol k are orthologs, and E. coli pol V and human Pol η are orthologs. Journal of Molecular Graphics and Modelling. 2006;25:87–102. doi: 10.1016/j.jmgm.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 48.Chandani S, Loechler EL. Y-Family DNA Polymerases May Use Two Different dNTP Shapes for Insertion: A Hypothesis and Its Implications. Journal of Molecular Graphics and Modelling. 2009;27:759–769. doi: 10.1016/j.jmgm.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujii S, Gasser V, Fuchs RP. The biochemical requirements of DNA polymerase V-mediated translesion synthesis revisited. J Mol Biol. 2004;341:405–17. doi: 10.1016/j.jmb.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 50.Wells JA. Additivity of Mutational Effects in Proteins. Biochem. 1990;29:8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- 51.Lawrence CW, Borden A, Banerjee SK, LeClerc JE. Mutation frequency and spectrum resulting from a single abasic site in a single-stranded vector. Nucleic Acids Res. 1990;18:2153–2157. doi: 10.1093/nar/18.8.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delaney JC, Essigmann JM. Context-dependent mutagenesis by DNA lesions. Chem Biol. 1999;6:743–753. doi: 10.1016/s1074-5521(00)80021-6. [DOI] [PubMed] [Google Scholar]
- 53.Delaney JC, Henderson PT, Helquist SA, Morales JC, Essigmann JM, Kool ET. High-fidelity in vivo replication of DNA base shape mimics without Watson-Crick hydrogen bonds. Proc Natl Acad Sci. USA. 2003;100:4469–4473. doi: 10.1073/pnas.0837277100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Opperman T, Murli S, Walker GC. The genetic requirements for UmuDC-mediated cold sensitivity are distinct from those for SOS mutagenesis. J. Bacteriol. 1996;178:4400–4411. doi: 10.1128/jb.178.15.4400-4411.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sommer S, Knezevic J, Bailone A, Devoret R. Induction of only one SOS operon, umuDC, is required for SOS mutagenesis in Escherichia coli. Mol. Gen. Genet. 1993;239:137–144. doi: 10.1007/BF00281612. [DOI] [PubMed] [Google Scholar]
- 56.Woodgate R, Ennis DG. Levels of chromosomally encoded Umu proteins and requirements for in vivo UmuD cleavage. Mol. Gen. Genet. 1991;229:10–16. doi: 10.1007/BF00264207. [DOI] [PubMed] [Google Scholar]
- 57.Peterson J, Phillips GJ. New pSC101-derivative cloning vectors with elevated copy numbers. Plasmid. 2008;59:193–201. doi: 10.1016/j.plasmid.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frank EG, Ennis DG, Gonzalez M, Levine AS, Woodgate R. Regulation of SOS mutagenesis by proteolysis. Proc. Natl. Acad. Sci. USA. 1996;93:10291–10296. doi: 10.1073/pnas.93.19.10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Ac. Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fujii S, Fuchs RP. Interplay among replicative and specialized DNA polymerases determines failure or success of translesion synthesis pathways. J. Mol. Biol. 2007;372:883–893. doi: 10.1016/j.jmb.2007.07.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.