

Abstract
Recent advances in sequencing strategies have made it feasible to rapidly obtain high-coverage genomic profiles of single individuals, and soon it will be economically feasible to do so with hundreds to thousands of individuals per population. While offering unprecedented power for the acquisition of population-genetic parameters, these new methods also introduce a number of challenges, most notably the need to account for the binomial sampling of parental alleles at individual nucleotide sites and to eliminate bias from various sources of sequence errors. To minimize the effects of both problems, methods are developed for generating nearly unbiased and minimum-sampling-variance estimates of a number of key parameters, including the average nucleotide heterozygosity and its variance among sites, the pattern of decomposition of linkage disequilibrium with physical distance, and the rate and molecular spectrum of spontaneously arising mutations. These methods provide a general platform for the efficient utilization of data from population-genomic surveys, while also providing guidance for the optimal design of such studies.
Keywords: genome scans, heterozygosity, linkage disequilibrium, maximum likelihood estimation, mutation rate, mutation spectrum, nucleotide diversity
Introduction
Past estimates of molecular variation at the population level typically relied on assays of moderate numbers of individuals at a small number of loci (Nei 1987; Weir 1996). This situation is now rapidly changing with the advent of very high-throughput methods for genomic sequencing (Margulies et al. 2005; Bentley 2006; Mardis 2008), which present unprecedented opportunities for procuring highly reliable measurements of nucleotide diversity within single individuals, global patterns of linkage disequilibrium, mutation rates per nucleotide site, and many other key population-genetic parameters. For random-mating populations, assays of massive numbers of largely unlinked sites from fully sequenced genomes can be highly informative with respect to the population-wide average nucleotide diversity, and the correlation of heterozygosity among linked sites can provide insight into spatial patterns of genomic disequilibrium. Moreover, observations on the complete genomes of multiple individuals harbor information on the variance of heterozygosity among sites, and surveys of experimental lines with known ancestry and relaxed selection can yield precise information on mutation rates and spectra (e.g., the frequencies of the 12 types of nucleotide changes). For nonrandom-mating populations, individual-based estimates of heterozygosity may also provide a basis for determining relative levels of inbreeding. All these observable features are functions of the evolutionary forces operating at the molecular level—mutation, recombination, random genetic drift, and selection, and thus by indirect inference can yield considerable insight into the processes molding patterns of molecular and genomic evolution (Kimura 1983; Lynch 2007).
Despite the promise of high-throughput sequencing strategies for population-genomic analysis, the most appropriate methods for extrapolating information from genome-sequencing projects remain to be determined. Two problems stand out in particular. First, in most studies involving random or “shotgun” sequencing, individual nucleotide sites are subject to variable sequence coverage. For sites with low coverage, there is then a relatively high probability that all sequences will be derived from just one of the two parental chromosomes in a diploid individual, which if unaccounted for would lead to downwardly biased estimates of nucleotide diversity. Although it is tempting to apply a minimum-coverage criterion to reduce the likelihood of such problems, such an approach will generally discard substantial amounts of information, particularly in light-coverage sequencing surveys.
Second, sequencing errors can mimic polymorphisms and are collectively more likely to arise at sites with high coverage (Clark and Whittam 1992; Hellmann et al. 2008; Johnson and Slatkin 2008). Although quality scores can be used to eliminate some unreliable reads (Ewing and Green 1998; Ewing et al. 1998), such filtering does not eliminate problems arising prior to or during sample preparation, and the remaining background error variance can still rise to levels exceeding true variation in species with low levels of nucleotide diversity such as humans. To guard against the assignment of false-positive heterozygosity, analyses might focus on high-coverage sites, with single aberrant reads being discarded as errors, but again the cutoffs for such treatments are arbitrary and lead to the loss of information. In principle, empirical estimates of the error frequency might be directly applied to the problem, but the optimal procedure for estimating the error frequency itself is unresolved, and because individual sequencing runs can vary substantially in quality (Richterich 1998; Huse et al. 2007), the use of predetermined (external) error rate estimates will often be problematical.
The most dramatic example of the insufficiency of quality scores as a means for eliminating problematical sequences concerns the use of ancient DNA samples. There is now considerable interest in deciphering past human population-genetic history from genomic fragments residing in bones and teeth up to tens of thousands of years old, but such DNA is subject to extremely high levels of in situ base modification, with the C→T damage rate often exceeding 1% (Briggs et al. 2007; Gilbert et al. 2008). A project to sequence a Neanderthal genome is underway, but as much as half of the apparent divergence from modern man appears to be an artifact of single-template errors (Green et al. 2006; Noonan et al. 2006). A rigorous statistical framework for dealing with such matters will be required if population-genomic approaches are to ever be applied to ancient DNA.
In the following sections, alternative methods for obtaining estimates of average levels of nucleotide diversity, linkage disequilibrium, and mutation rates are developed and their relative merits evaluated, for situations in which massive amounts of sequence data are available from a small number of individuals. Although only the simplest of applications are presented, these will be shown to be quite rich with respect to the insights that they yield. The general approach can be readily modified to investigate more complex problems as well as to provide guidance in the optimal design of sequencing strategies for future population-genomic analyses.
Nucleotide Diversity Within Single Diploid Individuals
We start with a pool of data acquired from a single diploid individual, making the reasonable assumption that both parental sets of chromosomes have been sequenced “on average” to equivalent depths of coverage. If an accurate estimate of the per-site sequence error rate, ϵ, is available, the mean nucleotide heterozygosity within the individual, π, can then be obtained by a method-of-moments (MM) approach, but the problem may also be solved without an external estimate of ϵ by using a maximum likelihood (ML) procedure to obtain joint estimates of π and ϵ.
No assumptions are made here with respect to the method of sequence acquisition, and the raw sequence reads may be subject to various levels of trimming and quality control prior to analysis. However, it is assumed that all remaining read fragments are properly aggregated, either by de novo assembly in the case of long reads or by guidance from a reference genome in the case of short reads, with potentially problematical regions involving paralogs and mobile elements having been masked out. To keep the general approach transparent, it will also be assumed that the error structure of the data is homogeneous, with each nucleotide having the same probability of misassignment to all others.
MM Analysis
A site that has been sequenced n times within an individual will have a sequence profile (n1, n2, n3, n4), where the integers refer to nucleotides A, C, G, and T and n = n1 + n2 + n3 + n4 is the depth of coverage of the site. For n > 1, any site with at least two observed nucleotide types is potentially heterozygous, but some such observations will be simple consequences of sequence errors (here broadly interpreted as being due to any mechanism that causes a deviation from the true genotype). For the total set of sites with depth-of-coverage n, the apparent heterozygosity (i.e., the fraction of sites at which two or more nucleotides are observed), H, has expected value
| (1) |
where π is the true average genome-wide heterozygosity per nucleotide site. The term in curly brackets following π denotes the probability that a true heterozygote is sampled as such. This condition will be violated if only one allele is sampled and no false heterozygosity is produced by a sequence error, with probability for or if both alleles are sampled but an error (specifically back to the nucleotide at the site on the homologous chromosome) causes the false appearance of homozygosity, with probability ∼2n(1/2)n(ϵ/3). The latter correction term assumes that obscured sampling configurations involve only single errors, confined to situations in which one of the parental alleles is sampled just once, probability 2n(1/2)n. This assumption is reasonable for error levels encountered in most sequencing projects (where ϵ is generally ) but may need to be modified with new-generation techniques that sacrifice quality for quantity of reads. The term nϵ following (1 − π) is the probability that a homozygous site falsely appears to be heterozygous as a consequence of a sequence error, again assuming no more than one error per site (). Rearranging equation (1), an MM estimator of the average nucleotide heterozygosity using sites with n-fold coverage is
| (2a) |
where ∧ denotes an estimate. The variance of 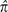 n associated with the sampling of N nucleotide sites,
obtained by the Delta method (Lynch and Walsh
1998), is estimated by
n associated with the sampling of N nucleotide sites,
obtained by the Delta method (Lynch and Walsh
1998), is estimated by
| (2b) |
Computer simulations of genomes with a wide array of values for π and n, and ϵ assumed to be known without error, demonstrate that equation (2a) yields essentially unbiased estimates of the parameter π and that equation (2b) yields an unbiased estimate of the variance of estimates from equation (2a) (fig. 1). For low enough levels of nucleotide diversity that because almost all observed variation is associated with read errors (false positives) and the sampling variance approaches an asymptotic lower bound that is independent of π,
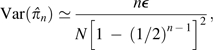 |
(3) |
which further simplifies to nϵ/N at high-coverage levels. This shows that with the MM method, there is little to be gained from increasing the sequence coverage per site beyond a few fold and actually something to be lost with highly homozygous genomes.
FIG. 1.—
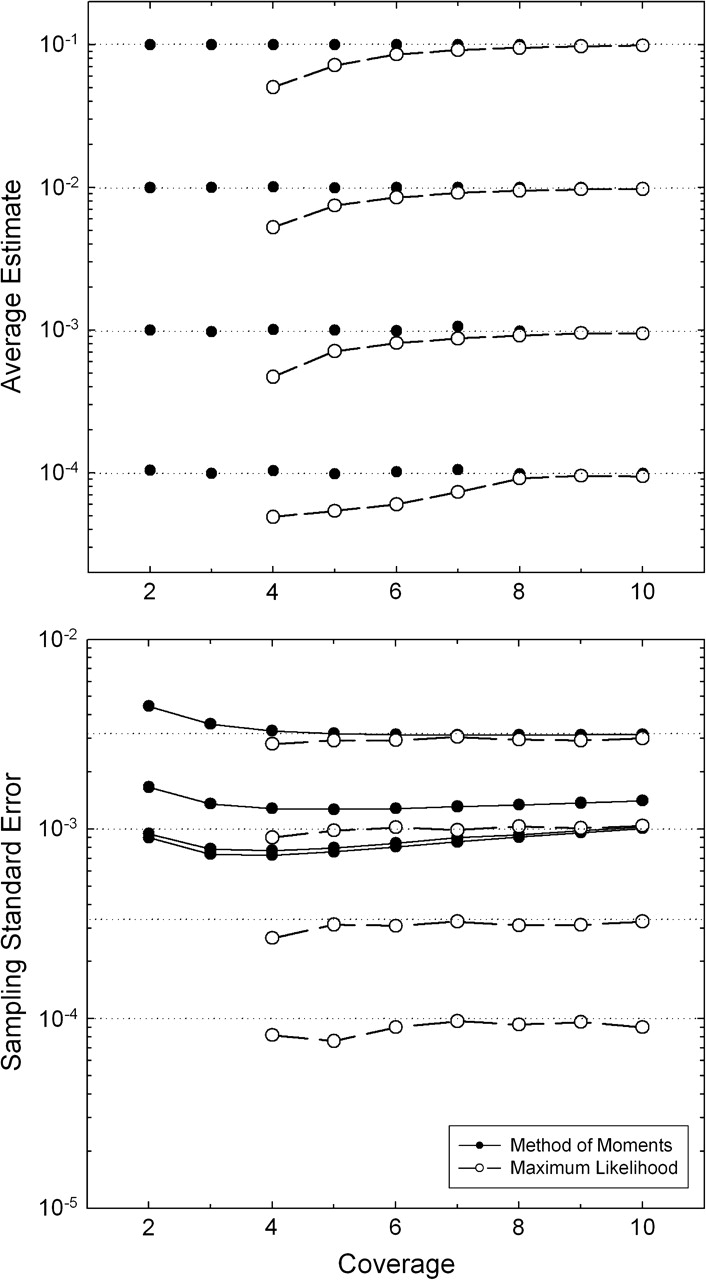
Behavior of the MM (solid circles) and ML (open circles) estimators of π, given for four values of the true nucleotide heterozygosity, π = 0.1, 0.01, 0.001, and 0.0001, with all four nucleotides assumed to have equal genome-wide frequencies. In all cases, each of N = 10,000 sites was assumed to be sequenced to the same depth of coverage (n), and simulations were performed on 500–2,000 stochastic samples. In the upper panel, the horizontal dotted lines denote the true value of π, whereas in the lower panel, they denote the true within-individual sampling SE of mean heterozygosity, . The assumed error rate is ϵ = 0.001.
ML Analysis
Under the MM approach, the use of an inaccurate estimate of ϵ can lead to biased estimates of π. Moreover, the precision of estimates must be less than optimal because each nucleotide site is viewed as being equally informative, whereas sites with multiple appearances of two nucleotides are much more reliable indicators of heterozygosity than sites with just one odd nucleotide, which at high coverage are indicative of errors. An alternative approach is to weight each site by its information content in order to obtain joint estimates of π and ϵ that maximize the likelihood of the full set of data. Such analysis requires as additional input measures of the genome-wide nucleotide frequencies (p1, p2, p3, p4), but with large genome-sequencing projects, these can be estimated with high precision from the full pool of sequence data.
Under the ML approach, for the full range of candidate values of π and ϵ, the likelihood of the data at each site can be obtained by considering the probabilities of the observed data conditional on all possible genotypic states. Here we assume that the probabilities of alternative allelic states are defined by the average nucleotide frequencies in the region of analysis. Thus, conditional on the site being homozygous, the likelihood of the observed data is obtained by summing over the likelihoods conditional on all four possible homozygous types (AA, CC, GG, and TT, with respective relative probabilities p1, p2, p3, and p4),
| (4a) |
where b(n − ni;n, ϵ) is the probability of n − ni errors in n reads given the error rate ϵ. For heterozygous sites, the likelihood must incorporate the sampling distribution of the two alternative parental alleles as well as the probability of read errors to alternative nucleotide states. Accounting for all possible heterozygous types, the conditional likelihood is
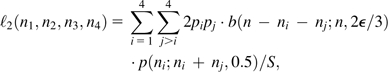 |
(4b) |
where p(x;y, 0.5) denotes the binomial probability of x events, each with independent probability 0.5, out of y trials, and the term is necessary to normalize the sum of the frequencies of expected heterozygote types to one. This expression follows from the fact that, conditional on the individual being genotype ij, b(n − ni − nj;n, 2ϵ/3) is the probability of errors to nucleotides other than i and j, whereas p(ni;ni + nj, 0.5) is the probability of sampling the ith nucleotide ni times from the remaining pool of ni + nj nonerroneous reads. Although there may be errors within the latter pool, this does not alter the usual binomial sampling probability, provided the errors are equal in both directions.
The total likelihood for the observed data at the site is then
| (5) |
Letting N(n1, n2, n3, n4) denote the number of times the sampling configuration (n1, n2, n3, n4) is observed over all sites, the log likelihood of the total data set is
| (6) |
where the summation is over all observed nucleotide configurations. The ML solution, given by the joint estimates of π and ϵ that maximize L, can be readily obtained by a grid survey of the relevant range of parameter space.
The analysis of computer-simulated data indicates that the ML method
asymptotically yields nearly unbiased estimates of π
with increasing coverage of sites n (fig. 1). For 2× and 3× coverage, with
no possibility of both nucleotides at a heterozygous site being sequenced at
least two times, there is insufficient information to distinguish between true
genotypic variation and that generated by read errors, and the ML approach is
ill-behaved, with the estimates of π always converging
on zero. However, for all other coverages, the sampling variance of the ML
estimator (among replicate samples) is always lower than that of the MM
estimator, despite the fact that the ML procedure generates its own estimate of
ϵ. Indeed, provided the coverage is
>3×, the ML estimator behaves nearly optimally in that the
sampling variance of approaches the true within-individual sampling variance of the
mean heterozygosity
π(1 − π)/N.
Thus, the asymptotic sampling coefficient of variation (ratio of the standard
error [SE] to the expected parametric value) of the ML
estimator of π is which because π is generally is where πN is the expected number of
heterozygous sites in the sample.
As can be seen in figure 1, if π is on the order of the error rate or smaller, the ML estimator is much more reliable than the MM estimator, as a consequence of the asymptotic lower bound of the sampling variance of the latter. On the other hand, at low coverages, the ML estimates are downwardly biased, the extreme being a 50% reduction at 4× coverage. An ad hoc but intuitive correction factor to eliminate this bias can be arrived at by recalling that the ML estimator fails to yield nonzero estimates of π when (1, n − 1) allelic configurations are the most extreme that can be achieved at a site (i.e., with 2× and 3× coverage). Reasoning that the bias in the ML estimates is largely caused by heterozygotes with (1, n − 1) configurations, and letting c = n(1/2)n−1 be the expected frequency of such configurations, an improved estimator of π is achieved by dividing the ML estimate by (1 − c). This modification completely eliminates the bias provided the error rate is <10−3 or so (fig. 2), although the sampling standard deviation will be inflated by the factor 1/(1 − c).
FIG. 2.—

Average ML estimates of π given for three values of the true nucleotide heterozygosity, π = 0.01, 0.001, and 0.0001 (denoted by the three horizontal dotted lines), with all four nucleotides assumed to have equal genome-wide frequencies and correction for sampling bias as described in the text. In all cases, each of N = 100, 000 sites is assumed to be sequenced to the same depth of coverage (n). The assumed error rate is ϵ = 0.001.
However, once the error rate exceeds the true level of heterozygosity, further
bias is introduced (independent of the number of sites sampled), the moreso at
lower coverages. Although I have been unable to obtain a simple means for
eliminating this shortcoming, the results in figure 2 provide guidance as to when such issues are likely to
arise, and the bias can be estimated computationally (through simulations with
the relevant n, π, and
ϵ). However, the salient point here is that the
conditions under which the ML estimates of π are biased
closely reflect those where the sampling variance of is already swamped by that of 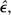 , rendering such estimates quite unreliable.
, rendering such estimates quite unreliable.
Combined Analysis
Given the disparities in the sampling variances of with the alternative approaches, the nonfunctionality of the
ML approach at 2× and 3× coverage, and the variation in
coverage that will generally exist among sites, a hybrid method that makes
optimal use of all the data is desirable. One deficiency of the MM approach is
its requirement for an accurate, external estimate of the read-error rate
(ϵ). However, a useful feature of the ML approach
is its ability to generate estimates of ϵ. Provided
the depth of coverage is sufficiently high that
(n − 2)ϵ > π,
the ML estimates of the error rate are nearly unbiased, with sampling variance
close to
ϵ(1 − ϵ)/[N(n − 1)],
although at lower coverages, these estimates are upwardly biased. Thus, under
appropriate sampling conditions, it should be possible to utilize the ML
approach to derive an estimate of ϵ, which can then be
applied to the MM method for conditions in which the latter estimator is
preferred. A near minimum-sampling-variance estimator of
π might then be achieved by using the ML approach
for coverages above a specific cutoff and the MM estimator for lower coverages.
Obtaining a pooled high-coverage ML estimate is straightforward, as by equation (6), one simply sums the
likelihoods over all configurations at all coverage levels.
Suppose, for example, that one wished to use the ML approach for all coverages >3×. After obtaining separate MM estimates of π for sites with n = 2 and 3, the pooled estimate would be
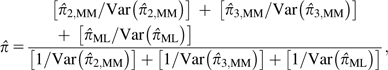 |
(7) |
where each estimate is weighted by the inverse of its sampling
variance. The sampling variance for each MM estimate can be obtained directly
from equation (2b), whereas
given the relative constancy of the variance of at all coverages with the ML approach, 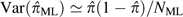 where NML is the total number of
sites used in the ML analysis.
where NML is the total number of
sites used in the ML analysis.
One major caveat with respect to this approach, and indeed any application of the MM method, concerns the assumption that the ML estimate of ϵ obtained at high coverages is applicable to lower-n sites. If, for example, a substantial fraction of low-coverage sites results from poor assembly of error-laden fragments, upwardly biased estimates of π would be generated by the MM method, as not enough variation resulting from sequence errors would be eliminated. Thus, prior to any attempt at using a pooling method, it would be prudent to evaluate whether estimates of ϵ generated by the ML approach are stable with respect to n.
Linkage Disequilibrium for Homozygosity Within Single Diploid Individuals
With only two chromosomes sampled, a single individual provides little insight into the overall level of linkage disequilibrium between any particular pair of nucleotide sites. However, with thousands to millions of pairs of sites along a chromosome, it is possible to extract information on the pattern of zygosity disequilibrium, that is, to evaluate whether individuals that are heterozygous (homozygous) at a particular site are more likely to be heterozygous (homozygous) at neighboring sites. Considering all pairs of sites a specific distance apart, the genome-wide expected frequencies of double homozygotes and double heterozygotes are, respectively, (1 − π)2 + Δπ(1 − π) and π2 + Δπ(1 − π), where Δ is the correlation of zygosity across all pairs of sites.
Following the general approach outlined in the previous section, after taking into account the random sampling of parental chromosomes and the loss of information associated with read errors, the expected frequencies of apparent doubly homozygous, doubly heterozygous, and homozygous/heterozygous pairs are, respectively
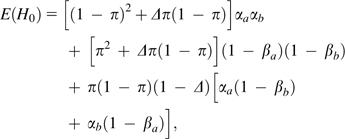 |
(8a) |
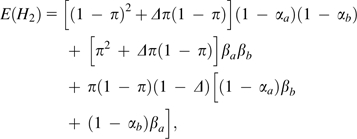 |
(8b) |
| (8c) |
where for locus a,
| (9a) |
| (9b) |
denote, respectively, the probabilities that true homozygotes are revealed as such (because only a single nucleotide is sequenced) and that true heterozygotes are revealed as such (because two or more nucleotide types are observed), with na denoting the coverage of site a, and similar expressions applying for the other member of the nucleotide pair (locus b).
Considering the sum of observed double homozygote and double heterozygote
frequencies, 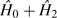 , the MM estimator for the zygosity correlation involving pairs of
sites with coverage (na, nb)
is
, the MM estimator for the zygosity correlation involving pairs of
sites with coverage (na, nb)
is
| (10a) |
where
c1 = 1 + 2αaαb − αa − αb,
c2 = 1 + 2βaβb − βa − βb,
and
c3 = αa + αb + βa + βb − 2αaβb − 2αbβa,
with being obtained by single-site analysis as described above. Note
that at high coverage, as the error rate approaches zero, this MM estimator for  converges on
converges on 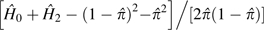 . The large sample–variance expression for obtained by the Delta method (Lynch and Walsh 1998), is given here relative to the observed estimate
(i.e., as the squared coefficient of sampling variation),
. The large sample–variance expression for obtained by the Delta method (Lynch and Walsh 1998), is given here relative to the observed estimate
(i.e., as the squared coefficient of sampling variation),
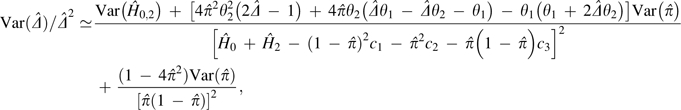 |
(10b) |
where 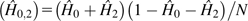 is the sampling variance for the summed frequency of pairs of
double homozygotes and double heterozygotes, with N being the
number of pairs of loci in the analysis,
θ1 = c3 − 2c1,
θ2 = c1 + c2 − c3,
and Var() defined by equation
(2b).
is the sampling variance for the summed frequency of pairs of
double homozygotes and double heterozygotes, with N being the
number of pairs of loci in the analysis,
θ1 = c3 − 2c1,
θ2 = c1 + c2 − c3,
and Var() defined by equation
(2b).
Analysis of computer-simulated data indicates that the MM estimator of Δ is essentially unbiased, again provided that the correct error rate is available. The large sample–variance estimator also performs quite well under a range of circumstances (fig. 3), although it does overestimate the sampling variance when π is very low (in which case the power of disequilibrium analysis is already greatly compromised as a consequence of the rarity of polymorphic sites).
FIG. 3.—
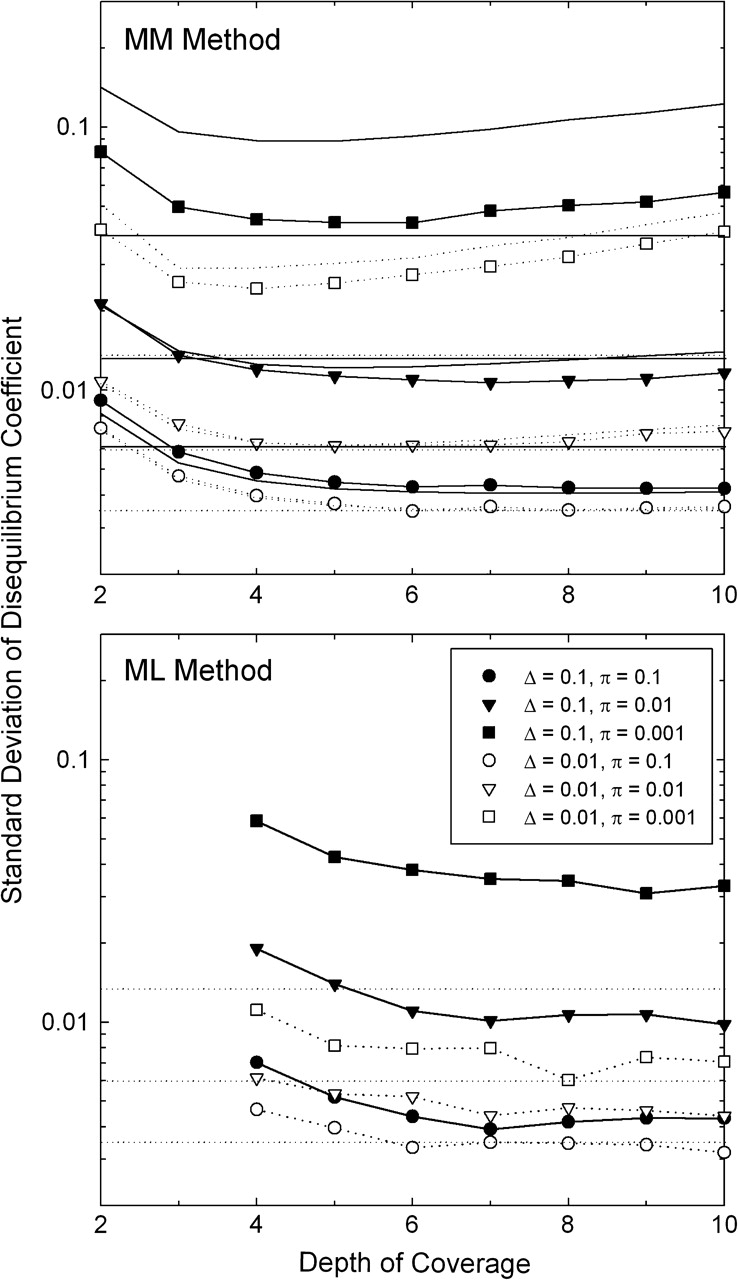
Sampling standard deviations associated with estimates of the disequilibrium coefficient Δ. Symbols refer to results obtained by stochastic simulations assuming 100,000 sites, with 2,500 replications performed for each condition with the MM method and 250–500 with the ML method. Curved lines without points in the upper panel give the results from the large sample–variance approximation for the MM estimates, equation (10b); and horizontal lines give the first-order high-coverage approximation, equation (10c). In both these latter cases, solid and dotted lines refer to situations with Δ = 0.1 and 0.01, respectively. To ease the comparison of results, the dotted lines are repeated in the lower panel. The assumed error rate is ϵ = 0.001.
Some sense of the baseline sampling properties of can be achieved by considering the limiting situation in which the
coverage is high enough and the error rate low enough that the estimation error is
dominated by the sampling of the two-locus genotypes, in which case as a first-order
approximation equation (10b)
reduces to
| (10c) |
This shows that the sampling variance of scales inversely with the expected number of heterozygous loci in
the sample (Nπ). Because it ignores the loss of information
from sequence errors, the latter expression will generally underestimate the actual
sampling variance of although it generally yields values close to those from computer
simulations at high coverage (fig. 3). For the sampling variance of using the MM estimator is in accordance with the large-sample variance of a correlation
coefficient being (Lynch and Walsh 1998),
with r = 0 in this limiting
case.
It is fairly straightforward, albeit tedious, to extend the single-locus ML approach to pairs of loci. Letting the sets of observations for the four nucleotides at a pair of sites, a and b, be (na1, na2, na3, na4) and (nb1, nb2, nb3, nb4), equations (4a) and (4b) can be used to derive the likelihoods of observations conditional on the sites being homozygous (ℓ1a and ℓ1b) or heterozygous (ℓ2a and ℓ2b). The likelihood for the pair of loci, given π, Δ, and ϵ, analogous to equation (5), is then
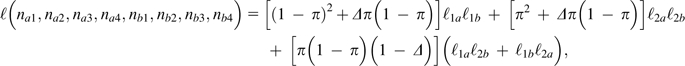 |
(11) |
The overall likelihood, summed over all pairs of loci, is
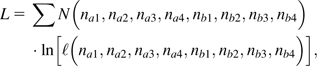 |
(12) |
where the N(na1, na2, na3, na4, nb1, nb2, nb3, nb4) denote the numbers of pairs of loci with each of the observed configurations of observations.
Application of the ML approach to computer-simulated data indicates that this method
generates joint, nearly unbiased estimates of Δ, and
ϵ, again provided the sample sizes at sites exceed
three. In general, the SEs of the ML estimates are similar to or slightly better
than those arising with the MM method (assuming known ϵ in
the latter case). Thus, because the MM method will yield biased results unless
ϵ is known with certainty, it appears preferable to
rely on the ML method for pairs of sites at which na,
nb > 4, resorting
to the MM method only at lower coverages (using an estimate of
ϵ derived via ML) if at all and obtaining a pooled
average estimate using the methods outlined above for analogous to equation
(7).
For the sampling variances of necessary to obtain a weighted estimate of
Δ, equation
(10b) applies to all terms involving the MM method. Equation (10c) provides a fairly good
approximation of the sampling variance of ML estimates of
Δ at high coverage (fig. 3), although the sampling variance of an ML estimate can also be
obtained directly from the curvature of the likelihood surface. Denoting the maximum
of the log-likelihood surface as L(, , ) and the maximum log likelihood when Δ
is constrained to equal zero as L(,) the likelihood ratio is defined as LR = −
2[L(,) − L(, , )]. With the large samples involved in genome sequencing, LR is
expected to be χ2 distributed with one degree of
freedom so that approximate 95% support boundaries for can be obtained by evaluating LR at values deviating above and
below until the drop in LR exceeds 3.84. As the width of this range,
W, is expected to be approximately four SEs, Var(ML) ≃ W2/16.
Extension to Pairs of Individuals
When high-coverage sequence data are available for more than a single individual,
opportunities exist for deriving genome-wide estimates of higher order moments of
the distribution of heterozygosity across sites. For example, the joint analysis of
the same sites in two individuals is conceptually analogous to the procedure
outlined above for pairs of sites within an individual. In this case, however,
Δ is equivalent to the correlation of heterozygosity
within sites. Because the covariance within sites is equal to the variance among
sites (a general feature of variance components; Lynch and Walsh 1998), the variance of heterozygosity among sites is
estimated by 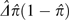 . This interpretation can be arrived at by noting that the expected
frequencies of doubly homozygous, doubly heterozygous, and homozygous/heterozygous
pairs of genotypes are, respectively, equal to
. This interpretation can be arrived at by noting that the expected
frequencies of doubly homozygous, doubly heterozygous, and homozygous/heterozygous
pairs of genotypes are, respectively, equal to 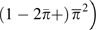 ,
, 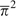 , and
, and 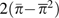 where
where 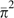 is the mean squared site-specific heterozygosity (i.e., the second
moment of π). Setting these expressions equal to the
respective three terms in brackets in equation (8a) demonstrates that
is the mean squared site-specific heterozygosity (i.e., the second
moment of π). Setting these expressions equal to the
respective three terms in brackets in equation (8a) demonstrates that 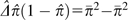 is an estimate of the variance of heterozygosity among sites.
is an estimate of the variance of heterozygosity among sites.
Likewise, extension of equations (8)–(12) to three individuals to account for single, double, and
triple heterozygotes would yield an estimate of the third moment of
π, that is, 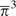 , providing information on the skewness of heterozygosity. By
generating an estimate of the fourth moment of π, a
four-individual analysis would yield insight into the kurtosis of the distribution
of π across loci.
, providing information on the skewness of heterozygosity. By
generating an estimate of the fourth moment of π, a
four-individual analysis would yield insight into the kurtosis of the distribution
of π across loci.
Mutation-Rate Estimation
Because of the rarity of new mutations and the past reliance on reporter constructs of uncertain sensitivity, the rate at which mutations arise at the nucleotide level and the spectra of their effects are among the most poorly understood genetic features of most organisms. However, with the feasibility of sequencing entire genomes from individuals of known relationship, rapid progress in this area is now possible (Lynch et. al 2008). In the following, we will assume a classically designed mutation–accumulation (MA) experiment, whereby multiple lines with initially identical genomes are passed through single-individual bottlenecks each generation. Such treatment eliminates the power of selection to remove anything other than mutations causing complete sterility or lethality (Lynch and Walsh 1998), which themselves generally constitute no more than ∼1% of all mutations. It will be assumed that the lines are either haploid (e.g., yeast and a number of other microbial organisms) or habitually self-fertilizing (as is possible with the nematode Caenorhabditis elegans, many plants, and ciliates undergoing regular autogamy). This simplifies the analysis as segregating (heterozygous) mutations can essentially be ignored provided the timescale of the experiment is at least several dozens of generations. For example, under self-fertilization, the mean time to loss of heterozygosity for a locus bearing a new mutation is just two generations. However, the methods presented below can be readily modified to allow for transient phases of heterozygosity for mutations en route to fixation/loss; for example, in full-sib mated lines, as well as for clonal diploids in which new mutations are essentially permanently heterozygous.
A likelihood framework is adhered to here, as it has been shown above that the ML
method is far superior to the MM method in estimating low variation levels (which
will almost always be the situation in MA experiments). Focusing on base
substitutions only, we will assume that the genome-wide usages of the four
nucleotides are essentially known without error, again designating them as
p1, p2,
p3, and p4 for
nucleotides A, C, G, and T, respectively. The likelihood of any configuration of
observed data across L sequenced lines is a function of the
mutation rate per site per generation (u), the number of
generations of MA for each line (Tk for the
kth line), and the error frequency
(ϵ). Here, we will assume that no more than a single line
carries a mutation at a particular site, which is quite reasonable because
u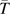 L will almost always be in an MA experiment extending for fewer than 10,000 or so
generations.
L will almost always be in an MA experiment extending for fewer than 10,000 or so
generations.
Under the above assumptions, the likelihood of the observed data for a particular configuration of reads can be partitioned into two components: the likelihoods conditional on there being no mutation or there being a single mutation in a single line at the site. The joint likelihood of the data under the first condition is
| (13a) |
where b(nk − nki;nk, ϵ) is the binomial probability that line k has (nk − nki) sequence errors conditional on the line actually carrying nucleotide i and is the probability that the line is nonmutant at the site. This likelihood is weighted over the full spectrum of possible nucleotides at the site, as we assume that the ancestral state of the site is not known at the outset. The likelihood of the observed data conditional on a mutation having occurred is
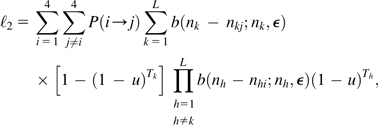 |
(13b) |
where is the probability that a mutation is of type Assuming mutation types are simply proportional to genome-wide nucleotide usage, then where is the normalization constant to ensure that the probabilities of the 12 mutation types sum to one.
Denoting the four-element arrays of nucleotide counts for each line at the site as n1, …, nL, the total log likelihood (summed over all sites) is
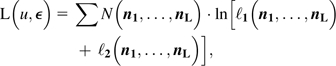 |
(14) |
where N(n1,
…, nL) is the number of sites
observed with configuration (n1, …,
nL) (a 4L-element
array). The ML estimates  and are obtained by evaluating L(u,
ϵ) over the full range of feasible mutation rates and
error frequencies, searching for the pair that maximizes the likelihood of the data.
Following the logic outlined above for , evaluation of the likelihood ratio statistic around can be used to construct upper and lower confidence limits for the
estimate.
and are obtained by evaluating L(u,
ϵ) over the full range of feasible mutation rates and
error frequencies, searching for the pair that maximizes the likelihood of the data.
Following the logic outlined above for , evaluation of the likelihood ratio statistic around can be used to construct upper and lower confidence limits for the
estimate.
Ascertainment of the Mutational Spectrum from Consensus Sequences
With experiments extending for at least a few hundred generations and genomes of moderate size, several hundreds to thousands of mutations can be expected to be harbored in any particular MA line, raising the possibility of estimating the full molecular spectrum of spontaneously arising mutations (including their contextual settings). A straightforward way to identify putative mutations, for further validation by conventional follow-up sequencing, is to determine whether the consensus sequence at a site in a particular focal line deviates from the consensus for the pooled sample from the remaining lines. The existence of a consensus sequence requires that the majority of the base calls at a nucleotide site be of the same type, for example, for a 5×-covered site, either three to five base calls must be of the same type or in the very rare occasion in which just two are of the same type, the remaining three must be different from each other. For a reasonable degree of reliability, this approach requires at least two reads in the focal and control samples.
The probability of incorrectly inferring a mutation by this approach (the probability of a false positive) is a function of the error frequency, here assumed to be available from the ML analysis noted above. A false positive can arise when read errors at either the focal line or the composite control lead to a false-consensus sequence. Letting b(x;n, r) denote the binomial probability of x errors in n reads within a line given an error frequency of r, the probability of a false-consensus sequence for a line with two reads at a site is
| (15a) |
This follows from the fact that with a sample size of only two, a false consensus arises only when both reads erroneously converge to the same base (three possible bases can be converged on, with the error rate to any particular base being ϵ/3 under the assumption of randomly distributed error types). For all odd values of n,
| (15b) |
whereas for all other even values of n,
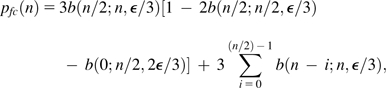 |
(15c) |
The extra leading term in equation (15c) accounts for the probability that with even coverage, a false consensus can arise when half of the reads converge on the same error and the remaining half contains at least two different read types. Denoting the numbers of reads for the focal line and the composite control as nf and nc, respectively, the probability of a false-positive mutation at the site in the focal line is
| (16) |
The probability of a false negative at a site (i.e., the probability of failing to reveal a true mutation), pfn, is simply pfp/3 as this requires that errors cause either the consensus sequence for mutant line itself to converge back to the ancestral state or the composite control to converge on the mutant state, both of which can only occur by one specific mutation.
For nf = 2, the false-positive rate is quite unresponsive with respect to the sample size for the control, as almost all false consensuses reside in the focal line (fig. 4). However, for all higher nf, there is a dramatic decline in pfp with increasing nc, until an asymptotic lower value is reached when nc is again large enough that virtually all false consensuses are a consequence of errors in the focal line. These results show that for moderate coverage and moderate error rates (ϵ = 0.001 in the figure), the consensus-sequence approach yields very low false-positive rates (well below the minimum expected mutation probability per site, ∼10−9 times the number of experimental generations).
FIG. 4.—

Probability of a false-positive mutation call from a consensus-sequence comparison, given as a function of the number of reads at the site in the focal line and the composite control (the sum of the pooled samples from the remaining L − 1 lines). The error rate (ϵ) is assumed to equal 0.001.
The false-consensus probability at a site is independent of the specific reads actually perceived and is primarily useful for experimental design purposes. However, using Bayes theorem, with the control reads observed at a particular site as a reference, one can also compute the approximate probability that the site carries a mutation in a particular focal line. The probability that a focal line is fixed for nucleotide i is
| (17) |
where pi is again the genome-wide frequency of usage of the ith nucleotide. Ignoring the multinomial coefficients, which cancel out in the above expression,
| (18a) |
| (18b) |
where n = n1 + n2 + n3 + n4. For the composite control, based on the data from all but the focal line,
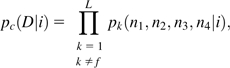 |
(19a) |
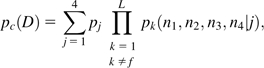 |
(19b) |
where D refers to the full set of configurations across all control lines. Applying equations (19a,b) to equation (17), the probabilities that the composite control is fixed for the alternative nucleotides are obtained. The approximate probability that the focal line carries a mutation at the site is then
| (20) |
Discussion
The preceding analyses demonstrate that despite the uneven coverage and presence of sequence errors, accurate information can be extracted from whole-genome analyses of single diploid individuals. Neither arbitrary coverage cutoffs nor external measures of the base call error rate are necessary, or even desirable, to obtain meaningful estimates of average within-individual heterozygosity, linkage disequilibrium among sites, or mutation rates. This is an obviously preferred situation as the former can discard substantial amounts of data and the latter can involve extrapolations from extrinsic studies with uncertain justification. There are, however, limitations to what can be accomplished. In particular, completely unbiased estimates of population-genetic parameters may not be possible at very low coverages.
Any approach of the sort developed above does require that, prior to analysis, the investigator utilizes a rigorous protocol for the alignment and concatenation of individual sequence reads. As almost all genomes contain small to moderate numbers of young duplicate genes as well as numerous mobile elements, both of which can mimic allelic variation, sequences at ambiguous paralogous positions should be removed prior to analysis, and usual practices of eliminating poorly resolved sequences should be adhered to as well. Erroneous alignments may be particularly problematical for some of the recent sequencing methodologies that generate short (<50 bp) reads, and the identification of paralogs in poorly assembled genomes might only be accomplished by adhering to high depth-of-coverage cutoffs as indicators of problematical sites. Nevertheless, it is notable that the influence of most remaining sources of errors can be factored out in an unbiased fashion with the ML methods introduced above. Such background inaccuracies need not be confined to machine-read errors but may include true sequences of somatic mutations, errors incurred during sample storage or preparation, and perhaps some misalignment errors. Whereas the methods developed above might be refined by explicitly incorporating a quality score for each individual base read (Johnson and Slatkin 2008), this would not eliminate the need to generate a separate error-rate estimate associated with all these additional sources of uncertainty and may be unnecessary for the types of analyses outlined herein.
The preceding approaches may be quite informative with respect to patterns of molecular evolution when the full collection of sites within a genome are partitioned into various subcategories, for example, synonymous versus nonsynonymous sites within coding regions, introns, untranslated regions, and intergenic DNA. Individual chromosomes may also be subdivided into segments for purposes of locating regions with unusually high or low levels of nucleotide diversity or disequilibria, which may provide insight into loci experiencing unusual patterns of purifying or balancing selection or the indirect consequences of selection on linked sites.
Such analyses should provide a potential basis for testing a number of evolutionary
hypotheses, while also yielding measures of population-genetic parameters central to
our understanding of molecular and genomic evolution. For example, under the
assumption of neutrality and drift–mutation equilibrium, the expected
value of π for a diploid population is
θ = 12Neu/[3 + 16Neu],
where Ne is the effective population size and
u is the mutation rate per nucleotide site, assuming a
symmetrical mutation model (Kimura 1983).
This expression is approximately twice the ratio of the power of mutation to the
power of random genetic drift, 4Neu,
provided (a condition that is essentially always met in multicellular
species; Lynch 2007), an interpretation
that applies even with unequal mutation rates among nucleotides. In addition, the
expected equilibrium variance of nucleotide heterozygosity among unlinked neutral
sites is (Tajima 1983). Thus,
substitution of for θ in the preceding formula provides
a means of testing whether the joint assumptions of neutrality and
mutation–drift equilibrium are met with the set of sites used to estimate
π. Although methods are available for testing for
neutrality among small to moderate numbers of sites within individual loci (e.g.,
Tajima 1989a, 1989b; Fu and Li
1993), the above summary statistics may prove useful in the genomics era
where smaller numbers of individuals but much larger numbers of sites are surveyed.
It should be realized, however, that the expression for
σ2(π) given above
assumes that the vast majority of pairs of sites in the region of analysis are
unlinked. Modifications required for narrow regions with restricted recombination
are provided by Pluzhnikov and Donnelly
(1996).
The preceding measure of the variance of heterozygosity is equivalent to the
“evolutionary variance,” estimated by in that it refers to stochastic variation in
π that develops among loci due to the vagaries of drift
and mutation. Such variation is distinct from the “sampling
variance” of π defined by design limitations,
described above as Var(), which is only a function of the number
of sites sampled within the focal individual and the read-error variance. The
expected value of the evolutionary coefficient of variation of site-specific
heterozygosities, estimated by the square root of [(1−)/], is which is closely approximated by
(3θ)−1/2 when
θ < 0.05.
A reparameterization of the model outlined above for the correlation of zygosity also
yields useful insight into the relative power of recombination and random genetic
drift, assuming the sites involved are not under direct selection. Letting AB, Ab,
aB, and ab denote the four alternative gametic states at two linked loci, their
expected frequencies are conventionally expressed as
pApB + D,
pApb − D,
papB − D,
and
papb + D,
where the terms involving p denote allele frequencies within loci
and D is the coefficient of linkage disequilibrium. For random
pairs of loci taken over the entire genome, the expected value of D
is zero as half of the disequilibria are expected to be positive and the other half
negative. However, the expected value of D2 is
equivalent to
Δπ(1 − π)/4
in the two-site model outlined above. An estimate of the average value of
D2 over sites, 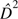 , is then given by
, is then given by 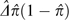 /4.
/4.
This rescaling is useful in the context of understanding the forces driving linkage disequilibrium because the expected value of D2 for pairs of neutral sites under mutation–drift equilibrium is
where θ = 4Neu, M = θ2/[(θ + 1)(18 + 13ρ + 54θ2 + ρ2 + 19ρθ + 40θ2 + 6ρθ2 + 8θ)], ρ = 4Nec, and c is the rate of recombination between sites (Hill 1975). Thus, provided the sites involved are neutral and in equilibrium, given estimates of π (as an estimator or θ) and D2, an estimate of ρ can be obtained by solving the preceding equation. Such estimates may be obtained for sets of nucleotide pairs separated by a range of physical distances (e.g., 0, 1, 2, etc., sites apart). A regression of these estimates on physical distance will then reveal the degree to which the rate of recombination increases with physical distance, with the estimated value for adjacent sites providing a measure of twice the power of recombination per site relative to the power of drift (4Nc0), where c0 denotes the recombination rate between adjacent sites. With the substantial data available from whole-genome-sequencing projects, this approach may provide a viable alternative to the current methods for estimating ρ from population samples of narrow genomic regions (Wall 2000; Stumpf and McVean 2003).
It should be noted that none of the above approaches involve the use of preexisting sequences from a reference strain, which will often be available for well-studied species. In principle, a reference sequence can provide a useful scaffold for assembling a new collection of shotgun sequence, and some reference strains themselves may provide useful information on average heterozygosity and linkage disequilibrium, provided they themselves were not subject to intentional inbreeding. However, reference strains will typically contain some sequencing errors, with rates deviating from those in a downstream study, and most species contain considerable numbers of presence/absence polymorphisms for young duplicate genes and mobile elements (Lynch 2007), which will complicate their complete elimination from novel genomes with incomplete assemblies. Thus, the application of reference sequences to studies of natural variation should be approached with caution.
Finally, although the methods developed above, particularly those involving the ML approach, appear to provide a solid basis for the analysis of high-throughput genomic data, there is still room for considerable expansion of these methods, just four of which are noted here. First, the assumption of homogeneous error rates can be relaxed by incorporating into the likelihood functions multiple terms for alternative nucleotide changes. Second, additional complexity can also be incorporated into the estimation of heterozygosity and/or mutation rates by distinguishing alternative types of heterozygotes (e.g., transitions vs. transversions). The utility of both these modifications can be evaluated by testing for the significance of the model fit by using conventional likelihood ratio test statistics. Third, the estimators for linkage disequilibrium might be substantially improved by taking into consideration the phase information that exists when sites have been recorded within the same read fragments. Finally, as data become available for large numbers of individuals within populations, it will be possible to go beyond summary statistics such as π to refined estimates of allele frequencies at individual nucleotide sites. Ordinarily, when it is assumed that records are error free, the estimation of allele frequencies is a straightforward exercise (Weir 1996), but the incursion of errors into high-throughput (but low coverage) sequencing surveys will introduce new challenges, particularly for low-frequency (and normally highly informative) alleles.
Acknowledgments
This work was funded by National Institutes of Health grant GM36827 to the author and W. Kelley Thomas. I am grateful to Abe Tucker and Way Sung for inspiration; Xiang Gao for computational assistance; Matt Hahn and Phil Nista for helpful discussion; and Elizabeth Housworth, Ignacio Lucas Lledo, and especially Philip Johnson for critical insights.
References
- Bentley DR. Whole-genome re-sequencing. Curr Opin Genet Dev. 2006;16:545–552. doi: 10.1016/j.gde.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Briggs AW, et al. (11 co-authors) Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl Acad Sci USA. 2007;104:14616–14621. doi: 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Whittam TS. Sequencing errors and molecular evolutionary analysis. Mol Biol Evol. 1992;9:744–752. doi: 10.1093/oxfordjournals.molbev.a040756. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Fu Y-X, Li W-H. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert MT, et al. (13 co-authors) DNA from pre-Clovis human coprolites in Oregon, North America. Science. 2008;320:786–789. doi: 10.1126/science.1154116. [DOI] [PubMed] [Google Scholar]
- Green RE, et al. (11 co-authors) Analysis of one million base pairs of Neanderthal DNA. Nature. 2006;444:330–336. doi: 10.1038/nature05336. [DOI] [PubMed] [Google Scholar]
- Hellmann I, Mang Y, Gu Z, Li P, De La Vega FM, Clark AG, Nielsen R. Population genetic analysis of shotgun assemblies of genomic sequence from multiple individuals. Genome Res. 2008;18:1020–1029. doi: 10.1101/gr.074187.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG. Linkage disequilibrium among multiple neutral alleles produced by mutation in finite population. Theor Popul Biol. 1975;8:117–126. doi: 10.1016/0040-5809(75)90028-3. [DOI] [PubMed] [Google Scholar]
- Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PL, Slatkin M. Accounting for bias from sequencing error in population genetic estimates. Mol Biol Evol. 2008;25:199–206. doi: 10.1093/molbev/msm239. [DOI] [PubMed] [Google Scholar]
- Kimura M. The neutral theory of molecular evolution. Cambridge (UK): Cambridge University Press; 1983. [Google Scholar]
- Lynch M. The origins of genome architecture. Sunderland (MA): Sinauer Assocs., Inc.; 2007. [Google Scholar]
- Lynch M, et al. (11 co-authors) A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci USA. 2008;105:9272–9277. doi: 10.1073/pnas.0803466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Walsh B. Genetics and analysis of quantitative traits. Sunderland (MA): Sinauer Assocs., Inc.; 1998. [Google Scholar]
- Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Margulies M, et al. (56 co-authors) Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987. [Google Scholar]
- Noonan JP, et al. (11 co-authors) Sequencing and analysis of Neanderthal genomic DNA. Science. 2006;314:1113–1118. doi: 10.1126/science.1131412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluzhnikov A, Donnelly P. Optimal sequencing strategies for surveying molecular genetic diversity. Genetics. 1996;144:1247–1262. doi: 10.1093/genetics/144.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richterich P. Estimation of errors in “raw” DNA sequences: a validation study. Genome Res. 1998;8:251–259. doi: 10.1101/gr.8.3.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf MP, McVean GA. Estimating recombination rates from population-genetic data. Nat Rev Genet. 2003;4:959–968. doi: 10.1038/nrg1227. [DOI] [PubMed] [Google Scholar]
- Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. The effect of change in population size on DNA polymorphism. Genetics. 1989a;123:597–601. doi: 10.1093/genetics/123.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989b;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD. A comparison of estimators of the population recombination rate. Mol Biol Evol. 2000;17:156–163. doi: 10.1093/oxfordjournals.molbev.a026228. [DOI] [PubMed] [Google Scholar]
- Weir BS. Genetic data analysis II. Sunderland (MA): Sinauer Assocs., Inc.; 1996. [Google Scholar]