

Abstract
Infection with Simian Immunodeficiency Virus (SIV) leads to high viral loads and progression to Simian AIDS (SAIDS) in rhesus macaques. The viral accessory protein Nef is required for this phenotype in monkeys as well as in HIV-infected humans. Previously, we determined that HIVNef binds HIVGagPol and Alix for optimal viral replication in cells. In this study, we demonstrated that these interactions could correlate with high viral loads leading to SAIDS in the infected host. By infecting rhesus macaques with a mutant SIVmac239, where sequences in the nef gene that are required for these interactions were mutated, we observed robust viral replication and disease in two out of four monkeys, where they reverted to the wild type genotype and phenotype. These two rhesus macaques also died of SAIDS. Two other monkeys did not progress to disease and continued to harbor mutant nef sequences. We conclude that interactions between Nef, GagPol and Alix contribute to optimal viral replication and progression to disease in the infected host.
Keywords: SIV, HIV, Nef, monkey infection, pathogenesis, disease progression, GagPol, Alix
INTRODUCTION
Primate lentiviruses are the only members of the Retroviridae family that encode the misnamed NEgative Factor (Nef). It is a 27–36 kDa myristylated protein that localizes to cellular membranes (Bentham, Mazaleyrat, and Harris, 2006; Kaminchik et al., 1994) and is also incorporated into viral particles (Pandori et al., 1996; Welker et al., 1996). Nef is a multi-functional protein with several activities in cells. They include the internalization of cell surface receptors CD4 (Iafrate et al., 2000; Lock et al., 1999), CD3 (Munch et al., 2002), MHC class I (Schwartz et al., 1996) and II (Stumptner-Cuvelette et al., 2001), modulation of cellular activation and increased viral infectivity (Goldsmith et al., 1995; Miller, Feinberg, and Greene, 1994; Miller et al., 1994; Munch et al., 2007; Saksela, Cheng, and Baltimore, 1995). Indeed, Nef increases the infectivity of HIV-1 (HIV) even in the absence of CD4 on the producer cell (Goldsmith et al., 1995; Saksela, Cheng, and Baltimore, 1995). Previously, we correlated this increased viral infectivity with direct interactions between Nef from HIV-1 (HIVNef) and the viral structural GagPol polyprotein (HIVGagPol) via the transframe p6* protein and protease (HIVp6*PR) and the cellular Alix protein. Furthermore, we mapped the binding to HIVp6*PR to the C-terminal flexible-loop (FL) domain and to Alix to the YPL sequence (tyrosine-based motif) in HIVNef, respectively (Costa et al., 2006; Costa et al., 2004). Unlike many studies on Nef alone, we could correlate our structural studies to function and HIV replication in transformed and primary cells in culture (Costa et al., 2006; Costa et al., 2004).
In monkey models of AIDS (Simian AIDS, SAIDS), Nef (SIVNef) is essential for high viral loads and the progression of disease (Kestler et al.). Animals infected with SIVmac239 (SIV) harboring mutations or truncations of the nef gene become chronically infected with markedly reduced symptoms (Daniel et al., 1992; Kestler et al., 1991; Marthas et al., 1993). Such changes in Nef have also been reported in HIV long-term nonprogressors or elite controllers among infected individuals (Deacon et al., 1995; Kirchhoff et al., 1999; Kirchhoff et al., 1995).
In this study, we extended our studies with HIV in cells to SIV infection in rhesus macaques. To this end, we mutated conserved residues in Nef from SIV that interact with SIVGagPol and Alix and then followed viral replication and disease progression in four infected monkeys. After long periods of low levels of viral replication, only the two rhesus macaques that reverted these binding surfaces developed high viral loads and succumbed to SAIDS.
RESULTS
Mutant SIV (SIVM8) containing 8 changes in the nef gene (NefM8) attenuates viral replication in infected rhesus macaques
Previously, we demonstrated that interactions between HIVNef, HIVGagPol and the cellular Alix protein correlated with its ability to increase viral infectivity in cells (Costa et al., 2006; Costa et al., 2004). Additionally, this binding was required for the dominant negative HIVNefF12 phenotype, which blocked HIV replication by retaining HIVGagPol near the endoplasmic reticulum (ER) (Costa et al., 2004). This functional interaction was confirmed with a mutant HIVNefNL4-3 protein, where the ER-retention signal KKXX was placed at its C-terminus (Costa et al., 2004). Finally, the binding to Alix and thus the ESCRT machinery was required for the proliferation of multivesicular bodies (MVBs) and intraluminal vesicles (ILVs) by HIVNef in cells (Costa et al., 2006). Since these binding motifs are conserved among Nef proteins from all primate lentiviruses, we wanted to interrogate these interactions with SIVNef not only biochemically and functionally in cells, but also in the infected host. To this end, we utilized the mutant NefM8 protein (Fig. 1, top panel), which came from SIVmac239 and bore mutations not only in the putative binding surfaces for Alix and SIVGagPol but in additional residues that were implicated in other functional interactions, such as CD4 internalization and binding to armadillo (ARM) repeat-containing proteins that include adaptor protein complexes, βCOP and V-ATPase, alone or in the context of the virus (SIVM8) (Geyer, Fackler, and Peterlin, 2002; Mandic et al., 2001). Indeed, with NefM8, both tyrosines at positions 28 and 39 (Y28XXL and Y39SQS) and the FL play a role in CD4 internalization and viral infectivity in cells (Mandic et al., 2001). Since Alix should only bind the first bona fide tyrosine-based motif (Y28XXL) and SIVGagPol should bind the central and C-terminal portions of the FL but not require more N-terminal sequences that engage ARM repeats, we took advantage of these additional introduced mutations in NefM8 (Fig. 1, SIVM8, top) to interrogate possible reversions in infected rhesus macaques (Mandic et al., 2001). Thus, if these binding partners are relevant for disease progression in vivo, residues that do not interact with Alix and GagPol, such as Y39 and D184D185 should not revert to the wild type (WT) sequence (see below).
Figure 1. Disease progression of the mutant SIVM8 virus bearing mutations in the nef gene.
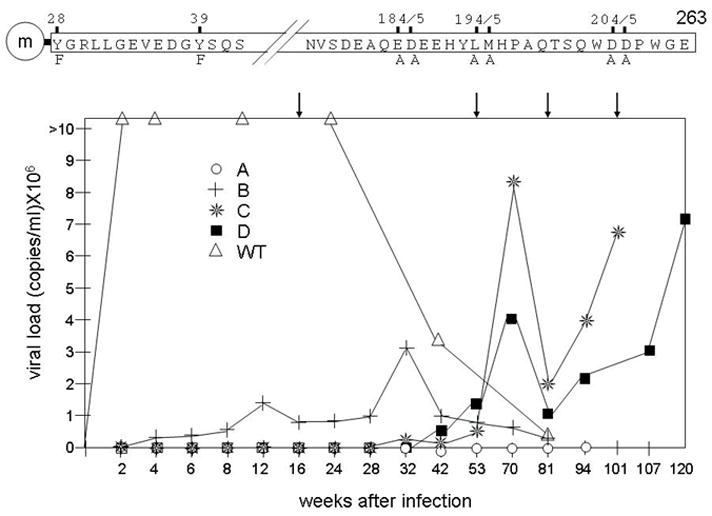
Eight residues in SIVNef were targeted by clustered point mutations (top panel). Only relevant sequences are presented in the single letter amino acid code. The myristylation signal (at G2) is denoted by an“m” inside the circle (not drawn to scale). This mutant nef gene was introduced into the SIVmac239 (SIV) infectious clone (SIVM8). Wild type (WT) SIV and mutant SIVM8 viruses were inoculated into 2 and 4 rhesus macaques, respectively. Blood was taken periodically throughout a 120 weeks period (times are indicated on the X axis, not drawn to scale) and viral loads were measured by RT-qPCR. Arrows point to informative time points on the graph. Open triangles, WT: two rhesus macaques infected with WT SIV; open circles, A; crosses, B; stars, C; and black squares, D: rhesus macaques infected with mutant SIVM8.
To this end, SIVM8 viral stocks were inoculated intravenously into four rhesus macaques. Two control monkeys were inoculated with the WT SIV under the same conditions (although viral loads from only one representative monkey are presented, the second monkey had similarly elevated viral loads and progressed quickly to SAIDS). After the inoculation, viral loads in plasma were determined at frequent intervals for a period of 81 to 120 weeks. Beginning at two weeks, we noted a dramatic difference in viral loads between animals infected with WT and mutant viruses (Fig. 1, bottom panel). In two of four SIVM8-infected monkeys (Fig. 1, bottom panel, monkeys C and D), viral loads started to increase after 53 weeks, reaching peak values at 70 weeks. In animals C and D, opportunistic infections and other signs of disease progression accompanied sustained viral replication. Symptoms included progressive weight loss (up to 25%), diarrhea as well as hematological abnormalities and severe lymph node involution (Table 1). One infected monkey (B) presented different kinetics with a less pronounced increase in viral loads around 12 weeks, which dropped and rose again to reach a peak at 32 weeks after inoculation (Fig. 1. bottom panel). Importantly, these viral loads were not sustained after 32 weeks of infection. Rather, monkey B had recurrent infections with M. avium. However, at 81 weeks, this animal was euthanized with no signs of SAIDS (Fig. 1). One infected monkey (A) remained healthy throughout the course of infection with undetectable viral loads (Fig. 1, bottom panel). These results demonstrate that eight mutations introduced into the nef gene alter the ability of SIVNef to contribute to the progression of SAIDS in rhesus macaques.
Table 1.
Clinical course and viral titers in 4 rhesus macaques inoculated with SIVM8
| Monkey | TCID50 | Clinical course | Viral load (70 wks) | Euthanasia (wks) | Pathology |
|---|---|---|---|---|---|
| A | 5000 | Healthy | <50 | 106 | None |
| B | 5000 | OI: M. Avium1 | 1.3 × 105 | 81 | None |
| C | 5000 | SAIDS | 8 × 106 | 101 | SAIDS |
| D | 5000 | SAIDS | 4 × 106 | 120 | SAIDS |
Opportunistic infections with mycobacterium avium
Some nef genes from SIVM8 revert during viral replication and disease progression in infected rhesus macaques
To investigate changes in the nefM8 gene that could have accounted for the rebound in viral loads and progression to disease in monkeys C and D, the nef gene was analyzed. At each time point, at least five clones from each rhesus macaque were sequenced. Sequences were compared to those from WT SIVNef and mutant NefM8 proteins (Table 2). Interestingly, for two monkeys with high viral loads after 53 weeks of infection and clear progression of disease, mutated central and C-terminal sites in the FL reverted back to encode residues found in the WT sequence (see positions 194 and 204 in monkeys C and D), or to a residue with the same characteristics (see position 195 in monkeys C and D). Exceptions were at positions 184 and 185 in monkeys C and D that reverted partially (monkey C) or not at all (monkey D) (Table 2). Monkey C, which developed the highest viral loads, also reverted the first (F28Y), but not the second (F39) mutated tyrosine-based motif. Whereas this reversion was observed already at week 53 in monkey C, it did not occur in monkey D until 101 weeks, but it correlated with a significant increase in viral loads (Fig. 1). Apart from position 205, which reverted back to encode aspartic acid in all rhesus macaques after 16 weeks, monkey A had no other reversions. In addition, we did not observe any additional reversions in NefM8 in monkey B at 53 and 81 weeks. Indeed, one half of sequences recovered from this monkey had only minor deletions in residues around position 70 near the N-terminus of Nef. These data point out the contribution of the first tyrosine-based motif and the FL in SIVNef for sustained viral replication and disease progression in SIV-infected rhesus macaques.
Table 2.
Reversions in Nef from SIVM8 in infected rhesus macaques
| Infections | Sequences in Nef | ||||||
|---|---|---|---|---|---|---|---|
| SIV clone | Monkey | Weeks1 | Y-based motif | Flexible loop | |||
| SIV | WT | - | Y282 | Y39 | D184 E185 | L194 M195 | D204 D205 |
| Y | Y | DE | LM | DD | |||
| 1 | 81 | Y | Y | DE | LM | DD | |
| 2 | 81 | Y | Y | DE | LM | DD | |
| SIVM8 | SIVM8 | - | F | F | AA | AA | AA |
| A | 16 | F | F | AA | AA | AD | |
| B | 53/81 | F | F | AA | AA | AD | |
| C | 53 | Y | F | AA | VA | AD | |
| C | 53.2 | F | F | AA | AM | AD | |
| C | 81 | Y | F | DA | LV | DD | |
| D | 81 | F | S | AT | LL | DD | |
| D | 101 | Y | S | AT | LL | DD | |
Time of informative sequences
Positions of residues in Nef from SIVmac239
SIVNef binds SIVGagPol via SIVp6*PR
Previous studies demonstrated that the FL in HIVNef binds HIVGagPol (Costa et al., 2004). As presented in Fig. 2A, SIVNef also binds its cognate SIVGagPol. GST pull-downs were performed with GST.SIVNef and SIVGag, fsSIVGagPR- (containing frameshifted p6* and inactive PR) or SIVp6*PR-. No protein bound to GST alone (Fig. 2A, lanes 2, 5 and 8). In contrast, we observed efficient binding between fsSIVGagPR- (Fig. 2A, lane 3), SIVp6*PR- (Fig. 2A; lane 6) and GST.SIVNef. In these pull-downs, we used fsSIVGagPR- and not fsSIVGagPolPR- because of greater degradation of the larger protein, when they was expressed from the coupled transcription and translation reaction using the rabbit reticulocyte lysate in vitro (IVT). Indeed, the use of the smaller peptide (fsSIVGagPR-) circumvented this problem. Additionally, no binding was observed between GST.SIVNef and SIVGag (Fig. 2A, lane 9). We conclude that as with HIV, interactions between Nef and GagPol occur via SIVp6*PR-. These results confirm that the binding between Nef and GagPol is conserved among primate lentiviruses.
Figure 2. SIVNef binds SIVGagPol via its p6*PR peptide.
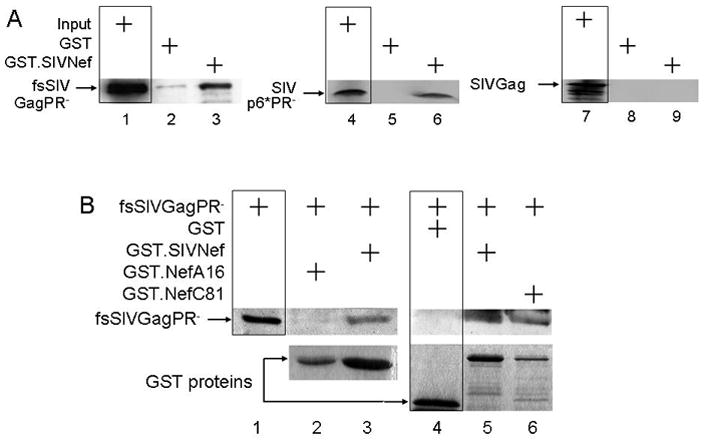
A. GST pull-downs. They were performed with GST or GST.SIVNef and [35S]-labeled Gag, the frameshifted, protease negative fsSIVGagPR- and SIVp6*PR- peptides. Lanes 1, 4 and 7 represent 10% input for fsSIVGagPR-, SIVp6*PR- and SIVGag, respectively (boxed). Inputs for GST and GST.Nef are related to panel B. B. Reverted mutant SIVNef proteins restore the binding to SIVGagPol. GST pull-downs were performed with GST alone or GST.NefM8 and reverted GST.SIVNef and the Myc epitope-tagged fsSIVGagPR- proteins. Proteins were separated by 10% SDS-PAGE followed by western blotting with anti-Myc antibodies. Lane 1 represents 10% of the input for fsSIVGagPR-.
Reverted SIVNef but not mutant NefM8 proteins bind SIVGagPol
Further specificity of this interaction came from studies of mutations introduced into SIVM8 in Fig. 2B. GST pull-downs were performed with IVT fsSIVGagPR- and GST.SIVNef or GST.SIVNefA16 (this nef was recovered from monkey A that did not progress to disease and used as a surrogate for NefM8). Whereas WT SIVNef bound fsSIVGagPR-, no fsSIVGagPR- was pulled-down by GST alone or by GST.SIVNefA16 (Fig. 2B, compare lanes 2 and 4 with lanes 3 and 5, respectively), demonstrating that mutations introduced into the FL of SIVNef disrupted the binding to SIVGagPol. Since the expression of GST.NefC81 was the lowest and it still pulled down fsSIVGagPR- efficiently (Fig. 2B, upper and lower panels, compare lanes 5 and 6), differences in input levels in western blots do not explain the lack of binding between GST.NefA16 and fsSIVGagPR-. To prove this point more convincingly, we also examined GST.NefB53 with the same sequence in the FL, which, despite being expressed at higher levels than the WT GST.SIVNef, still failed to bind fsSIVGagPR- (Supplemental Fig. 1, lane 4). Next, we examined if reverted sequences would restore this binding. Indeed, GST.SIVNef from monkey C at 81 weeks (NefC81) pulled-down IVT fsSIVGagPR- (Fig. 2B, lane 6). An additional isolate from monkey D at 81 weeks (GST.NefD81) also pulled down fsSIVfsGagPR- (Supplemental Fig. 1, lane 6). As found also with HIVNef, the N-terminal residues D184E185, which did not revert to WT sequence in monkey C by 81 weeks and in monkey D by 101 weeks, were not required for interactions with GagPol (Geyer, Fackler, and Peterlin, 2001). These results were confirmed by co-immunoprecipitation experiments (co-IPs) in cells. 293T cells co-expressed SIVGag or fsSIVGagPolPR- with SIVNef. Immunoprecipitations of Nef with an antibody against the C-terminal V5 epitope-tag co-immunoprecipitated fsSIVGagPolPR- (Fig. 3, lanes 2), but not SIVGag (data not presented). As with the WT SIVNef, NefC81 and NefD81 also bound fsSIVGagPolPR- (Fig. 3, lanes 3 and 5). As expected, NefB53 and NefA16 did not bind fsSIVGagPolPR- (Fig. 3, lanes 4 and 6). We conclude that the FL in SIVNef binds SIVp6*PR. Thus, we correlated the appearance of functional nef alleles with the restoration of binding between SIVNef and SIVGagPol.
Figure 3. Reverted mutations restore interactions between mutant SIVNef proteins and SIVGagPol in cells.
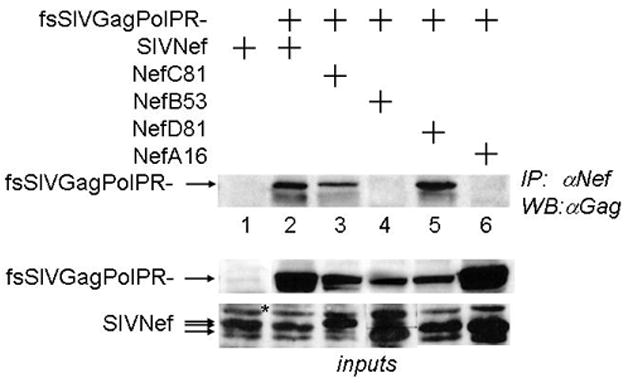
293T cells co-expressed SIVGag or fsSIVGagPolPR- and the V5 epitope-tagged SIVNef and reverted mutant Nef proteins. Cell lysates were immunopreciptated with anti-V5 antibodies. Upper panel presents co-IPs of SIVGagPolPR- with WT SIVNef and Nef proteins recovered from monkeys A, B, C and D at different weeks of the infection (numbers after capital letters)(IP, WB). Lower panels present expression levels of all proteins used in these experiments (inputs) (WB). Asterisk indicates an unspecific band that is recognized by the anti-V5 antibody. Note differences in the apparent molecular weight of Nef proteins from monkeys B and C.
SIVNef binds Alix
The V domain of Alix binds tyrosine-based motifs (YPX(n)L) in late domains (LDs) from HIVGag and SIVGag (Fisher et al., 2007; Strack et al., 2003; Zhai et al., 2008). Previously, we demonstrated that the YPL sequence in HIVNef binds Alix (Costa et al., 2006). The N-terminal tyrosine-based motif in SIVNef (Y28GRL) also binds Alix (Fig. 4A). For these binding studies, we used two different HIVNef alleles (from HIV-1NL4-3 and HIV-1SF2) and the cellular TSG101 protein, which is the binding partner of Alix, as positive controls (Fig. 4A, lanes 1, 3 and 5, respectively). Indeed, we observed that GST.Alix also pulled-down SIVNef (Fig. 4A, lane 4). As with SIVNef, mutations of the tyrosine-based motif in HIVNef abolished this interaction (Fig. 4A, lane 2). Inputs for all IVT proteins were equivalent (Fig. 4A, lower panel). We used two mutant SIVNef proteins, which changed tyrosine 28 and tyrosine 39 to phenylalanines (Y28F and Y39F). Whereas WT and SIVNefY39F bound Alix (Fig. 4B, lanes 2 and 4, where lanes 1 and 3 are inputs for these proteins), this interaction was absent with SIVNefY28F (Fig. 4B, lane 6, where lane 5 represents the input for this protein). We also performed GST pull-downs using GST.Nef as bait (Fig. 5A) and confirmed this binding between SIVNef and Alix (Fig. 5AC, lanes 2 and 5). Thus, SIVNef also binds Alix and the tyrosine at position 28 is required for this interaction, which establishes the importance of specific reversions in SIVM8 (see below).
Figure 4. WT SIVNef protein binds Alix.
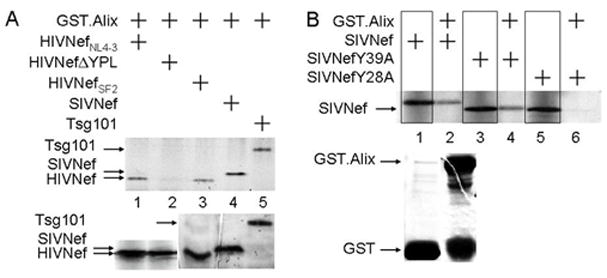
A and B. HIVNef and SIVNef bind Alix, and the N-terminal tyrosine-based motif in SIVNef is required for this interaction. GST pull-downs were performed with the GST.Alix and [35S]-labeled WT or mutant HIVNef and SIVNef or Tsg101 proteins. A. Inputs for all proteins are presented in the lower panel. B. Lanes 1, 3 and 5 contain 50% input of each of the SIVNef proteins. Inputs for GST and GST.Alix proteins for all pull-downs are presented in the lower panel.
Figure 5. Reverted mutant Nef proteins from SIVM8 bind Alix.
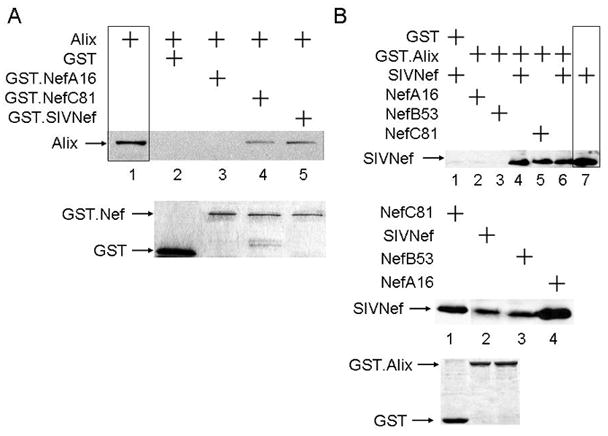
A. Some reverted SIVNefM8 proteins also bind Alix. GST pull-downs were performed with GST alone or with WT GST.SIVNef, GST.NefA16 or GST.NefC81 and the HA epitope-tagged Alix protein. Lane 1 represents 20% input of Alix. Inputs for GST and GST.Nef are presented in the lower panel. B Analysis of further reverted SIVNefM8 proteins with GST.Alix. Experiments were performed as in A. Lane 7 represents 20% input of one representative Nef protein. The expression levels of all Nef proteins, GST and GST.Alix are presented in the middle and lower panels.
Reversion of the first tyrosine-based motif in SIVNef restores its binding to Alix
Since Y28 in SIVNef was required for binding (Fig. 4B), NefA16 and NefB53 also did not bind Alix (Fig. 5A, lane 3 and Fig. 5B, lanes2 and 3). In contrast, like the WT SIVNef (Fig. 5B, lanes 4, 5 and 6, where lane 7 is a representative input for SIVNef), the recovered Nef protein from monkey C (NefC81), which reverted phenylalanine at position 28 back to tyrosine, restored this binding (Fig. 5A, lane 4 and Fig. 5B, lane 5). Thus, Y28 is required for interactions between SIVNef and Alix. To confirm this interaction in cells, we co-expressed the N-terminal HA epitope-tagged Alix and C-terminal V5 epitope-tagged Nef proteins and performed co-IPs from transfected 293T cells. We observed that only WT and reverted SIVNef proteins, NefC53 and NefC81, co-immunoprecipitated Alix (Fig. 6, lanes 4, 5 and 6). As expected, NefA16 and NefB53 did not interact with Alix in cells (Fig. 6, lanes 2 and 3). Importantly, since NefC53 reverted F28 to Y28 but not the relevant residues in the FL (Table 2), only the first tyrosine-based motif (Y28GRL) in SIVNef is required for binding to Alix as well as contributing to high viral loads and the progression to SAIDS in rhesus macaques.
Figure 6. WT SIVNef and reverted mutant SIVNefM8 proteins bind Alix in cells.
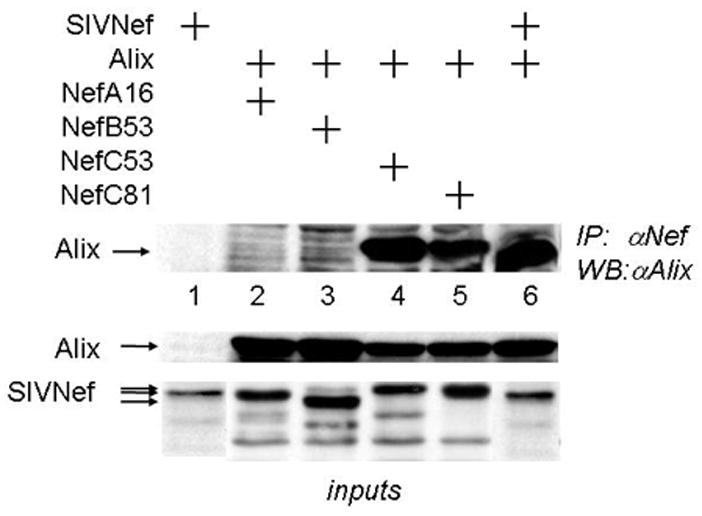
293T cells co-expressed the HA epitope-tagged Alix protein and V5 epitope-tagged WT SIVNef, NefA16 or reverted mutant Nef proteins. Cell lysates were immunopreciptated with anti-V5 antibodies, followed by western blotting with antibodies against HA (IP, WB). Lower panels present expression levels for the proteins used in these experiments (inputs)(WB).
Fusion proteins between HIVGagΔp6 and WT SIVNef or reverted NefM8 proteins support the release of mutant viral particles lacking LD from cells
To confirm that the binding to Alix is an important feature of SIVNef, we used an assay described by Martin-Serrano et. al. (Martin-Serrano, Zang, and Bieniasz, 2001). In this system, the budding of new viruses from a LD-deleted (HIVΔp6) provirus harboring a stop codon at the beginning of p6 (this mutation does not affect p6*) can be rescued by adding minimal LDs to a mutant Gag that lacks p6 and pol (GagΔp6). The requirement for budding is the recruitment of the host protein Tsg101 to the site of viral assembly. Since Alix binds Tsg101 and SIVNef, we hypothesized that linking Nef to the C-terminus of GagΔp6 would perform the same function. Therefore, we fused GagΔp6 in frame with SIVNef, NefM8 or its revertants. As expected, the expression of HIVΔp6 in 293T cells did not release VLPs into the supernatant (Fig. 7, top panel, lane 1). In contrast, the co-expression of HIVΔp6 with HIVGag fully restored VLP release (Fig. 7, top panel, lane 2). Similar results were obtained by co-expressing HIVΔp6 with GagΔp6.SIVNef or GagΔp6.NefC81 but not with GagΔp6.NefA16 or GagΔp6.NefD81 (Fig. 7, top panel, lanes 5 and 6). Viral particles released from 293T cells co-expressing GagΔp6.SIVNef or GagΔp6.NefC81, but not GagΔp6.NefA16 or GagΔp6.NefD81 were also as infectious as those from 293T cells expressing HIVΔp6 with HIVGag in TZM-Bl indicator cells (Fig. 7, lower panel, lanes 2 – 4). The expression of viral proteins was similar in all producer cells (Fig. 7, middle panel, lanes 1 – 6). These results confirm functional interactions between SIVNef and Alix in cells.
Figure 7. GagΔp6.SIVNef restores the release of viruses from the LD-deficient HIVΔp6 provirus.
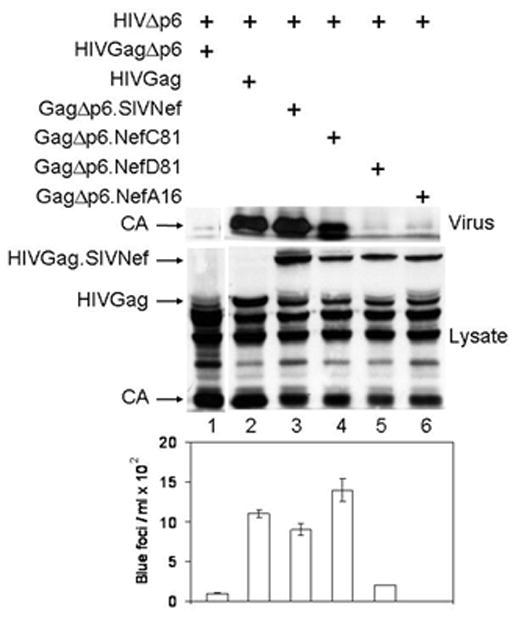
293T cells co-expressed the LD-deficient NLHXΔp6 (HIVΔp6) virus together with the WT Gag, mutant GagΔp6 or GagΔp6.SIVNef proteins. Protein content in cell (Lysate) and viral (Virus) lysates was analyzed by western blotting with antibodies against p24 (HIVCA). Supernatants of transfected cells were examined for viral infectivity in TZM-bl cells. Values are presented as numbers of blue foci/ml in the lower panel. Data are representative of three experiments performed in duplicate with variations depicted by error bars.
Reverted NefM8 proteins restore the infectivity of SIVΔNef in cells
Next, we wanted to correlate different nef sequences recovered from rhesus macaques, especially those that progressed to disease, with viral infectivity in cells. First, we expressed SIVΔNef alone or together with different SIVNef proteins in 293T cells. Amounts of cell-associated and released viral particles were determined with antibodies against p27 (SIVCA) by western blotting and ELISA, respectively. They were found to be equivalent for all cells that expressed SIVΔNef (Fig. 8, middle and right panels, lanes 1 – 8). Expression levels of different Nef proteins were also equivalent (Fig. 8, lower panel, lanes 2 to 8. Look also at Figs. 3 and 5, lower panels). Next, we examined the infectivity of these viral particles in indicator cells. Levels of infectivity of the SIVΔNef co-expressed with SIVNef or NefC81 were up to 8-fold higher than those of SIVΔNef alone or co-expressed with NefB53, NefB81 or NefC53.2, where all critical residues for binding to GagPol and Alix had reverted (Table 2) (Fig. 8, upper panel, compare bars 2 and 3 to bars 1, 4, 6 and 7). Importantly, NefC53, which only reverted F28 back to Y28, and NefD81, which reverted critical residues in the FL but not the F28, had intermediate infectivity (Fig. 8, middle panel, bars 5 and 8). These results demonstrate that interactions between SIVNef, GagPol and Alix are important for viral infectivity. Of note, in our previous study, the infectivity levels of SIVΔNef and SIVM8 viruses were 5-fold lower than those of the WT SIV (Mandic et al., 2001). This disparity is due to different cell lines used in our infectivity assays. Thus, Nef is important for SIV infectivity, which could be complemented in the case of SIV Nef with our reverted but not mutant Nef proteins. This finding also correlates viral loads in the host with SIV infectivity in cells.
Figure 8. Reverted mutant SIVNefM8 proteins restore the infectivity of SIVΔNef in trans-complementation assays.
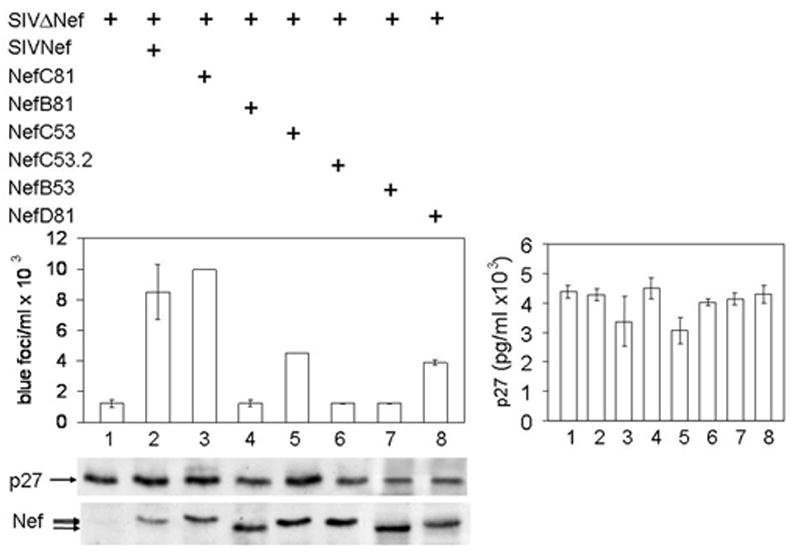
293T cells co-expressed equal amounts of SIVΔNef and WT or reverted mutant Nef proteins (lower panel). Cell-free viral particles were quantified by p27 ELISA and these values are presented in the right panel. Cell lysates were also analyzed with anti-p27 and Nef antibodies by western blotting in the lower panels. Equivalent amounts of viral particles were used to infect TZM-Bl cells and analyzed as in Fig. 7. Data are representative of three experiments performed in duplicate with variations depicted by error bars.
DISCUSSION
In this study, we evaluated the role/s of two proteins that interact with Nef in the context of SIV infection in cells and in the host. Previously, we established that HIVNef binds HIVGagPol and the cellular Alix protein and correlated these findings with optimal viral replication in cells (Costa et al., 2006; Costa et al., 2004). In this study, we extended these observations to SIV in rhesus macaques. First, we introduced informative mutations into SIVNef and infected four rhesus macaques with the mutant SIV. Whereas rhesus macaques infected with the WT virus developed high viral loads and progressed rapidly to SAIDS, only two of four monkeys inoculated with the mutant virus displayed a similar picture, albeit with much delayed kinetics. Importantly, in both cases, we observed reversions in SIVNef that restored its binding to SIVGagPol and Alix. In two other rhesus macaques, mutant Nef sequences persisted and they developed neither high viral loads nor SAIDS. Further studies in cells provided additional support for these findings. We conclude that interactions between SIVNef, SIVGagPol and Alix contribute to optimal viral replication and progression to disease in the monkey model of AIDS. Since these interactions are preserved in HIVNef, they are also pertinent to the human disease.
SIVNef is similar to HIVNef in most respects. Besides the highly conserved core domain, it also contains critical FL and tyrosine-based motifs. Moreover, we mapped interactions with p6*PRs to FLs in HIVNef and SIVNef. Their tyrosine-based motifs bound Alix, most likely via direct contact between the tyrosine, proline residues in p6 and the hydrophobic V pocket of Alix (Fisher et al., 2007; Zhai et al., 2008). Importantly, only residues that are absolutely required for these interactions reverted in SIVM8. Since surfaces that contact Alix and p6*PR are distinct and separated by the core domain, Nef could act as an adaptor between GagPol and the ESCRT machinery. Importantly, these N- and C-terminal sequences in Nef were also restored in frame in previous studies with SIVΔNef, which contained large deletions in the nef ORF. Thus, before those mutant SIVΔNef viruses could replicate in rhesus macaques, although lacking the core sequence, reading frames for FL and tyrosine-based motifs were restored in the truncated Nef (tNef) (Chakrabarti et al., 2003; Sawai et al., 2000). Importantly, WT SIVNef bound SIVGagPol and Alix before these specific residues were mutated and after they reverted to the WT sequence. The binding selectivity came from lack of reversions of irrelevant F39 and N-terminal A184A184 residues in the FL of NefM8. Since Y38 was still important for CD4 internalization in cells (Mandic et al., 2001) and other (ARM)-repeat-containing proteins require these additional residues in the FL (Geyer, Fackler, and Peterlin, 2002), it is less likely that these other interactions played a major role in our observed phenotype in infected rhesus macaques. This finding is confirmed by another independent study, where a deletion from positions 64 to 67 in SIVNef that abrogated all effects on CD4, still supported high levels of viral replication and progression to SAIDS in rhesus macaques without any reversion of this mutation (Brenner et al., 2006).
Although reversions in our NefM8 also restored these critical interacting domains, we cannot formally rule out that other compensatory mutations also occurred elsewhere in the viral genome. However, since changes were so precise and exempted the residues mentioned above, we feel that such a scenario is unlikely. Nevertheless, the FL had been implicated in other functional interactions in cells. Most of these involve the (ARM)-repeat-containing proteins mentioned above. These repeats contain three αhelices and if interactions with p6*PR require a similar secondary structure, only one or two αhelices could be contributed by p6* and the others by PR (Beissinger et al., 1996; Geyer, Fackler, and Peterlin, 2002). Indeed, PR should provide the major contribution as limited primary data reveal that p6* alone does not form any structure in solution (Beissinger et al., 1996). As PR also cleaves Nef, this association must be intimate at least during the assembly and early budding of new viral particles. In this regard, the recent publications by Leiherer et al might be instructive (Leiherer, Ludwig, and Wagner, 2009). Indeed, their extensive mutations in p6* did not abrogate the ability of HIVNefF12 to block HIV replication, which depends on interactions between Nef and GagPol (Costa et al., 2004). Thus, Nef most likely interacts with several surfaces on GagPol, which are centered on the p6*PR peptide. However, this finding does not exclude the possibility that other HIVNef alleles or Nef proteins from other primate lentiviruses interact with other parts of their cognate GagPol polyprotein precursors. Of interest, retroviruses (other than lentiviruses) that do not encode p6* also lack Nef.
Although the number of infected monkeys was small, our findings were convincing. When viral loads increased and monkeys developed immunodeficiency and opportunistic infections, sequences in SIVNef reverted to those that interact with SIVGagPol and Alix. Of interest, levels of viral replication never approached those of WT SIV, implying that SIVM8 could have acted as an attenuated strain and immunized partially these monkeys against the virus (Iafrate et al., 2000). In two monkeys, where we observed no reversions, we also observed only asymptomatic infections. The only spikes in viral titers were observed in one of these two monkeys that had concurrent infections, especially those with M. avium. When they cleared, viral titers subsided. These monkeys were euthanized at 81 days and extensive necropsy results revealed no SAIDS. Importantly, we were able to correlate viral loads in vivo with SIV infectivity in cells.
What do these interactions mean for Nef and primate lentiviruses? First, they could facilitate connections between GagPol, which lacks an LD, and the ESCRT machinery. Together with Nef oligomers, these interactions could help in the assembly and incorporation of cognate and irrelevant retroviral Envs into new viral particles (Sandrin and Cosset, 2006; Schiavoni et al., 2004). These Nef oligomers could also contribute to the shape and/or improve the composition of progeny virions (Brugger et al., 2007; Zheng et al., 2003). Moreover, since p6* has also been implicated in the inhibition of PR, Nef could also contribute to the optimal spatial and temporal regulation of Gag and GagPol processing (Ludwig, Leiherer, and Wagner, 2008). To this end, it is important to note that one of the main determinants of viral infectivity is the proper processing of Gag and GagPol (Hill et al., 2002; Zhou et al., 2004). However, as no gross defects in these processes had been detected to date, these effects could be subtle. To this end, a mutant p6* protein, where 40 out of the 56 residues were changed led to significantly decreased viral infectivity without gross effects on Gag and GagPol processing in the presence or absence of Nef (Leiherer, Ludwig, and Wagner, 2009). Thus, in addition to roles of free Nef in the internalization of cell surface receptors and intracellular signaling, Nef bound to GagPol plays an important role for the virus in its difficult passage out of the cell and for the infection of new target cells. Our studies provide a new framework for thinking about this enigmatic accessory protein of primate lentiviruses.
METHODS
Cell Lines and transfections
293T and TZM-bl cells were obtained from Dr. Bergman (Federal University of Brasilia, Brazil) and the NIH AIDS Research and Reference Reagent Program (NARRRP, Division of AIDS, NIAID, NIH, Bethesda, MD). Both cell lines were maintained in DMEM/10% FCS and antibiotics. Transfections were performed with Lipofectamine2000 (Invitrogen, Carlsbad, CA) or Fugene6 (Roche Applied Sciences, Indianapolis, IN) according to the manufactures’ instructions. Anti-p27 SIVGag hybridoma was obtained from the NARRRP (Higgins et al., 1992), and was maintained in RPMI1640/10% FCS and antibiotics.
Antibodies
Policlonal anti-hemagglutinin (anti-HA), anti-myc (anti-c-Myc) epitope (Sigma-Aldrich Corp., St. Louis, MO), monoclonal anti-V5 epitope (Invitrogen), p24 HIV antiserum (NARRRP) and monoclonal anti-p27 SIVGag antibodies were used as first antibodies. Secondary anti-mouse and anti-rabbit IgG horseradish peroxidase-linked species-specific F(ab′)2 fragments (GE Healthcare Bio-Sciences Corp., Piscatways, NJ) were detected by the enhanced chemiluminescence reagent (GE Healthcare Bio-Sciences Corp.).
Construction of SIVM8 and preparation of viral stocks
Cloning of the nef gene from the SIVmac239 (SIV) into an expression vector, the introduction of eight mutations, and the generation of SIVM8 containing this mutant nef gene, were described previously (Mandic et al., 2001). In contrast with which is described previously for this mutant M8, we mutated tyrosines 28 and 39 to phenylalanines to preserve the correct amino acid residues in the gp41 gene in which region overlaps with the nef gene. WT SIV and mutant SIVM8 infectious viral particles were recovered from transfections of 293T cells. Supernatants from transfected cells were filtered and used to inoculate rhesus macaques.
Viral studies in rhesus macaques
The 50% Tissue Culture Infective Dose (TCID50) of virus stocks and their verification were determined as previously described (Shacklett et al., 2002). Animals were colony-bred juvenile or newborn rhesus macaques (Macaca mulatta) housed at the California Regional Primate Research Center (UC Davis, Davis, CA) and determined to be free of SIV as well as simian T-lymphotropic and type D retroviruses. Rhesus macaques were maintained in accordance with the standards of the Institutional Animal Care and Use Committee. Physical examinations were performed at regular intervals to detect lymphadenopathy, splenomegaly, and opportunistic infections. Clinical criteria for euthanasia consisted of three or more of the following: (i) more than 10 % weight loss within 2 weeks or more than 20 % in 2 months; (ii) chronic diarrhea unresponsive to treatment; (iii) infections unresponsive to antibiotic treatment; (iv) inability to maintain body heat or fluids without supplementation; and (v) persistent, marked hematological abnormalities (persistent, marked splenomegaly or hepatomegaly). At defined time points after inoculation, 8 to 15 ml of blood was collected by venipuncture. Plasma was stored for virus loads. Lymph nodes were obtained by surgical biopsy. Viral RNA in plasma samples was quantified by TaqMan real-time PCR as described previously (Shacklett et al., 2002). The TaqMan real-time PCR can detect as few as 50 copies of SIV RNA/ml. Viral RNA was extracted from plasma (Qiagen viral RNA kit) at different time points from each infected monkey. SIVnef sequences were amplified in a nested PCR as described (Shacklett et al., 2002), cloned into the pCR2.1 vector (Invitrogen) and at least five clones from each macaque at each time point were sequenced on an ABI automated sequence analyzer (Applied Biosystems-Perkin-Elmer, Foster City, CA).
Plasmid constructions
To express the GST.Alix fusion protein, a pGEX.alix vector was obtained. The full-length human alix gene was obtained by PCR-amplification of the alix cDNA (cloned into the pSPORT vector) and introduced the into the pGEX2TK-KG plasmid (GE Healthcare Bio-Sciences Corp.). To expresses the WT, mutant, and reverted GST.Nef fusion proteins, pGEX.nef vectors were obtained. WT nef gene was PCR-amplified from the pSIVmac239 infectious clone previously described. Mutant and reverted nef sequences were amplified from the pCR2.1 clones obtained previously. The amplification products were introduced into the pGEX2TK-KG plasmid. The HIVNefNL4-3, HIVNefSF2; HIVNefΔYPL; Tsg101 and Alix.HA expression plasmids are described elsewhere (Costa et al., 2006). The mutant SIVNefY28F and SIVNefY39F expression plasmids were obtained by site direct mutagenesis by using the QuikChange site-direct mutagenesis kit (Stratagene, Carlsbad, CA) using the pcDNA3.1SIVNef plasmid as template. For the expression of the V5 epitope-tagged Nef proteins from WT, mutant and reverted SIV in eukaryotic cells, nef genes were amplified and cloned into pcDNA3.1D.V5 TOPO (Invitrogen). For in vitro translation, SIVfsGagPR- was generated by PCR amplification from SIV, PCR fragment was introduced into the T7-plink c-Myc and a constitutive frameshift mutation at the ribosome slippery region between SIVGag and SIVGagPol, to ensure the synthesis of SIVGagPR-, and a point mutation in the active site of PR (D25N) to ensure the expression of unprocessed SIVGagPol, were introduced by standard PCR mutagenesis by using the QuikChange site-direct mutagenesis kit (Stratagene). The T7-c-Myc.SIVp6*PR- plasmid was obtained by PCR amplification of the p6* transframe and protease region from the T7-c-Myc.SIVfsGagPR-. The Rev-independent, codon-optimized SIVGagPol in pcDNA3 was kindly provided by Dr. Ralf Wagner (University of Regensburg, Regensburg, Germany) (Wagner et al., 2000). A constitutive frameshift mutation at the ribosome slippery region between SIVGag and SIVGagPol, and a point mutation in the active site of PR (D25N) were introduced into this plasmid by using the QuikChange site-direct mutagenesis kit (Stratagene). For the expression of SIVGag alone, the SIVgag was amplified from the original plasmid and cloned into pcDNA3.1. The plasmids pNLHX (HIV), pSTOP (HIVΔp6), pENX (GagΔp6) and pENX.p6 (Gag) were generous gifts from Dr. Paul Bieniasz (The Rockefeller University, New York, NY) (Martin-Serrano, Zang, and Bieniasz, 2001). To generate the expression plasmid for the WT and reverted pGagΔp6.SIVNef fusion proteins, the nef gene from SIV or pCR2.1SIVNef were amplified and cloned into the Eco RI and Xho I restriction sites of the pENX polylinker. The SIVΔNef infectious clone is described elsewhere (Sawai et al., 2000).
Protein purification, in vitro translation, and glutathione S-transferase (GST) pull-down assays
GST.Nef fusion proteins or the GST.Alix fusion protein were expressed in BL21 E. coli. Cells were lysed with a modified lysis buffer (50 mM HEPES pH 7.8, 100 mM KCl, 1% Triton X-100, 2 mM EDTA, 0.1 mM PMSF, and 1 mg/ml of lysozyme). Proteins were purified by affinity chromatography by using glutathione-Sepharose beads (GE Healthcare Bio-Sciences Corp.). The purity and integrity of GST fusion proteins were verified by Coomassie-blue staining after separation in 12% SDS-PAGE. SIVfsGagPR- and SIVp6*PR- or WT and mutant HIVNef and SIVNef proteins were transcribed and translated according to the manufacturer’s instructions, using the rabbit reticulocyte system (TNT, Promega Biotech, Madison, WI). Alix.HA and WT, mutant and reverted SIVNef.V5 proteins were produced in 293T cells. All expressed proteins were examined by SDS-PAGE followed by western blotting. For in vitro binding assays, immobilized GST or GST fusion proteins were incubated with approximately 0.5 μg of IVT translated or cell lysates of expressed proteins for 4 h at 4 °C in 1 ml of binding buffer (40 mM Tris-HCL pH7.5, 1% CHAPS and 100 mM NaCl). After incubation, beads were washed three times in the binding buffer and twice in binding buffer containing 500 mM NaCl, resuspended in Laemmli sample buffer, boiled and subjected to 12 % SDS-PAGE and western blotting or autoradiography.
Co-immunoprecipitations
To examine the binding between Nef and Alix, 293T cells were co-transfected with a proportion of 1:9 of Alix.HA and of WT, mutant or revertant Nef.V5 expression plasmids. 24 hours later, cells were lysed in HEPES/CHAPS buffer (50 mM Hepes pH 7.4; 1 % CHAPS; 150 mM NaCl) for 30 minutes on ice, cell lysates were collected, clarified by centrifugation and incubated for 4 h at 4 °C with anti-V5 antibody, followed by incubation with protein G-Sepharose (GE Healthcare Bio-Sciences Corp.) for 90 min at 4 °C. Immune complexes were then precipitated by centrifugation and washed three times in HEPES/CHAPS buffer containing 150 mM NaCl and two times in HEPES/CHAPS buffer containing 500 mM NaCl. Precipitates were resuspended in Laemmli sample buffer, boiled for 5 min, and subjected to 8 % SDS-PAGE, followed by western blotting with anti-HA antibodies. To examine the binding between SIVNef and SIVGagPol, 293T cells were co-transfected with a proportion of 1:3 of the SIVGag or SIVfsGagPolPR- and of the WT, mutant or revertant Nef.V5 expression plasmids. 24 hours later, cells were obtained as described above. Cell lysates were incubated for 4 h at 4 °C with anti-V5 antibodies and processed as above. Precipitates were resuspended in Laemmli sample buffer, boiled for 5 min, and subjected to 8 % SDS-PAGE, followed by western blotting with the monoclonal antibody against p27. Western blotting was performed with anti-V5 antibodies to determine levels of expression of Nef proteins.
Production of viral particles
To assess the effect of Nef on virion budding, 293T cells were transfected with the HIVΔp6 proviral DNA alone or co-transfected with the HIVΔp6 proviral DNA together with the GagΔp6, Gag or the GagΔp6.SIVNef fusion proteins in a 1:6 proportion. At 48 h post-transfection, cells and cell-free supernatants were harvested. Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris pH 7.2, 1 % Triton X-100, 0.1 % SDS) and analyzed by western blotting for p24 HIVGag. Culture supernatants were clarified by low-speed centrifugation and filtered through a 0.22 μm pore-size filter, followed by ultracentrifugation through a 20 % sucrose cushion at 44000 rpm for 1.5 h. Pellets were suspended in 1X PBS overnight at 4 °C. Viral particles were lysed in Laemmli sample buffer, and viral protein content was analyzed by western blotting for p24 HIVGag.
Trans-complementation assay
To assess the role of Nef proteins on SIV infectivity, 293T cells were transfected with the SIV or SIVΔNef proviral DNAs alone or co-transfected with SIVΔNef proviral DNA and WT, mutant or reverted SIVNef expression plasmids. 48 hours after transfection, cells and cell-free supernatants were harvested. Cells were lysed in RIPA buffer and analyzed by western blotting with anti-p27 SIVGag and anti-V5 antibodies. Culture supernatants were treated as above. Viral particles were lysed in Laemmli sample buffer, and viral protein content was analyzed by western blotting for p27 SIVGag.
Infectivity assay
Infectivity assays conducted in TZM-bl cells are described elsewhere (Derdeyn et al., 2000; Kimpton and Emerman, 1992; Platt et al., 1998; Wei et al., 2002). Since this cell line expresses the viral CD4 and CCR5 receptors, it is suitable for SIV infection. Briefly, assays were performed in 96-well plates. On day one, 2×105 cells were plated. The next day cells were infected with undiluted or serially diluted suspensions of viral stocks. 48 after infection cells were fixed and incubated with the X-Gal substrate. Blue nuclei were counted with a microscope and graphed. Each viral stock was always tested in triplicate.
Supplementary Material
Figure S1. Reverted but not mutant SIVNef proteins bind SIVGagPol. GST pull-downs were performed with GST or GST.SIVNef, GST.NefM8 and reverted GST.SIVNef proteins and fsSIVGagPR-. Lane 1 represents 10% input for fsSIVGagPR- (boxed). Lower panel presents inputs for GST and GST.Nef. Whereas reverted mutant SIVNef proteins from monkeys C and D at 81 weeks (GST.NefC81 and GST.NefD81, lanes 5 and 6) bound fsSIVGagPR-. GST.NefB53, which did not revert, did not interact with fsSIVGagPR-. Since NefD81 reverted only residues in the FL, we conclude that Nef binds GagPol via this motif. Proteins were separated by 10% SDS-PAGE followed by western blotting with anti-Myc antibodies. Inputs for fsSIVGagPR-, GST and GST.Nef are presented in top panel, lane1 and bottom panel, lanes 2 – 6.
Abbreviations
- FL
C-terminal flexible-loop
- ARM
armadillo repeat
- LD
Late Domain
- IVT
in vitro transcribed and translated
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beissinger M, Paulus C, Bayer P, Wolf H, Rosch P, Wagner R. Sequence-specific resonance assignments of the 1H-NMR spectra and structural characterization in solution of the HIV-1 transframe protein p6. Eur J Biochem. 1996;237(2):383–92. doi: 10.1111/j.1432-1033.1996.0383k.x. [DOI] [PubMed] [Google Scholar]
- Bentham M, Mazaleyrat S, Harris M. Role of myristoylation and N-terminal basic residues in membrane association of the human immunodeficiency virus type 1 Nef protein. J Gen Virol. 2006;87(Pt 3):563–71. doi: 10.1099/vir.0.81200-0. [DOI] [PubMed] [Google Scholar]
- Brenner M, Munch J, Schindler M, Wildum S, Stolte N, Stahl-Hennig C, Fuchs D, Matz-Rensing K, Franz M, Heeney J, Ten Haaft P, Swigut T, Hrecka K, Skowronski J, Kirchhoff F. Importance of the N-distal AP-2 binding element in Nef for simian immunodeficiency virus replication and pathogenicity in rhesus macaques. J Virol. 2006;80(9):4469–81. doi: 10.1128/JVI.80.9.4469-4481.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugger B, Krautkramer E, Tibroni N, Munte CE, Rauch S, Leibrecht I, Glass B, Breuer S, Geyer M, Krausslich HG, Kalbitzer HR, Wieland FT, Fackler OT. Human immunodeficiency virus type 1 Nef protein modulates the lipid composition of virions and host cell membrane microdomains. Retrovirology. 2007;4:70. doi: 10.1186/1742-4690-4-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti LA, Metzner KJ, Ivanovic T, Cheng H, Louis-Virelizier J, Connor RI, Cheng-Mayer C. A truncated form of Nef selected during pathogenic reversion of simian immunodeficiency virus SIVmac239Deltanef increases viral replication. J Virol. 2003;77(2):1245–56. doi: 10.1128/JVI.77.2.1245-1256.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa LJ, Chen N, Lopes A, Aguiar RS, Tanuri A, Plemenitas A, Peterlin BM. Interactions between Nef and AIP1 proliferate multivesicular bodies and facilitate egress of HIV-1. Retrovirology. 2006;3:33. doi: 10.1186/1742-4690-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa LJ, Zheng YH, Sabotic J, Mak J, Fackler OT, Peterlin BM. Nef binds p6* in GagPol during replication of human immunodeficiency virus type 1. J Virol. 2004;78(10):5311–23. doi: 10.1128/JVI.78.10.5311-5323.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel MD, Kirchhoff F, Czajak SC, Sehgal PK, Desrosiers RC. Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science. 1992;258(5090):1938–41. doi: 10.1126/science.1470917. [DOI] [PubMed] [Google Scholar]
- Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C, Lawson VA, Crowe S, Maerz A, Sonza S, Learmont J, Sullivan JS, Cunningham A, Dwyer D, Dowton D, Mills J. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science. 1995;270(5238):988–91. doi: 10.1126/science.270.5238.988. [DOI] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O’Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol. 2000;74(18):8358–67. doi: 10.1128/jvi.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RD, Chung HY, Zhai Q, Robinson H, Sundquist WI, Hill CP. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell. 2007;128(5):841–52. doi: 10.1016/j.cell.2007.01.035. [DOI] [PubMed] [Google Scholar]
- Geyer M, Fackler OT, Peterlin BM. Structure--function relationships in HIV-1 Nef. EMBO Rep. 2001;2(7):580–5. doi: 10.1093/embo-reports/kve141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer M, Fackler OT, Peterlin BM. Subunit H of the V-ATPase involved in endocytosis shows homology to beta-adaptins. Mol Biol Cell. 2002;13(6):2045–56. doi: 10.1091/mbc.02-02-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith MA, Warmerdam MT, Atchison RE, Miller MD, Greene WC. Dissociation of the CD4 downregulation and viral infectivity enhancement functions of human immunodeficiency virus type 1 Nef. J Virol. 1995;69(7):4112–21. doi: 10.1128/jvi.69.7.4112-4121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JR, Sutjipto S, Marx PA, Pedersen NC. Shared antigenic epitopes of the major core proteins of human and simian immunodeficiency virus isolates. J Med Primatol. 1992;21(5):265–9. [PubMed] [Google Scholar]
- Hill MK, Shehu-Xhilaga M, Crowe SM, Mak J. Proline residues within spacer peptide p1 are important for human immunodeficiency virus type 1 infectivity, protein processing, and genomic RNA dimer stability. J Virol. 2002;76(22):11245–53. doi: 10.1128/JVI.76.22.11245-11253.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Carl S, Bronson S, Stahl-Hennig C, Swigut T, Skowronski J, Kirchhoff F. Disrupting surfaces of nef required for downregulation of CD4 and for enhancement of virion infectivity attenuates simian immunodeficiency virus replication in vivo. J Virol. 2000;74(21):9836–44. doi: 10.1128/jvi.74.21.9836-9844.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminchik J, Margalit R, Yaish S, Drummer H, Amit B, Sarver N, Gorecki M, Panet A. Cellular distribution of HIV type 1 Nef protein: identification of domains in Nef required for association with membrane and detergent-insoluble cellular matrix. AIDS Res Hum Retroviruses. 1994;10(8):1003–10. doi: 10.1089/aid.1994.10.1003. [DOI] [PubMed] [Google Scholar]
- Kestler HW, 3rd, Ringler DJ, Mori K, Panicali DL, Sehgal PK, Daniel MD, Desrosiers RC. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell. 1991;65(4):651–62. doi: 10.1016/0092-8674(91)90097-i. [DOI] [PubMed] [Google Scholar]
- Kimpton J, Emerman M. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J Virol. 1992;66(4):2232–9. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff F, Easterbrook PJ, Douglas N, Troop M, Greenough TC, Weber J, Carl S, Sullivan JL, Daniels RS. Sequence variations in human immunodeficiency virus type 1 Nef are associated with different stages of disease. J Virol. 1999;73(7):5497–508. doi: 10.1128/jvi.73.7.5497-5508.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC. Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N Engl J Med. 1995;332(4):228–32. doi: 10.1056/NEJM199501263320405. [DOI] [PubMed] [Google Scholar]
- Leiherer A, Ludwig C, Wagner R. Influence of extended mutations of the HIV-1 transframe protein p6 on Nef-dependent viral replication and infectivity in vitro. Virology. 2009 doi: 10.1016/j.virol.2009.01.042. [DOI] [PubMed] [Google Scholar]
- Lock M, Greenberg ME, Iafrate AJ, Swigut T, Muench J, Kirchhoff F, Shohdy N, Skowronski J. Two elements target SIV Nef to the AP-2 clathrin adaptor complex, but only one is required for the induction of CD4 endocytosis. Embo J. 1999;18(10):2722–33. doi: 10.1093/emboj/18.10.2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig C, Leiherer A, Wagner R. Importance of protease cleavage sites within and flanking human immunodeficiency virus type 1 transframe protein p6* for spatiotemporal regulation of protease activation. J Virol. 2008;82(9):4573–84. doi: 10.1128/JVI.02353-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandic R, Fackler OT, Geyer M, Linnemann T, Zheng YH, Peterlin BM. Negative factor from SIV binds to the catalytic subunit of the V-ATPase to internalize CD4 and to increase viral infectivity. Mol Biol Cell. 2001;12(2):463–73. doi: 10.1091/mbc.12.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthas ML, Ramos RA, Lohman BL, Van Rompay KK, Unger RE, Miller CJ, Banapour B, Pedersen NC, Luciw PA. Viral determinants of simian immunodeficiency virus (SIV) virulence in rhesus macaques assessed by using attenuated and pathogenic molecular clones of SIVmac. J Virol. 1993;67(10):6047–55. doi: 10.1128/jvi.67.10.6047-6055.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J, Zang T, Bieniasz PD. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med. 2001;7(12):1313–9. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- Miller MD, Feinberg MB, Greene WC. The HIV-1 nef gene acts as a positive viral infectivity factor. Trends Microbiol. 1994;2(8):294–8. doi: 10.1016/0966-842x(94)90007-8. [DOI] [PubMed] [Google Scholar]
- Miller MD, Warmerdam MT, Gaston I, Greene WC, Feinberg MB. The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J Exp Med. 1994;179(1):101–13. doi: 10.1084/jem.179.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch J, Janardhan A, Stolte N, Stahl-Hennig C, Ten Haaft P, Heeney JL, Swigut T, Kirchhoff F, Skowronski J. T-cell receptor:CD3 down-regulation is a selected in vivo function of simian immunodeficiency virus Nef but is not sufficient for effective viral replication in rhesus macaques. J Virol. 2002;76(23):12360–4. doi: 10.1128/JVI.76.23.12360-12364.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch J, Rajan D, Schindler M, Specht A, Rucker E, Novembre FJ, Nerrienet E, Muller-Trutwin MC, Peeters M, Hahn BH, Kirchhoff F. Nef-mediated enhancement of virion infectivity and stimulation of viral replication are fundamental properties of primate lentiviruses. J Virol. 2007;81(24):13852–64. doi: 10.1128/JVI.00904-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandori MW, Fitch NJ, Craig HM, Richman DD, Spina CA, Guatelli JC. Producer-cell modification of human immunodeficiency virus type 1: Nef is a virion protein. J Virol. 1996;70(7):4283–90. doi: 10.1128/jvi.70.7.4283-4290.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72(4):2855–64. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saksela K, Cheng G, Baltimore D. Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down-regulation of CD4. Embo J. 1995;14(3):484–91. doi: 10.1002/j.1460-2075.1995.tb07024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandrin V, Cosset FL. Intracellular versus cell surface assembly of retroviral pseudotypes is determined by the cellular localization of the viral glycoprotein, its capacity to interact with Gag, and the expression of the Nef protein. J Biol Chem. 2006;281(1):528–42. doi: 10.1074/jbc.M506070200. [DOI] [PubMed] [Google Scholar]
- Sawai ET, Hamza MS, Ye M, Shaw KE, Luciw PA. Pathogenic conversion of live attenuated simian immunodeficiency virus vaccines is associated with expression of truncated Nef. J Virol. 2000;74(4):2038–45. doi: 10.1128/jvi.74.4.2038-2045.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavoni I, Trapp S, Santarcangelo AC, Piacentini V, Pugliese K, Baur A, Federico M. HIV-1 Nef enhances both membrane expression and virion incorporation of Env products. A model for the Nef-dependent increase of HIV-1 infectivity. J Biol Chem. 2004;279(22):22996–3006. doi: 10.1074/jbc.M312453200. [DOI] [PubMed] [Google Scholar]
- Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. 1996;2(3):338–42. doi: 10.1038/nm0396-338. [DOI] [PubMed] [Google Scholar]
- Shacklett BL, Shaw KE, Adamson LA, Wilkens DT, Cox CA, Montefiori DC, Gardner MB, Sonigo P, Luciw PA. Live, attenuated simian immunodeficiency virus SIVmac-M4, with point mutations in the Env transmembrane protein intracytoplasmic domain, provides partial protection from mucosal challenge with pathogenic SIVmac251. J Virol. 2002;76(22):11365–78. doi: 10.1128/JVI.76.22.11365-11378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack B, Calistri A, Craig S, Popova E, Gottlinger HG. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114(6):689–99. doi: 10.1016/s0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- Stumptner-Cuvelette P, Morchoisne S, Dugast M, Le Gall S, Raposo G, Schwartz O, Benaroch P. HIV-1 Nef impairs MHC class II antigen presentation and surface expression. Proc Natl Acad Sci U S A. 2001;98(21):12144–9. doi: 10.1073/pnas.221256498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner R, Graf M, Bieler K, Wolf H, Grunwald T, Foley P, Uberla K. Rev-independent expression of synthetic gag-pol genes of human immunodeficiency virus type 1 and simian immunodeficiency virus: implications for the safety of lentiviral vectors. Hum Gene Ther. 2000;11(17):2403–13. doi: 10.1089/104303400750038507. [DOI] [PubMed] [Google Scholar]
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. 2002;46(6):1896–905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker R, Kottler H, Kalbitzer HR, Krausslich HG. Human immunodeficiency virus type 1 Nef protein is incorporated into virus particles and specifically cleaved by the viral proteinase. Virology. 1996;219(1):228–36. doi: 10.1006/viro.1996.0240. [DOI] [PubMed] [Google Scholar]
- Zhai Q, Fisher RD, Chung HY, Myszka DG, Sundquist WI, Hill CP. Structural and functional studies of ALIX interactions with YPX(n)L late domains of HIV-1 and EIAV. Nat Struct Mol Biol. 2008;15(1):43–9. doi: 10.1038/nsmb1319. [DOI] [PubMed] [Google Scholar]
- Zheng YH, Plemenitas A, Fielding CJ, Peterlin BM. Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV-1 progeny virions. Proc Natl Acad Sci U S A. 2003;100(14):8460–5. doi: 10.1073/pnas.1437453100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Yuan X, Dismuke D, Forshey BM, Lundquist C, Lee KH, Aiken C, Chen CH. Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J Virol. 2004;78(2):922–9. doi: 10.1128/JVI.78.2.922-929.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Reverted but not mutant SIVNef proteins bind SIVGagPol. GST pull-downs were performed with GST or GST.SIVNef, GST.NefM8 and reverted GST.SIVNef proteins and fsSIVGagPR-. Lane 1 represents 10% input for fsSIVGagPR- (boxed). Lower panel presents inputs for GST and GST.Nef. Whereas reverted mutant SIVNef proteins from monkeys C and D at 81 weeks (GST.NefC81 and GST.NefD81, lanes 5 and 6) bound fsSIVGagPR-. GST.NefB53, which did not revert, did not interact with fsSIVGagPR-. Since NefD81 reverted only residues in the FL, we conclude that Nef binds GagPol via this motif. Proteins were separated by 10% SDS-PAGE followed by western blotting with anti-Myc antibodies. Inputs for fsSIVGagPR-, GST and GST.Nef are presented in top panel, lane1 and bottom panel, lanes 2 – 6.