

Abstract
Carrier proteins are central to the biosynthesis of primary and secondary metabolites in all organisms. Here we describe metabolic labeling and manipulation of native acyl carrier proteins in both type I and II fatty acid synthases. Utilizing natural promiscuity in the CoA biosynthetic pathway in combination with synthetic pantetheine analogues, we demonstrate metabolic labeling of endogenous carrier proteins with reporter tags in Gram-positive and Gram-negative bacteria and in a human carcinoma cell line. The highly specific nature of the posttranslational modification utilized for tagging allows for simple visualization of labeled carrier proteins, either by direct fluorescence imaging or after chemical conjugation to a fluorescent reporter. In addition, we demonstrate the utility of this approach for the isolation and enrichment of carrier proteins by affinity purification. Finally, we use these techniques to identify a carrier protein from an unsequenced organism, a finding which validates this proteomic approach to natural product biosynthetic enzyme discovery.
Keywords: proteomic, fatty acid, natural product, carrier protein, protein labeling
Introduction
The carrier protein (CP) is central to the biosynthetic pathways of many important primary and secondary metabolites.[1] These small proteins, found either as discrete polypeptides or as small domains within larger synthases, facilitate biosynthesis by acting as a scaffold for growing natural products. Due to their key role in the biosynthesis of fatty acids,[2] polyketides,[3] and nonribosomal peptides,[4] methods for the specific labeling of CPs in vivo would be useful in order to visualize the localization and dynamics of these biosynthetic lynchpins, as well as to facilitate the proteomic identification of CPs from unsequenced natural product producing organisms.
In CP-mediated biosynthesis, the growing acyl-chain is linked to the enzyme through a thioester bond to the terminal thiol of a coenzyme A (CoA) derived 4′-phosphopantetheine (4′-PPant) prosthetic group. This posttranslational modification is introduced by a phosphopantetheinyltransferase (PPTase, E.C. 2.7.7.7), which transfers the moiety from CoA to a conserved serine of the apo-CP.[5] The finding that many PPTases are capable of accepting functionalized CoA analogues as substrates offers an opportunity for selective labeling of CP domains with fluorescence and affinity reporters.[6, 7] However, in order for this approach to succeed, reporter-labeled CoA analogues must be available to the PPTase, or else the PPTase will utilize endogenous CoA to produce holo-CPs which are not easily visualized or detected. Due to the highly charged nature of CoA and its analogues, which precludes their use in intracellular labeling approaches,[8] our previous work has focused on studying the ability of reporter-labeled CoA precursors to cross the cell membrane and be transformed into fully formed CoA analogues in vivo via the endogenous CoA biosynthetic pathway (Figure 1).[9, 10] Past studies have applied similar metabolic delivery strategies to the labeling of posttranslationally lipidated and glycosylated proteins.[11–13] However, while our earlier studies were essential to the identification of potentially useful CoA precursors, this work was only explored in the context of a single Gram-negative bacterial organism, Escherichia coli, and only proved capable of labeling CPs when they were heterologously co-overexpressed with a promiscuous PPTase.
Figure 1.

In vivo labeling strategy. Cells are grown in the presence of azido-pantetheine (1) or fluorescent pantetheine (2). After uptake, the native CoA biosynthetic enzymes convert the pantetheine analogues to CoA analogues which then are appended to endogneous CPs via the native PPTase enzyme. After cell lysis azido-modified CPs can be detected by reaction with alkyne probes (3–5).
Here we report a significant advance in CP-labeling methodology, describing for the first time the labeling of endogenous CP domains in native bacterial systems by azide-labeled CoA precursor 1. We demonstrate its utility in both Gram-positive and Gram-negative bacterium, as well as in a number of knockout strains which probe its requirements for uptake and PPTase type. We also explore the activity of this analogue in a human carcinoma cell line, and perform an in-depth analysis of labeling of the human FAS in this cell line by 2, a previously reported fluorescent CoA precursor. Finally, to evaluate the utility of this method for proteomic identification of CP-containing enzymes from unsequenced organisms, we demonstrate its application towards affinity purification of CPs, as well as for the de novo sequencing and genetic identification of a fatty acid biosynthetic CP from an unsequenced bacterium.
Results
In Vivo Labeling of Native ACP
In our previous studies, pantetheine analogues 1 and 2 demonstrated cellular uptake and processing by the endogenous E. coli CoA biosynthetic enzymes pantothenate kinase (PanK, E.C. 2.7.1.33), phosphopantothenoyl adenyltransferase (PPAT, E.C. 2.7.7.3), and dephospho-CoA kinase (DPCK, E.C. 2.7.1.24) to form reporter-labeled CoA in vivo.[9, 10] The formation of these CoA analogues was demonstrated by their ability to undergo reaction with a heterologously expressed CP and PPTase, resulting in labeling of the overexpressed CP with a fluorescence or affinity tag. While valuable for method development, the heterologous PPTase and CP were present within the cells at levels far exceeding those of endogenous proteins. To explore the utility of this method for the metabolic labeling of endogenous CPs in a native organism, we directed our efforts at the fatty acid ACP of E. coli. Fatty acid and CoA metabolism are well understood in this organism, and our previous reports indicated that the CoA biosynthetic pathway in BL-21 strains of E. coli is permissive enough to allow for the in vivo conversion of pantetheine analogues 1 and 2 to CoA analogues.
Using the wild-type K12 strain of E. coli as a model system, we set out to demonstrate in vivo modification of the native fatty acid CP AcpP. Overnight growth with 1 mM compound 1 in LB media followed by cell lysis and copper-catalyzed cycloaddition reaction with fluorescent alkyne (3 or 4) allowed visualization of the modified CP via SDS-PAGE (Figure 2). Fluorescent pantetheine 2 showed no detectable labeling of native E. coli CPs under identical growth conditions. In addition, control cultures grown with vehicle DMSO showed no similar fluorescence after incubation with 3 or 4 under cycloaddition reaction conditions. The protein modified by 1 was found to run at the same apparent molecular weight as recombinantly purified and labeled AcpP on both denaturing and native PAGE gels, a compelling finding given that AcpP is readily separated from most other proteins on high percentage native gels due to its small size and charge.[14] In order to definitively identify the labeled protein, the fluorescent band was excised, proteolytically digested, and analyzed by LC-MS/MS. The results confirmed that the labeled band was indeed the AcpP protein from E. coli (Supplementary Figure S1).
Figure 2.

Detection of in vivo modified carrier proteins. (A) Cultures were grown with or without compound 1 and reacted with RRX-alkyne 4 after lysis. Distinct CP labeling is seen in E. coli (1), B subtilis 168 (2), B. subtilis 6051 (3), and S. oneidensis (4) as compared to negative controls (B) Labeling of E. coli ACP by 1 with visualization by DMAC-alkyne 3 was effective in both LB (lane 1) and M9 minimal media (lane 2) as compared to negative controls (lanes 2,4).
Since a traditional obstacle to the use of in vivo labeling techniques has been the ability of the small molecule to penetrate the cell, we also examined in detail the method of uptake of 1 by E. coli. While E. coli are capable of synthesizing pantothenate de novo, they also express a pantothenate transport system. The panF gene encodes pantothenate permease, a sodium dependant panothenate symporter.[15] If implicated in the uptake 1, PanF activity could represent a caveat to the general applicability of this labeling method to organisms lacking PanF homologues. In order to decouple this dedicated pantothenate uptake system from the metabolic labeling protocol, we performed metabolic labeling in E. coli strains containing an in frame deletion in the panF gene to knock-out activity.[16] The result of these labeling experiments were qualitatively indistinguishable from those performed in wild type E. coli (Supplementary Figure S6), suggesting that pantetheine analogues such as 1 enter E. coli by passive diffusion or an alternate uptake mechanism. Additionally, strains of E. coli with pantothenate biosynthetic genes panD and panC knocked out showed no difference in CP-labeling by 1 relative to wild type E. coli, indicating the presence or absence of active pantothenate biosynthesis has no effect on labeling efficiency.
Finally we examined the toxicity of metabolic labeling by 1 to growing E. coli. An ideal metabolic label would allow for the study of CPs in live organisms with minimal perturbation to their natural growth and metabolism. However, past studies have associated the in vivo modification of the fatty acid acyl carrier protein (ACP) by CoA analogues with growth inhibition in a number of bacteria, including E. coli.[17, 18] To examine the toxicity of the metabolic labeling procedure we determined minimum inhibitory concentrations for 1, 2, and N5-Pan, a prototypical bacteriostatic pantetheine analogue (see Figure S15 for structure), towards E. coli grown in nutrient rich and minimal media. As opposed to N5-Pan, which inhibits E. coli growth in minimal media at 30 μM, the compounds used in this study (1, 2) showed no growth inhibition of E. coli at concentrations up to 500 μM. This lack of growth inhibition was not due to a lack of ACP-modification under these conditions, as growth of E. coli supplemented with 1 in minimal media showed modification of ACP at similar levels to those observed in LB (Figure 2B). To reconcile the non-toxic ACP-labeling of 1 with the proposed mode of action of bacteriostatic pantetheine analogues, we have since performed a full study of the effects secondary structural characteristics have on the toxicity of CoA precursors.[19] These findings indicated that the more polar reporter-labels incorporated in 1 and 2 may result in decreased toxicity relative to N5-Pan modified ACP due to an inability of ACPs modified by 1 and 2 to interact with partner enzymes of the fatty acid biosynthetic pathway, an optimal property for non-toxic, in vivo labeling of CPs.
In Vivo Modification of ACPs from Other Bacteria
Having demonstrated in vivo CP modification in wild-type E. coli, we sought to next test the generality of this methodology for labeling of CP domains in other bacterial species whose CoA and fatty acid biosynthetic pathways have been less well-studied (Figure 2). Accordingly Bacillus subtilis 6051 and Shewanella oneidensis MR-1, and Bacillus brevis 8246 were grown with 1 mM pantetheine azide 1 overnight. Lysis, followed by chemical conjugation with fluorescent alkyne 4 allowed low molecular weight proteins corresponding to the fatty acid ACPs in each of these strains to be visualized by SDS-PAGE (Figure 2A and 5). Once again, this tentative identification was verified by band excision and LC-MS/MS analysis. Database searching allowed identification of the fatty acid ACPs from B. subtilis and S. oneidensis. This demonstrates 1 possesses properties compatible with cellular uptake and processing by the CoA biosynthetic pathways in both Gram positive and Gram negative bacterial species, which together constitute a large number of known natural product producing organisms.
Figure 5.
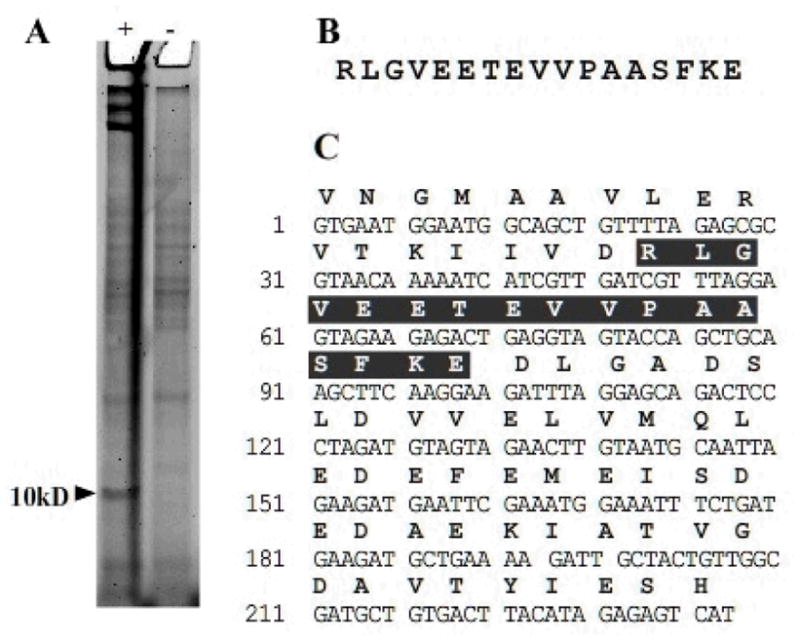
Identification of a carrier protein from and unsequenced organism. (A) Metaboilc labeling of B. brevis 8246 by 1 gives a band around 10 kDa not present in the negative control. (B) Tryptic MS analysis of this band using a database of 200 known bacterial ACP sequences identified a peptide fragment matching an ACP sequence. (C) Degenerate and arbitrary PCR allowed for sequencing of the gene encoding this peptide and identification as the fatty acid ACP.
In addition to demonstrating metabolic labeling of the fatty acid ACPs from these organisms, we explored in further detail the biosynthetic capabilities necessary for transfer of the 4′-phosphopantetheine arm from CoA to the conserved serine residue of ACP by a PPTase (Figure 1). There are two classes of bacterial PPTase enzymes: the AcpS type, associated with primary metabolism and necessary for modification of the fatty acid ACP; and the Sfp type, associated with secondary metabolism and involved in the modification of CPs in NRPS and PKS systems.[6, 20, 21] While some cross-reactivity exists, AcpS type PPTases are less permissive of variation in CP or CoA analogue identity.[22] Conversely, the Sfp type shows relaxed specificity in terms of CP or CoA identity.[6] We had previously observed CP labeling in B. subtilis 6051, which carries a functional copy of Sfp. To ensure that CP labeling by 1 would not be hindered at the last step in organisms lacking a Sfp type PPTase we also tested this method in B. subtilis strain 168, which contains an in frame deletion in its Sfp gene and therefore contains only the less permissive AcpS type PPTase. Metabolic labeling by 1 followed by chemoselective ligation to fluorescent alkyne 4 demonstrated labeling at similar levels to those observed using strain 6051 (Figure 2A). This indicates the azido-CoA analogue formed from 1 is compatible with less promiscuous AcpS type PPTases, a desired feature for the labeling of CPs from disparate biosynthetic systems.
Labeling of Human Type I FAS in Living Cells
Fatty acid, polyketide, and nonribosomal peptide synthases can be classified as type I or II depending on whether the active domains required for product biosynthesis are located on discrete polypeptides (type II) or are housed on multimodular megasynthases (type I).[1] Bacterial fatty acid synthases are of type II and as such have discrete active domains, including the ACP. In animals, most fatty acids are biosynthesized by type I synthases. Our success labeling ACPs of type II fatty acid biosynthesis in bacterial organisms with 1 lead us to test whether this approach would also be feasible in eukaryotic cells, particularly in the labeling of type I fatty acid ACP domains.
We chose as a model system the SKBR3 cell line, which is derived from human breast cancer cells.[23] SKBR3 cells were plated at 25% confluency and allowed to grow for 24 hours at 37 °C before introduction of pantetheine analogue. Incubation of the cells with compound 1 for up to 60 hours did not result in detectable labeling of the FAS after cell lysis and reaction with fluorescent alkyne 4. This was surprising, as we had expected labeling in eukaryotic cells to be enhanced compared to bacteria, since mammalian cells cannot produce endogenous pantothenate.[24] The hypothesis that this difference could be due to differential pantothenate import mechanisms in eukaryotic and prokaryotic cells lead us to test another pantetheine analogue which had shown good compatibility with the CoA biosynthetic pathway in other studies, fluorescent pantetheine 2.[25, 26] Interestingly, this compound, which had proven ineffective in modifying fatty acid ACPs from endogenous bacterial systems, effectively labeled the type I FAS in human cells (Figure 3A). Growth of SKBR3 cells with 2 for between 40 and 60 hours resulted in visibly bright staining of the cells, and an increased amount of observable cell death compared to incubation with 1 or DMSO. In order to quantify the level of uptake, cell cultures grown with 2 or vehicle DMSO were subjected to analysis by flow cytometry (FACS, Figure 3B). Of cells grown with 2, 100% of the cells could be identified via FACS as containing significant fluorescence. Lysis of the cells and visualization of the fluorescent labeled FAS via SDS-PAGE gel confirmed labeling of the FAS protein (Figure 3A). However, this labeling was relatively modest compared to expectations based on the qualitative observation of strong uptake. In order to reconcile these results, lysate from the cells was examined by HPLC along with chemoenzymatically prepared standards of the four possible intermediates in the processing of pantetheine 2 by the CoA biosynthetic pathway. The major fluorescent peak found in SKBR3 lysate was observed to co-elute with the product of 2 and PanK, leading us to identify it as the singly processed phosphopantetheine analogue. (Figure 3C) This is consistent with previous reports on the processing of pantetheine prodrugs by human cells, which indicated the bifunctional PPAT/DPCK as the major point of blockage for formation of CoA analogues in vivo.[27] While we have previously shown that recombinant human PPAT/DPCK can be used to convert fluorescent pantetheine analogues into CoA analogues for CP labeling in vitro,[26] in living cells these enzymes are most likely present at far lower levels and only process the most efficient 4′-phosphopantetheine substrates. Additionally the human FAS is cytosolic, while CoA is known to be sequestered at its highest intracellular levels in the mitochondria, making localization of the CoA analogue of 2 another complicating factor.[28] Efforts are currently ongoing to develop an analogue that shows uptake similar to 2 and demonstrates improved compatibility with the human CoA biosynthetic enzymes.
Figure 3.

Analysis of type I FAS ACP labeling in SKBR3 cells (A) In vivo modification of the human type I FAS. Lysate from cultures grown with compound 2 (+) show fluorescent modification of the FAS megasynthase as compared to negative controls (left). Blot with anti-FAS antibody confirms the presence and location of FAS on the gel (right). (B) FACS analysis of SKBR3 cells grown with compound 2 shows 99% of the SKBR3 cells grown with 2 took up the compound. (C) (panel 1) Compound 2 (black) incubated with PanK + ATP for 15 minutes results in addition of a phosphate on the 4′ hydroxyl and a shift to lower retention time (grey). (panel 2) After 120 minutes the reaction is complete, with all of 2 converted to the phospho-analogue. (panel 3) Lysate from SKBR3 cells grown with 2 contains mostly phospo-analogue. No further conversion to the CoA analogue in the SKBR3 cells could be detected (Supplementary Info).
Affinity Purification of a Metabolically Labeled Bacterial Carrier Protein
One of the major goals of this research has been to isolate and identify new modular biosynthetic enzymes with these techniques. The studies described above had shown that metabolic labeling in genetically unmodified cultures by 1 or 2 allowed for the visualization and identification of fatty acid ACPs from human, E. coli, B. subtilis, S. oneidensis, and B. brevis lysates. Here, labeled CPs from sequenced organisms were readily identified by proteolytic digest and MS analysis of excised gel bands, followed by comparison of the observed peptides to genomic databases. However, these analyses also commonly identified background proteins which migrated at the same molecular weight on SDS-PAGE. In unsequenced or unannotated organisms, this phenomenon could complicate de novo sequencing efforts and hinder identification of novel synthases by diluting the pool of labeled CP peptides or identifying false positives based on sequence homology. In order to remove contaminating proteins and also to verify the specificity of CP modification by metabolic labeling, we tested the utility of a biotin-streptavidin affinity purification method to isolate modified fatty acid ACPs.
This technique was demonstrated using S. oneidensis MR-1, a bacterial strain noted for its production of polyunsaturated fatty acid natural products.[29] The finding that metabolic labeling by 1 is not toxic to growing bacteria suggested to us that it modifies only a fraction of the total cellular ACP content, since it allows for at least a basal level of vital cellular processes such as fatty acid biosynthesis. Therefore in order to facilitate the recovery of labeled CPs we performed the metabolic labeling on a large scale using one-liter cultures. S. oneidensis MR-1 was grown overnight in the presence of 1 mM pantetheine analogue 1, pelleted, washed, and lysed to yield azide-labeled CPs within crude cell lysate. The lysate was then subjected to copper-catalyzed [3+2] cycloaddition reaction with the biotin alkyne 5. After removal of excess 5 by desalting column, followed by incubation with streptavidin agarose beads, the affinity purified proteins were recovered by the addition of SDS-PAGE running buffer and boiling. Figure 4 shows the results of enrichment of lysate from S. oneidensis metabolically labeled with 1 (lanes 1–6) in contrast to a DMSO treated control (7–9). Comparison by SDS-PAGE shows enrichment of an approximately 10 kDa protein from the lysate of MR-1 grown with 1 and reacted with biotin 5 (lane 3). This protein is not enriched when alkyne 5 is not added (lane 6), and is also not present in non-metabolically labeled, DMSO-treated cultures of S. oneidensis (lane 9). This indicates that the enriched band does not result from endogenous biotinylation or non-specific reaction of biotin-probe 5 in cell lysates. To our satisfaction, excision, tryptic digest, and MS analysis of this band resulted in the identification of the fatty acid ACP. This experiment shows that in vivo CP labeling techniques can be used to specifically recover labeled CPs from their native systems.
Figure 4.
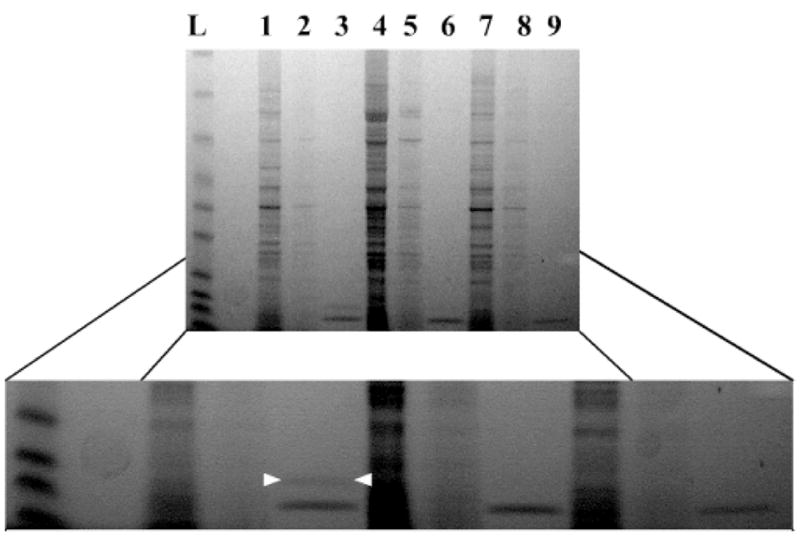
Affinity purification of ACP. Lysate from cultures of S. oneidensis MR1 grown with compound 1 and then treated with biotin-alkyne 5 were incubated with streptavidin agarose allowing for purification of modified CP (crude lysate lane 1, desaltting column flow lane 2, recovered ACP band—between white arrows—10 kDa lane 3). Cultures grown with compound 1 but not reacted with biotin-alkyne 5 (lanes 4,5,6) or grown without compound 1 (lanes 7,8,9) showed no protein enrichment after incubation with streptavidin agarose. The strong band below ACP is a contaminate from the resin and was seen under all elution condtions.
Identification of a Carrier Protein from an Unsequenced Organism
B. brevis is a Gram positive bacterium responsible for the production of the natural products gramicidin and tyrocidine.[30] We chose the non-producing strain (ATCC 8246) of this unsequenced organism to test whether this chemical proteomic approach was capable of identifying CPs without the aid of genomic sequence data. As shown above with S. oneidensis, native CPs labeled by 1 and 2 can be reliably identified throughdatabase searches of known genomes after tryptic digest and MS. The ability to extend this methodology to the large number of unsequenced natural product producing organisms would further validate this approach for studying CP-based natural product biosynthetic enzymes.
Accordingly, B. brevis 8246 was grown overnight in the presence or absence of compound 1. The cultures were lysed and prepared as in the other in vivo labeling experiments, followed by reaction with cognate fluorescent alkyne 4. As can be seen in Figure 5, B. brevis 8246 cultures grown in the presence of 1 show distinct labeling of a protein with an apparent MW of 15 kDa as compared to control cultures. Although this organism is unsequenced, many other Bacillus species have been well studied, and these lead us to the expectation that this labeled protein was the type II fatty acid ACP, as we had seen in previous work with B. subtilis. In order to verify this hypothesis, we again excised the fluorescent band and submitted it to MS/MS analysis (Figure 5B). Since no database was available for this unsequenced organism, we constructed a data set comprising all known bacterial fatty acid ACPs against which to search. Searching the observed peptide fragments against this database returned a single hit matching a tryptic fragment from the ACP in B. cereus. Because of the small size of type II fatty acid ACPs, only a small number of tryptic peptides are generated on proteolytic digest, and a single matching peptide represents a significant percentage of sequence coverage.
To complete the identification of the CP from B. brevis 8246 we constructed a set of degenerate primers from the identified peptide. Using degenerate and arbitrary PCR techniques, we were able to generate PCR fragments from genomic DNA [31]. Sequencing confirmed that we had amplified the ACP gene from 8246. As seen in Figure 6, the sequence is nearly identical to the ACP gene from B. cereus and has high homology to the ACP genes from B. thuringenes and B. anthrasis. It differs considerably from the sequence for B. subtilis ACP. The taxonomy of B. brevis has been reclassified a number of times, but this result concurs with recent reports indicating B. brevis and B. cereus are closely related.[32] While this analysis relied on the relatively good sequence homology among small CP domains, the application of this method in combination with affinity tags targeting alternate active sites on PKS and NRPS multienzymes[33] should facilitate de novo sequencing of a variety CP-containing systems from unsequenced organisms.
Figure 6.

Alignment of Bacillus ACP sequences. Alignment of the new ACP sequence from B. brevis shows high homology with known Bacillus ACP sequences.
DISCUSSION
Previous attempts to label CPs in vivo with functionally useful labels have been hindered by the inability of charged CoA analogues to permeate the cell membrane. In our prior studies we addressed this issue by developing uncharged pantetheine analogues and studying the ability of the native CoA biosynthetic pathway to convert them into reporter-labeled CoA analogues in living cells.[25, 34] While these investigations provided insight into the structural features necessary for in vivo production of CoA analogues from synthetic pantetheine analogues, all of these experiments were conducted with heterologously expressed CPs and PPTases, in a single Gram negative bacteria, E. coli. Here we have extended this technique to metabolically deliver CoA analogues to apo-CPs at native levels. Using this strategy, we have modified CPs from both Gram positive and Gram negative bacteria. The modified type II fatty acid ACPs in these organisms are detectable after cell lysis by bioconjugation of fluorescently tagged alkynes with azido-pantetheine 1. After visualization by PAGE, these labeled proteins can be readily identified from LC-MS/MS analysis of excised gel fragments. Alternatively, cell lysate from organisms grown with 1 can be subjected to copper-catalyzed reaction with biotin alkyne 5 and their CPs isolated. The metabolic incorporation of 1 shows minimal toxicity and is compatible with CoA and PPTase pathways from a variety of different natural product producing organisms. This new ability to selectively label and affinity purify endogenous CPs from native organisms should allow for the discovery of novel CPs as well as their associated biosynthetic systems.
We have also demonstrated the metabolic labeling of an ACP from a type I FAS. This indicates our methodology is also applicable to type I modular biosynthetic systems and could provide a means for isolation and identification of the analogous type I PKS and NRPS megasynthases, which are responsible for the production of diverse natural products. Interestingly, while 1 was effective in modifying type II fatty acid ACPs in prokaryotes, it did not appear to be efficiently processed by the same pathway in the human SKBR3 cell line. Our previous work has demonstrated that turnover of an analogue in both the CoA biosynthetic pathway and the phosphopantetheinylation reaction can be greatly influenced by small changes in the structure of the particular analogue, and it has been observed previously that the human PanK isoform shows a very low sequence homology to those used by many prokaryotes.[35] One explanation for the extremely robust uptake of fluorescent pantetheine 2, as observed by FACS and HPLC analysis, could be that 2 possesses greater activity with the human PanK enzyme than 1. Efficient processing of 2 by the PanK enzyme would lead to production of a phosphorylated, cell impermeable intermediate. As there are no currently annotated import or export mechanisms for pantetheine analogues in human cells, continued passive diffusion of the compound into the human cells could result in increasing accumulation of this cell impermeable intermediate, a finding consistent with the increased cell death observed in cells grown with 2 for longer periods of time. With such a concentration mechanism in place, it seems credible that despite its low turnover by the PPAT/DPCK enzyme, a small amount of 2 is converted to CoA and shuttled to the ACP by the human PPTase, leading to the observed modest labeling of human FAS. This preliminary observation of the differential activity of pantetheine analogues 1 and 2 in bacteria and humans could eventually prove useful for the specific tuning of the CP labeling method to different organisms, or for the in vivo labeling of specific CPs in complex mixtures of organisms. With further refinement this technique may prove practical for the study of symbiotic natural product producing communities, by allowing for time-dependent CP-labeling in an organism specific fashion.[36]
In addition to possessing compatibility with several different biosynthetic enzymes, a useful metabolic label must also compete with the natural substrate without inducing cell death. In this study pantetheine analogues 1 and 2 have been shown to be compatible with three CoA biosynthetic enzymes and the CP posttranslational modification process in a full spectrum of prokaryotic and eukaryotic organisms, including Gram positive and Gram negative prokaryotes and eukaryotes. These analogues show model properties in terms of their ability to successfully compete with the natural substrates for the CoA biosynthetic enzymes and PPTases without any genetically or media-based advantages. One disadvantage of this technique from the perspective of its use in proteomic identification of CPs is that this competition likely results in only intermediate to low-level labeling of CPs by our unnatural CoA precursors. However, metabolic labeling of CPs offers an advantage compared to genetic methods (RT-PCR) or post-lysis protein identification techniques in its ability to detect proteins throughout all stages of growth, rather than representing a single temporal snapshot of the dynamic proteome.[33] This characteristic should prove valuable in study of the synthesis and degradation of biosynthetic enzymes.
Finally, the use of native metabolic pathways as a means of in vivo modification of CPs has enabled the identification of a previously unknown CP. Especially in systems with large or unsequenced genomes, a protein-based approach allows for a directed search for CP dependent biosynthetic pathways without the use of laborious techniques such as genomic library construction and screening. No genetic information is required, and complications arising from silent biosynthetic gene clusters are removed. Furthermore, since this method depends only on the activity of a protein to be phosphopantetheinylated, it should hold no obvious bias against CP containing systems which are not readily identifiable by homology alone, including those belonging to orphan gene clusters.[37] We have initially developed this methodology using type I and II fatty acid biosynthetic CPs which are constitutively expressed in growing cultures. In these studies no effort was made to correlate polyketide or nonribosomal peptide production with CP labeling by 1 or 2. However, the demonstrated compatibility of this method with uptake, CoA biosynthesis, and PPTase activity in a wide range of organisms should allow for its similar application to PKS and NRPS producing organisms when correlated with small-molecule production or antibiotic activity. In the future, we believe that in vivo labeling of native CPs should prove highly complementary to existing methods and add greatly to the knowledge of natural product producing synthases.
Conclusion
A significant number of these biologically relevant natural products are produced in systems that tether the growing product to a 4′-phosphopantetheine modified CP. Selective labeling of these enzymes could allow for a better understanding of these proteins in their native environments and provide a platform for discovery of natural product synthases from unsequenced organisms. Here we have demonstrated a new set of tools for investigation and discovery of natural product producing systems. Using native metabolic pathways as a means of in vivo modification of endogenous CPs has allowed for the identification of a previously unknown CP. Because CPs and CP domains are found in fatty acid, polyketide, nonribosomal peptide, and other metabolite biosynthetic pathways, the ability to specifically modify these peptides in vivo allows probing of numerous biochemical pathways. We have demonstrated for the first time the modification of native CPs by reporter-labeled CoA precursors in both prokaryotic and eukaryotic organisms, giving access to the full range of CP dependent systems.
Experimental Section
EXPERIMENTAL PROCEDURES
Primers used in this study
ARB1 5′-GGCCACGCGTCGACTAGTACNNNNNNNNNNGATAT -’3
ARB2 5′-GGCCACGCGTCGACTAGTAC -’3
ARB6 5′-GGCCACGCGTCGACTAGTACNNNNNNNNNNACGCC -’3
Internal Frag 5′-CGCCTGGGCGTAGAAGAAACGGAA -’3
External Frag 5′-CGCCTGGGCGTAGAAGAAACGGAA -’3
B. brevis acpp F 5′-ATGGAATGGCAGCTGTTTTAGAGCGCGT -’3
B. brevis acpp R 5′ TAGATGACTCTCTATGTAAGTCACAGCATCGC -’3
In primers ARB1 and ARB6 N is an equalmolar mix of A/T/G/C bases.
Media
Luria Broth (LB) was purchased from Fisher Scientific (Waltham, MA). M9 was made according to the standard procedure.[42] Rich media (RM) contained 1% bactopeptone, 1% beef extract, 0.25% NaCl. McCoy’s 5a Media (Invitrogen) was modified to contain a final concentration of 10% fetal bovine serum. E. coli knockout strains were grown in LB.
In Vivo Carrier Protein Labeling
Bacteria were grown in the media noted above with or without 1 (1 mM) for 12–17 hrs in an orbital shaker at 37°C. Cells were harvested by centrifugation at 20,000 rpm for 5 minutes, and resuspended in lysis buffer (100 mM NaCl, 25 mM potassium phosphate pH 7.0). For small cultures (<10 mL), cells were lysed by addition of 3 mg/mL lysozyme (Worthington Biochemical Corporation, Lakewood NJ) incubation at 25°C for one hour and sonication (3 × 30s with a microtip at low power). For large cultures (>10 mL) cells were resuspended in 10 mL of lysis buffer per liter of culture with 0.1 mg/mL lysozyme. After one hour incubation with shaking on ice, cells were lysed in by two passes through a French pressure cell. Lystate was then subjected to reaction with fluorescent alkyne 3 or 4 as previously reported. [34, 43]
Determination of Kinetic Parameters of Pantetheine Analogs with PanK
Analogs were assayed for turnover with E. coli PanK in a coupled enzyme assay as previously described. [34]
Identification of B. brevis ACP
Cultures of B. brevis were grown in the presence of 1 as described above. MS identification of a labeled protein gave a single peptide matching the B. cereus AcpP protein. Using this sequence primers were designed(ARB1,2,6, Internal Frag, External Frag) and arbitrary PCR was carried out following the procedure of Caetano-Anolles.[31] Amplified bands were ligated into TA vectors (Invitrogen) and sent for sequencing. The resulting sequence allowed for the design of specific primers and the cloning of the full acpp gene (B. brevis F & R). (Table 1)
Table 1.
Strains and Cell Lines Used in this Study
Tissue Culture
SKBR3 human breast cancer cells were grown in McCoy’s 5A media (Invitrogen) supplemented with 10% FBS (HyClone) and 2% penicillin-streptomycin-glutamine (Invitrogen). Cells were plated at ~25% confluency on Day 0. On Day 1, the media was changed to media containing 1 mM fluorescent pantetheine 2, or 0.5% DMSO control. On Day 3, cells were trypsinized and washed twice with cold PBS. Cells were lysed by incubation in lysis buffer and 0.1% Triton-X 100 on ice. Protein was recovered by centrifugation.
FACS Analysis
106 cells were trypsinized and washed once in cold PBS + 1% FBS (HyClone). Cells were resuspened in 1mL PBS + 1% FBS and analyzed by flow cytometry. Live cells, as determined by forward and side scatter profile, were analyzed using a FACScalibur (Becton Dickinson) for uptake of compound 2.
Large Scale In vivo Carrier Protein Labeling and Affinity Purification
Lysate from 1 L of MR-1 culture grown with or without 1 mM compound 1 was subjected to the bioconjugation reaction as previously reported with alkyne 5 as the conjugate probe. [34, 43] 1 mL reactions were diluted to 3 mL in RIPA buffer (10 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, .5% deoxycholate, 0.1 % SDS, 1 % TritonX 100) and run over two desalting columns (Econo-Pac 10 DG, Bio-Rad) to remove unreacted 5. The reaction was eluted in lysis buffer and incubated with streptavidin agarose resin (Thermo Sci) for 2 hrs at room temperature. The resin was collected by centrifugation and washed 5 times in RIPA buffer. The bound protein was then recovered from the resin by addition of SDS-PAGE loading buffer and incubation at 95°C for 10 minutes. After brief centrifugation to remove the melted agarose, the supernatant was loaded onto a 12% SDS-PAGE gel for analysis.
Mass Spectroscopic Analysis, 1-D SDS-PAGE, and In-Gel Digestion
The proteins were separated by 1-D SDS-PAGE using 10% Bis-Tris NuPAGE gels (Invitrogen). The gel was fixed overnight in 50% MeOH/43% H2O/7% AcOH, washed twice with 200 mL water for 10 minutes, and stained overnight with Gel Code Blue (Pierce). Subsequently, the gel was washed with 200 mL of water for 1 hr. Excised gel bands were washed twice with 200 μL of 50% acetonitrile (ACN) and 50% 5 mM DTT/25 mM NH4HCO3 with vortexing for 10 min, and finally washed with 200 μL ACN. The dehydrated gel piece was rehydrated by addition of 20 μL of ice-cold 10 ng/μL trypsin (Promega) in 5 mM DTT/25 mM NH4HCO3, and incubated on ice for 30 min and the remaining trypsin solution was removed and replaced with fresh 5 mM DTT/25 mM NH4HCO3. The digestion was allowed to continue at 37°C overnight. The peptide mixture was then acidified with 2 μL of 2% trifluoroacetic acid (TFA) (Sigma), vortexed for 30 min, and the supernate extracted. Finally, 20 μL of 20% ACN/0.1% TFA was added followed by vortexing to extract the remaining peptides and combined with the previous fraction. The combined extractions are analyzed directly by nanobore LC-MS/MS.
Mass Spectrometry
The samples were analyzed by electrospray ionization using a QSTAR-Elite hybrid mass spectrometer (Applied Biosystems/MDS Sciex) interfaced to a Tempo nanoscale reverse-phase HPLC (Eksigent/Applied Biosystems) using a 75 μm × 15 cm column (Grace Davison) packed with Vydac MS C18 (300 A, 5 μm) packing material. The buffer compositions were as follows: buffer A: 98% H2O, 2% ACN, 0.1% acetic acid (Fluka), 0.005% heptafluorobutyric acid (Fluka); buffer B: 98% ACN, 2% H2O, 0.1% acetic acid, 0.005% heptafluorobutyric acid. Samples of 10 μL were injected by the Tempo autosampler onto a C18 PepMap pre-column (5 mm × 300 μm, LC Packings) using the Channel 1 loading pump at a flow rate of 15 μL/min buffer A. After washing for 5 min, the peptides were transferred onto the analytical column and eluted directly into the mass spectrometer with a 25-min linear gradient from 5 to 40% buffer B at a flow rate of 300 nL/min using Channel 2. LC-MS/MS data were acquired in a data-dependent fashion by selecting the most intense peak with charge state 2–4 that exceeds 40 counts with exclusion of former target ions set to always and the mass tolerance for exclusion set to 100 ppm. TOF MS were acquired at m/z 500–1800 Da for 0.5 sec with 20 time bins to sum. MS/MS are acquired from m/z 65 – 2000 Da using “enhance all” and 20 time bins to sum, Dynamic Background Subtract, Automatic Collision Energy, and Automatic MS/MS Accumulation with the Fragment Intensity Multiplier set to 12 and Maximum Accumulation set to 3 sec before returning to the survey scan.
Database Search
The MS/MS were analyzed by Analyst 2.0 (Applied Biosystems) and subjected to database search using Mascot 2.2.1 (Matrix Science) with Mascot Daemon 2.2 (Matrix Science) data import filter parameters set as follows: default precursor charge state 2–4; precursor and MS/MS data centroiding using 50 % height and 0.05 amu merge distances. MS/MS peaks with intensity less than 1% of the base peak were discarded, as were MS/MS spectra with less than 22 peaks remaining. Data were searched against the Swissprot database obtained at ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/ containing 237,168 sequences. The search identified tryptic peptides with up to 2 missed cleavages and used mass tolerances of 100 ppm (MS) and 0.10 Da (MS/MS), with variable modifications as follows: deamidation (NQ), oxidation (M), pyro-Glu (N-term Q). The search results indicated that individual ion scores > 42 indicate identity or extensive homology (P < 0.05).
Growth Inhibition Assay
Assays were modified from reported procedure. [35] A single colony of K12 was picked into M9 media for overnight growth and the resulting culture was diluted 1:10000. This diluted culture was plated into 96 well tissue culture plates containing dilutions of pantetheine analogues in M9 media. The plate was incubated overnight at 37 and the wells assessed for growth.
HPLC Analysis of SKBR3 Lysate
Cytosolic extract from the growth of SKBR3 cells with 2 was precipitated by trichloroacetic acid, centrifuged, and the supernatant analyzed by reverse-phase HPLC using a Burdick and Jackson OD5 column (4.6 mm by 25 cm) with monitoring at 254 and 360 nm. Solvents used were 0.05% TFA in H2O (Solvent A) and 0.05% TFA in ACN (solvent B). Compounds were eluted at a flow rate of 1 mL/minute. The method utilized an isocratic step from 0 to 5 minutes with 100% A, followed by a linear gradient to 45% B over 15 minutes, followed by an increasing gradient with solution B until at 25 minutes the solvent composition was 100% solution B. Under these conditions 2 and its CoA analogue intermediates eluted between 17 and 21 minutes. For comparison of the fluorescent compounds observed in SKBR3 lysate to authentic CoA analogue intermediates of 2, 2 was incubated in a stepwise manner with the recombinant E. coli biosynthetic enzymes PanK, PPAT, and DPCK as previously described.[25] The product of the reaction of 2 and PanK was combined with a small amount of SKBR3 lysate to verify co-elution of the two peaks.
General Synthetic Procedures and Materials
All commercial reagents (Sigma-Aldrich, Alfa Aesar, TCI America) were used as provided unless otherwise indicated. The 5-isomer of Rhodamine-Red X NHS Ester was purchased from Molecular Probes. Azido-pantetheine 1, fluorescent pantetheine 2, biotin alkyne 5, and 7-dimethylaminocoumarin-4-acetic acid were prepared as previously described.[25, 34] All reactions were carried out under argon atmosphere in dry solvents with oven-dried glassware and constant magnetic stirring unless otherwise noted. Triethylamine (TEA), and ethyl-N,N-diisopropylamine (DIPEA) were dried over sodium and freshly distilled. 1H-NMR spectra were taken at 300, 400, or 500 MHz and 13C-NMR spectra were taken at 100.6 or 75.5 MHz on Varian NMR spectrometers and standardized to the NMR solvent signal as reported by Gottlieb. [44] Multiplicities are given as s=singlet, d=doublet, t=triplet, q=quartet, p=pentet, dd=doublet of doublets, bs=broad singlet, bt=broad triplet, m=multiplet using integration and coupling constant in Hertz. TLC analysis was performed using Silica Gel 60 F254 plates (EM Scientific) and visualization was accomplished with UV light (λ=254 nm) and/or the appropriate stain (iodine, 2,4-dinitrophenylhydrazine, cerium molybdate, ninhydrin). Silica gel chromatography was carried out with Silicycle 60 Angstrom 230–400 mesh according to the method of Still. [45] TLC prep plate purification was performed with EMD Silica Gel 60 F254 pre-coated plates. Electrospray (ESI) and fast atom bombardment (FAB) mass spectra were obtained at the UCSD Mass Spectrometry Facility by Dr. Yongxuan Su using a Finnigan LCQDECA mass spectrometer and a ThermoFinnigan MAT- 900XL mass spectrometer, respectively.
Synthesis of Fluorescent Alkyne 3
7-dimethylaminocoumarin-4-acetic acid (250 mg, 1.01 mmol), DIPEA (440 μL, 2.53 mmol), propargyl amine (70 μL, 1.01 mmol), and HOBt (484 mg, 3.16 mmol) were dissolved in DMF (25 mL) with stirring and cooled to 0°C. EDC (485 mg, 2.53 mmol) was added in one portion and the reaction was allowed to slowly warm to RT and followed by TLC. After 20 hrs the solvent was removed under reduced pressure and the sparingly soluble crude reaction mixture was taken up in EtOAc with heating. After filtration the soluble portion was purified by column chromatography (1:1 EtOAc/hexanes to EtOAc to 5% MeOH/DCM). This procedure was repeated several times on the EtOAc resuspended filter cake to afford propargyl-DMC 3 (87 mg, 30%) as an orange solid. 1H-NMR (500MHz, CDCl3) δ 7.43 (d, J=9.0 Hz, 1H), 6.61 (dd, J=2, 9.0 Hz, 1H), 6.51 (d, J=2 Hz, 1H), 6.05 (s, 1H), 5.92 (bs, 1H), 4.02 (dd, J=2.0, 4.5 Hz, 2H), 3.64 (s, 2H), 3.06 (m, 7H). 13C-NMR (75.5 MHz, (CD3)2SO) δ 168.4, 161.4, 156.1, 153.5, 151.7, 126.7, 110.1, 109.7, 108.8, 98.2, 81.5, 74.0, 41.0, 39.1, 28.9. HRMS (EI) (m/z): [M]+ calcd for C16H16N2O3, 284.1155 found 284.1154.
Synthesis of Fluorescent Alkyne 4
Rhodamine Red-X NHS Ester (10 mg, 0.013 mmol), DIPEA (10 μL, 0.06 mmol), and propargyl amine (5 μL, 0.07 mmol), were dissolved in DMF (0.5 mL), covered with aluminum foil, and allowed to stir 1 hr. After removal of the solvent under reduced pressure the bright red residue was redissolved in 1.4 mL ACN and purified by RP-HPLC. Solvents used were 0.05% TFA in H2O (Solvent A) and 0.05% TFA in ACN (solvent B). Compounds were eluted at a flow rate of 1 mL/minute with monitoring at 560 nm. The method used utilized an isocratic step from 0 to 5 minutes with 40% B, followed by a linear gradient to 58% B over 15 minutes, followed by an increasing gradient with solution B until at 16 minutes the solvent composition was 100% solution B. Under these conditions 4 eluted around 11.3 minutes. Pooling and lyophilization of multiple HPLC runs yielded 4 (6.5 mg, 72%) as a red solid. 1H-NMR (500MHz, (CD3)2SO) δ 8.39 (s, 1H), 8.20 (bt, J=5.0 Hz, 1H), 7.91, (d, J=8.0 Hz, 1H), 7.90 (bs, 1H), 7.45 (d, J=8.0 Hz, 1H), 7.03 (d, J=9.5 Hz, 2H), 6.94 (d, J=9.5 Hz, 2H), 6.91 (s, 2H), 3.79 (d, J=2.5 Hz, 2H), 3.62 (q, J=7.5 Hz, 8H), 3.01 (s, 1H), 2.83 (q, J=6.5 Hz, 2H), 2.04 (t, J= 7.5 Hz, 2H), 1.42 (p, J=8.0 Hz, 2H), 1.37 (p, J=7.5 Hz, 2H), 1.19 (t, J=7.0 Hz, 12H), 1.15 (t, J=7.0 Hz, 2H). MS (ESI) (m/z): [M]+ calcd for C36H44N4O7S2, 708.26 found 731.25 [M+Na]+
Supplementary Material
Acknowledgments
The authors thank Elinore Mercer for assistance with FACS analysis. This work was funded by NIH R01GM075797 and NSF CAREER MCB-0347681.
References
- 1.Mercer AC, Burkart MD. Natural Product Reports. 2007;24:750. doi: 10.1039/b603921a. [DOI] [PubMed] [Google Scholar]
- 2.White SW, Zheng J, Zhang YM, Rock CO. Annual Review of Biochemistry. 2005;74:791. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 3.Hill AM. Natural Product Reports. 2006;23:256. doi: 10.1039/b301028g. [DOI] [PubMed] [Google Scholar]
- 4.Schwarzer D, Finking R, Marahiel MA. Natural Product Reports. 2003;20:275. doi: 10.1039/b111145k. [DOI] [PubMed] [Google Scholar]
- 5.Lambalot RH, Gehring AM, Flugel RS, Zuber P, LaCelle M, Marahiel MA, Reid R, Khosla C, Walsh CT. Chemistry & Biology. 1996;3:923. doi: 10.1016/s1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]
- 6.Quadri LEN, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT. Biochemistry. 1998;37:1585. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 7.La Clair JJ, Foley TL, Schegg TR, Regan CM, Burkart MD. Chemistry & Biology. 2004;11:195. doi: 10.1016/j.chembiol.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 8.Meyer BH, Segura JM, Martinez KL, Hovius R, George N, Johnsson K, Vogel H. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2138. doi: 10.1073/pnas.0507686103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clarke KM, Mercer AC, LaClair JJ, Burkart MD. Journal of the American Chemical Society. 2005;127:11234. doi: 10.1021/ja052911k. [DOI] [PubMed] [Google Scholar]
- 10.Meier JL, Mercer AC, Rivera H, Burkart MD. Journal of the American Chemical Society. 2006;128:12174. doi: 10.1021/ja063217n. [DOI] [PubMed] [Google Scholar]
- 11.Laughlin ST, Agard NJ, Baskin JM, Carrico IS, Chang PV, Ganguli AS, Hangauer MJ, Lo A, Prescher JA, Bertozzi CR. Methods Enzymol. 2006;415:230. doi: 10.1016/S0076-6879(06)15015-6. [DOI] [PubMed] [Google Scholar]
- 12.Hang HC, Geutjes EJ, Grotenbreg G, Pollington AM, Bijlmakers MJ, Ploegh HL. J Am Chem Soc. 2007;129:2744. doi: 10.1021/ja0685001. [DOI] [PubMed] [Google Scholar]
- 13.Foley TL, Burkart MD. Current Opinion in Chemical Biology. 2007;11:12. doi: 10.1016/j.cbpa.2006.11.036. [DOI] [PubMed] [Google Scholar]
- 14.Jackowski S, Rock CO. Journal of Biological Chemistry. 1984;259:1891. [PubMed] [Google Scholar]
- 15.Jackowski S, Alix JH. Journal of Bacteriology. 1990;172:3842. doi: 10.1128/jb.172.7.3842-3848.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Molecular Systems Biology. 2006 doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang YM, Frank MW, Virga KG, Lee RE, Rock CO, Jackowski S. J Biol Chem. 2004;279:50969. doi: 10.1074/jbc.M409607200. [DOI] [PubMed] [Google Scholar]
- 18.Strauss E, Begley TP. Journal of Biological Chemistry. 2002;277:48205. doi: 10.1074/jbc.M204560200. [DOI] [PubMed] [Google Scholar]
- 19.Mercer AC, Meier JL, Hur GH, Smith AR, Burkart MD. Bioorganic & Medicinal Chemistry Letters. 2008;18:5991. doi: 10.1016/j.bmcl.2008.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambalot RH, Walsh CT. J Biol Chem. 1995;270:24658. doi: 10.1074/jbc.270.42.24658. [DOI] [PubMed] [Google Scholar]
- 21.Marshall CG, Burkart MD, Meray RK, Walsh CT. Biochemistry. 2002;41:8429. doi: 10.1021/bi0202575. [DOI] [PubMed] [Google Scholar]
- 22.Mercer AC, Clair JJL, Burkart MD. ChemBioChem. 2005;6:1335. doi: 10.1002/cbic.200500051. [DOI] [PubMed] [Google Scholar]
- 23.Thompson BJ, Stern A, Smith S. Biochimica Et Biophysica Acta. 1981;662:125. doi: 10.1016/0005-2744(81)90232-1. [DOI] [PubMed] [Google Scholar]
- 24.Hwang Y, Ganguly S, Ho AK, Klein DC, Cole PA. Bioorganic and Medicinal Chemistry. 2007;15:2147. doi: 10.1016/j.bmc.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clarke KM, Mercer AC, LaClair JJ, Burkart MD. J Am Chem Soc. 2005;127:11234. doi: 10.1021/ja052911k. [DOI] [PubMed] [Google Scholar]
- 26.Worthington AS, Burkart MD. Organic & Biomolecular Chemistry. 2006;4:44. doi: 10.1039/b512735a. [DOI] [PubMed] [Google Scholar]
- 27.Cebrat M, Kim CM, Thompson PR, Daugherty M, Cole PA. Bioorganic & Medicinal Chemistry. 2003;11:3307. doi: 10.1016/s0968-0896(03)00265-7. [DOI] [PubMed] [Google Scholar]
- 28.Leonardi R, Zhang YM, Rock CO, Jackowski S. Progress in Lipid Research. 2005;44:125. doi: 10.1016/j.plipres.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 29.Lovley DR, Holmes DE, Nevin KP. Advances in Microbial Physiology. 2004;49:219. doi: 10.1016/S0065-2911(04)49005-5. [DOI] [PubMed] [Google Scholar]
- 30.Okuda K, Edwards GC, Winnick T. Journal of Bacteriology. 1963;85:329. doi: 10.1128/jb.85.2.329-338.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caetanoanolles G. Pcr-Methods and Applications. 1993;3:85. doi: 10.1101/gr.3.2.85. [DOI] [PubMed] [Google Scholar]
- 32.Xu D, Cote JC. International Journal of Systematic and Evolutionary Microbiology. 2003;53:695. doi: 10.1099/ijs.0.02346-0. [DOI] [PubMed] [Google Scholar]
- 33.Meier JL, Mercer AC, Burkart MD. Journal of the American Chemical Society. 2008;130:5443. doi: 10.1021/ja711263w. [DOI] [PubMed] [Google Scholar]
- 34.Meier JL, Mercer AC, Rivera H, Burkart MD. J Am Chem Soc. 2006;128:12174. doi: 10.1021/ja063217n. [DOI] [PubMed] [Google Scholar]
- 35.Virga KG, Zhang YM, Leonardi R, Ivey RA, Hevener K, Park HW, Jackowski S, Rock CO, Lee RE. Bioorganic & Medicinal Chemistry. 2006;14:1007. doi: 10.1016/j.bmc.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 36.Angell S, Bench BJ, Williams H, Watanabe CMH. Chemistry & Biology. 2006;13:1349. doi: 10.1016/j.chembiol.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 37.Gross H. Applied Microbiology and Biotechnology. 2007;75:267. doi: 10.1007/s00253-007-0900-5. [DOI] [PubMed] [Google Scholar]
- 38.Blattner FR, Plunkett G, III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. Science. 1997;277:1453. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 39.Mootz HD, Finking R, Marahiel MA. J Biol Chem. 2001;276:37289. doi: 10.1074/jbc.M103556200. [DOI] [PubMed] [Google Scholar]
- 40.Bretschger O, Obraztsova A, Sturm CA, Chang IS, Gorby YA, Reed SB, Culley DE, Reardon CL, Barua S, Romine MF, Zhou J, Beliaev AS, Bouhenni R, Saffarini D, Mansfeld F, Kim BH, Fredrickson JK, Nealson KH. Appl Environ Microbiol. 2007;73:7003. doi: 10.1128/AEM.01087-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson BJ, Smith S. Biochimica Et Biophysica Acta. 1982;712:217. doi: 10.1016/0005-2760(82)90105-9. [DOI] [PubMed] [Google Scholar]
- 42.Paliy O, Gunasekera TS. Applied Microbiology and Biotechnology. 2007;73:1169. doi: 10.1007/s00253-006-0554-8. [DOI] [PubMed] [Google Scholar]
- 43.Weerapana E, Speers AE, Cravatt BF. Nature Protocols. 2007;2:1414. doi: 10.1038/nprot.2007.194. [DOI] [PubMed] [Google Scholar]
- 44.Gottlieb HE, Kotlyar V, Nudelman A. Journal of Organic Chemistry. 1997;62:7512. doi: 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- 45.Still WC, Kahn M, Mitra A. Journal of Organic Chemistry. 1978;43:2923. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.