

Abstract
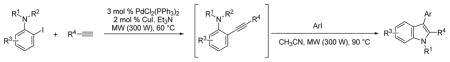
A microwave-assisted, one-pot, three-component coupling reaction for the synthesis of indoles has been developed. The reaction is carried out in two steps under standard Sonogashira coupling conditions from an N-substituted/N,N-disubstituted 2-iodoaniline and a terminal alkyne, followed by the addition of acetonitrile and an aryl iodide. A variety of polysubstituted indoles have been prepared in moderate to excellent yields using the present method.
Keywords: Microwave-assisted, One-pot, Three-component coupling, Indoles, Sonogashira coupling
1. Introduction
The indole nucleus is a ubiquitous heterocyclic structure found in numerous natural and synthetic compounds with a wide variety of biological activities and considerable pharmaceutical importance.1 The synthesis of indoles, therefore, has attracted enormous attention from synthetic organic chemists and a substantial number of methods for the preparation of indoles have been developed.2 Among the methods developed so far, palladium-catalyzed indole syntheses have received extraordinary attention due to the relatively mild reaction conditions employed in these processes and the fact that they usually tolerate a wide variety of functional groups, thus avoiding protecting group chemistry. High regioselectivities and chemical yields are also generally achieved.2b-d,3 Flynn previously demonstrated a one-pot, two-step synthesis of indoles by consecutive Sonogashira4 and Cacchi5 reactions. However, only one example of this process was reported.6 Lu and co-workers later on reported a one-pot, three-component synthesis of indoles by the same Sonogashira/Cacchi process in which they replaced the aryl iodide in the Cacchi cyclization with an aryl bromide.7 However, a significant substituent effect in the three starting components was observed on the rate of reaction. Sluggish reactions were observed, especially when an electron-withdrawing group was present at the para-position of either the iodide or the amide moiety of the starting material as in 2′-iodo-trifluoroacetanilide.
It is noteworthy that microwave technology has recently attracted more and more attention from synthetic organic chemists due to the many advantages microwave irradiation affords over conventional heating in chemical transformations, particularly the enormous acceleration of the reaction rate, significant energy savings, as well as high chemical yields and cleaner reactions.8 Our group has been interested in developing new methodologies for the synthesis of functionalized indoles for almost two decades. We have previously developed a palladium-catalyzed heteroannulation reaction of internal alkynes and 2-iodoanilines known as the Larock indole synthesis;9 and the electrophilic cyclization of N,N-dialkyl-2-(1-alkynyl)anilines induced by halide,10 sulfur or selenium electrophiles to generate indoles.11 As a continuation of our long-term interest in indole synthesis, we hereby report a microwave-assisted, one-pot, three-component reaction to synthesize 2,3-disubstituted indoles under Sonogashira coupling conditions.
2. Results and discussion
Our group previously developed synthetic protocols for the preparation of 3-iodo-,10 3-sulfenyl-, and 3-selenylindoles11 by the electrophilic cyclization of N,N-dialkyl-2-(1-alkynyl)anilines by iodine or sulfenyl/selenyl chlorides. While preparing the starting N,N-dialkyl-2-(1-alkynyl)anilines for this process, we discovered an interesting solvent effect during the Sonogashira coupling process. When the coupling of N,N-dialkyl-2-iodoanilines and terminal alkynes was carried out in Et3N, the corresponding internal alkynes were generally obtained as a single product in high chemical yield. On the other hand, in the presence of a polar solvent, such as CH3CN or DMF, with only 10 equiv of Et3N present, a significant amount of an indole was obtained, alongside the desired N,N-dialkyl-2-(1-alkynyl)anilines. The indole is apparently generated by the palladium-catalyzed cyclization of the Sonogashira coupling product and any unreacted N,N-dialkyl-2-iodoaniline.
Cacchi has previously developed a similar cyclization between 2-(1-alkynyl)trifluoroacetanilides and aryl iodides in the presence of inorganic bases, such as K2CO3 or Cs2CO3.5a,d,e,g In the Cacchi reaction, the reaction outcome was influenced by both the base and the nature of the nitrogen nucleophile. Employing Et3N as the base gave only low yields. On the other hand, a trifluoroacetamido group plays a key role in this cyclization. When a free amino or acetamido group is used, no cyclization occurs and only the starting alkynes are recovered. In our case, due to the high nucleophilicity of the N,N-dialkylamino moiety, intramolecular cyclization takes place more readily.
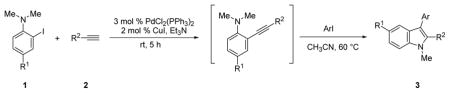
In our view, this one-pot cyclization approach provides an ideal protocol for parallel library synthesis. Thus, a one-pot, three-component coupling reaction was carried out using N,N-dimethyl-2-iodoaniline, phenylacetylene and ethyl 4-iodobenzoate (Table 1, entry 1). The Sonogashira coupling took place smoothly in Et3N at room temperature, while efficient further cyclization required a higher reaction temperature (60 °C) and the addition of a polar solvent, such as CH3CN. When a more bulky alkyne, such as 3,5-dimethoxyphenylacetylene, and an electron-rich aryl iodide, such as 2-iodothiophene, were employed in this coupling, a considerably longer reaction time was needed for complete cyclization (Table 1, entry 2).
Table 1.
One-pot synthesis of indoles under Sonogashira coupling conditions.a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | 1 | R1 | R2 | Ar | time (h) |
3 | % yieldb | |
| Step 1 | Step 2 | |||||||
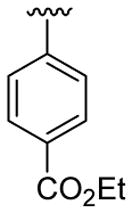
| 1 | 1a | H | C6H5 | 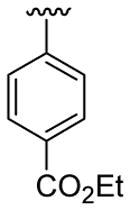 |
5 | 4 | 3a | 82 |
| 2 | 1b | Br | 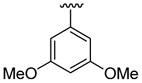 |
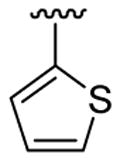 |
5 | 12 | 3j | 83 |
Representative procedure: Step 1) 2-Iodoaniline 1 (0.500 mmol), terminal alkyne 2 (0.525 mmol), PdCl2(PPh3)2 (0.015 mmol), CuI (0.010 mmol), and 3 mL of Et3N were mixed in a sealed 4-dram vial. The reaction was stirred at room temperature for the indicated time. Step 2) Aryl iodide (0.550 mmol) and 3 mL of CH3CN were added to the reaction mixture of Step 1. The resulting mixture was stirred at 60 °C for the indicated time.
Isolated yields of indole product after column chromatography.
In order to enhance the reaction rate of this one-pot coupling/cyclization process for the purpose of developing a high-throughput parallel synthetic protocol, microwave technology has been employed. To our delight, the entire process was dramatically accelerated by microwave irradiation. Both of the reactions were completed in less than an hour in yields comparable to those obtained previously.
Encouraged by these results, we next explored the scope of this one-pot, two-step approach to substituted indoles. Both the Sonogashira coupling and cyclization take place smoothly when electron-rich aryl acetylenes are used (Table 2; entries 2, 4 and 6). A longer reaction time is necessary for complete conversion for both the Sonogashira and cyclization steps, when an electron-deficient aryl acetylene is employed (Table 2, entry 3). Smooth couplings were also observed when aliphatic acetylenes are employed (Table 2; entries 5, 7 and 8). When 2-methoxyphenylacetylene is used, the steric bulkiness induced by the 2-methoxy group requires a longer reaction time for cyclization (Table 2, entry 9). A free hydroxyl group in the alkyne is not well accommodated by this coupling process as only a 33% yield of the desired indole product was obtained (Table 2, entry 16).
Table 2.
Microwave-assisted, one-pot synthesis of indoles under Sonogashira coupling conditions.a
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| entry | 1 | R1 | R2 | R3 | R4 | Ar | time (min) |
3 | % yieldb | |
| Step 1 | Step 2 | |||||||||
| 1 | 1a | Me | Me | H | C6H5 | 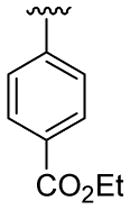 |
20 | 30 | 3a | 86 |
| 2 | 1a | Me | Me | H | 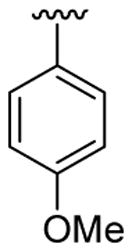 |
 |
20 | 20 | 3b | 86 |
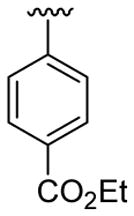
| 3 | 1a | Me | Me | H | 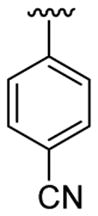 |
 |
30 | 50 | 3c | 77 |
| 4 | 1a | Me | Me | H | 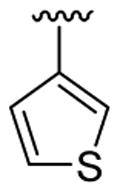 |
 |
20 | 30 | 3d | 78 |
| 5 | 1a | Me | Me | H | 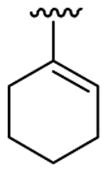 |
 |
20 | 30 | 3e | 91 |
| 6 | 1a | Me | Me | H | 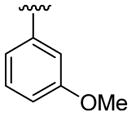 |
C6H5 | 20 | 30 | 3f | 91 |
| 7 | 1a | Me | Me | H | 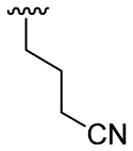 |
 |
20 | 30 | 3g | 72 |
| 8 | 1a | Me | Me | H | 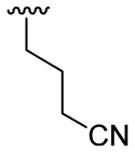 |
 |
20 | 30 | 3h | 63 |
| 9 | 1a | Me | Me | H | 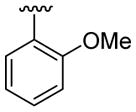 |
 |
20 | 50 | 3i | 87 |
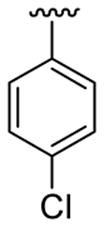
| 10c | 1b | Me | Me | 4-Br | 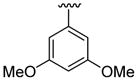 |
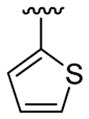 |
20 | 30 | 3j | 74 |
| 11c | 1b | Me | Me | 4-Br | 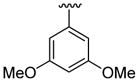 |
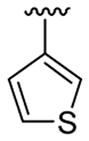 |
20 | 30 | 3k | 76 |
| 12c | 1b | Me | Me | 4-Br | 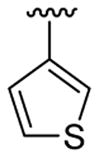 |
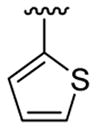 |
20 | 30 | 3l | 85 |
| 13c | 1b | Me | Me | 4-Br | C6H5 | 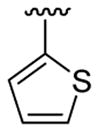 |
20 | 30 | 3m | 79 |
| 14 | 1c | Me | Me | 4-Me | C6H5 | 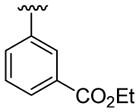 |
30 | 30 | 3n | 68 |
| 15 | 1c | Me | Me | 4-Me | 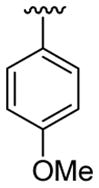 |
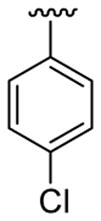 |
30 | 30 | 3o | 94 |
| 16 | 1c | Me | Me | 4-Me | 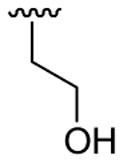 |
 |
30 | 30 | 3p | 33 |
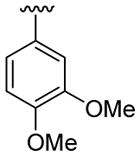
| 17 | 1d | Me | Me | 4-CO2Me | 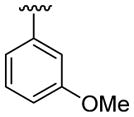 |
 |
30 | 30 | 3q | 70 |
| 18 | 1e | H | 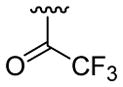 |
H | C6H5 | 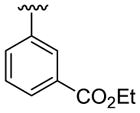 |
30 | 30d | 3r | 93 |
| 19 | 1e | H | 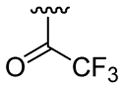 |
H | 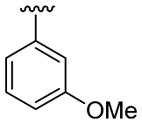 |
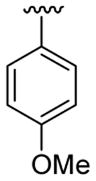 |
30 | 30d | 3s | 82 |
| 20 | 1f | H | 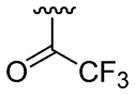 |
4-Me | 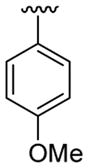 |
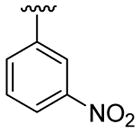 |
30 | 30d | 3t | 88 |
| 21 | 1f | H | 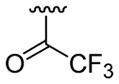 |
4-Me | 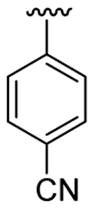 |
C6H5 | 30 | 30d | 3u | 66 |
| 22 | 1f | H | 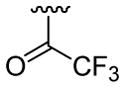 |
4-Me | 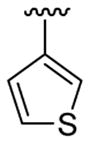 |
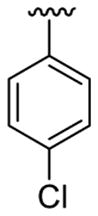 |
30 | 30d | 3v | 67 |
| 23 | 1g | H | 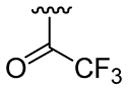 |
4-CO2Me | C6H5 | 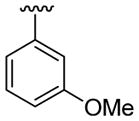 |
30 | 30d | 3w | 76 |
| 24 | 1g | H | 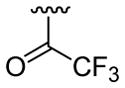 |
4-CO2Me | 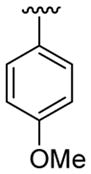 |
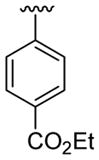 |
30 | 30d | 3x | 60 |
Representative procedure: Step 1) 2-Iodoaniline 1 (0.500 mmol), terminal alkyne 2 (0.525 mmol), PdCl2(PPh3)2 (0.015 mmol), CuI (0.010 mmol), and 3 mL of Et3N were mixed in a sealed 20 mL microwave vial. The reaction was stirred at 60 °C under microwave (300 W) irradiation for the indicated time. Step 2) Aryl iodide (0.550 mmol) and 3 mL of CH3CN were added to the reaction mixture of Step 1. The resulting mixture was stirred at 90 °C under microwave irradiation (300 W) for the indicated time.
Isolated yields of indole product after column chromatography.
An extra equivalent of aryl iodide was employed in Step 2).
Step 2) was carried out at 100 °C with the addition of Cs2CO3 (3 equiv)
No significant electronic effect has been observed in either the 2-iodoanilines or the aryl iodides employed. Both electron-withdrawing and electron-releasing groups are readily accommodated in these two components. An extra equivalent of aryl iodide was employed in the coupling processes utilizing N,N-dimethyl-4-bromo-2-iodoaniline in order to suppress any interference by the bromo moiety in the cyclization step (Table 2, entries 10–13). Both benzyl bromide and allyl acetate have been examined in this coupling process in place of the aryl iodide. However, none of the desired cyclization product was obtained in either case. The two alkyl groups present on the aniline nitrogen play a crucial role in the success of the overall process. Only Sonogashira coupling product was obtained when either 2-iodoaniline or N-methyl-2-iodoaniline were employed, which is in good agreement with our previous experience with such Sonogashira processes.10,11
Besides N,N-dialkyl-2-iodoanilines, 2′-iodo-trifluoroacetanilides can also be employed in the current microwave-irradiated process (Table 2, entries 18–24). As described earlier using conventional heating, the addition of an inorganic base is necessary for the success of this cyclization. In addition, a slightly higher reaction temperature is needed for efficient cyclization.
As mentioned above, this overall process involves two steps (Scheme 1). The first step is a Sonogashira coupling to generate the N,N-dialkyl-2-(1-alkynyl)aniline A. The aryl iodide is added upon completion of the Sonogashira coupling. Oxidative addition of the aryl iodide to Pd(0) affords an electrophilic ArPdI species, which activates the alkyne triple bond of A by coordination to form a π-palladium complex B, which subsequently undergoes intramolecular trans-aminopalladation by a 5-endo-dig cyclization, affording the indolium species C. The latter undergoes methyl group removal via SN2 displacement by the in situ generated iodide anion, leading to the indole-containing Pd(II) intermediate D. The 2,3-disubstituted indole is generated after reductive elimination.
Scheme 1.

A proposed mechanism for the one-pot, two-step indole synthesis.
3. Conclusion
In summary, an efficient, microwave-assisted, one-pot, three-component reaction for the synthesis of polysubstituted indoles has been developed. A variety of functionalities, such as nitro, ester, hydroxyl, cyano, and halide groups are tolerated in this coupling/cyclization process. The desired indoles have been obtained in moderate to excellent overall yields. This protocol provides an ideal synthetic approach for the parallel synthesis of an indole library. Due to the limited capacity of our current microwave equipment, these reactions have not been carried out on a larger scale. However, we believe that the current method should be easily extended to gram scale syntheses when appropriate microwave equipment is employed. Further examination of the current reaction conditions for a one-pot, four-component synthesis of indoles, as well as other biologically interesting heterocycles, is underway in our laboratory.
4. Experimental
4.1. General comments
All microwave irradiation reactions were carried out on a Biotage-EXP Microwave synthesis system, operating at a frequency of 2450 MHz with continuous irradiation power from 0–300 W. All reactions were carried out in 20 mL oven-dried Biotage microwave vials sealed with an aluminum/Teflon® crimp top, which can be exposed to a maximum of 250 °C and 20 bar internal pressure. The reaction temperature was measured by an IR sensor on the outer surface of the process vial. All commercially obtained chemicals were used as received without further purification unless otherwise indicated. The 1H NMR and 13C NMR spectra were recorded at 400 MHz and 100 MHz respectively, using CDCl3, acetone-d6 or DMSO-d6 as solvents. The chemical shifts of the 1H NMR and 13C NMR spectra are reported relative to the residual signal of CDCl3 (δ 7.26 ppm for the 1H NMR and δ 77.23 ppm for the 13C NMR), acetone-d6 (2.05 ppm for the 1H NMR and δ 29.92 ppm for the 13C NMR) or DMSO-d6 (2.50 ppm for the 1H NMR and δ 39.51 ppm for the 13C NMR). The high resolution mass spectra were recorded on a double focusing magnetic sector mass spectrometer using EI at a voltage of 70 eV. The melting points are uncorrected.
4.2. General procedure for preparation of the N,N-dimethyl-2-iodoanilines
These compounds were prepared according to a procedure reported by Cadogan.12 To a solution of the corresponding 2-iodoaniline (2.0 mmol) and iodomethane (0.85 g, 6.0 mmol) in DMF (10 mL) was added K2CO3 (0.55 g, 4.0 mmol). The resulting mixture was stirred at room temperature for 48 h. Water (10 mL) was added to the reaction mixture and the resulting solution was extracted with diethyl ether (3 × 10 mL). The organic layers were combined and washed with water to remove any remaining DMF and dried over anhydrous MgSO4. The solvent was removed under vaccum and the residue was purified by flash column chromatography on silica gel using ethyl acetate/hexanes as the eluent.
4.2.1. N,N-Dimethyl-2-iodoaniline (1a)
This compound was obtained as a yellow oil in an 81% yield: 1H NMR (400 MHz, CDCl3) δ 2.76 (s, 6H), 6.77 (dt, J = 7.6, 1.5 Hz, 1H), 7.09 (dd, J = 7.8, 1.5 Hz, 1H), 7.31 (dt, J = 7.6, 1.5 Hz, 1H), 7.84 (dd, J = 7.8, 1.5 Hz, 1H). The 1H NMR spectral data are in good agreement with the literature data.10a
4.2.2. N,N-Dimethyl-4-bromo-2-iodoaniline (1b)
This compound was obtained as a light red oil in an 81% yield: 1H NMR (400 MHz, CDCl3) δ 2.72 (s, 6H), 6.92 (d, J = 8.5 Hz, 1H), 7.40 (dd, J = 8.5, 2.4 Hz, 1H), 7.94 (d, J = 2.4 Hz, 1H). The 1H NMR spectral data are in good agreement with the literature data.11
4.2.3. N,N,4-Trimethyl-2-iodoaniline (1c)
This product was obtained as an orange liquid in a 91% yield: 1H NMR (400 MHz, CDCl3) δ 2.26 (s, 3H), 2.72 (s, 6H), 6.99 (d, J = 8.1 Hz, 1H), 7.12 (d, J = 8.1 Hz, 1H), 7.68 (s, 1H). The 1H NMR spectral data are in good agreement with the literature data.10b
4.3. Preparation of methyl 4-dimethylamino-3-iodobenzoate (1d)
This compound was prepared according to a procedure reported by Larock.13 The product was obtained as a colorless oil in a 44% yield: 1H NMR (400 MHz, CDCl3) δ 2.82 (s, 6H), 3.85 (s, 3H), 6.98 (d, J = 8.4 Hz, 1H), 7.92 (dd, J = 8.4, 2.0 Hz, 1H), 8.46 (d, J = 2.0 Hz, 1H). The 1H NMR spectral data are in good agreement with the literature data.13
4.4. General procedure for preparation of the N-trifluoroacetyl-2-iodoanilines
These compounds were prepared according to a procedure reported by Srinivasan. 14 To a solution of the corresponding 2-iodoaniline (4.3 mmol) and triethylamine (0.63 mL, 4.55 mmol) in THF (11 mL) at −15 °C was slowly added trifluoroacetic anhydride (0.6 mL, 4.3 mmol) in 6.5 mL of THF. The resulting mixture was stirred for 1 h and then allowed to warm to room temperature and stirred for 16 h. The reaction mixture was then poured into a separatory funnel containing water (115 mL) and extracted with ethyl acetate (3 × 50 mL). The organic layers were dried over anhydrous MgSO4. The solvent was removed under vacuum and the residue was purified by flash column chromatography on silica gel using ethyl acetate/hexanes as the eluent.
4.4.1. N-Trifluoroacetyl-2-iodoaniline (1e)
This product was obtained as a white solid in a 96% yield: mp 105–107 °C; 1H NMR (400 MHz, CDCl3) δ 6.98 (t, J = 7.0 Hz, 1H), 7.42 (t, J = 7.4 Hz, 1H), 7.84 (d, J = 7.9 Hz, 1H), 8.21 (d, J = 8.2 Hz, 1H), 8.29 (s, 1H). The 1H NMR spectral data are in good agreement with the literature data.15
4.4.2. N-Trifluoroacetyl-2-iodo-4-methylaniline (1f)
This product was obtained as a pink solid in a 95% yield: mp 84–85 °C; 1H NMR (400 MHz, CDCl3) δ 2.32 (s, 3H), 7.19 (d, J = 8.1 Hz, 1H), 7.66 (s, 1H), 8.01 (d, J = 8.3 Hz, 1H), 8.21 (s, 1H).
4.4.3. Methyl 4-(N-trifluoroacetamino)-3-iodobenzoate (1g)
This product was obtained as a white solid in an 88% yield: mp 87–88 °C; 1H NMR (400 MHz, CDCl3) δ 3.92 (s, 3H), 8.06 (dd, J = 1.9, 8.6 Hz, 1H), 8.34 (d, J = 8.6 Hz, 1H), 8.50 (m, 2H). The 1H NMR spectral data are in good agreement with the literature data.16
4.5. General procedure for the microwave-assisted, one-pot synthesis of 1-methylindoles
The 2-iodoaniline 1 (0.500 mmol), a terminal alkyne 2 (0.525 mmol), PdCl2(PPh3)2 (0.015 mmol), CuI (0.010 mmol), and 3 mL of Et3N were mixed in a sealed 20 mL microwave vial. The reaction mixture was stirred at 60 °C under microwave (300 W) irradiation for 20 min or until disappearance of the starting material as monitored by thin layer chromatography. To the reaction mixture was added the aryl iodide (0.550 mmol) and 3 mL of CH3CN at room temperature. The resulting mixture was then stirred at 90 °C under microwave irradiation for 30 min or until disappearance of the starting material as monitored by thin layer chromatography. The reaction mixture was diluted by 10 mL of diethyl ether and washed with brine (10 mL). The aqueous phase was extracted with diethyl ether (2 × 5 mL). The organic layers were combined and dried over anhydrous MgSO4. The solvent was removed under vacuum and the residue was purified by flash column chromatography on silica gel using ethyl acetate/hexanes as the eluent.
4.5.1. Ethyl 4-(1-methyl-2-phenylindol-3-yl)benzoate (3a)
This product was obtained as a light yellow oil in an 86% yield: 1H NMR (400 MHz, CDCl3) δ 1.48 (t, J = 7.1 Hz, 3H), 3.73 (s, 3H), 4.47 (q, J = 7.1 Hz, 2H), 7.31–7.35 (m, 1H), 7.39–7.45 (m, 3H), 7.47–7.51 (m, 6H), 7.95 (d, J = 7.8 Hz, 1H), 8.08 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.5, 31.0, 60.8, 109.9, 114.2, 119.4, 120.7, 122.5, 126.6, 127.3, 128.4, 128.6, 129.5, 129.6, 131.1, 131.6, 137.5, 138.6, 140.5, 166.7; HRMS (EI) calcd for C24H21NO2 355.1572, found 355.1570.
4.5.2. Ethyl 4-[2-(4-methoxyphenyl)-1-methylindol-3-yl]benzoate (3b)
This product was obtained as a white solid in an 86% yield: mp 168–170 °C; 1H NMR (400 MHz, CDCl3) δ 1.44 (t, J = 7.1 Hz, 3H), 3.70 (s, 3H), 3.88 (s, 3H), 4.42 (q, J = 7.1 Hz, 2H), 6.97 (d, J = 8.8 Hz, 2H), 7.25–7.29 (m, 3H), 7.35–7.39 (m, 1H), 7.44–7.46 (m, 3H), 7.88 (d, J = 7.8 Hz, 1H), 8.03 (d, J = 8.5 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.6, 31.0, 55.4, 60.8, 109.9, 113.9, 114.2, 119.3, 120.7, 122.4, 123.7, 126.7, 127.2, 129.4, 129.6, 132.4, 137.4, 138.6, 140.7, 159.8, 166.9; HRMS (EI) calcd for C25H23NO3 385.1678, found 385.1681.
4.5.3. Ethyl 4-[2-(4-cyanophenyl)-1-methylindol-3-yl]benzoate (3c)
This product was obtained as a light yellow solid in a 77% yield: mp 183–185 °C; 1H NMR (400 MHz, CDCl3) δ 1.41 (t, J = 7.1 Hz, 3H), 3.72 (s, 3H), 4.40 (q, J = 7.1 Hz, 2H), 7.23–7.27 (m, 1H), 7.32 (d, J = 8.2 Hz, 2H), 7.36–7.40 (m, 1H), 7.42–7.47 (m, 3H), 7.68 (d, J = 8.2 Hz, 2H), 7.79 (d, J = 8.0 Hz, 1H), 8.00 (d, J = 8.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.5, 31.4, 61.0, 110.1, 112.0, 115.8, 118.6, 119.8, 121.2, 123.4, 126.6, 128.1, 129.7, 129.9, 131.8, 132.4, 136.1, 136.4, 138.0, 139.5, 166.6; HRMS (EI) calcd for C25H20N2O2 380.1525, found 380.1524.
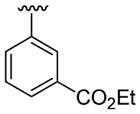
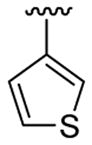
4.5.4. Ethyl 3-[1-methyl-2-(thiophen-3-yl)indol-3-yl]benzoate (3d)
This product was obtained as a yellow oil in a 78% yield: 1H NMR (400 MHz, CDCl3) δ 1.34 (t, J = 7.1 Hz, 3H), 3.69 (s, 3H), 4.33 (q, J = 7.1 Hz, 2H), 7.00 (dd, J = 1.2, 4.9 Hz, 1H), 7.17–7.22 (m, 2H), 7.28–7.35 (m, 3H), 7.39 (d, J = 8.2 Hz, 1H), 7.45 (dd, J = 6.2, 1.4 Hz, 1H), 7.76 (d, J = 7.9 Hz, 1H), 7.88 (d, J = 7.7 Hz, 1H), 8.08 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.5, 31.1, 60.9, 109.8, 114.8, 119.4, 120.6, 122.6, 126.0, 126.3, 126.8, 126.9, 128.4, 129.6, 130.7, 130.9, 131.76, 133.2, 134.1, 135.8, 137.4, 166.9; HRMS (EI) calcd for C22H19NO2S 361.11370, found 361.11422.
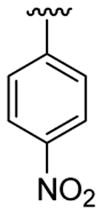
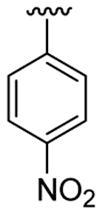
4.5.5. 2-(Cyclohex-1-enyl)-1-methyl-3-(4-nitrophenyl)indole (3e)
This product was obtained as a yellow solid in a 91% yield: mp 127–129 °C; 1H NMR (400 MHz, CDCl3) δ 1.71–1.75 (m, 4H), 2.11–2.12 (m, 2H), 2.24–2.27 (m, 2H), 3.73 (s, 3H), 5.92–5.95 (m, 1H), 7.20–7.24 (m, 1H), 7.29–7.33 (m, 1H), 7.39 (d, J = 8.2 Hz, 1H), 7.73 (d, J = 8.9 Hz, 2H), 7.78 (d, J = 8.9 Hz, 1H), 8.25 (d, J = 8.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.9, 23.0, 25.9, 29.8, 30.5, 110.0, 111.3, 118.9, 120.9, 122.4, 123.8, 126.2, 128.8, 129.4, 134.2, 137.1, 141.9, 143.7, 145.2; HRMS (EI) calcd for C21H20N2O2 332.1525, found 332.1534.
4.5.6. 2-(3-Methoxyphenyl)-1-methyl-3-phenylindole (3f)
This product was obtained as a colorless oil in a 91% yield: 1H NMR (400 MHz, CDCl3) δ 3.73 (s, 3H), 3.75 (s, 3H), 6.90 (s, 1H), 6.93–6.97 (m, 2H), 7.20–7.26 (m, 2H), 7.30–7.38 (m, 6H), 7.45 (d, J = 8.2 Hz, 1H), 7.84 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 31.1, 55.3, 109.8, 113.9, 115.3, 116.8, 119.8, 120.4, 122.4, 123.7, 125.7, 127.1, 128.3, 129.6, 130.0, 133.3, 135.4, 137.5, 137.6, 159.5; HRMS (EI) calcd for C22H19NO 313.1467, found 313.1471.
4.5.7. 4-[1-Methyl-3-(4-nitrophenyl)indol-2-yl]butyronitrile (3g)
This product was obtained as a yellow solid in a 72% yield: mp 157–159 °C; 1H NMR (400 MHz, CDCl3) δ 1.96 (quintet, J = 7.8, 7.0 Hz, 2H), 2.33 (t, J = 7.0 Hz, 2H), 3.10 (t, J = 7.8 Hz, 2H), 3.82 (s, 3H), 7.17–7.21 (m, 1H), 7.29–7.33 (m, 1H), 7.39 (d, J = 8.2 Hz, 1H), 7.60–7.62 (m, 3H), 8.34 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 17.0, 23.9, 25.9, 30.3, 109.6, 113.7, 118.8, 119.0, 121.0, 122.8, 124.4, 126.5, 130.0, 135.9, 137.2, 142.9, 146.1; HRMS (EI) calcd for C19H17N3O2 319.1321, found 319.1310.
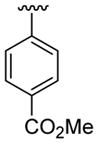
4.5.8. Methyl 4-[2-(3-cyanopropyl)-1-methylindol-3-yl]benzoate (3h)
This product was obtained as a yellow oil in a 63% yield: 1H NMR (400 MHz, CDCl3) δ 1.93 (quintet, J = 7.6, 7.0 Hz, 2H), 2.27 (t, J = 7.0 Hz, 2H), 3.06 (t, J = 7.6 Hz, 2H), 3.79 (s, 3H), 3.97 (s, 3H), 7.15–7.19 (m, 1H), 7.27–7.31 (m, 1H), 7.38 (d, J = 8.2 Hz, 1H), 7.54 (d, J = 8.3 Hz, 2H), 7.63 (d, J = 7.9 Hz, 1H), 8.16 (d, J = 8.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.7, 23.8, 25.8, 30.0, 52.2, 109.3, 114.5, 119.0, 119.1, 120.5, 122.3, 126.7, 127.9, 129.5, 130.2, 135.3, 137.1, 140.5, 167.1; HRMS (EI) calcd for C21H20N2O2 332.1525, found 332.1531.
4.5.9. 3-(4-Chlorophenyl)-2-(2-methoxyphenyl)-1-methylindole (3i)
This product was obtained as a colorless oil in an 87% yield: 1H NMR (400 MHz, CDCl3) δ 3.63 (s, 3H), 3.78 (s, 3H), 6.98 (t, J = 7.6 Hz, 1H), 7.05 (d, J = 8.2 Hz, 1H), 7.14 (dd, J = 7.4, 1.6 Hz, 1H), 7.22–7.31 (m, 5H), 7.35 (dt, J = 7.4, 0.8 Hz, 1H), 7.43–7.47 (m, 2H), 7.85 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 30.7, 55.6, 109.7, 111.2, 114.2, 119.4, 120.2, 120.7, 121.0, 122.1, 126.7, 128.4, 130.5, 130.6, 131.0, 133.3, 134.4, 135.2, 137.3, 158.5; HRMS (EI) calcd for C22H18ClNO 347.1077, found 347.1079.
4.5.10. 5-Bromo-2-(3,5-dimethoxyphenyl)-1-methyl-3-(thiophen-2-yl)indole (3j)
This product was obtained as a yellow solid in a 74% yield: mp 181–183 °C; 1H NMR (400 MHz, CDCl3) δ 3.63 (s, 3H), 3.77 (s, 6H), 6.53–6.55 (m, 3H), 6.95–6.96 (m, 1H), 6.99–7.01 (m, 1H), 7.17 (d, J = 5.0 Hz, 1H), 7.23–7.26 (m, 1H), 7.37–7.39 (m, 1H), 8.03 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 31.1, 55.5, 101.0, 108.0, 109.2, 111.2, 113.8, 122.3, 123.9, 125.2, 127.0, 128.4, 132.8, 135.6, 136.2, 139.2, 160.8; HRMS (EI) calcd for C21H18BrNO2S 427.0242, found 427.0251.
4.5.11. 5-Bromo-2-(3,5-dimethoxyphenyl)-1-methyl-3-(thiophen-3-yl)indole (3k)
This product was obtained as a yellow solid in a 76% yield: mp 172–174 °C; 1H NMR (400 MHz, CDCl3) δ 3.66 (s, 3H), 3.75 (s, 6H), 6.48–6.52 (m, 3H), 6.94 (d, J = 4.0 Hz, 1H), 7.15–7.16 (m, 1H), 7.23–7.26 (m, 2H), 7.36–7.38 (m, 1H), 7.93 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 31.3, 55.6, 100.8, 109.1, 110.0, 111.2, 113.7, 121.5, 122.4, 124.9, 125.1, 128.6, 128.7, 133.4, 134.7, 136.0, 138.8, 160.8; HRMS (EI) calcd for C21H18BrNO2S 427.0242, found 427.0251.
4.5.12. 5-Bromo-1-methyl-3-(thiophen-2-yl)-2-(thiophen-3-yl)indole (3l)
This product was obtained as a light yellow solid in an 85% yield: mp 151–153 °C; 1H NMR (400 MHz, CDCl3) δ 3.65 (s, 3H), 6.96–6.97 (m, 1H), 7.01–7.03 (m, 1H), 7.10 (d, J = 5.0 Hz, 1H), 7.20 (d, J = 5.0 Hz, 1H), 7.23–7.26 (m, 1H), 7.36–7.38 (m, 2H), 7.42–7.44 (m, 1H), 8.01 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 31.2, 108.7, 111.2, 113.9, 122.4, 124.1, 125.2, 125.3, 126.2, 127.16, 127.20, 128.4, 129.5, 131.0, 134.5, 135.8, 136.3; HRMS (EI) calcd for C17H12BrNS2 372.9595, found 372.9604.
4.5.13. 5-Bromo-1-methyl-2-phenyl-3-(thiophen-2-yl)indole (3m)
This product was obtained as a yellow solid in a 79% yield: mp 190–192 °C; 1H NMR (400 MHz, CDCl3) δ 3.61 (s, 3H), 6.90 (d, J = 2.8 Hz, 1H), 6.98 (t, J = 4.4 Hz, 1H), 7.15 (d, J = 4.6 Hz, 1H), 7.25–7.27 (m, 1H), 7.38–7.40 (m, 3H), 7.45–7.46 (m, 3H), 8.03 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 31.2, 108.3, 111.2, 114.0, 122.5, 123.9, 125.28, 125.32, 127.1, 128.6, 128.7, 129.0, 131.23, 131.26, 135.9, 136.4, 139.5; HRMS (EI) calcd for C19H14BrNS 367.0030, found 367.0040.
4.5.14. Ethyl 3-(1,5-dimethyl-2-phenylindol-3-yl)benzoate (3n)
This product was obtained as a yellow oil in a 68% yield: 1H NMR (400 MHz, CDCl3) δ 1.33 (t, J = 7.1 Hz, 3H), 2.48 (s, 3H), 3.63 (s, 3H), 4.31 (q, J = 7.1 Hz, 2H), 7.13 (dd, J = 8.3, 1.1 Hz, 1H), 7.29 (d, J = 8.3 Hz, 4H), 7.36 (m, 3H), 7.40 (dt, J = 7.7, 1.3 Hz, 1H) 7.56 (s, 1H), 7.84 (dt, J = 7.8, 1.3 Hz, 1H), 8.07 (t, J = 1.3 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.5, 21.8, 31.1, 60.9, 109.5, 113.8, 119.0, 124.1, 126.7, 127.1, 128.30, 128.33, 128.6, 129.9, 130.6, 131.0, 131.2, 131.9, 134.3, 135.9, 138.4, 166.9; HRMS (EI) calcd for C25H23NO2 369.1729, found 369.1732.
4.5.15. 3-(4-Chlorophenyl)-2-(4-methoxyphenyl)-1,5-dimethylindole (3o)
This product was obtained as a colorless solid in a 94% yield: mp 165–167 °C; 1H NMR (400 MHz, CDCl3) δ 2.46 (s, 3H), 3.61 (s, 3H), 3.82 (s, 3H), 6.90 (d, J = 8.3 Hz, 2H), 7.11 (d, J = 8.2 Hz, 1H), 7.19–7.22 (m, 6H), 7.27 (d, J = 8.3 Hz, 1H), 7.51 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 21.8, 31.1, 55.5, 109.5, 113.3, 114.2, 118.9, 123.9, 124.0, 127.1, 128.5, 129.8, 131.0, 131.1, 132.4, 134.3, 135.9, 138.1, 159.7; HRMS (EI) calcd for C23H20ClNO 361.1233, found 361.1241.
4.5.16. 2-([3-(3,4-Dimethoxyphenyl)]-1,5-dimethylindol-2-yl)ethanol (3p)
This product was obtained as a light brown oil in a 33% yield: 1H NMR (400 MHz, CDCl3) δ 1.66 (br s, 1H), 2.42 (s, 3H), 3.11 (t, J = 6.8 Hz, 2H), 3.74 (s, 3H), 3.81 (t, J = 6.8 Hz, 2H), 3.88 (s, 3H), 3.92 (s, 3H), 6.96 (d, J = 8.2 Hz, 1H), 7.01 (dd, J = 8.1, 1.8 Hz, 1H), 7.05 (d, J = 8.2 Hz, 2H), 7.22 (d, J = 8.3 Hz, 1H), 7.36 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 21.6, 28.5, 30.2, 56.1, 62.4, 108.9, 111.6, 113.6, 115.4, 118.8, 122.2, 123.3, 127.7, 128.3, 129.2, 133.4, 135.4, 147.7, 149.0; HRMS (EI) calcd for C20H23NO3 325.1678, found 325.1685.
4.5.17. Methyl 2-(3-methoxyphenyl)-1-methyl-3-(thiophen-3-yl)indole-5-carboxylate (3q)
This product was obtained as a yellow oil in a 70% yield: 1H NMR (400 MHz, CDCl3) δ 3.67 (s, 3H), 3.75 (s, 3H), 3.94 (s, 3H), 6.87 (s, 1H), 6.86 (m, 3H), 7.18–7.27 (m, 1H), 7.30–7.42 (m, 3H), 8.00 (d, J = 8.6 Hz, 1H), 8.57 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 31.3, 52.0, 55.4, 109.4, 111.8, 114.5, 116.5, 121.8, 122.3, 122.8, 123.4, 123.8, 124.9, 126.6, 128.8, 129.8, 132.8, 134.5, 138.9, 139.7, 159.7, 168.2; HRMS (EI) calcd for C22H19NO3S 377.1086, found 377.1095.
4.6. General procedure for the microwave-assisted, one-pot synthesis of 1H-indoles
In an oven-dried 20 mL microwave vial, N-trifluoroacetyl-2-iodoaniline (0.6 mmol) was dissolved in Et3N (4 mL), then PdCl2(PPh3)2 (12.6 mg, 0.018 mmol, 3 mol %), CuI (2.3 mg, 0.012 mmol, 2 mol %) and the alkyne (0.63 mmol) were added. The vial was flushed with Ar, sealed and stirred at 60 °C under microwave irradiation for 20–30 min. The resulting mixture was dissolved in CH3CN (4 mL), ArI (0.66 mmol) and Cs2CO3 (586 mg, 1.8 mmol) were added, and the vial was flushed with Ar, sealed and stirred at 100 °C under microwave irradiation for 30 min. To the reaction mixture were added ethyl acetate (10 mL) and brine (10 mL) and the aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic layers were dried over anhydrous MgSO4 and concentrated under vacuum to afford the crude product, which was purified by flash chromatography on silica gel using ethyl acetate/hexanes as eluent.
4.6.1. Ethyl 3-(2-phenylindol-3-yl)benzoate (3r)
This product was obtained as a yellow solid in a 93% yield: mp 145–147 °C; 1H NMR (400 MHz, CDCl3) δ 1.34 (t, J = 7.1 Hz, 3H), 4.34 (q, J = 7.1 Hz, 2H), 7.16 (t, J = 7.5 Hz, 1H), 7.22–7.29 (m, 4H), 7.37–7.43 (m, 4H), 7.54 (d, J = 7.7 Hz, 1H), 7.66 (d, J = 7.9 Hz, 1H), 7.97 (d, J = 7.8 Hz, 1H), 8.21 (s, 1H), 8.41 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.5, 61.1, 111.2, 114.1, 119.6, 120.8, 123.0, 127.6, 128.0, 128.4, 128.7, 128.8, 128.9, 131.0, 131.3, 132.5, 134.8, 134.9, 135.7, 136.1, 167.0; HRMS (EI) calcd for C23H19NO2 341.1416, found 341.1426.
4.6.2. 2-(3-Methoxyphenyl)-3-(4-methoxyphenyl)indole (3s)
This product was obtained as a yellow oil in an 82% yield: 1H NMR (400 MHz, CDCl3) δ 3.62 (s, 3H), 3.79 (s, 3H), 6.79 (ddd, J = 8.3, 2.6, 0.9 Hz, 1H), 6.89–6.99 (m, 4H), 7.09–7.22 (m, 3H), 7.32–7.37 (m, 3H), 7.62 (d, J = 7.9 Hz, 1H), 8.19 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 55.3, 55.4, 111.1, 113.5, 113.6, 114.2, 115.0, 119.8, 120.5, 122.8, 127.5, 129.1, 129.9, 131.4, 133.7, 134.2, 135.9, 158.3, 159.7; HRMS (EI) calcd for C22H19NO2 329.1416, found 329.1425.
4.6.3. 2-(4-Methoxyphenyl)-5-methyl-3-(3-nitrophenyl)indole (3t)
This product was obtained as an orange oil in an 88% yield: 1H NMR (400 MHz, CDCl3) δ 2.43 (s, 3H), 3.77 (s, 3H), 6.83 (d, J = 8.7 Hz, 2H), 7.06 (d, J = 7.8 Hz, 1H), 7.20–7.28 (m, 3H), 7.40–7.46 (m, 2H), 7.65 (d, J = 7.7 Hz, 1H), 8.06 (d, J = 8.2 Hz, 1H), 8.23 (s, 1H), 8.30 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 21.8, 55.4, 110.9, 111.3, 114.6, 118.3, 120.9, 124.5, 124.6, 124.7 128.4, 129.4, 129.7, 130.4, 134.2, 135.6, 136.4, 137.7, 148.7, 159.7; HRMS (EI) calcd for C22H18N2O3 358.1317, found 358.1325.
4.6.4. 4-(5-Methyl-3-phenylindol-2-yl)benzonitrile (3u)
This product was obtained as a yellow solid in a 66% yield: mp 219–221 °C; 1H NMR (400 MHz, acetone-d6) δ 2.37 (s, 3H), 7.02 (d, J = 8.2 Hz, 1H), 7.29–7.39 (m, 7H), 7.59 (s, 4H), 10.53 (s, 1H); 13C NMR (100 MHz, acetone-d6) δ 21.6, 110.7, 111.8, 116.7, 117.9, 119.0, 119.5, 125.5, 127.1, 128.9, 129.2, 129.4, 130.6, 132.5, 132.6, 135.6, 135.8, 138.0; HRMS (EI) calcd for C22H16N2 308.1313, found 308.1323.
4.6.5. 3-(4-Chlorophenyl)-5-methyl-2-(thiophen-3-yl)indole (3v)
This product was obtained as a light brown oil in a 67% yield: 1H NMR (400 MHz, CDCl3) δ 2.41 (s, 3H), 7.00 (d, J = 4.7 Hz, 1H), 7.05 (d, J = 8.0 Hz, 1H), 7.22–7.27 (m, 3H), 7.34 (s, 1H), 7.37 (s, 4H), 8.14 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 21.7, 110.7, 113.5, 119.0, 122.1, 124.6, 126.3, 127.1, 128.9, 130.1, 130.4, 130.5, 131.7, 132.3, 133.5, 133.9, 134.1; HRMS (EI) calcd for C19H14ClNS 323.0535, found 323.0542.
4.6.6. Methyl 3-(3-methoxyphenyl)-2-phenylindole-5-carboxylate (3w)
This product was obtained as an ivory solid in a 76% yield: mp 211–213 °C; 1H NMR (400 MHz, DMSO-d6) δ 3.70 (s, 3H), 3.83 (s, 3H), 6.87 (s, 1H), 6.93 (d, J = 7.9 Hz, 2H), 7.34–7.42 (m, 4H), 7.49 (d, J = 7.0 Hz, 2H), 7.53 (d, J = 8.5 Hz, 1H), 7.81 (d, J = 8.5 Hz, 1H), 8.15 (s, 1H), 12.01 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 51.7, 54.9, 111.5, 111.9, 114.2, 114.8, 115.4, 121.1, 122.1, 122.9, 127.6, 127.9, 128.2, 128.5, 129.9, 131.7, 135.7, 135.8, 138.5, 159.4, 167.0; HRMS (EI) calcd for C23H19NO3 357.1365, found 357.1374.
4.6.7. Methyl 3-[4-(ethoxycarbonyl)phenyl]-2-(4-methoxyphenyl)indole-5-carboxylate (3x)
This product was obtained as an ivory solid in a 60% yield: mp 264–266 °C; 1H NMR (400 MHz, DMSO-d 6) δ 1.33 (t, J = 7.0 Hz, 3H), 3.78 (s, 3H), 3.83 (s, 3H), 4.33 (q, J = 7.0 Hz, 2H), 6.98 (d, J = 8.6 Hz, 2H), 7.38 (d, J = 8.6 Hz, 2H), 7.49 (d, J = 8.1 Hz, 2H), 7.53 (d, J = 8.6 Hz, 1H), 7.81 (d, J = 8.4 Hz, 1H), 8.01 (d, J = 8.1 Hz, 2H), 8.16 (s, 1H), 12.04 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 14.2, 51.7, 55.2, 60.6, 111.4, 112.3, 114.2, 114.8, 120.5, 121.3, 122.9, 127.2, 127.5, 129.6, 129.7, 136.8, 138.6, 139.9, 159.3, 165.6, 166.9; HRMS (EI) calcd for C26H23NO5 429.1576, found 429.1588.
Acknowledgments
We thank the National Institute of General Medical Sciences (GM070620 and GM079593) and the National Institutes of Health Kansas University Center of Excellence for Chemical Methodologies and Library Development (P50 GM069663) for financial support of this project. We also thank Johnson Matthey, Inc. and Kawaken Fine Chemicals Co., Ltd. for donations of palladium catalysts.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.For selected recent reviews on the biological and pharmaceutical importance of indoles, see: Weng JR, Tsai CH, Kulp SK, Chen CS. Cancer Lett. 2008;262:153. doi: 10.1016/j.canlet.2008.01.033.Rieck GC, Fiander AN. Mol Nutr Food Res. 2008;52:105. doi: 10.1002/mnfr.200700138.Brancale A, Silvestri R. Med Res Rev. 2007;27:209. doi: 10.1002/med.20080.
- 2.For selected recent reviews on the synthesis of indoles, see: Humphrey GR, Kuethe JT. Chem Rev. 2006;106:2875. doi: 10.1021/cr0505270.Cacchi S, Fabrizi G. Chem Rev. 2005;105:2873. doi: 10.1021/cr040639b.Li JJ, Gribble GW. Palladium in Heterocyclic Chemistry. Pergamon. 2000;Chapter 3:73.Gribble GW. J Chem Soc, Perkin Trans 1. 2000:1045.
- 3.Zeni G, Larock RC. Chem Rev. 2004;104:2285. doi: 10.1021/cr020085h. [DOI] [PubMed] [Google Scholar]
- 4.(a) Sonogashira K. In: Metal-Catalyzed Cross-Coupling Reactions. Diederich F, Stang PJ, editors. Chapter 5. Wiley-VCH; Weinheim, Germany: 1998. p. 203. [Google Scholar]; (b) Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975;50:4467. [Google Scholar]
- 5.(a) Arcadi A, Cacchi S, Marinelli F. Tetrahedron Lett. 1992;33:3915. [Google Scholar]; (b) Cacchi S, Fabrizi G, Pace P. J Org Chem. 1998;63:1001. [Google Scholar]; (c) Arcadi A, Cacchi S, Fabrizi G, Marinelli F. Synlett. 2000:394. [Google Scholar]; (d) Arcadi A, Cacchi S, Cassetta A, Fabrizi G, Parisi LM. Synlett. 2001:1605. [Google Scholar]; (e) Battistuzzi G, Cacchi S, Fabrizi G. Eur J Org Chem. 2002:2671. [Google Scholar]; (f) Cacchi S, Fabrizi G, Lamba D, Marinelli F, Parisi LM. Synthesis. 2003:728. [Google Scholar]; (g) Cacchi S, Fabrizi G, Parisi LM. Synthesis. 2004:1889. [Google Scholar]; (h) Arcadi A, Cacchi S, Fabrizi G, Marinelli F, Parisi LM. J Org Chem. 2005;70:6213. doi: 10.1021/jo050517z. [DOI] [PubMed] [Google Scholar]
- 6.(a) Chaplin JH, Flynn BL. Chem Commun. 2001:1594. doi: 10.1039/b104624c. [DOI] [PubMed] [Google Scholar]; (b) Flynn BL, Hamel E, Jung MK. J Med Chem. 2002;45:2670. doi: 10.1021/jm020077t. [DOI] [PubMed] [Google Scholar]
- 7.Lu BZ, Zhao W, Wei HX, Dufour M, Farina V, Senanayake CH. Org Lett. 2006;8:3271. doi: 10.1021/ol061136q. [DOI] [PubMed] [Google Scholar]
- 8.For selected recent reviews on the topic of microwave-assisted organic synthesis, see: Kappe CO. Chem Soc Rev. 2008;37:1127. doi: 10.1039/b803001b.Polshettiwar V, Varma RS. Chem Soc Rev. 2008;37:1546. doi: 10.1039/b716534j.Polshettiwar V, Varma RS. Pure Appl Chem. 2008;80:777.Polshettiwar V, Varma RS. Acc Chem Res. 2008;41:629. doi: 10.1021/ar700238s.Dallinger D, Kappe CO. Chem Rev. 2007;107:2563. doi: 10.1021/cr0509410.
- 9.(a) Larock RC, Yum EK. J Am Chem Soc. 1991;113:6689. [Google Scholar]; (b) Larock RC, Yum EK, Refvik MD. J Org Chem. 1998;63:7652. [Google Scholar]
- 10.(a) Yue D, Larock RC. Org Lett. 2004;6:1037. doi: 10.1021/ol0498996. [DOI] [PubMed] [Google Scholar]; (b) Yue D, Yao T, Larock RC. J Org Chem. 2006;71:62. doi: 10.1021/jo051549p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Cho CH, Larock RC. Org Lett. 2009;11:173. doi: 10.1021/ol8021287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cadogan JIG, Hickson CL, Husband JB, McNab H. J Chem Soc, Perkin Trans 1. 1985:1891. [Google Scholar]
- 13.Yao T, Yue D, Larock RC. J Comb Chem. 2005;7:809. doi: 10.1021/cc050062r. [DOI] [PubMed] [Google Scholar]
- 14.Palimkar SS, More VS, Kumar PH, Srinivasan KV. Tetrahedron. 2007;63:12786. [Google Scholar]
- 15.Kabalka GW, Wang L, Pagni RM. Tetrahedron. 2001;57:8017. [Google Scholar]
- 16.Harper S, Pacini B, Avolio S, Di Filippo M, Migliaccio G, Laufer R, De Francesco R, Rowley M, Narjes F. J Med Chem. 2005;48:1314. doi: 10.1021/jm049122i. [DOI] [PubMed] [Google Scholar]