

Summary
Hematopoiesis is maintained by stem cells (HSCs) that undergo fate decisions while integrating intrinsic and extrinsic signals, the latter principally derived from the bone marrow (BM) microenvironment. Cell cycle regulation can modulate stem cell fate. Whether this represents an intrinsic or extrinsic effector of fate decisions is unknown. We have investigated the role of the retinoblastoma protein (Rb), a central regulator of the cell cycle, in hematopoiesis. Widespread inactivation of Rb in the murine hematopoietic system resulted in profound myeloproliferation. HSCs were lost from the BM due to mobilization to extramedullary sites and differentiation. This phenotype was not intrinsic to HSCs, but rather the consequence of an Rb dependent interaction between myeloid-derived cells and the microenvironment. These findings demonstrate that myeloproliferation may result from perturbed interactions between hematopoietic cells and the niche. Therefore, Rb extrinsically regulates HSCs by maintaining the capacity of the BM to support normal hematopoiesis and HSCs.
Introduction
Under homeostatic conditions, the adult hematopoietic system is maintained by a small number of stem cells (HSCs) that reside in the bone marrow in a specialized microenvironment, termed the niche (Adams and Scadden, 2006; Schofield, 1978). It is within the niche that HSCs undertake fate decisions, including differentiative divisions to generate progenitor cells, and self-renewal divisions necessary to sustain HSCs throughout life. Both intrinsic and extrinsic cues are integrated within the niche to maintain effective control over HSCs, ensuring contribution to hematopoiesis without aberrant proliferation (Fuchs et al., 2004; Moore and Lemischka, 2006). Whereas the majority of HSCs are in a slowly dividing state, termed relative quiescence, with a cell division cycle in the mouse in the range of 2-4 weeks, progenitor cells exhibit rapid cycling (Bradford et al., 1997; Passegue et al., 2005). HSCs can also be stimulated to rapidly enter the cell cycle and contribute to hematopoiesis (Li and Johnson, 1994). In part, the dramatic contrast in cell cycle status between stem and progenitor cells has led to the hypothesis that cell cycle regulation plays a fundamentally important role in stem cell fate determination.
Decisions to enter the cell cycle are regulated by the G1-S phase restriction point (Sherr and Roberts, 2004). The sequential phosphorylation and subsequent inactivation of the retinoblastoma protein (Rb) is an important part of this transition (Weinberg, 1995). Rb is phosphorylated by cyclin-cyclin dependent kinase (Cdk) complexes. Several negative regulators of Cdk activity have been studied in the context of HSC biology. Loss of the Cdk2-inhibitors p21Cip1 and p27Kip1 revealed a divergent role in HSC regulation with loss of p21Cip1 resulting in a subtle increase in sensitivity to stress induced exhaustion apparent in vivo after quaternary transplant (Cheng et al., 2000). Loss of p27Kip1 resulted in a 2-fold increase in the number of long-term repopulating HSCs, in addition to an enlarged progenitor compartment (Walkley et al., 2005). Loss of both Cdk4/6-inhibitors p16Ink4a and p19ARF revealed a small increase in serial transplant potential (Stepanova and Sorrentino, 2005), with a similar phenotype observed in p16Ink4a single mutant HSCs (Janzen et al., 2006). Loss of p18Ink4c resulted in increased HSC repopulation and frequency (Yuan et al., 2004).
Collectively, these studies suggest that negative cell cycle regulators that impact directly on Rb-family protein function may influence HSC fate. It is indeterminate if these phenotypes reflect intrinsic or extrinsic effects on HSCs and hematopoiesis, as all studies to date have utilized non-conditional mutant alleles that are not hematopoietic-restricted in their effects. The analysis of HSCs from germ-line deficient animals does not allow for the clear delineation of intrinsic and extrinsic contribution to the observed HSC phenotype. Such studies have largely not accounted for effects on HSC genesis or potentially defective niche support that affect HSCs prior to transplantation analysis. While serial transplant studies are suggestive of an intrinsic role for Cdkis in HSC biology, they do not exclude a role for the environment from which these cells were removed, necessitating analysis utilizing hematopoietic restricted deletion. Indeed, a recent study demonstrated that the p27Kip1-/- microenvironment mediates lymphoid expansion observed in the p27Kip1-/-animals, possibly indicating that the HSC expansion observed in p27Kip1-/- bone marrow is extrinsic in nature (Chien et al., 2006; Walkley et al., 2005). This result suggests that cell cycle regulators may play a role in regulating the competence of the hematopoietic niche, in addition to intrinsic roles in HSC fate determination.
Recent studies have begun to characterize the adult bone marrow niche (Schofield, 1978). Osteoblasts appear to comprise an important component of the HSC niche, as modulation of osteoblast number and function influences hematopoiesis and HSC fate via extrinsic mechanisms (Calvi et al., 2003; Visnjic et al., 2004; Zhang et al., 2003). Additionally, numerous extrinsic factors modulate HSC function. These factors include retinoic acid, extracellular calcium, osteopontin, angiopoietins and Notch ligands (Adams et al., 2006; Arai et al., 2004; Purton et al., 2000; Stier et al., 2005; Varnum-Finney et al., 1998; Zhang et al., 2006a). Extrinsic regulation of homeostatic HSC numbers can be dominant to intrinsic cues in vivo. For example, HSCs engineered to overexpress HoxB4 expand in vivo only to the level of normal HSCs, despite markedly enhanced in vitro self-renewal and proliferative capacity (Krosl et al., 2003). Additionally, systemic factors contained in the peripheral blood of young animals may reactivate self-renewal associated pathways in progenitors of older animals, suggesting an important role for extrinsic signalling in stem cell regulation (Conboy et al., 2005). While these studies have begun to define the bone marrow niche, little is currently known regarding molecular regulators of the niche and their role in influencing HSC fate decisions. Regulatory interactions between the hematopoietic cells and the non-hematopoietic derived microenvironment are largely unknown. Moreover, the regulators of these potential interactions, and how they affect hematopoiesis and HSC function, are unexplored.
Here we have utilized a conditional deletion strategy to investigate the role of the Rb in the regulation of adult HSC fate. We found that widespread inactivation of Rb resulted in the development of a myeloproliferative disease, characterized by extramedullary hematopoiesis and mobilization of primitive cells into the periphery. HSCs were lost from the BM as a result of increased differentiation and mobilization from the BM. The phenotype is not HSC intrinsic, as it was not recapitulated upon inactivation of Rb in HSCs maintained in a wild-type environment (Walkley and Orkin, 2006). Strikingly, however, concomitant deletion of Rb from myeloid-derived cells and the microenvironment generated the myeloproliferative disorder, thereby demonstrating that Rb is an essential regulator of the interaction between myeloid-derived cells and the BM microenvironment. Thus, Rb extrinsically controls HSCs by maintaining the competence of the BM to support normal HSCs and hematopoiesis.
Results
Rb Deletion Leads to Myleoproliferation
Rb was inactivated in hematopoietic cells, including HSCs, using the interferon inducible Mx-Cre transgene and pRbfl/fl animals (Kuhn et al., 1995; Sage et al., 2003; Walkley and Orkin, 2006). We performed PCR on both genomic DNA and cDNA from whole BM samples to confirm Rb deletion (BM, Fig 1A, 1B). Rb was quantitatively and stably deleted from hematopoietic cells, and expression of the related p130 and p107 was not altered as a result of Rb loss. Thus, with this conditional system, we achieve specific loss of Rb without compensatory gain of expression of other genes coding for pocket proteins.
Figure 1.
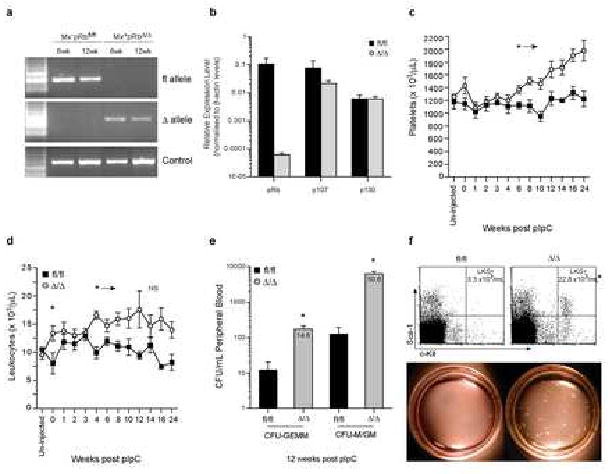
Rapid Mobilization of Primitive Cells into the Peripheral Blood following Deletion of Rb.
A) Genomic PCR on whole BM from control (Mx-pRbfl/fl) and Rb deficient animals (Mx+pRbΔ/Δ) at 6 and 12 wks post-pIpC. B) qRT-PCR for pRb, p130 and p107 on cDNA of control and Rb deficient animals (n=3 independent samples) 12 wks post-pIpC. C) Platelets and D) Leukocytes in PB following Rb deletion (time 0 = final dose of pIpC); n≥4/time point; *P<0.05. e) Day 12 CFU-GEMM and CFU-GM/M from the PB of at 12 wks post-pIpC; n≥9/genotype; *P<0.01. Value inside bars represents fold increase. F) FACS profile and mean number of Lin-c-Kit+Sca-1+ (LKS+) in the PB; n>4/genotype; *P<0.01. Methylcellulose plates from day 12 of culture. Data expressed as mean ± SEM.
Analysis of the peripheral blood of control (Mx-Cre-pRbfl/fl, pIpC injected) and RbΔ/Δ animals following pIpC treatment revealed that RbΔ/Δ animals developed a mild but stable anemia immediately following Rb deletion (unpublished data C.R.W and S.H.O) and by 6 weeks developed thrombocytosis (Fig 1C). By 4 weeks, RbΔ/Δ animals developed pan-leukocytosis (Fig 1D) that was accompanied by elevated levels of circulating progenitor cells, as determined by in vitro colony forming capacity (CFU-GEMM and CFU-G/GM) and phenotypic staining (lin-c-Kit+Sca-1+, LKS+; Fig 1e, 1f (Okada et al., 1992)). Although leukocytosis was apparent by 4 weeks post Rb deletion, increased circulating progenitors could be detected as early as 2 weeks after pIpC (LKS+ increased 3.7-fold, P≤0.01, n=7 per genotype; CFU-GEMM increased 2.4-fold, P≤0.01, CFU-M/GM increased 3.9-fold, P≤0.01, n=6 per genotype). Surprisingly, the levels of circulating progenitors were comparable to those achieved during pharmacologically induced mobilization of stem and progenitors in the C57Bl/6 strain background (Ghiaur et al., 2006). However this was a chronic, rather than an acute, response in the RbΔ/Δ mutant.
BM cellularity was not initially altered, however at 12-weeks post pIpC it was increased by 40% in RbΔ/Δ animals (Fig 2A). We observed the rapid development of a myeloproliferative-like disease within the bone marrow. The phenotype was fully penetrant and characterized by myeloid hyperplasia (predominantly neutrophilia) and suppression of both B-lymphopoiesis and erythropoiesis (Fig 2B, Supp Fig 1 and 2). Phenotypic stem and primitive progenitor populations (LKS- and LKS+) were increased significantly in the BM of RbΔ/Δ animals (Fig 2C), however the number of phenotypic HSCs per femur was not significantly altered (LKS+CD34-/lo) (Osawa et al., 1996; Yang et al., 2005). In addition, RbΔ/Δ animals exhibited striking changes in the architecture of the bone, evidenced by loss of trabecular bone (Fig 2D). Trabecular bone is thought to represent an important niche for HSCs within the BM (Calvi et al., 2003). Quantitative histomorphometric analysis of the bone at 2 weeks post pIpC, a time point correlating with the presence of progenitors in the peripheral blood and spleen, demonstrated a significant reduction in trabecular volume as a proportion of total marrow volume, a ∼40% reduction in the number of trabeculae, and a doubling of the separation of trabeculae (Fig 2E-2H).
Figure 2.
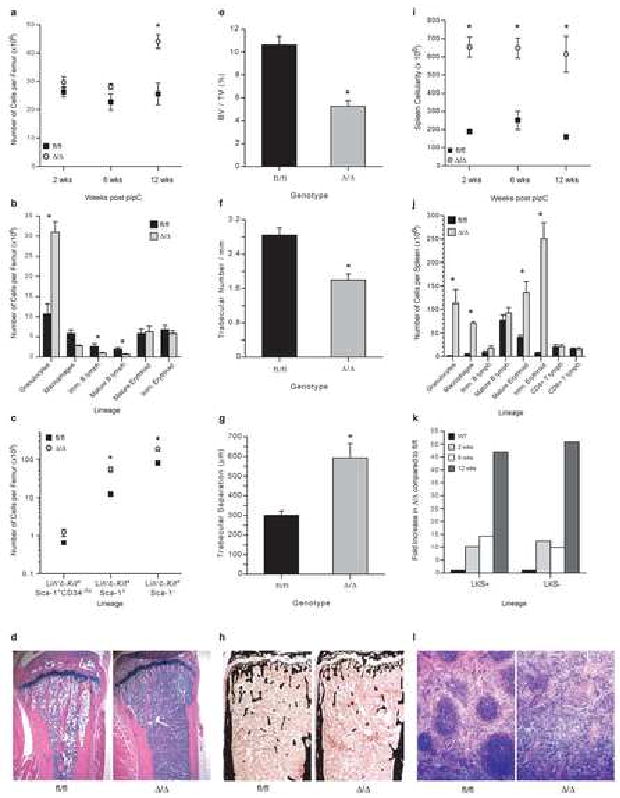
Myeloproliferation following Rb deletion.
A) Femoral cellularity, n≥3/genotype/time point. B) Number of cells of each lineage/femur at 12 wks post pIpC; Granulocytes CD11b+Gr-1+, Macrophages CD11b+F4/80+, Immature B lymphoid IgM-B220+, Mature B lymphoid IgM+B220+, Mature Erythroid CD71-Ter119+, Immature Erythroid CD71+Ter119+; n≥8/genotype; *P<0.01. C) Number of phenotypic HSCs (LKS+CD34-/lo) and primitive progenitors/femur; 12 wks post pIpC; n≥5/genotype; *P<0.05. D) Representative sections of tibiae at 12 wks post pIpC. E) Volume of marrow space occupied by bone (BV/TV); 2 wks post pIpC; n≥13/genotype; *P<0.05. F) Trabecular number/mm; *P<0.05. G) Separation of trabeculae; *P<0.05. H) Representative longitudinal sections of tibiae stained with Von Kossa technique (mineralised bone stained black). I) Spleen cellularity; n≥3/genotype/time point; *P<0.01. J) Number of cells of each lineage/spleen; 12 wks post pIpC; n≥8/genotype; *P<0.01. K) Fold change in phenotypic LKS+ and LKS- in the spleen; n≥3/genotype/time point; P<0.05. L) Representative spleen sections (12 wks post pIpC). Unless noted all data expressed as mean ± SEM.
In parallel with BM myeloproiferation, RbΔ/Δ animals developed extensive extramedullary hematopoiesis. Spleen weight increased rapidly by 5.5-fold relative to controls due to expanded numbers of myeloid cells, megakaryocytes and erythroid cells (Fig 2I and 2J). B- and T-cell lymphopoiesis was present at comparable levels in RbΔ/Δ and control spleens. Phenotypic stem and progenitor populations (LKS+ and LKS-) increased progressively in the spleens of RbΔ/Δ animals, and by 12 weeks were increased 45-50-fold (Fig 2K, Supp Fig 3 and 4). Splenic architecture was effaced as a result of myeloid and erythroid elements (Fig 2J). Hematopoietic foci were also observed in the liver, but not in the kidney (data not shown). Despite chronic myeloproliferation, no hematopoietic tumors have developed during the lifespan of RbΔ/Δ mutant animals. RbΔ/Δ animals survive for approximately 8 months post pIpC; heterozygous animals are normal (Supp Fig 5 and 6). Eight month old RbΔ/Δ animals present with a phenotype reminiscent of hematopoietic failure, characterized by a significant reduction in spleen weight and replacement of BM by granulocytes; however, pituitary tumors are also observed (Supp Figure 5 and data not shown).
HSCs are Lost From BM Following Rb Deletion
Rb and other negative cell cycle regulators have been postulated to play an important role in the regulation of HSCs, and subsequent hematopoiesis. However, neither the myeloproliferative disease nor defective HSC function was observed when Rb was deleted from HSCs in the context of a wild-type microenvironment (Walkley and Orkin, 2006). Given the striking phenotype we observed when Rb was deleted from both hematopoietic cells and the BM microenvironment, as occurs with Mx-Cre (Zhang et al., 2003), we sought to determine the consequences of Rb loss on HSCs in these animals.
Within the BM we observed a significant increase in the frequency of mature day 7 colony-forming cells, but a decrease in the frequency of the more primitive in vivo day 12 colony-forming unit-spleen (CFU-S12, Fig 3A, 3B). The numbers of both in vitro colony-forming cells (68-fold increase in CFU-GEMM) and CFU-S8 in the spleen were markedly increased (Fig 3C, 3D). As we observed high levels of circulating progenitors and substantial levels of progenitor activity in the spleen, we sought to determine if latent HSC activity was also present in extramedullary sites. Whole spleen cells from RbΔ/Δ animals (either 1×106 or 2×106) were transplanted with competitor whole BM (2×105) into congenic recipients. At 17-weeks post transplant, significant multilineage repopulating activity derived from the spleens of RbΔ/Δ animals was present, demonstrating that functional HSCs were present in the periphery of RbΔ/Δ mice (Fig 3E, 3F).
Figure 3.
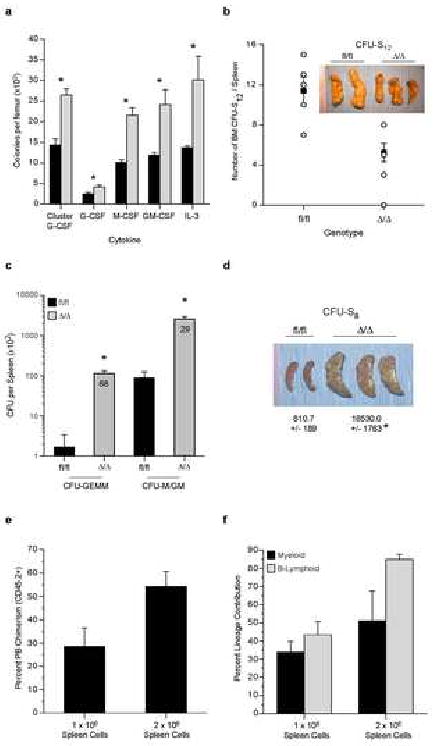
Increase in Progenitors in the Bone Marrow and HSCs/Progenitors in the Spleen of RbΔ/Δ Animals.
A) Number of day 7 CFC per femur; n≥5/genotype/time point; *P<0.05. B) Number of CFU-S12 from 1 × 105 whole BM cells; n=5/genotype. C) Splenic Day 12 CFU-GEMM and CFU-GM/M at 12 wks post-pIpC; n≥9/genotype; *P<0.01. Value inside bars is fold increase. D) CFU-S8/spleen; n=5/genotype; P<0.05. E) Percent PB chimerism at 17 wks post transplant from either 1 × 106 or 2 × 106 whole spleen cells from RbΔ/Δ animals 8 wks post pIpC with 2 × 105 WT BM cells; n=5/genotype. F) Lineage contribution of spleen derived HSCs at 17 wks. Data expressed as mean ± SEM.
To determine the HSC content of the BM, we performed limit-dilution competitive repopulation analysis with whole BM from donor animals treated 12-weeks earlier with pIpC (Purton et al., 2006; Szilvassy et al., 1990; Walkley et al., 2005). Whole BM from control (Rbfl/fl) or Rb deficient (RbΔ/Δ, both CD45.2+) mice was mixed at varying doses with a fixed number of competitor BM cells (CD45.1+/CD45.2+) and transplanted into congenic recipient animals (CD45.1+). The frequency of long-term repopulating HSCs in the RbΔ/Δ BM at 6 months post transplant was reduced by 8-fold (P=0.0005). When normalized to reflect the increased cellularity of the RbΔ/Δ BM, this represents a 5-fold decrease in the absolute number of HSCs per femur (Fig 4A). Secondary transplantation demonstrated that RbΔ/Δ HSCs were serially transplantable and capable of stable multilineage contribution for at least 3 months. Importantly, we did not observe a progressive decline in contribution from RbΔ/Δ HSCs to hematopoiesis, thereby demonstrating that self-renewal mediated maintenance of HSCs over time is not affected by the absence of Rb (Supp Fig 7).
Figure 4.
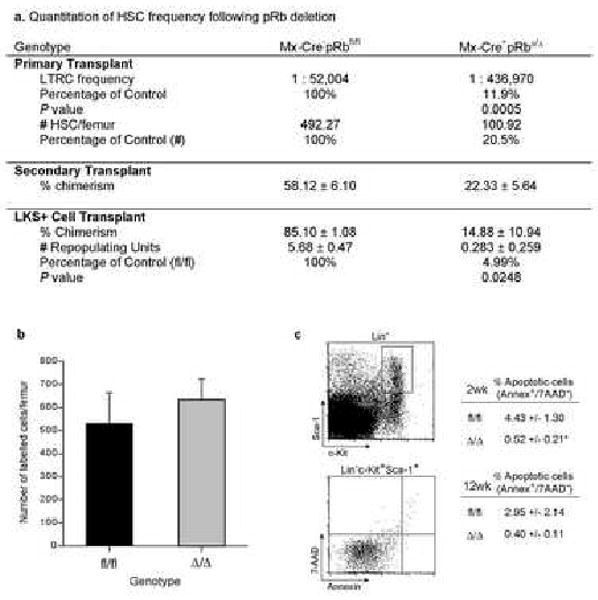
Loss of HSCs from the Bone Marrow Following Rb Deletion.
A) HSC frequency and absolute number/femur; n=4-5 recipients/cell dose/genotype; experiment performed twice; data pooled from 2 independent experiments for calculation of HSC frequency. Primary transplant data from 6 mths post transplant. Secondary transplant data from 3 mths post transplant. 1000 freshly isolated LKS+ were transplanted and RU calculated 6 mths post transplant; n=5 recipients/cell dose/genotype. B) Number of CFDA-SE labelled cells in the BM of recipients 16 hours after injection; BM from 12 wks post pIpC; n=5 recipients/genotype. C) Apoptotic cells in the LKS+ and LKS- populations; n=9/genotype; P<0.05. Data expressed as mean ± SEM.
When 1000 freshly isolated lin-c-Kit+Sca-1+ cells were competitively transplanted, RbΔ/Δ LKS+ displayed a 20-fold reduction in long-term repopulating potential on a per cell basis (Fig 4A). The reduced repopulating potential of the RbΔ/Δ LKS+ fraction, despite a marked increase in this phenotypic population observed in the bone marrow (see Fig 2), demonstrates that the surface phenotype of the cells does not faithfully reflect their functional potential. We, and others, have previously observed a lack of fidelity of phenotypic makers both in mutant mice and following perturbation of homeostasis in wild-type animals (Purton et al., 2006; Spangrude et al., 1995; Tajima et al., 2000; Walkley et al., 2005).
HSCs may be lost from the BM for several reasons, including a failed capacity of RbΔ/Δ HSCs to home and engraft following transplantation, an increased rate of apoptosis, or a mobilization/redistribution to extramedullary sites. As cell cycle status correlates with engraftment capacity of HSCs (Gothot et al., 1998; Passegue et al., 2005), we directly assessed the cell cycle status of phenotypic RbΔ/Δ progenitors (LKS-), primitive progenitors (LKS+) and HSCs (LKS+CD34-/lo). All three populations displayed a comparable cell cycle profile, with HSCs from RbΔ/Δ displaying the same distribution of cells in the G0/G1 or S-phase of the cell cycle as control cells (fl/fl: G0/G1= 86.2 ± 5.7%; Δ/Δ: G0/G1= 85.1 ± 2.4%, P=0.85, fl/fl: S= 6.7 ± 1.9%; Δ/Δ: S= 7.8 ± 0.7%, P=0.54, n=4 fl/fl, 7 Δ/Δ; expressed as mean ± sem). There was no difference in the rate of cell cycle entry of LKS+ cells between control and RbΔ/Δ cells as determined by BrdU incorporation rates at either 2 or 4 weeks post pIpC (Supp Fig 9). Furthermore, analysis of the in vivo homing of RbΔ/Δ BM did not reveal a difference compared to control BM at either 2 or 12-weeks post pIpC (Fig 4B and data not shown). RbΔ/Δ LKS+ cells exhibited decreased apoptosis, as assessed by annexin-V staining, at 2-weeks and normal levels at 12-weeks post pIpC compared to control LKS+ cells (Fig 4C). As we had observed significantly increased progenitors and HSCs in extramedullary sites, these data are consistent with the loss of HSCs from the BM as a result of both enhanced differentiation of HSCs within the BM and a redistribution to extramedullary sites as a consequence of the changes in the niche.
Myeloid-Restricted Inactivation of Rb Does Not Result in Myeloproliferation
To determine the contribution of myeloid-derived cells (granulocytes, macrophages and osteoclasts) to the myeloproliferation observed in Rb mutants, we generated Lysozyme-M-Cre pRbfl/fl mice to achieve myeloid-restricted deletion of Rb (Fig 5). Deletion of Rb with Lys-M-Cre did not lead to myeloproliferation or extramedullary hematopoiesis, consistent with the results obtained by deletion of Rb from hematopoietic cells in a wild-type environment (Fig 5A; (Walkley and Orkin, 2006)). We observed a subtle increase in the numbers of granulocytes in the BM and slight reduction in erythroid cells, but no change in either lymphoid or phenotypic progenitor and HSC enriched fractions (Fig 5A, 5B). Lineage distribution within the spleen or PB was largely comparable to controls (Sup Table 1 and 2; and data not shown). Thus, deletion of Rb from myeloid-derived populations does not recapitulate the phenotype observed in the Mx-Cre model. Collectively, these results suggest that Rb may regulate HSCs and hematopoiesis in an extrinsic manner, possibly through regulating the competence of the bone marrow niche.
Figure 5.
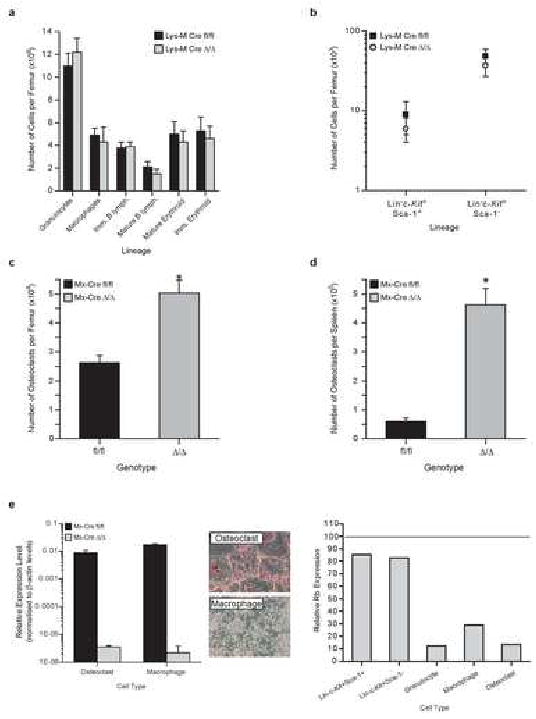
Myeloid Restricted Rb deletion Does Not Result in a Myeloproliferation.
A) BM analysis; n≥6/genotype. B) Phenotypic primitive progenitors/femur; n≥6/genotype. C) Number of osteoclasts/femur; 6 wks post pIpC; n=3/genotype. D) Number of osteoclasts/spleen; 6 wks post pIpC; n=3/genotype. E) Rb excision analysis of osteoclasts and macrophages derived from either Mx-Cre mutants of Lysozyme-M-Cre-pRbfl/fl mutants; n≥3/genotype. Representative photographs of osteoclast and macrophage cultures (original magnification 10×).
An Rb Dependent Interaction Between Myeloid-Derived Cells and the BM Microenvironment Results in Myeloproliferation
We next sought to determine the relative contributions of the hematopoietic cells and the non-hematopoietic (non-transplantable) elements of the BM microenvironment to the observed phenotype. Hematopoietic cells alone were not capable of inducing either myeloproliferation or the loss of HSCs from the BM that we observed in the Mx-CrepRbΔ/Δ model (Walkley and Orkin, 2006). Consistent with this conclusion we did not observe myeloproliferation when previously excised RbΔ/Δ HSCs were supported by a wild-type microenvironment, even at high cell doses (described in Fig 3).
To ascertain if Rb loss from the niche was responsible for the myeloproliferation and loss of BM HSCs, reciprocal transplants of wild-type hematopoietic cells into lethally irradiated Mx-Cre-pRbfl/fl and Mx-Cre+pRbfl/fl recipients were performed, and following establishment of hematopoiesis, recipients were injected with pIpC to delete Rb from the hematopoietic microenvironment. This strategy was successfully used to demonstrate a role for BMP receptor type 1 in the regulation of the HSC niche (Zhang et al., 2003), and a similar approach demonstrated that a RARγ-/- microenvironment alone could induce myeloproliferation (Purton et al, accomp manuscript). Following inactivation of Rb, recipients were monitored and analyzed at 8 and 20 weeks post transplant. We failed to observe significant changes in hematological parameters of these recipients (Fig 6A). The data show that loss of Rb uniquely in either hematopoietic cells or niche cells alone is insufficient to account for the findings in Mx-Cre+pRbΔ/Δ mice.
Figure 6.
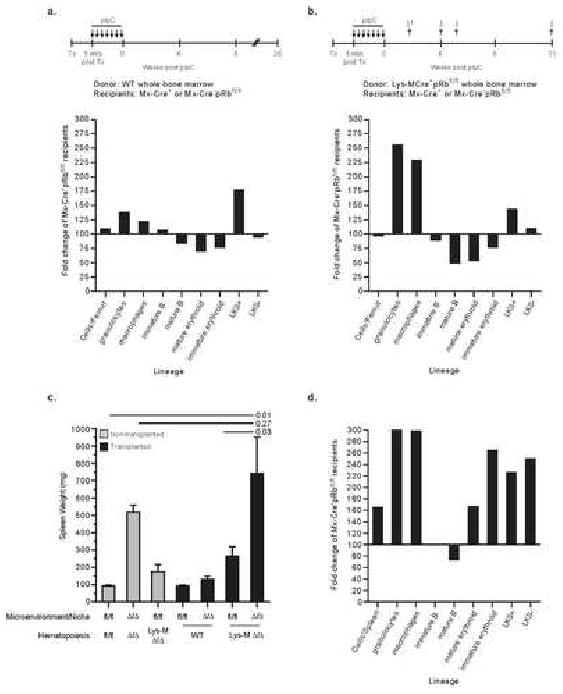
A pRb dependent interaction between myeloid cells and the bone marrow microenvironment causes myeloproliferation
A) Wild-type BM was transplanted into un-excised Mx-Cre-pRbfl/fl and Mx-Cre+pRbfl/fl recipients and 5 wks post transplant recipients received pIpC. Recipients were analyzed at 8- (n=3/genotype) and 20-wks (n=4/genotype) post pIpC. Data expressed as fold change of Mx+ recipients compared to Mx-Cre-pRbfl/fl recipients (normalized to 100%). Data shown from 8-wks post pIpC (comparable results at 20-wks). B) Sex mismatched Lysozyme-M-Cre+pRbfl/fl BM was transplanted into un-excised Mx-Cre-pRbfl/fl and Mx-Cre+pRbfl/fl recipients and 5 wks post transplant pIpC was administered. Data expressed as fold change of Mx+ recipients compared to Mx-Cre-pRbfl/fl recipients (normalized to 100%). † = approximate time of analysis post pIpC; n=11 Mx-Cre+ recipients in three independent experiments (2 found dead 2 weeks post pIpC); n=10 Mx-Cre- control recipients. C) Comparison of spleen weights amongst groups. D) Splenic hematopoiesis in Mx-Cre+pRbfl/fl recipients of Lysozyme-M-Cre+pRbfl/fl bone marrow. Data expressed as fold change compared to Mx-Cre-pRbfl/fl recipients (normalized to 100%).
Bone homeostasis is maintained through balanced activities of mesenchymal derived osteoblasts and myeloid-derived osteoclasts (Martin and Sims, 2005). As we observed a rapid loss of trabecular bone following Mx-Cre–mediated deletion, we quantitated the numbers of osteoclasts present in the BM and spleen. The numbers of osteoclasts in both the BM and spleen by 6 weeks post pIpC were markedly increased (Fig 5C and 5D). Osteoclasts and macrophages derived from pIpC-treated Mx-Cre mice showed efficient deletion of Rb, as did osteoclasts and macrophages derived from Lysozyme-M-Cre pRbfl/fl mice (Fig 5E). Osteoclasts have been proposed to contribute to the release of HSCs from the bone marrow during mobilization (Kollet et al., 2006). Having observed significantly increased neutrophils and monocytic derived osteoclasts in the Mx-Cre+pRbΔ/Δ model (Fig 5C), we hypothesized that deletion of Rb from myeloid-derived cells together with an Rb deficient microenvironment might recapitulate the phenotype observed in the Mx-Cre+pRbΔ/Δ model.
Hematopoietic cells from sex-mismatched Lysozyme-M-Cre+pRbfl/fl animals were transplanted into lethally irradiated Mx-Cre-pRbfl/fl and Mx-Cre+pRbfl/fl recipients, and following establishment of hematopoiesis, recipients were injected with pIpC to delete Rb from the BM microenvironment. Analysis of Y chromosome levels by quantitative PCR on peripheral blood leukocytes confirmed engraftment and high-level chimerism of all recipients prior to pIpC and at the time of analysis (data not shown). This transplant strategy resulted in Rb-deficiency in myeloid-derived cells (granulocytes, macrophages and osteoclasts) and an Rb-deficient niche. In addition, Rb expression is retained within the HSC compartment.
We observed synergistic interaction between the pRbΔ/Δ myeloid cells and the pRbΔ/Δ microenvironment (Fig 6). Mx-Cre+pRbΔ/Δ recipients rapidly developed signs of distress, as early as 2 weeks after the completion of pIpC. No Mx-Cre+pRbΔ/Δ recipient survived beyond 11 weeks post pIpC. Greater than 70% of recipients were moribund by 5 weeks, in contrast to recipients of wild-type BM cells that survived at least 20 weeks post pIpC with no significant changes in hematopoiesis (Fig 6). We observed rapid development of a completely penetrant myeloproliferative disorder in the BM, characterized by myeloid cell hyperplasia and suppression of lymphopoiesis and erythropoiesis. Splenomegaly, accompanied by myeloid and erythroid hyperplasia and extramedullary hematopoiesis, was also observed, demonstrating a striking similarity to the full Mx-Cre model (see Fig 2, Sup Table 1 and 2). The increased spleen size of Mx-Cre- recipients of LysM-Cre pRbfl/fl BM can be accounted for by the LysM-Cre pRbfl/fl BM itself, rather than a contribution from the recipient environment (Sup Table 1 and 2). These data demonstrate that the observed myeloproliferation is the consequence of an Rb-dependent interaction between myeloid-derived cells and the BM microenvironment, and develops independent of the HSC Rb status. Our data provide direct experimental evidence that myeloproliferation may ensue from aberrant interactions between myeloid-derived cells and the BM microenvironment, revealing hematopoietic extrinsic contribution to myeloproliferation.
Discussion
We sought to determine the role Rb plays in the regulation of hematopoiesis and stem cell function. Recent studies suggest that cell cycle regulation is an important determinant of stem cell fate; however, none has discriminated between intrinsic or extrinsic contributions (Cheng et al., 2000; Janzen et al., 2006; Walkley et al., 2005; Yuan et al., 2004). Rb was implicated as an important regulator of stem cell maintenance in Arabidopsis, however the limitations of the experimental system did not allow for the clear demonstration of a stem cell intrinsic role for Rb (Wildwater et al., 2005). Here we demonstrate that Rb extrinsically regulates HSCs by maintaining the competence of the adult bone marrow to support HSCs and, in turn, normal homeostatic hematopoiesis.
Rb and Stem Cell Self-Renewal
Understanding the regulation of cell cycle in stem cells is important from several perspectives. Stem cells must enter the cell cycle to self-renew; hence, induction of cycling may be desirable to achieve HSC expansion. Engraftment of transplanted HSCs is cell cycle dependent (Gothot et al., 1998; Passegue et al., 2005). The slow cycling of HSCs may spare them from acute toxicity (such as chemotherapy), but may also prevent neoplastic cells from eradication (Hodgson and Bradley, 1979; Lerner and Harrison, 1990). Our understanding of the normal regulation of self-renewal will also provide insight into tumorigenesis, where self-renewal pathways are thought to be active (Krivtsov et al., 2006).
The importance of cell cycle regulation in HSC fate decisions has been suggested by the analysis of animals deficient in negative cell cycle regulators such as p21Cip1, p27Kip1 and p16INK4a/p19ARF (Cheng et al., 2000; Stepanova and Sorrentino, 2005; Walkley et al., 2005). However, these studies have not revealed if such HSC defects are cell intrinsic or extrinsic in nature. The “Rb pathway” has also been implicated in phenotypes observe in both the Bmi1-/- and ATM-/- HSCs (Ito et al., 2004; Lessard and Sauvageau, 2003; Park et al., 2003). Surprisingly, we did not observe an intrinsic requirement for Rb in HSCs. If provided with a wild-type niche, RbΔ/Δ HSCs contribute normally to multilineage hematopoiesis and display serial transplant potential comparable to wild-type HSCs. Furthermore, we failed to detect alterations in numerous cell cycle or self-renewal associated genes in Rb-deficient HSCs and progenitors isolated from both a wild-type and mutant microenvironment, consistent with our interpretation that Rb is dispensable in the HSCs (Supp Fig 8). Our observations, taken together with those from analysis of p27Kip1 mutant mice, reveal that cell cycle regulation is a novel extrinsic regulator of hematopoiesis (Chien et al., 2006; Walkley and Orkin, 2006). The loss of BM HSCs in the Mx-Cre model is a secondary consequence of the disrupted environment within the BM and was not observed in the context of a wild-type niche, demonstrating that myeloproliferative-like disorders may deplete HSCs from the BM. A reanalysis of cell cycle mutants proposed to harbor HSC defects is needed to clarify the intrinsic and extrinsic roles that cell cycle regulation plays in these phenotypes.
It has been documented that self-renewal of embryonic stem cells occurs in an Rb independent manner (Stead et al., 2002); however, the “Rb pathway” is thought to be near universally targeted in human cancer cells (Hanahan and Weinberg, 2000). We have described that self-renewal of non-transformed HSCs occurs independent of Rb, highlighting the important question of what role Rb plays in the regulation of the process of cellular self-renewal, in both normal and oncogenic settings. It may be that the requirement for Rb and the “Rb pathway” in self-renewal is developmentally and lineage dependent, with progenitor cells having a greater dependence on Rb for their division than bona fide stem cells. One prediction of such a hypothesis is that mutation of the “Rb pathway” is of greater benefit to a progenitor cell than a stem cell during tumor formation.
Rb and Hematopoiesis
Previous studies examining the role of Rb in hematopoiesis have raised conflicting evidence regarding intrinsic and extrinsic effects, particularly in erythropoiesis (Clark et al., 2004; Iavarone et al., 2004; Spike et al., 2004; Whyatt and Grosveld, 2002). Our study utilized compartment restricted somatic mutagenesis to analyze the role of Rb in adult hematopoiesis and HSCs. Compartment restricted deletion enables a direct assessment of the contribution of hematopoietic and non-hematopoietic cells to the observed phenotype. We have not observed a progressive failure of hematopoiesis as reported by Spike et al. when Rb-deficient HSCs were supported by a wild-type environment, nor was this observed in a separate study utilizing germ-line deficient fetal liver hematopoietic cells (Hu et al., 1997). In contrast to in vitro findings (Iavarone et al., 2004), deletion of Rb from myeloid-derived cells using Lys-M-Cre did not result in anemia in vivo. Further studies utilizing lineage restricted deletion of Rb will be required to clarify the role of Rb in erythropoiesis.
Interactions Between Hematopoietic Cells and their Microenvironment Regulates HSCs and Hematopoiesis
The phenotype of Mx-Cre+pRbΔ/Δ animals is due to an Rb-dependent interaction between myeloid-derived cells (most probably macrophages and osteoclasts) and the bone marrow microenvironment (summarized in Table 1 and Supp Table 1). Evidence supports a direct role for the bone marrow microenvironment, but we cannot entirely exclude a contribution from other sites of Cre activity in the Mx-Cre model. Myeloproliferation is generally considered to be hematopoietic intrinsic, and evidence from the overexpression of activated kinase receptors in mouse models is consistent with this view (Araki et al., 2004; Chan et al., 2004; Le et al., 2004). In light of the data derived from Mx-Cre+pRbΔ/Δ mice, the BM microenvironment may play an active role in the promotion and/or maintenance of myeloproliferative disorders. Additional studies are required to define the cell(s) within the BM niche that are responsible for this interaction. The BM microenvironment is composed of numerous non-hematopoietic cell types including osteoblasts, endothelial cells, adipocytes and nerve cells. Histomorphometry demonstrated a significant disruption to bone homeostasis in the Rb-deficient animals, correlating with the observed mobilization and extramedullary hematopoiesis (Fig 2E-2H). Myeloproliferation in Mx-Cre+pRbΔ/Δ mice is dependent on concomitant deletion of Rb from both myeloid-derived cells and the environment. In other situations, myeloproliferation may result directly from an aberrant niche and may be independent on mutation(s) within hematopoietic cells (Purton et al, accomp manuscript).
Table 1.
Summary of the phenotype observed following loss of Rb.
| Bone Marrow | ||||||
|---|---|---|---|---|---|---|
| Condition | Hematopoietic Cells | Niche / Microenvironment | HSC | Myeloid | Lymphoid | Erythroid |
| Mx-Cre pRbfl/fl model | Δ/Δ | Δ/Δ | ↓↓↓ | ↑↑↑ | ↓ | ↔ |
| aΔ/Δ HSC into WT niche | Δ/Δb | WT | ↔ | ↑ | ↔ | ↔ |
| WT HSC into Δ/Δ niche | WT | Δ/Δb | ↔ | ↔ | ↔ | ↔ |
| Δ/Δ myeloid cells | Δ/Δ (myeloid) | WT | ↔ | ↔ | ↔ | ↔ |
| Δ/Δ myeloid cells into Δ/Δ niche | Δ/Δ (myeloid) | Δ/Δb | ↑↑↑ | ↓ | ↓ | |
| Spleen and Extramedullary Sites | ||||||
| Condition | Hematopoietic Cells | Niche / Microenvironment | cHSC / Progenitors | Myeloid | Lymphoid | Erythroid |
| Mx-Cre pRbfl/fl model | Δ/Δ | Δ/Δ | ↑↑↑ | ↑↑↑ | ↔ | ↑↑↑ |
| aΔ/Δ HSC into WT niche | Δ/Δb | WT | ↔ | ↔ | ↔ | ↔ |
| WT HSC into Δ/Δ niche | WT | Δ/Δb | ↔ | ↔ | ↔ | ↔ |
| Δ/Δ myeloid cells | Δ/Δ (myeloid) | WT | ↔ | ↔ | ↔ | ↑ |
| Δ/Δ myeloid cells into Δ/Δ niche | Δ/Δ (myeloid) | Δ/Δb | ↑↑↑ | ↑↑↑ | ↔ | ↑↑↑ |
Summary of data previously described (Walkley and Orkin, 2006)
Indicated compartment was non-deleted (Mx-Cre pRbfl/fl) at time of transplant and deleted 5 weeks after transplant with pIpC injection.
HSC and Progenitors as determined by flow cytometry (Lin-c-Kit+Sca-1+ and Lin-c-Kit+Sca-1-) and in vitro progenitor analysis.
Evidence of the role of stroma and the microenvironment in oncogenesis is accumulating, notably from analysis of solid tumors. Moreover, mathematical modelling of tumor behavior predicts that the environment is a major selective modifier of tumor morphology and phenotype (Allinen et al., 2004; Anderson et al., 2006; Balkwill, 2004; Hill et al., 2005; Kurose et al., 2002; Oh et al., 2004; St Croix et al., 2000). Somatic mutations divergent from those found in the tumor have been identified in stromal cells. In prostate cancer, results suggest that such mutations may contribute non-autonomously to tumor behaviour (Hill et al., 2005; Kurose et al., 2002). Understanding the interactions between hematopoietic cells and their microenvironment is directly relevant to hematopoietic disease. Mutations in the “Rb pathway” occur in ∼75% of cases of multiple myeloma (Kramer et al., 2002). Multiple myeloma clearly demonstrates that the interaction of hematopoietic cells, in this case B-cells, and the BM microenvironment is a major contributor to disease (Hideshima and Anderson, 2002; Mitsiades et al., 2006). These studies have focused on mutations present in established disease, but have not addressed the role of the microenvironment or stroma in the initiation of the disease process.
In addition to our data focused on Rb loss, the significance of the hematopoietic microenvironment to disease initiation has been suggested by recent studies. Mx-Cre+PtenΔ/Δ mice (Pten deficient hematopoietic cells and microenvironment) develop rapid and aggressive myeloproliferation that progresses to overt leukemia/lymphoma in 4-5 weeks post deletion (Yilmaz et al., 2006; Zhang et al., 2006b). However, when Pten deletion was activated in the context of a wild-type BM microenvironment, phenotypic and functional HSCs were lost without evidence of myeloproliferation or transformation (Yilmaz et al., 2006). This striking result suggests that PtenΔ/Δ hematopoietic cells alone are not intrinsically susceptible to myeloproliferation and subsequent malignant transformation in the presence of a wild-type microenvironment. Mutations in PTEN have been reported in the stroma of human breast tumors, suggesting a broader role for this pathway in the microenvironment and stroma of diverse organ systems (Kurose et al., 2002). Intriguingly JunB, Bmi-1 and ATM, implicated in HSC regulation and myeloproliferation, also have roles in regulating the bone marrow microenvironment (Kenner et al., 2004; Oguro et al., 2006; Passegue et al., 2004; Rasheed et al., 2006). JunB deficient mice develop severe osteopenia due to intrinsic defects in osteoclasts and osteoblasts, cellular constitutents of the HSC niche, while ATM mutants develop osteoporosis as a result of defective osteoblast differentiation. The contribution of these microenvironmental defects to the HSC phenotypes in these mutants has yet to be described. Such results demonstrate the need for further analysis of the interaction between the hematopoietic cells and their environment. This reconsideration will further our understanding of normal homeostatic hematopoiesis and the development of hematopoietic disease.
Our finding that the myeloproliferative-like disorder in the Rb mutants is the result of an interaction between myeloid-derived cells and the bone marrow microenvironment, together with the microenvironment-induced myeloproliferative-like disorder that develops in the RARγ-/- mice (Purton et al, accomp manuscript), underscores a previously unrecognized role for the hematopoietic microenvironment in the development of myeloid disease. These data further suggest that mutations within the hematopoietic niche might also serve as initiating events in the development of hematopoietic disease. In contrast to previous reports of the importance of cell cycle regulation in HSC fate determination, we find scant evidence for an intrinsic requirement for Rb in HSCs and that, indeed, Rb is a novel extrinsic regulator of hematopoietic stem cells. As our findings underscore, interactions between hematopoietic cells and the bone marrow niche/microenvironment profoundly affect hematopoietic homeostasis and the behavior of HSCs.
Supplementary Material
Acknowledgments
The authors thank Tyler Jacks for the generous provision of pRbfl/fl mice and Ingrid Poulton for bone histology. We thank David Williams, Kevin Shannon, Jack Martin, Hans Widlund and Ernestina Schipani for discussion, helpful suggestions and critical comment; DFCI and Children's Animal Facility Staff for care of experimental animals; John Daley and Suzan Lazo-Kallanian of DFCI HemNeo Flow facility for assistance with FACS sorting; David Dombkowski for assistance with FACS analysis; DFCI/Harvard Cancer Centre Rodent Histology Core.
Research Grants and Financial Support: This work was supported in part by a Center of Excellence in Molecular Hematology Award from the NIH-NIDDK (S.H.O). C.R.W is a Special Fellow of the Leukemia & Lymphoma Society and S.H.O is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Author Contribution Statement: C.R.W designed and performed experiments, analyzed and interpreted data and wrote paper; J.M.S performed experiments; N.A.S performed experiments and interpreted data; L.E.P analyzed and interpreted data; S.H.O. analyzed and interpreted data and wrote paper.
Conflict of Interest Statement: Authors have no conflicts of interest to declare pertaining to this research.
Supplemental Data: Supplemental Data include Supplemental Experimental Procedures, Supplemental References, and 11 supplemental Figures/Tables.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, Poznansky MC, Kos CH, Pollak MR, Brown EM, Scadden DT. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature. 2006;439:599–603. doi: 10.1038/nature04247. [DOI] [PubMed] [Google Scholar]
- Adams GB, Scadden DT. The hematopoietic stem cell in its place. Nat Immunol. 2006;7:333–337. doi: 10.1038/ni1331. [DOI] [PubMed] [Google Scholar]
- Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, Porter D, Hu M, Chin L, Richardson A, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Anderson ARA, Weaver AM, Cummings PT, Quaranta V. Tumor Morphology and Phenotypic Evolution Driven by Selective Pressure from the Microenvironment. Cell. 2006;127:905–915. doi: 10.1016/j.cell.2006.09.042. [DOI] [PubMed] [Google Scholar]
- Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, Ito K, Koh GY, Suda T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–161. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, Yang W, Pao LI, Gilliland DG, Epstein JA, Neel BG. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med. 2004;10:849–857. doi: 10.1038/nm1084. [DOI] [PubMed] [Google Scholar]
- Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Bradford GB, Williams B, Rossi R, Bertoncello I. Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp Hematol. 1997;25:445–453. [PubMed] [Google Scholar]
- Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- Chan IT, Kutok JL, Williams IR, Cohen S, Kelly L, Shigematsu H, Johnson L, Akashi K, Tuveson DA, Jacks T, Gilliland DG. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J Clin Invest. 2004;113:528–538. doi: 10.1172/JCI20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- Chien WM, Rabin S, Macias E, Miliani de Marval PL, Garrison K, Orthel J, Rodriguez-Puebla M, Fero ML. Genetic mosaics reveal both cell-autonomous and cell-nonautonomous function of murine p27Kip1. Proc Natl Acad Sci U S A. 2006;103:4122–4127. doi: 10.1073/pnas.0509514103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AJ, Doyle KM, Humbert PO. Cell-intrinsic requirement for pRb in erythropoiesis. Blood. 2004;104:1324–1326. doi: 10.1182/blood-2004-02-0618. [DOI] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Tumbar T, Guasch G. Socializing with the neighbors: stem cells and their niche. Cell. 2004;116:769–778. doi: 10.1016/s0092-8674(04)00255-7. [DOI] [PubMed] [Google Scholar]
- Ghiaur G, Lee A, Bailey J, Cancelas JA, Zheng Y, Williams DA. Inhibition of RhoA GTPase activity enhances hematopoietic stem and progenitor cell proliferation and engraftment. Blood. 2006;108:2087–2094. doi: 10.1182/blood-2006-02-001560. [DOI] [PubMed] [Google Scholar]
- Gothot A, van der Loo JC, Clapp DW, Srour EF. Cell cycle-related changes in repopulating capacity of human mobilized peripheral blood CD34(+) cells in non-obese diabetic/severe combined immune-deficient mice. Blood. 1998;92:2641–2649. [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hideshima T, Anderson KC. Molecular mechanisms of novel therapeutic approaches for multiple myeloma. Nat Rev Cancer. 2002;2:927–937. doi: 10.1038/nrc952. [DOI] [PubMed] [Google Scholar]
- Hill R, Song Y, Cardiff RD, Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005;123:1001–1011. doi: 10.1016/j.cell.2005.09.030. [DOI] [PubMed] [Google Scholar]
- Hodgson GS, Bradley TR. Properties of haematopoietic stem cells surviving 5-fluorouracil treatment: evidence for a pre-CFU-S cell? Nature. 1979;281:381–382. doi: 10.1038/281381a0. [DOI] [PubMed] [Google Scholar]
- Hu N, Gulley ML, Kung JT, Lee EY. Retinoblastoma gene deficiency has mitogenic but not tumorigenic effects on erythropoiesis. Cancer Research. 1997;57:4123–4129. [PubMed] [Google Scholar]
- Iavarone A, King ER, Dai XM, Leone G, Stanley ER, Lasorella A. Retinoblastoma promotes definitive erythropoiesis by repressing Id2 in fetal liver macrophages. Nature. 2004;432:1040–1045. doi: 10.1038/nature03068. [DOI] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- Kenner L, Hoebertz A, Beil T, Keon N, Karreth F, Eferl R, Scheuch H, Szremska A, Amling M, Schorpp-Kistner M, et al. Mice lacking JunB are osteopenic due to cell-autonomous osteoblast and osteoclast defects. J Cell Biol. 2004;164:613–623. doi: 10.1083/jcb.200308155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, Tesio M, Samstein RM, Goichberg P, Spiegel A, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12:657–664. doi: 10.1038/nm1417. [DOI] [PubMed] [Google Scholar]
- Kramer A, Schultheis B, Bergmann J, Willer A, Hegenbart U, Ho AD, Goldschmidt H, Hehlmann R. Alterations of the cyclin D1/pRb/p16(INK4A) pathway in multiple myeloma. Leukemia. 2002;16:1844–1851. doi: 10.1038/sj.leu.2402609. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Krosl J, Beslu N, Mayotte N, Humphries RK, Sauvageau G. The competitive nature of HOXB4-transduced HSC is limited by PBX1: the generation of ultra-competitive stem cells retaining full differentiation potential. Immunity. 2003;18:561–571. doi: 10.1016/s1074-7613(03)00090-6. [DOI] [PubMed] [Google Scholar]
- Ku H, Yonemura Y, Kaushansky K, Ogawa M. Thrombopoietin, the ligand for the mpl receptor, synergizes with steel factor and other early acting cytokines in supporting proliferation of primitive hematopoietic progenitors of mice. Blood. 1996;87:4544–4551. [PubMed] [Google Scholar]
- Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet. 2002;32:355–357. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
- Le DT, Kong N, Zhu Y, Lauchle JO, Aiyigari A, Braun BS, Wang E, Kogan SC, Le Beau MM, Parada L, Shannon KM. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood. 2004;103:4243–4250. doi: 10.1182/blood-2003-08-2650. [DOI] [PubMed] [Google Scholar]
- Lerner C, Harrison DE. 5-Fluorouracil spares hemopoietic stem cells responsible for long-term repopulation. Exp Hematol. 1990;18:114–118. [PubMed] [Google Scholar]
- Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- Li CL, Johnson GR. Stem cell factor enhances the survival but not the self-renewal of murine hematopoietic long-term repopulating cells. Blood. 1994;84:408–414. [PubMed] [Google Scholar]
- Martin TJ, Sims NA. Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med. 2005;11:76–81. doi: 10.1016/j.molmed.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Mitsiades CS, Mitsiades NS, Munshi NC, Richardson PG, Anderson KC. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma: interplay of growth factors, their receptors and stromal interactions. Eur J Cancer. 2006;42:1564–1573. doi: 10.1016/j.ejca.2005.12.025. [DOI] [PubMed] [Google Scholar]
- Moore KA, Lemischka IR. Stem cells and their niches. Science. 2006;311:1880–1885. doi: 10.1126/science.1110542. [DOI] [PubMed] [Google Scholar]
- Oguro H, Iwama A, Morita Y, Kamijo T, van Lohuizen M, Nakauchi H. Differential impact of Ink4a and Arf on hematopoietic stem cells and their bone marrow microenvironment in Bmi1-deficient mice. J Exp Med. 2006;203:2247–2253. doi: 10.1084/jem.20052477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh P, Li Y, Yu J, Durr E, Krasinska KM, Carver LA, Testa JE, Schnitzer JE. Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy. Nature. 2004;429:629–635. doi: 10.1038/nature02580. [DOI] [PubMed] [Google Scholar]
- Okada S, Nakauchi H, Nagayoshi K, Nishikawa S, Miura Y, Suda T. In vivo and in vitro stem cell function of c-kit- and Sca-1-positive murine hematopoietic cells. Blood. 1992;80:3044–3050. [PubMed] [Google Scholar]
- Osawa M, Hanada K, Hamada H, Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273:242–245. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegue E, Wagner EF, Weissman IL. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell. 2004;119:431–443. doi: 10.1016/j.cell.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Purton LE, Bernstein ID, Collins SJ. All-trans retinoic acid delays the differentiation of primitive hematopoietic precursors (lin-c-kit+Sca-1(+)) while enhancing the terminal maturation of committed granulocyte/monocyte progenitors. Blood. 1999;94:483–495. [PubMed] [Google Scholar]
- Purton LE, Bernstein ID, Collins SJ. All-trans retinoic acid enhances the long-term repopulating activity of cultured hematopoietic stem cells. Blood. 2000;95:470–477. [PubMed] [Google Scholar]
- Purton LE, Dworkin S, Olsen GH, Walkley CR, Fabb SA, Collins SJ, Chambon P. RAR{gamma} is critical for maintaining a balance between hematopoietic stem cell self-renewal and differentiation. J Exp Med. 2006;203:1283–1293. doi: 10.1084/jem.20052105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasheed N, Wang X, Niu QT, Yeh J, Li B. Atm-deficient mice: an osteoporosis model with defective osteoblast differentiation and increased osteoclastogenesis. Hum Mol Genet. 2006;15:1938–1948. doi: 10.1093/hmg/ddl116. [DOI] [PubMed] [Google Scholar]
- Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma is sufficient for cell cycle re-entry. Nature. 2003;424:223–228. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells. 1978;4:7–25. [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- Spangrude GJ, Brooks DM, Tumas DB. Long-term repopulation of irradiated mice with limiting numbers of purified hematopoietic stem cells: in vivo expansion of stem cell phenotype but not function. Blood. 1995;85:1006–1016. [PubMed] [Google Scholar]
- Spike BT, Dirlam A, Dibling BC, Marvin J, Williams BO, Jacks T, Macleod KF. The Rb tumor suppressor is required for stress erythropoiesis. Embo J. 2004;23:4319–4329. doi: 10.1038/sj.emboj.7600432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, Lal A, Riggins GJ, Lengauer C, Vogelstein B, Kinzler KW. Genes expressed in human tumor endothelium. Science. 2000;289:1197–1202. doi: 10.1126/science.289.5482.1197. [DOI] [PubMed] [Google Scholar]
- Stead E, White J, Faast R, Conn S, Goldstone S, Rathjen J, Dhingra U, Rathjen P, Walker D, Dalton S. Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene. 2002;21:8320–8333. doi: 10.1038/sj.onc.1206015. [DOI] [PubMed] [Google Scholar]
- Stepanova L, Sorrentino BP. A limited role for p16Ink4a and p19Arf in the loss of hematopoietic stem cells during proliferative stress. Blood. 2005;106:827–832. doi: 10.1182/blood-2004-06-2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stier S, Ko Y, Forkert R, Lutz C, Neuhaus T, Grunewald E, Cheng T, Dombkowski D, Calvi LM, Rittling SR, Scadden DT. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med. 2005;201:1781–1791. doi: 10.1084/jem.20041992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilvassy SJ, Humphries RK, Lansdorp PM, Eaves AC, Eaves CJ. Quantitative assay for totipotent reconstituting hematopoietic stem cells by a competitive repopulation strategy. Proc Natl Acad Sci U S A. 1990;87:8736–8740. doi: 10.1073/pnas.87.22.8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F, Sato T, Laver JH, Ogawa M. CD34 expression by murine hematopoietic stem cells mobilized by granulocyte colony-stimulating factor. Blood. 2000;96:1989–1993. [PubMed] [Google Scholar]
- Taswell C. Limiting dilution assays for the determination of immunocompetent cell frequencies. I. Data analysis. J Immunol. 1981;126:1614–1619. [PubMed] [Google Scholar]
- Till J, McCulloch E. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat Res. 1961;14:213. [PubMed] [Google Scholar]
- Varnum-Finney B, Purton LE, Yu M, Brashem-Stein C, Flowers D, Staats S, Moore KA, Le Roux I, Mann R, Gray G, et al. The Notch ligand, Jagged-1, influences the development of primitive hematopoietic precursor cells. Blood. 1998;91:4084–4091. [PubMed] [Google Scholar]
- Visnjic D, Kalajzic Z, Rowe DW, Katavic V, Lorenzo J, Aguila HL. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood. 2004;103:3258–3264. doi: 10.1182/blood-2003-11-4011. [DOI] [PubMed] [Google Scholar]
- Walkley CR, Fero ML, Chien WM, Purton LE, McArthur GA. Negative cell-cycle regulators cooperatively control self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol. 2005;7:172–178. doi: 10.1038/ncb1214. [DOI] [PubMed] [Google Scholar]
- Walkley CR, Orkin SH. Rb is dispensable for self-renewal and multilineage differentiation of adult hematopoietic stem cells. Proc Natl Acad Sci U S A. 2006;103:9057–9062. doi: 10.1073/pnas.0603389103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Whyatt D, Grosveld F. Cell-nonautonomous function of the retinoblastoma tumour suppressor protein: new interpretations of old phenotypes. EMBO Rep. 2002;3:130–135. doi: 10.1093/embo-reports/kvf033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildwater M, Campilho A, Perez-Perez JM, Heidstra R, Blilou I, Korthout H, Chatterjee J, Mariconti L, Gruissem W, Scheres B. The RETINOBLASTOMA-RELATES Gene Regulates Stem Cell Maintenance in Arabidopsis Roots. Cell. 2005;123:1337–1349. doi: 10.1016/j.cell.2005.09.042. [DOI] [PubMed] [Google Scholar]
- Yang L, Bryder D, Adolfsson J, Nygren J, Mansson R, Sigvardsson M, Jacobsen SE. Identification of Lin-Sca1+kit+CD34+Flt3- short-term hematopoietic stem cells capable of rapidly reconstituting and rescuing myeloablated recipients. Blood. 2005;105:2717–2723. doi: 10.1182/blood-2004-06-2159. [DOI] [PubMed] [Google Scholar]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat Cell Biol. 2004;6:436–442. doi: 10.1038/ncb1126. [DOI] [PubMed] [Google Scholar]
- Zhang CC, Kaba M, Ge G, Xie K, Tong W, Hug C, Lodish HF. Angiopoietin-like proteins stimulate ex vivo expansion of hematopoietic stem cells. Nat Med. 2006a;12:240–245. doi: 10.1038/nm1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, Haug JS, Rupp D, Porter-Westpfahl KS, Wiedemann LM, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006b;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.