

SUMMARY
Dendritic cells (DCs) produce type I interferons (IFNs) in greater amounts than other cells, but the mechanisms remain elusive. Here we studied the role of a transcription factor, IRF-8 in DC induction of type I IFNs. Upon NDV infection, bone marrow derived plasmacytoid and conventional DCs induced IFN transcripts, exhibiting two phase kinetics. The second, amplifying phase represented an IFN feedback response that accounted for much of IFN protein production. Induction of second phase transcription required IRF-8. CMV and Toll like receptor-mediated IFN induction in DCs also required IRF-8. Chromatin immunoprecipitation analysis showed that IRF-7, IRF-8 and RNA polymerase II were recruited to the IFN promoters upon stimulation. Moreover, sustained RNA polymerase II recruitment to the promoters critically depended on IRF-8. Together, IRF-8 magnifies the second phase of IFN transcription in DCs by prolonging binding of basic transcription machinery to the IFN promoters, thereby playing a role in innate immunity.
INTRODUCTION
Type I interferons (IFNs) are produced immediately after pathogen infection, profoundly influencing the nature of innate and adaptive immunity (Biron, 2001). Although many types of cells produce type I IFNs, dendritic cells (DCs) are the highest IFN producers (Asselin-Paturel et al., 2001; Siegal et al., 1999), accounting for the early establishment of innate immunity (Asselin-Paturel and Trinchieri, 2005; Banchereau et al., 2004). Type I IFN genes belong to a multigene family consisting of more than 10 IFNα genes and a single IFNβ gene, all clustered in a ~ 500 kb long region in chromosome 4 in the mouse (Pestka et al., 2004). IFN induction is mediated through Toll like receptor (TLR) signaling, that simulates sequential activation of adaptors and kinases of the IRAK, IKK and TRAF families, ultimately leading to the activation of transcription factors, IRF-3, IRF-7 and NFkB (Honda and Taniguchi, 2006; Kawai and Akira, 2006). Besides TLR signaling, activation of RNA helicase pathways such as RIG I leads to activation of IRF-3 and type I IFN induction (Honda and Taniguchi, 2006; Yoneyama et al., 2005).
Mechanisms underlying type I IFN gene induction have been extensively studied in non-DCs, including fibroblasts (Civas et al., 2002; Marie et al., 1998; Sato et al., 2000). These studies showed that IFN production involves initial induction of IFNβ and IFNα4 transcripts that are mediated by IRF-3 and IRF-7. These IRFs are phosphorylated, dimerized and translocated into the nucleus to activate the two IFN genes (Sharma et al., 2003). This initial event then leads to the next stage of transcription where additional IFNα genes and IRF-7 are induced in an IFN dependent manner (Marie et al,1998; Tailor et al., 2006). A recent study indicates that IRF-7, not IRF-3 is essential for the initial IFN induction both in DCs and non-DCs (Honda et al., 2005b).
DCs are a heterogeneous population of cells with diverse functions. Although it was previously thought that plasmacytoid DCs (pDCs) are responsible for much of type I IFN production, more recent studies showed that conventional DCs (cDCs) also produce considerable amounts of type I IFNs (Diebold et al., 2003; Kato et al., 2005). pDCs seem to have several distinct characteristics that might be relevant to high IFN production. For example, the TLR complexes containing MyD88/IRF-7 are retained in pDCs longer than in other cells (Honda et al., 2005a). In addition pDCs express IRF-7 at high levels, possibly enabling enhanced IFN gene activation (Dai et al., 2004). Furthermore, the RNA helicase-RIG I pathway appears dispensable in pDCs (Kato et al., 2005).
Despite intense interests, molecular mechanisms by which pDCs and in some cases cDCs express high levels of type I IFNs have remained poorly understood. While several reports indicated an IFN feedback as a significant mechanism of IFN production in DCs, evidence is inconsistent, making it uncertain as to the significance of the feedback loop (Gautier et al., 2005; Honda et al., 2005b; Kerkmann et al., 2003). Another unsolved question is the role of IRF-8 in DC induction of type I IFN genes. IRF-8 is an immune system specific member of the IRF family (Levi et al., 2002; Tailor et al., 2006; Tamura and Ozato, 2002). It is a nuclear protein expressed at high levels in pDC and other DC subtypes (Tamura et al., 2005a). We previously showed that IRF-8−/− DCs produced little type I IFNs and that reintroduction of IRF-8 rescued type I IFN induction in −/− DCs (Schiavoni et al., 2002; Tamura et al., 2005a; Tsujimura et al., 2003b). In addition, IRF-8 is essential for the development of DC subsets, particularly pDCs and CD8α+ DCs, although another member IRF-4 also contributes to pDC development, albeit to a minor extent (Aliberti et al., 2003; Schiavoni et al., 2002; Tamura et al., 2005a; Tsujimura et al., 2003a; Tsujimura et al., 2003b). Because of its prominent role in DC development, the contribution of IRF-8 to IFN induction in differentiated DCs has not been fully elucidated so far.
The present study reveals two notable aspects of IFN transcription that are different between DCs and other cell types. First, many IFNα subtypes are induced immediately in the first phase prior to the initiation of IFN feedback response. IRF-8 is required for the second, amplifying phase of IFN induction both in pDCs and cDCs, although IRF-8 is not required for the activation of IRF-3 or IRF-7. Our results indicate that the role of IRF-8 is to help prolong the recruitment of basal transcription machinery to the IFN promoters, a role not shared by IRF-7 or IRF-3, which supports characteristically high IFN transcription in DCs.
RESULTS
Two Phase Kinetics of Type I IFN Induction in DCs: The Absence of the Second Phase Peak in IRF-8−/− DCs
To study the mechanism of type I IFN gene induction in DCs, bone marrow derived IRF-8+/+ and −/− DCs generated in the presence of Flt3L were exposed to New Castle Disease Virus (NDV) for 1 h. Cells were washed and then incubated in fresh media for up to 24 h. IFNα transcripts induced after viral exposure were measured by quantitative (q) RT-PCR using a primer set that detected all IFNα subtypes (Figure 1A). The transcript induction in +/+ DCs followed two phase kinetics, exhibiting two peaks. The first relatively small peak was seen at 1 h, followed by the greater second peak at 7 h. Strikingly, IRF-8−/− DCs did not elicit the second peak, although they elicited the first peak similar to that in +/+ cells in terms of timing and transcript levels. In Figure 1B, the amount of IFNα proteins secreted from IRF-8+/+ DCs rose sharply during initial 11 h and plateaued thereafter. In contrast, IFNα produced in IRF-8−/− DCs remained near a background level, in line with previous reports (Schiavoni et al., 2002; Tamura et al., 2005a; Tsujimura et al., 2003b). Given that IFN protein production was virtually absent in −/− DCs, despite the presence of the first transcript peak, it is likely that much of IFN protein production in DCs is accounted for by the second phase of IFN transcript expression. In Figure 1C, IFNβ transcript expression displayed similar two phase kinetics in +/+ DCs. Again, the second peak was completely missing in IRF-8−/− DCs, although the first peak in −/− DCs was higher than that in +/+ DCs.
Figure 1.

Two Phase Kinetics of Type I IFN Transcript Induction in DCs.
A: DCs from IRF-8+/+ and IRF-8−/− mice were infected with NDV for 1 h, washed, and IFNα mRNA levels were measured at indicate times (h) by qRT-PCR using pan-IFNα primers.
B: IFNα protein production in above cells was measured by ELISA.
C: IFNβ mRNA levels in above cells were measured using IFNβ primers as in A.
D: Semi-quantitative RT-PCR was performed for IFNα4 and non-α4 IFNα transcripts in IRF-8+/+ and −/− cells at indicated times after NDV infection as in A.
The two phase kinetics observed above is reminiscent of two-step IFN induction described for non-DCs (Civas et al., 2002; Marie et al., 1998; Sato et al., 2000). In non-DCs, IFNβ and IFNα4 are the first subtypes expressed in the early stage, and other, non-α4 subtypes are expressed in the second stage. To assess whether IFNα subtypes were induced in a similar, hierarchical order in DCs, expression of non-α4 transcripts was examined. Semi-quantitative RT-PCR, rather than qRT-PCR was employed in these experiments, because selection of primers that could detect individual non-α4 transcripts in qRT-PCR was not practical. As seen in Figure 1D, non-α4 IFNα transcripts were induced at 1 h along with induction of IFNβ and IFNα4 both in +/+ and −/− DCs. In IRF-8 +/+ DCs, the levels of non-α4 mRNA markedly increased at 7 h, along with an increase in IFNα4 and IFNβ mRNAs. In −/− DCs, however, neither non-α4, IFNα4, nor IFNβ transcripts were detected at 7 h and thereafter. These data indicate that contrary to non-DCs, multiple IFNα subtypes are induced in the first phase in DCs, and their expression is enhanced in the second phase. Determination of 25 IFNα transcript sequences expressed at 1 h in DCs confirmed the abundant presence of non-α4 IFN species in the first phase (Table S1).
The Role of IRF-8 in Type I IFN Expression in cDCs
We noted that ~20% –30% of IRF-8+/+ DCs were pDCs carrying B220, Siglec-H and Ly49Q, whereas cells with these markers were virtually nonexistent in IRF-8−/− DCs (Figure S1). Since the failure of IRF-8−/− DCs to elicit the second IFN peak may be attributed to the absence of pDCs in the culture, we tested if IFNs were induced in cDCs after NDV stimulation. MACS fractionation of total DCs from IRF-8+/+ mice yielded a clean cDC population in which B220+ pDC were 0.1%. The other, remaining population was enriched with pDCs (~ 70% B220+) (Figure S2). IFNα transcripts were induced both in cDCs and pDC-enriched populations, both showing two peaks, similar to those in total DCs. Similarly, IFNβ transcripts were expressed in the both cDCs and pDC enriched cells, generating again two peaks resembling those in total DCs. These results show that type I IFN genes are induced both in cDC and pDC following NDV stimulation, and that the absence of the second peak in IRF-8−/− DCs was not due the absence of pDCs, but due to the absence of IFN mRNA expression.
Because Flt3L and GM-CSF promote development of different DC subsets (Gilliet et al., 2002; Tamura et al., 2005a), IFN induction was also studied in GM-CSF derived DCs (Figure S3). After NDV stimulation, +/+ DCs from GM-CSF culture expressed much lower IFNα transcripts and proteins than those from the Flt3L culture (~100 fold lower mRNA and 20 fold lower protein). More significantly, IFN induction in IRF-8−/− DCs from GM-CSF culture were at a background level (Figure S3). These results are in agreement with the previous observations that Flt3L, but not GM-CSF supports development of IFN producing cells (Tamura et al., 2005b).
It was of importance to test whether the requirement of IRF-8 is limited to the NDV system, or this requirement is a more general aspect of DC IFN induction. To address this question, we infected +/+ and −/− DCs with the murine cytomegalovirus (MCMV). Results in Figure 2A show that whereas +/+ DCs induced IFNα transcripts at high levels that peaked at 7 h, IRF-8−/− DCs expressed virtually no IFN transcripts. IFNβ expression exhibited essentially the same pattern, where the transcripts were almost undetectable in −/− DCs. Consequently, little IFN proteins were produced in −/− cells in contrast to +/+ cells that yielded a ng/ml order of IFNs. Vesicular stomatitis virus (VSV), another virus we tested led to much lower levels of IFN transcripts than NDV and MCMV. Nevertheless, +/+ DCs expressed measurable levels of IFN transcripts, while IRF-8−/− DCs induced IFNα transcripts at a level barely above detection (data not shown). In Figure 2B and 2C, ligands for TLR 3, 7, and 9 (poly IC, R848 and CpG, respectively) were tested for IFN induction. These ligands constitute major components of RNA and DNA viruses. In +/+ DCs, CpG DNA led to the highest induction of both IFNα and β transcripts and proteins, although other ligands also stimulated IFN transcript induction. In contrast, essentially no IFN transcripts were expressed in IRF-8−/− DCs in response to these TLR ligands. These results show that the defect in expressing type I IFN in IRF-8−/− DCs is not a specialized feature limited to certain stimuli, but it represents a fundamental, mechanistic deficit of the IRF-8 −/− DCs. Unlike what was observed with the NDV stimulation, the above stimuli elicited a single major IFN transcripts peak and did not display two clear peaks. It is likely that these stimuli generated two peaks in a shorter, overlapping interval, indicating that IFN induction kinetics may differ depending on types of stimuli.
Figure 2.

Absence of IFN Induction in IRF-8−/− DCs After MCMV Infection and TLR Signaling.
A: DCs were stimulated with MCMV for 1h and induction of IFNα and IFNβ transcripts and IFN α protein production was measured as in Figure 1.
B–C: DCs were stimulated with indicated TLR ligands and IFN transcripts and proteins were detected as above. The ligands were present for the entire incubation time. U D: IFN production was undetectable in IRF-8−/− DCs.
The Second Peak Represents an IFN Feedback Response
We surmised that the second peak elicited by NDV was an equivalent of the IFN feedback loop described for non-DCs. To ascertain whether this idea hold true, we tested IFN transcript induction in DCs from IFNAR−/− mice that would not generate the feedback loop due to the lack of type I IFN receptor. DCs generated from IFNAR+/+ and IFNAR−/− mice, both in the BALB/c background, contained similar percentages of pDCs (Figure 3A). In Figure 3B, while IFNAR+/+ DCs showed a typical two phase IFN mRNA induction upon NDV addition, IFNAR−/− DCs produced no detectable second peak. The first peak in IFNAR−/− DCs, although observed reproducibly, was lower than in IFNAR+/+ DCs, possibly due to prior IFN priming in IFNAR+/+ DCs. These data indicate that the second IFN peak, missing in IRF-8−/− DCs, is accounted for by an IFN feedback response, which provides a major mechanism of enhanced IFN induction in DCs.
Figure 3.

The Absence of the Second Peak in IFNAR−/− DCs after NDV infection.
A: Flow cytometry detection of pDCs in IFNAR+/+ and IFNAR−/− DCs. B220+, and CD11b+ (MacI) cells are boxed in each panel.
B: IFNα transcripts in above cells were measured as in Figure 1A.
C: cDCs and pDC-enriched cells from IRF-8+/+ mice, DCs from IRF-8−/− or IFNAR−/− mice were stimulated with 500 U/ml of IFNα and expression of indicated genes was monitored by qRT-PCR at indicated times.
The above results raised the possibility that IRF-8−/− DCs were deficient in responding to the IFN feedback signal. To test this possibility, we examined expression of several classical IFN responsive genes (Figure 3C). Upon IFNα addition, 2′–5′ oligo (A) synthetase (2–5OAS), interferon induced transmembrane protein 3 (IFITM3), and IRF-7 were expressed in IRF-8−/− DCs along with +/+ pDCs and +/+ cDCs, indicating that IRF-8−/− DCs respond to IFN signals. Only IFNAR−/− DCs failed to express these genes. Supporting the idea that IRF-8−/− DCs are capable of responding to the IFN feedback signal generated by viral stimulation, above genes were also induced following NDV infection in both IRF-8+/+ and −/− DCs (Figure S4).
To further delineate defects in IRF8−/− DCs, we asked whether IRF-3 and IRF-7 are activated in IRF-8−/− DCs. To this end, nuclear translocation of the two proteins was examined, since it represented an endpoint event of TLR signaling, initiating IFN gene transcription. For IRF-7, we analyzed DCs expressing IRF-7 cDNA fused to the green fluorescent protein (GFP), because available antibodies were not suitable for immunostaining of endogenous IRF-7. As seen in Figure 4A, IRF-7-GFP was distributed mostly in the cytoplasm prior to infection. One h after stimulation, intense fluorescent signals were observed in the nucleus (except for nucleoli), overlapping with DNA stain, both in +/+ and −/− DCs, attesting nuclear translocation in both DCs. In the majority of cells, there was residual IRF-7-GFP in the cytoplasm after NDV stimulation, suggesting incomplete translocation or rapid reshuffling. Nuclear translocation of IRF-3 was studied by immunoblot analysis of the cytoplasmic and nuclear fractions prepared before and 1 h after NDV stimulation (Figure 4B). There was little cross contamination between the two fractions, as verified by the selective presence of c-cbl and YY1 in the cytoplasmic and nuclear fractions, respectively. Prior to NDV addition, IRF-3 was detected mostly in the cytoplasmic fraction in +/+ DCs, although the nuclear fraction also contained some IRF-3. In −/− DCs, IRF-3 was almost exclusively in the cytoplasm prior to infection. After stimulation, the nuclear IRF-3 markedly increased both in +/+ and −/− DCs, along with the reduction in the cytoplasm. In agreement with these results, immunostaining of endogenous IRF-3 showed clear nuclear translocation following NDV stimulation (Figure 4C). These data indicate that the TLR signaling cascade functioned normally in −/− DCs to activate IRF-3 and IRF-7. It should be noted here that IRF-8 resided almost exclusively in the nucleus and its levels did not change before and after NDV stimulation for 7 h, as would have been expected (Figure 4B and 4D).
Figure 4.

Nuclear Translocation of IRF-7 and IRF-3 in IRF-8−/− DCs.
A: IRF-8+/+ and −/− DCs were transduced with a vector for IRF-7-GFP and stimulated without (control) or with NDV for 1 h. Cells were fixed and stained with anti-GFP antibody and counterstained with Hoechst 33342 for DNA.
B: IRF-8+/+ and IRF-8−/− DCs were infected with NDV for 1 h as above and nuclear and cytoplasmic fractions were tested for indicated proteins by immunoblot analysis. c-cbl and YY1 were used to monitor the quality of fractionation.
C: IRF-8+/+ and −/− DCs were stimulated with or without NDV and immunostained with antibody for endogenous IRF-3 as in A.
D: IRF-8+/+ DCs stimulated with NDV for 7 h and whole cell extracts were tested for IRF-8 by immunoblot.
Exogenous IRF-8 Amplifies IFN Gene Expression in Fibroblasts
Whereas type I IFNs are produced in many cells including fibroblasts, IRF-8 is expressed only in the cells of hematopoietic lineage. It was of interest to test whether ectopic expression of IRF-8 in fibroblasts enhances IFN expression. To this end NIH3T3 fibroblasts were transduced with a retroviral vector for IRF-8 and IFNα and β transcript induction was examined after NDV stimulation (Figure 5A and 5B). Cells transduced with control, empty vector generated a relatively large first IFNα peak at 2 h, followed by a much lower second peak seen between 10 h and 16 h. In contrast, cells transduced with IRF-8 generated a second IFNα peak of much greater magnitude, although the magnitude of the first peak was not affected by IRF-8. IRF-8 also led to a dramatic amplification of the second peak for IFNβ transcripts. IFNβ induction in control cells was largely limited to the first phase, yielding a negligible second peak under these conditions. We noted that IFNβ transcript levels were higher than those of all IFNα transcripts in NIH3T3 cells, suggesting a greater significance of this IFN subtype in fibroblasts. Together, in NIH3T3 cells type I IFN induction followed slower kinetics with a reduced second peak compared to those in DCs and IRF-8 introduction enhanced the second peak without altering the first peak.
Figure 5.

Amplification of the Second IFN Peak in Fibroblasts after IRF-8 Introduction.
A – B: NIH3T3 cells were transduced with control pMSCV vector or wild type IRF-8 vector and stimulated with NDV for 1 h and transcripts for IFNα and IFNβ were measured as in Figure 1A.
C: Diagram of IRF-8 mutants tested in D.
D: NIH3T3 cells transduced with indicated IRF-8 mutants were stimulated with NDV as above and production of IFNα was measured by ELISA.
To assess domains within IRF-8 required for enhancing IFN induction, a series of IRF-8 deletions and mutants were tested for IFN protein induction in NIH3T3 cells. Previous studies have shown that both the DNA binding domain (DBD, gray in Figure 5C) and the IRF association domain in the C-terminal region (IAD, solid) are required for IRF-8’s transcriptional activity. Particularly, lysine (K) at position 79 and arginine (R) at 289 are essential for activity of the DBD and IAD, respectively (Tamura et al., 2005b; Tsujimura et al., 2003a). In Figure 5D, whereas full length IRF-8 and 1–390 gave a ~6 fold increase in the total amount of IFNα, other deletions, delN, 1–253, 1–305, 1–356 gave only a background level of IFN. The mutants with substitutions in K79 and R289 also failed to enhance IFN production. These data are consistent with the idea that IRF-8 enhances IFN induction at the level of transcription, not of IFN signaling.
IRF-8 Binds to Type I IFN Promoters
Type I IFN transcription requires ordered binding of sequence specific factors such as IRF-3 and IRF-7 as well as cofactors that affect chromatin, leading to the recruitment of general transcription factors (Agalioti et al., 2000; Maniatis et al., 1998; McWhirter et al., 2004). IFNα and IFNβ promoters carry IRF binding sites that are variously called such as the virus response element (VRE) and PRDI (Figure 6A) (Civas et al., 2006; Maniatis et al., 1998; Yang et al., 2003). To ascertain whether IRF-8 participates in IFN transcription, we performed chromatin immunoprecipitation (ChIP) assays for several IFN promoters in IRF-8+/+ and −/− DCs. Figure 6B shows detailed kinetics of IRF-8 binding to the IFNα4 promoter after NDV addition. While low constitutive IRF-8 binding was seen before stimulation, the binding significantly increased after NDV stimulation, peaking at 4 h with a slight decline at 5 and 6 h. Afterwards IRF-8 binding increased slightly followed by a reduction again during the subsequent 11 h period, overall showing a mild, oscillating pattern of recruitment, reminiscent of the patterns reported for other transcription factors (Reid et al., 2003). Specificity of IRF-8 recruitment was substantiated by the lack of binding by control IgG to the promoter (Figure 6B) as well as by the lack of IRF-8 antibody binding to the unrelated gene HPRT (Figure S5B). Further confirming specificity of ChIP results, IRF-8 antibody did not bind to the promoter in IRF-8−/− DCs (Figure 6C). Moreover, IRF-8 was recruited to the IFNα5 and IFNβ promoters as well, with a pattern similar to that for IFNα4, i.e., low constitutive binding prior to NDV stimulation, followed by increased recruitment after NDV stimulation (Figure 6C and 6D). We also performed ChIP assays after TLR signaling, and found that IRF-8 was recruited to the IFNα4 promoter following CpG stimulation as well, supporting the view that IRF-8 recruitment to the promoter is a general event that takes place during IFN induction (Figure S5A).
Figure 6.
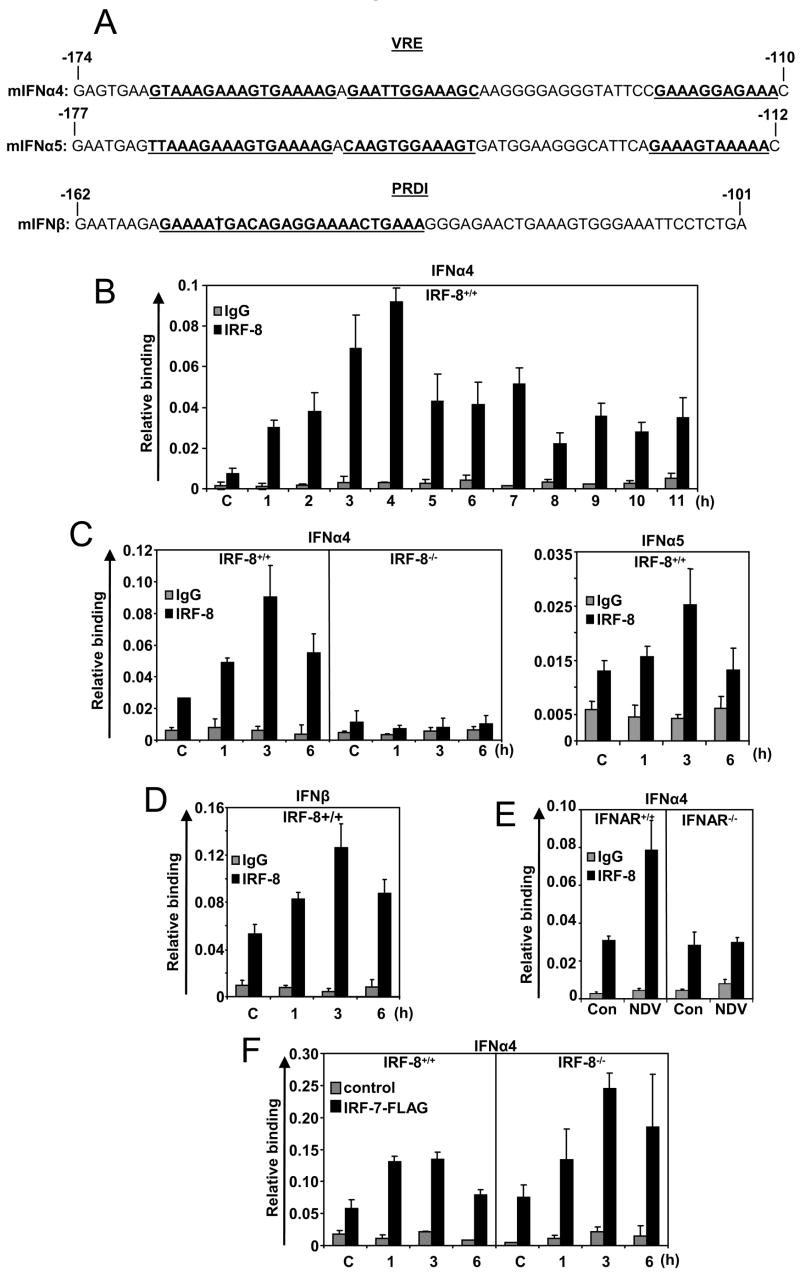
Recruitment of IRF-8 to the IFN Promoters.
A: Diagram of the IFNα4, IFNα5 and IFNβ promoters. IRF binding sites (VRE, PRDI) are underlined. The numbers indicate nucleotide positions relative to transcription start sites.
B: Kinetics of IRF-8 recruitment to the IFNα4 promoter. IRF-8+/+ DCs were stimulated with NDV and ChIP analysis was performed at indicated times using anti-IRF-8 antibody or control IgG.
C: IRF-8 ChIP analysis was performed with IRF-8+/+ and −/− DCs for the IFNα4 (left panel) and the IFNα5 promoters (right panel).
D: IRF-8 ChIP analysis for the IFNβ promoter with IRF-8+/+ DCs.
E: ChIP analysis was performed for the IFNα4 promoter with IFNAR+/+ and IFNAR−/− DCs.
F: IRF-8+/+ and IRF-8−/− DCs were transduced with pMSCV-IRF-7-GFP-Flag, and stimulated with NDV, ChIP analysis was performed using anti-Flag antibody.
In Figure 6E, ChIP was performed in IFNAR−/− DCs. Whereas IRF-8 binding to the IFNα4 promoter increased after NDV stimulation in IFNAR+/+ DCs, no increase in IRF-8 binding was detected in IFNAR−/− DCs. Similar results were seen for IFNα5 and IFNβ promoter (not shown). These data indicate that increased IRF-8 binding after NDV stimulation depends on IFN feedback signals.
ChIP assays were next performed to examine binding of IRF-7 to the IFNα4 promoter. To this end we tested IRF-8+/+ and −/− DCs expressing IRF-7-Flag (Figure 6F). IRF-7-Flag bound to the promoter at a low level prior to NDV addition and the binding increased after NDV stimulation, reaching a plateau at 1 or 3 h and then declined at 6 h both in IRF-8+/+ and −/− DCs. IRF-7-Flag binding was slightly higher in IRF-8−/− cells than +/+ DCs, which may be partly attributed to higher ectopic expression of IRF-7 noted in −/− compared to +/+ cells. ChIP with anti-IRF-7 antibody in above cells also showed IRF-7 binding both in IRF-8+/+ and −/− cells (Figure S5C and 5D). These results show that binding of IRF-7 to the IFN promoters does not require IRF-8.
IRF-8 Supports Sustained RNA Polymerase II Recruitment to the IFN Promoters
Transcription initiation begins after the assembly of basal transcription machinery at the promoter (Roeder, 2005; Sims et al., 2004). RNA polymerase II (RNAPII) is the central molecule in the machinery that catalyzes RNA synthesis. To address the mechanism by which IRF-8 binding impacts on type I IFN transcription, we performed ChIP assays for RNAPII. Data in Figure 7A show kinetics of RNAPII recruitment to the IFNα4 promoter in IRF-8+/+ DCs. RNAPII was not bound before stimulation, but strong binding was seen at 1h after NDV stimulation followed by a slight reduction at 2 h. RNAPII recruitment rose again at 3 h, after which RNAPII binding remained steady for the subsequent 6 h period. In Figure 7B-D, RNAPII recruitment to the IFNα4, IFNα5 and IFNβ promoters was compared between IRF-8+/+ and −/− DCs. RNAPII was recruited 1 h after NDV stimulation both in IRF-8+/+ and −/− cells on all promoters. In +/+ cells, the recruitment continued for the remaining period. However, in IRF-8 −/− DCs, RNAPII recruitment rapidly declined after an initial rise; a decline was seen as early as at 3 h with a further fall at 6 h. Thus, while RNAPII was recruited to the IFN promoters in the early phase in both +/+ and −/− DCs, its subsequent recruitment was prematurely attenuated in the absence of IRF-8. These data indicate that IRF-8 facilitates sustained recruitment of RNAPII, most likely responsible for amplification of IFN transcription in the second phase.
Figure 7.

Recruitment of RNAPII to the IFN Promoters after NDV Stimulation.
A: Kinetics of RNAPII recruitment to the IFNα4 promoter. IRF-8+/+ DCs were stimulated with NDV and ChIP analysis was performed at indicated times using antibody for RNAPII.
B–D: ChIP for RNAPII was performed with IRF-8+/+ and IRF-8−/− DCs for the indicated promoters.
E: Contribution of IRF-8 to DC Induction of Type I IFN Genes (a model).
TLR and viral stimulations lead to the activation of IRF-3 and IRF-7 that initiates the first phase of IFN transcription. This step involves multiple IFNα subtypes and IFNβ in DCs. The initial IFN induction creates an IFN feedback response that sets off the second phase of IFN transcription. IRF-8 participates in this step of IFN transcription. IRF-8 helps to amplify IFN transcription by prolonging RNAPII recruitment, resulting in enhanced IFN production in DCs.
DISCUSSION
This investigation revealed two unique features associated with type I IFN induction in DCs. First, IFN induction is composed of two responses, and elicitation of the second response, mediated by IFN feedback response, requires IRF-8. This conclusion is supported by the observations that (i) IRF-8−/− cDCs did not elicit the second IFN mRNA peak and (ii) ectopic IRF-8 conferred a large second peak upon fibroblasts. Amplification of the second peak is likely to be a major contributor of copious IFN production characteristic to DCs, since IRF-8−/− DCs, although generated the first response, produced only a background level of IFNs. Thus, IRF-8 appears to have a mechanism to augment IFN gene transcription in DCs in response to both viral and TLR signals. Our results indicate that IRF-8 functions in a post-induction phase to amplify IFN transcription and protein production. The notion that IRF-8 acts on a late phase of transcription is consistent with the previous reports that IRF-8 augments expression of some IFN responsive genes with a delayed time course in macrophages (Kanno et al., 2005).
The second aspect of DC specific IFN induction unraveled in this work is that multiple IFNα subtypes are induced during the first phase in DCs, unlike in non-DCs where the initial induction is restricted to the IFNβ and IFNα4 subtypes. Although the molecular basis of this difference is not fully clear at present, it is possible that the IFN gene cluster assumes an open chromatin configuration in DCs, allowing IRF-3 and IRF-7 to have immediate access to multiple IFNα promoters. Irrespective of the mechanism, early activation of multiple IFNα subtypes may have a large impact on the overall IFN production in DCs, since it would help provide a rapid onset and greater elicitation of second response. Supporting this view, the onset of the second peak was seen earlier and its magnitude markedly greater in DCs than in NIH3T3 fibroblasts. In view of DC specific mode of gene expression revealed here, the IFN induction pathways previously outlined for non-DCs could be modified to accommodate above characteristics (Figure. 7E).
Our data clearly show that IRF-8’s main role is to stimulate IFN gene transcription, rather than to act in the upstream signaling events. In line with a role in transcription rather than signaling, IRF-8 was not required for nuclear translocation of IRF-3 and IRF-7, nor was it required for establishment of an IFN feedback arm, as evidenced by induction of IFN responsive genes in IRF-8−/− DCs. The involvement of IRF-8 in transcription was demonstrated by the recruitment of IRF-8 to the IFNα and IFNβ promoters. In line with these data, Kawai T. et. al., recently identified IRF-8 as an activator of IFNβ promoter by screening a spleen cDNA library (Kawai et al., 2005). Significantly, IRF-8 recruitment was increased upon NDV addition, peaking at 4 h well after induction of the first phase, consistent with its action in the second phase of transcription. It is clear that increased IRF-8 recruitment was dependent on IFN feedback, since IFNAR−/− DCs revealed no increase in IRF-8 recruitment after NDV. In these ChIP assays a low level of IRF-8 appeared to be bound to the IFN promoters before and soon after NDV addition, suggesting that some fraction of IRF-8 was recruited to the promoter as a functionally inactive state. It is of note here that binding of IRF-8 (and that of IRF-7-Flag) was not very robust in these experiments, although reproducibly observed. We attribute the relatively low binding to the fact that our ChIP assays were performed with primary DC cultures composed of heterogeneous cell populations, some of which may not express IFN genes.
What is the mechanism by which IFN feedback signals increase IRF-8 recruitment? Given that IRF-8 expression levels remained unchanged before and after NDV stimulation, it is likely that IFN signals increase the affinity of existing IRF-8 for the promoters, presumably through a post-translational modification. Phosphorylation of IRF-8 may be a major mechanism of increased binding, in light of the fact that IFN signals activate kinases of the JAK and MAPK families (Darnell, 1996; Platanias, 2005). IFN signals may also stimulate IRF-8 to interact with additional regulatory factors not associated before, allowing IRF-8 to bind to the promoter more efficiently. Supporting this view, IRF-8 is shown to be under control of tyrosine phosphorylation and dephosphorylation and interact with a partner in an IFN signal dependent manner (Huang et al., 2006; Kuwata et al., 2002; Sharf et al., 1997). Ubiquitination of IRF-8, shown to occur through an IFN inducible E3 ligase may also play a role in modulating IRF-8’s transcriptional activity (Kong et al., 2007).
Specific transcription factors, when bound to the regulatory element, trigger assembly of many factors at/near the initiation site, culminating in the recruitment of the RNAPII complex and synthesis of new RNA chains (Roeder, 2005; Sims et al., 2004). RNAPII was not present on the IFN promoters prior to stimulation, but was recruited upon NDV stimulation. Our results are consistent with signal dependent recruitment of RNAPII previously reported for the promoters of inducible proinflammatory genes (Saccani and Natoli, 2002). Once recruited, RNAPII continued to occupy the IFN promoters up to 9 h in +/+ DCs. However, in IRF-8−/− cells RNAPII recruitment fell prematurely after 1 h in all three promoters. Our data indicate that IRF-8 is equipped with the capacity to retain RNAPII on the IFN promoters for an extended period of time. This capacity is not shared by IRF-7, since IRF-7 was bound to the promoter both in +/+ and −/− DCs. It has been shown that activities of RNAPII are regulated at many steps including those of pre-initiation, initiation, elongation, termination and recycling (Sims et al., 2004). Given that IFN transcription in DCs was typified by prolonged RNAPII recruitment, recycling of RNAPII and transcriptional re-initiation may be an important mechanism that constitutes DC specific IFN transcription. In light of our findings that IRF-8 functions during the second phase of IFN transcription, it is conceivable that its main role is to support efficient transcriptional re-initiation by facilitating repeated recruitment of RNAPII to the promoters. While mechanisms controlling RNAPII recycling are not fully understood, it has been shown that some DNA specific transcription factors play a role in this process (Liu et al., 2001).
In summary, IRF-8 plays a key role in amplifying the second phase of type I IFN gene transcription in DCs. It helps prolong RNAP II recruitment to the IFN promoters. Together, IRF-8 is a critical contributor of rapid and abundant type I IFN production typified in DCs, thereby participating in the establishment of innate immunity.
EXPERIMENTAL PROCEDURES
Mice and Cell Cultures
IRF-8−/− mice were maintained in the NICHD animal facility. IFNAR−/− mice were obtained from J. Durbin (Ohio State University) through H. Young (NCI). All animal work conformed to the NICHD animal care and use committee guidelines. Bone marrow mononuclear cells were cultured in the presence of Flt3L to generate DCs (Tamura et al., 2005a; Tsujimura et al., 2003b). DC surface markers were examined by flow cytometry using anti-CD11b, anti-B220 and anti-CD11c antibodies (BD Pharmingen), and biotin labeled anti-Siglec-H antibody (Hycult Biotechnology, Netherlands) and FITC labeled anti-Ly49Q antibody (MBL International). Data were analyzed using FlowJo software (Tree Star San Carlos, CA). To prepare cDC and pDC enriched DC populations, ~107 cells were incubated at 4°C for 15 min with FITC conjugated anti-B220 antibody (1:500 dilution) in 200μl of PBS containing 0.5% BSA and 2mM EDTA. Cells were washed and incubated with anti-FITC microbeads (10 μl) in total 100 μl buffer. Cells were then subjected to magnetic separation using MACS separation columns (Miltenyi Biotec, Germany). Cells that did not bind to column were collected as pDC free cDCs and the bound fraction was collected as a pDC-enriched population. For NDV viral stimulation, 106 DCs were infected with the Heartz stain of NDV at 640 hemagglutination units (Schiavoni et al., 2002) for 1 h. Similarly 106 DCs were infected with wild type MCMV, derived from pSM3fr, a Bac clone of the Smith strain (Klesza and Shenk, 2006) at a moi of 5 for 1 h. Cells were washed twice and incubated with complete media and harvested at indicated intervals starting from initial exposure of virus. For stimulation with TLR ligands DCs were incubated with 1 μg/ml of CpG 1826 (Lofstrand Labs), or 100 μg/ml of polyIC (Amersham Pharmacia) or 100 nM of R848 (Alexis Biochemicals) for indicated periods of time. NIH3T3 cells were maintained as in (Laricchia-Robbio et al., 2005). One × 105 cells in a well of 6 well plate of control or IRF-8 transduced NIH3T3 cells were infected with NDV at 100 hemaglutination units/ml for 1 h, cells were washed twice and incubated in the media for 24 h. Supernatants were analyzed for IFNα production by ELISA (Pestka Biological Laboratories). Total RNA was extracted using TRIzol reagent (Invitrogen) and cDNA was prepared using Superscript II enzyme (Invitrogen) according to manufacturer’s protocol. For qPCR, amplification of sample cDNA was monitored with the fluorescent DNA-binding dye SYBR Green (SYBR Green PCR master kit; Applied Biosystems) in combination with the ABI Prism 7000 Sequence Detection System (Applied Biosystems), according to the manufacturer’s instructions. Transcript levels were normalized by GAPDH levels. Primer sequences used for PCR are listed in Table S2. Semiquantitative RT-PCR was performed using primer sequence described in (Marie et al., 1998).
Retroviral Transduction
pMSCV retroviral vectors for IRF-8 and mutants were described (Tamura et al., 2005a; Tsujimura et al., 2003a). Construction of pMSCV vector for IRF-7-flag is described in Figure S5. DCs were transduced with the viral supernatants prepared in BOSC23 packaging cells by spinoculation (2400 rpm, 33°C, 1 h) with 4 μg/ml polybrene on day 5 and selected by 2 μg/ml of puromycin for 48 h (Tamura et al., 2005a). NIH3T3 cells were transduced as above and selected with 4 μg/ml puromycin for 4 days (Laricchia-Robbio et al., 2005).
Immunofluorescent Staining
DCs were placed on cytospin slides and fixed with 4% paraformaldehyde, permeabilized with 0.2 % Triton X-100 for 5 min and blocked by 3% BSA for 1 h. Cells were incubated with anti-GFP monoclonal antibody (Roche Applied Science) diluted at 1:200, or rabbit anti-IRF-3 antibody (Zymed) diluted at 1:100 followed by Biotin-conjugated anti-mouse or rabbit IgG (Amersham Biosciences) for 45 min and then incubated with Streptavidin Alexa-488 (Molecular Probes). Cells were counterstained with Hoechst 33342. Stained cells were viewed on a confocal microscope (Leica, Model TCS, SP2).
Immunoblot Analysis
Nuclear extracts and cytoplasmic fractions were prepared as follows. Ten million DCs were incubated in 0.3 ml of buffer containing 10 mM HEPES, pH 7.9, 2.5 mM MgCl2, 10 mM KCl, 0.5 % NP-40 with protease inhibitor cocktail (Roche Applied Science) on ice for 15 min, and centrifuged. Supernatants were used as cytoplasmic fractions. Nuclear pellets were incubated with 20 mM HEPES, pH 7.9, 2.5 mM MgCl2, 20 mM EDTA, 420 mM KCl, 25% glycerol supplemented with protease inhibitor cocktail on ice with occasional stirring and supernatants were collected by centrifugation. Proteins separated on 4–12 % SDS-PAGE were transferred onto PVDF membrane (Millipore) and reacted with indicated antibodies. Secondary detection was carried out with horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG (Amersham Bioscience), followed by chemiluminescence development with ECL reagent (Pierce Biotechnology).
ChIP Assays
This assay was performed with the protocol recommended by Upstate with slight modifications (Tamura et al., 2005a). Briefly, 2.5–10 × 106 cells were treated with 1 % formaldehyde for 30 min at room temperature. Cells were lysed in 650 μl of lysis buffer and sonicated on ice using an XL2007 sonicator (Misonix) to shear DNA into 300 bp–2 kb long fragments. After centrifugation, 100 μl of supernatants were precleared with 25 % of protein A/G agarose slurry (Santa Cruz Biotechnology) supplemented with 200 μg/ml of salmon sperm DNA for 1 h at 4°C. Chromatin was then incubated with 2 μg of normal goat IgG, or goat anti-IRF-8 (C-19, Santa Cruz Biotechnology) overnight with rotation. Normal mouse IgG (2μg), monoclonal anti-Flag antibody M2 (2μg, Sigma), Ig control, rabbit anti-RNAPII antibody (8WG16; Covance) were used for ChIP assays for IRF-7-Flag and RNAPII. This was followed by incubation with Protein A/G agarose slurry supplemented with salmon sperm DNA for additional 1 h, and precipitated. Precipitates were washed twice with low salt buffer, twice with high salt buffer, then with a LiCl buffer and finally with Tris–EDTA buffer, pH 8.0. After elution, complexes were digested with RNase and proteinase K. DNA was purified by phenol/chloroform, precipitated in ethanol and resuspended in 50 μl of buffer containing 5 mM Tris, pH. 8.0. Five μl of the final preparations were subjected to qPCR using primers in Table S2. Input DNA (1%) was used for normalization.
Supplementary Material
Acknowledgments
We thank Durbin, J. and Young, H. for IFNAR−/− mice, Trinchieri, G., Levy, D., Marie, I. and Levi, B. for suggestions. We also thank Shenk, T. and Kulesza, C. for the gift of MCMV virus and stimulating discussions. This work was supported by the Intramural Program of the NICHD, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D. Ordered Recruitment of Chromatin Modifying and General Transcription Factors to the IFN-β Promoter. Cell. 2000;103:667–678. doi: 10.1016/s0092-8674(00)00169-0. [DOI] [PubMed] [Google Scholar]
- Aliberti J, Schulz O, Pennington DJ, Tsujimura H, Reis e Sousa C, Ozato K, Sher A. Essential role for ICSBP in the in vivo development of murine CD8α + dendritic cells. Blood. 2003;101:305–310. doi: 10.1182/blood-2002-04-1088. [DOI] [PubMed] [Google Scholar]
- Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, Vicari A, O’Garra A, Biron C, Briere F, Trinchieri G. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. 2001;2:1144–1150. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- Asselin-Paturel C, Trinchieri G. Production of type I interferons: plasmacytoid dendritic cells and beyond. J Exp Med. 2005;202:461–465. doi: 10.1084/jem.20051395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, Pascual V, Palucka AK. Autoimmunity through cytokine-induced dendritic cell activation. Immunity. 2004;20:539–550. doi: 10.1016/s1074-7613(04)00108-6. [DOI] [PubMed] [Google Scholar]
- Biron CA. Interferons alpha and beta as immune regulators--a new look. Immunity. 2001;14:661–664. doi: 10.1016/s1074-7613(01)00154-6. [DOI] [PubMed] [Google Scholar]
- Civas A, Genin P, Morin P, Lin R, Hiscott J. Promoter organization of the interferon-A genes differentially affects virus-induced expression and responsiveness to TBK1 and IKKepsilon. J Biol Chem. 2006;281:4856–4866. doi: 10.1074/jbc.M506812200. [DOI] [PubMed] [Google Scholar]
- Civas A, Island ML, Genin P, Morin P, Navarro S. Regulation of virus-induced interferon-A genes. Biochimie. 2002;84:643–654. doi: 10.1016/s0300-9084(02)01431-1. [DOI] [PubMed] [Google Scholar]
- Dai J, Megjugorac NJ, Amrute SB, Fitzgerald-Bocarsly P. Regulation of IFN regulatory factor-7 and IFN-alpha production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J Immunol. 2004;173:1535–1548. doi: 10.4049/jimmunol.173.3.1535. [DOI] [PubMed] [Google Scholar]
- Darnell JE., Jr The JAK-STAT pathway: summary of initial studies and recent advances. Recent Prog Horm Res. 1996;51:391–403. discussion 403–394. [PubMed] [Google Scholar]
- Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, Sousa CRe. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. 2003;424:324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EEM, Trinchieri G, Caux C, Garrone P. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med. 2005;201:1435–1446. doi: 10.1084/jem.20041964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliet M, Boonstra A, Paturel C, Antonenko S, Xu XL, Trinchieri G, O’Garra A, Liu YJ. The development of murine plasmacytoid dendritic cell precursors is differentially regulated by FLT3-ligand and granulocyte/macrophage colony-stimulating factor. J Exp Med. 2002;195:953–958. doi: 10.1084/jem.20020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, Taniguchi T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005a;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005b;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- Huang W, Saberwal G, Horvath E, Zhu C, Lindsey S, Eklund EA. Leukemia-associated, constitutively active mutants of SHP2 protein tyrosine phosphatase inhibit NF1 transcriptional activation by the interferon consensus sequence binding protein. Mol Cell Biol. 2006;26:6311–6332. doi: 10.1128/MCB.00036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno Y, Levi BZ, Tamura T, Ozato K. Immune cell-specific amplification of interferon signaling by the IRF-4/8-PU.1 complex. J Interferon Cytokine Res. 2005;25:770–779. doi: 10.1089/jir.2005.25.770. [DOI] [PubMed] [Google Scholar]
- Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- Kerkmann M, Rothenfusser S, Hornung V, Towarowski A, Wagner M, Sarris A, Giese T, Endres S, Hartmann G. Activation with CpG-A and CpG-B Oligonucleotides Reveals Two Distinct Regulatory Pathways of Type I IFN Synthesis in Human Plasmacytoid Dendritic Cells. J Immunol. 2003;170:4465–4474. doi: 10.4049/jimmunol.170.9.4465. [DOI] [PubMed] [Google Scholar]
- Kong HJ, Anderson E, Lee CH, Jang MK, Tamura T, Tailor P, Cho HK, Cheong JH, Xiong H, Morse H, III , Ozato K. Autoantigen Ro52 is an interferon inducible E3 ligase that ubiquitinates IRF-8 and enhances cytokine expression in macrophages. Cuting Edge J Immunol. 2007 doi: 10.4049/jimmunol.179.1.26. In Press. [DOI] [PubMed] [Google Scholar]
- Kulesza KA, Shenk T. Murine cytomegalovirus encodes a stable intron that facilitates persistent replication in the mouse. Proc Nat Acad Sci, USA. 2006;103:18302–18307. doi: 10.1073/pnas.0608718103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwata T, Gongora C, Kanno Y, Sakaguchi K, Tamura T, Kanno T, Basrur V, Martinez R, Appella E, Golub T, Ozato K. Gamma interferon triggers interaction between ICSBP (IRF-8) and TEL, recruiting the histone deacetylase HDAC3 to the interferon-responsive element. Mol Cell Biol. 2002;22:7439–7448. doi: 10.1128/MCB.22.21.7439-7448.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laricchia-Robbio L, Tamura T, Karpova T, Sprague BL, McNally JG, Ozato K. Partner-regulated interaction of IFN regulatory factor 8 with chromatin visualized in live macrophages. Proc Natl Acad Sci U S A. 2005;102:14368–14373. doi: 10.1073/pnas.0504014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi BZ, Hashmueli S, Gleit-Kielmanowicz M, Azriel A, Meraro D. ICSBP/IRF-8 transactivation: a tale of protein-protein interaction. J Interferon Cytokine Res. 2002;22:153–160. doi: 10.1089/107999002753452764. [DOI] [PubMed] [Google Scholar]
- Liu Z, Wong J, Tsai SY, Tsai MJ, O’Malley BW. Sequential recruitment of steroid receptor coactivator-1 (SRC-1) and p300 enhances progesterone receptor-dependent initiation and reinitiation of transcription from chromatin. Proc Natl Acad Sci U S A. 2001;98:12426–12431. doi: 10.1073/pnas.231474798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Falvo JV, Kim TH, Kim TK, Lin CH, Parekh BS, Wathelet MG. Structure and function of the interferon-beta enhanceosome. Cold Spring Harb Symp Quant Biol. 1998;63:609–620. doi: 10.1101/sqb.1998.63.609. [DOI] [PubMed] [Google Scholar]
- Marie I, Durbin JE, Levy David E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci U S A. 2004;101:233–238. doi: 10.1073/pnas.2237236100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol Cell. 2003;11:695–707. doi: 10.1016/s1097-2765(03)00090-x. [DOI] [PubMed] [Google Scholar]
- Roeder RG. Transcriptional regulation and the role of diverse coactivators in animal cells. FEBS Lett. 2005;579:909–915. doi: 10.1016/j.febslet.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Saccani S, Natoli G. Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev. 2002;16:2219–2224. doi: 10.1101/gad.232502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. Distinct and Essential Roles of Transcription Factors IRF-3 and IRF-7 in Response to Viruses for IFN-α/β Gene Induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC, III, Belardelli F, Gabriele L. ICSBP Is Essential for the Development of Mouse Type I Interferon-producing Cells and for the Generation and Activation of CD8α+ Dendritic Cells. J Exp Med. 2002;196:1415–1425. doi: 10.1084/jem.20021263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharf R, Meraro D, Azriel A, Thornton AM, Ozato K, Petricoin EF, Larner AC, Schaper F, Hauser H, Levi BZ. Phosphorylation events modulate the ability of interferon consensus sequence binding protein to interact with interferon regulatory factors and to bind DNA. J Biol Chem. 1997;272:9785–9792. doi: 10.1074/jbc.272.15.9785. [DOI] [PubMed] [Google Scholar]
- Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the Interferon Antiviral Response Through an IKK-Related Pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ. The Nature of the Principal Type 1 Interferon-Producing Cells in Human Blood 10.1126/science.284.5421.1835. Science. 1999;284:1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- Sims RJ, 3rd, Belotserkovskaya R, Reinberg D. Elongation by RNA polymerase II: the short and long of it. Genes Dev. 2004;18:2437–2468. doi: 10.1101/gad.1235904. [DOI] [PubMed] [Google Scholar]
- Tailor P, Tamura T, Ozato K. IRF family proteins and type I interferon induction in dendritic cells. Cell Res. 2006;16:134–140. doi: 10.1038/sj.cr.7310018. [DOI] [PubMed] [Google Scholar]
- Tamura T, Ozato K. ICSBP/IRF-8: its regulatory roles in the development of myeloid cells. J Interferon Cytokine Res. 2002;22:145–152. doi: 10.1089/107999002753452755. [DOI] [PubMed] [Google Scholar]
- Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O’Shea JJ, Singh H, Ozato K. IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J Immunol. 2005a;174:2573–2581. doi: 10.4049/jimmunol.174.5.2573. [DOI] [PubMed] [Google Scholar]
- Tamura T, Thotakura P, Tanaka TS, Ko MSH, Ozato K. Identification of target genes and a unique cis element regulated by IRF-8 in developing macrophages. Blood. 2005b;106:1938–1947. doi: 10.1182/blood-2005-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimura H, Tamura T, Gongora C, Aliberti J, Reis e Sousa C, Sher A, Ozato K. ICSBP/IRF-8 retrovirus transduction rescues dendritic cell development in vitro. Blood. 2003a;101:961–969. doi: 10.1182/blood-2002-05-1327. [DOI] [PubMed] [Google Scholar]
- Tsujimura H, Tamura T, Ozato K. Cutting edge: IFN consensus sequence binding protein/IFN regulatory factor 8 drives the development of type I IFN-producing plasmacytoid dendritic cells. J Immunol. 2003b;170:1131–1135. doi: 10.4049/jimmunol.170.3.1131. [DOI] [PubMed] [Google Scholar]
- Yang H, Lin CH, Ma G, Baffi MO, Wathelet MG. Interferon regulatory factor-7 synergizes with other transcription factors through multiple interactions with p300/CBP coactivators. J Biol Chem. 2003;278:15495–15504. doi: 10.1074/jbc.M212940200. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Jr, Akira S, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.