

Abstract
A secreted chlamydial protease designated CPAF (Chlamydial Protease/proteasome-like Activity Factor) degrades host proteins, enabling Chlamydia to evade host defenses and replicate. The mechanistic details of CPAF action, however, remain obscure. We used a computational approach to search the protein data bank for structures that are compatible with the CPAF amino acid sequence. The results reveal that CPAF possesses a fold similar to that of the catalytic domains of the tricorn protease from Thermoplasma acidophilum, and that CPAF residues H105, S499, and E558 are structurally analogous to the tricorn protease catalytic triad residues H746, S965, and D1023. Substitution of these putative CPAF catalytic residues blocked the CPAF from degrading substrates in vitro, while the wild type and a noncatalytic control mutant of CPAF remained cleavage-competent. Substrate cleavage is also correlated with processing of CPAF into N-terminal (CPAFn) and C-terminal (CPAFc) fragments, suggesting that these putative catalytic residues may also be required for CPAF maturation.
Keywords: Chlamydia, CPAF, Hidden Markov Models, HHPRED, MODELLER, molecular modeling, tricorn protease, catalytic triad, site directed mutagenesis, protein structure prediction
Introduction
Infection with chlamydial organisms that have adapted an obligate intracellular growth life cycle [1] imposes severe health problems in both humans and animals [2-8]. However, the pathogenic mechanisms of Chlamydia-induced diseases remain unclear. It is hypothesized that inflammatory responses induced during chlamydial intracellular replication significantly contribute to the chlamydial pathogenesis [9-12]. We have previously identified a Chlamydia-secreted protease designated as CPAF (Chlamydial Protease/proteasome-like Activity Factor) that may participate in chlamydial immune evasion and promote chlamydial intracellular survival [13-17].
CPAF was initially discovered through its ability to degrade RXF5 and USF1, host transcriptional factors that are required for MHC antigen expression [13-15]. This activity also suggested a mechanism through which Chlamydia evades host immune detection. CPAF may also contribute to chlamydial inhibition of apoptosis by degrading various proapoptotic BH3-only proteins [16, 18-20]. To facilitate chlamydial vacuole expansion, CPAF can solubilize portions of intermediate filaments (IF) by cleaving cytokeratin 8 [17], a major component of IF in epithelial cells. Recently, it was shown that CPAF could also cleave cyclin B and PARP, which may contribute to the blockade of host cell replication and repairing efforts at the late stages of infection [21]. It thus seems clear that CPAF can promote chlamydial pathogenesis via multiple means ranging from evasion of host defense to facilitation of chlamydial vacuole expansion. CPAF therefore represents a bona fide virulence factor of Chlamydia.
Although full length CPAF is a 70 kDa protein, once secreted into the cytoplasm of the infected host cell, it is cleaved into two shorter polypeptides of MW ∼29 KDa (CPAFn) and of MW ∼35 KDa (CPAFc) [15, 17, 22, 23]. CPAFn and CPAFc remain associated as a complex designated CPAFn:CPAFc, Two CPAFc:CPAFn complexes subsequently come together to form the catalytically active “dimeric” molecule [17]. Interestingly, full length CPAF is partially processed and acquires measurable proteolytic activity when expressed as GST-fusion proteins in bacterial but not eukaryotic cell expression systems [22, 24], which has provided a platform for further characterization of CPAF proteolytic activity. Biochemical studies have led to the suggestion that both the cleavage and the subsequent dimerization events are required for CPAF to degrade its host substrates [22, 24]. However, the mechanistic details of CPAF proteolytic activity remain unknown.
Here, we used a computational approach in which the CPAF amino acid sequence was used to search for protease homologs of known structure in order to identify putative catalytic residues. The results of this in silico analysis reveal that in three-dimensional (3-D) space, the H105, S499, and E558 residues of CPAF map to the catalytic residues H746, S965, and D1023 of the structure of the tricorn protease, a 720 kDa serine proteolytic complex from Thermoplasma acidophilum [25], strongly suggesting that these CPAF residues also play a catalytic role in CPAF proteolytic activity. This suggestion was tested using a site-directed mutagenesis approach in which we confirmed that CPAF mutants with substitutions at these putative catalytic residues lost their ability to degrade substrate proteins, while the wild type or a control CPAF mutant retained the ability to competently cleave these substrates. Furthermore, the ability to cleave the substrate proteins was correlated with the processing of CPAF into CPAFn and CPAFc fragments, suggesting that the putative catalytic residues are also required for CPAF self-processing.
Materials and Methods
Identification of CPAF Catalytic Residues using HHPRED
We searched for possible structural homologs of CPAF using a variation of protein threading as implemented in the program HHPRED, which is available on a web server (http://toolkit.tuebingen.mpg.de/hhpred) [26, 27]. HHPRED uses pair-wise comparison of profile Hidden Markov Models (HMMs). Briefly, in the first step, an alignment of sequence homologs is built for the CPAF query sequence by multiple iterations of PSI-BLAST against the non-redundant sequence database from NCBI. In the second step, a single CPAF profile HMM is generated from the multiple sequence alignment, which contains a concise statistical description of the underlying alignment, including secondary structural information. For each column in the multiple alignment that has a residue in the query sequence, an HMM column is created that contains the probabilities of each of the 20 amino acids, plus 4 probabilities that describe how often amino acids are inserted and deleted at this position (insert open/extend, delete open/extend). These insert/delete probabilities are translated into position-specific gap penalties when an HMM is aligned to a sequence or to another HMM [26, 27].
These same two steps are also performed for each sequence corresponding to a known structure in the Protein Data Bank (PDB) in order to generate a library of profile HMMs to which the query profile HMM can be compared. In the third step, the query profile HMM is compared to each profile HMM in the structural database and scored [26, 27]. The HHPRED output, which consists of an alignment of a sequence to be modeled with known related structures is used as input for the program MODELLER, a program that automatically calculates a model of the query sequence containing all non-hydrogen atoms by satisfaction of spatial restraints [28].
Construction of CPAF Mutants
The gene encoding the full-length CPAF wild type (Wt) enzyme was cloned into a pGEX6p-2 vector (Amersham Biosciences Corp, Piscataway, NJ) using the Chlamydia trachomatis serovar D genomic DNA as template and the oligonucleotide sequences 5′-CGC.GGA.TCC.ATG.GGT.TTT.TGG.AGA.ACA.TCG (forward) and 5′-AAAAGGAAAAGCGGCCGC.TCA.AAA.ACT.ACC.ATC.TTC.CGC (reverse) as cloning primers. The putative leader sequence (residues 1-24; http://stdgen.northwestern.edu/) in CPAF was not excluded during primer design. This is because the GST is fused to the N-terminus, and constructs with or without the putative leader sequence displayed no differences in expression level. This vector allows genes of interest to be expressed as fusion proteins with a 26 kDa glutathione-S-transferase (GST) as a fusion partner at the N-terminus. A site-directed mutagenesis kit (cat# 200518, Stratagene, La Jolla, CA) was used to create various mutant CPAF proteins using the Wt full length CPAF gene as the template [24]. Briefly, complementary primers incorporating the desired nucleotide substitutions were used to introduce the corresponding mutations. After primer extension and amplification using PfuUltra DNA polymerase, the parental methylated and hemimethylated DNA was digested with the nuclease DpnI. The mutated molecules were then transformed into competent bacterial cells for nick repair and gene expression. The primers 5′-AAT.GAC.TTT.GCC.GCT.GGA.GTA (forward; the codon CAC coding for an H at 105 position in the Wt CPAF was changed to GCC that codes for an A and the altered nucleotides are underlined) and 5′-TAC.TCC.AGC.GGC.AAA.GTC.ATT (reverse) were used to create the mutant CPAF that carries the residue H105 to A mutation (designated as H105A), 5′-CAA.GAC.TTT.GCT.TGT.GCT.GAC (forward, TCT coding for S in Wt CPAF was mutated to GCT coding for A) and 5′-GTC.AGC.ACA.AGC.AAA.GTC.TTG (reverse) for S499A, 5′-GCC.TTC.ATT.GCC.AAC.ATC.GGA (forward, GAG coding for E in Wt CPAF was mutated to GCC coding for A) and 5′-TCC.GAT.GTT.GGC.AAT.GAA.GGC (reverse) for E558A, 5′-ACT.GGA.ATA.GCA.ACT.TGT.TCT (forward, AAA coding for K in Wt CPAF was mutated to GCA coding for A) and 5′-AGA.ACA.AGT.TGC.TAT.TCC.AGT (reverse) for the control mutant K540A.
Expression of CPAF and CPAF Mutants
All GST-CPAF constructs were expressed in E. coli XL1-Blue with IPTG as inducer as previously described [24, 29]. To reduce the amount of insoluble protein, all CPAF fusion constructs were induced at 30 °C for 3 hours. The GST-fusion proteins were released from bacteria by sonication on ice and purified using glutathione-conjugated beads (Amersham Biosciences Corp). The bead-immobilized fusion proteins were quantified, aliquoted and stored at -80°C till the digestion experiments.
Cell-free Degradation Assays
Cell-free degradation assays were carried out as described previously [15, 24]. Cytosolic extracts (CE) containing Puma and keratin 8 or nuclear extracts (NE) containing RFX5 and USF-1 was used as substrates. Each CE was prepared by following a protocol previously described [17, 19]. Briefly, 1-2 × 107 normal HeLa cells were pelleted and resuspended in 0.5 ml of NP-40 buffer containing 1 % NP-40 (v/v), 0.5 % Triton X-100 (v/v) and 0.15 M NaCI, in 50 mM Tris (pH 8.0) plus a protease inhibitor cocktail [1 mM PMSF (cat# P7626), 20 μM leupeptin (L2884), 1.6 μM pepstatin A (P5318) and 1.7 μg/ml of aprotinin (A6279), all from Sigma, St. Louis, MO]. After gentle mixing, the extraction was carried out on ice for 20 min followed by a microfuge centrifugation to pellet the cell ghosts. The supernatants were collected, aliquoted and stored at -80 °C until use. Each NE was prepared as previously described [22, 24] by using the pellets after CE extraction. The pellets were resuspended in 0.5 ml of high salt lysis buffer containing 0.5 M NaCI and 1 % Triton X-100 (v/v) in 20 mM Tris (pH 8.0) plus the same protease inhibitor cocktail used for preparing CE. The extraction was carried out on ice for 30 min followed by a high-speed centrifugation. The supernatant was collected as NE. In some cases, the extractions were repeated a few times and the supernatants were pooled, aliquoted and stored at -80 °C until use. The cytosolic fractions from cells infected with C. trachomatis serovar L2 (L2S100) were used as the source of enzyme. Each L2S100 was prepared using an established protocol [18]. Briefly, the infected cells were harvested via low speed centrifugation and the cell pellets were resuspended in a douncing buffer [(10 mM KCL, 1.5 mM MgCL2, 1 mM EDTA, 1 mM DTT, 250 mM sucrose in 20 mM Hepes-KOH (pH 7.5) with a protease inhibitor cocktail as described above]. After limited douncing to make sure that > 70% cells are broken without damage to either the nuclei or inclusions, the supernatants were harvested after a series of centrifugation including a final airfuge centrifugation at 100,000 × g and designated as L2S100. For digestion, the enzyme preps and substrates were mixed and incubated for 1 h at 37 °C. The residual substrates after digestion were detected using a Western blot assay as described below.
Western Blot
The Western blot assays were carried out as we previously described [30, 31]. Briefly, the bead-bound fusion proteins or reaction mixtures from cell-free degradation assays were subjected to protein separation in the SDS-polyacrylamide gels. After the resolved protein bands were blotted onto nitrocellulose membranes, primary antibodies were applied. These include mouse monoclonal antibodies (mAb) 100a for detecting CPAFc [15, 22], M20 for cytokeratin 8 (C5301, Sigma, Saint Louis, MO), rabbit polyclonal antibodies for RFX5 (cat# 200-401-191, Rockland Immunochemicals Inc. Gilbertsville, PA) and for USF-1 (sc-229, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and a rabbit mAb against Puma (EP512Y, ab33906, Abcam, Cambridge, MA). Primary antibody binding was probed with the corresponding secondary antibodies conjugated with horseradish peroxidase (Jackson ImmunoResearch Laboratories, Inc. West Grove, PA), followed by standard enhanced chemiluminescence (Amersham Biosciences Corp).
Results
Computational Identification of CPAF Catalytic Triad
Figure 1 shows a sequence alignment between CPAF and Tricorn protease that includes secondary structural information and highlights the residues in both proteins that form the catalytic triad. There amino acid identity overall between the two proteins shown in Figure 1 is only 17 %, and due to multiple insertions in the CPAF sequence relative to that of the tricorn protease catalytic domain (CPAF residues 159-183, 217-249, 274-321, 335-345, 401-474), the relationship between CPAF and tricorn protease was not obvious from sequence alignment methods alone. Table 1 shows the statistics for the five top-scoring hits coming from the HHPRED analysis using the query CPAF amino acid sequence. The tricorn protease, the structure of which (pdb code 1K32 [25] is shown in Figure 2A, scored highest for the query CPAF sequence. Tricorn protease consists of a trimer of dimers that associate to form a hexameric ring. The active sites of the tricorn protease are located at the dimer interfaces and are comprised of residues coming from the catalytic domains of each subunit (boxed in Figure 2A). The highest scoring portions of the CPAF molecule map only to the catalytic domains of tricorn protease and are shown in cartoon format in Figures 2A and 2B. An expanded view of the active site of tricorn protease and that predicted for CPAF based on the tricorn protease structure are shown in Figures 2C and 2D, respectively. The results of this computational analysis strongly suggest that CPAF amino acid residues H105, S499, and E558 correspond in 3-D space to H746, S965, and D1023, the catalytic triad of tricorn protease. These residues of the putative CPAF catalytic triad were chosen for further mutagenesis studies.
Figure 1.
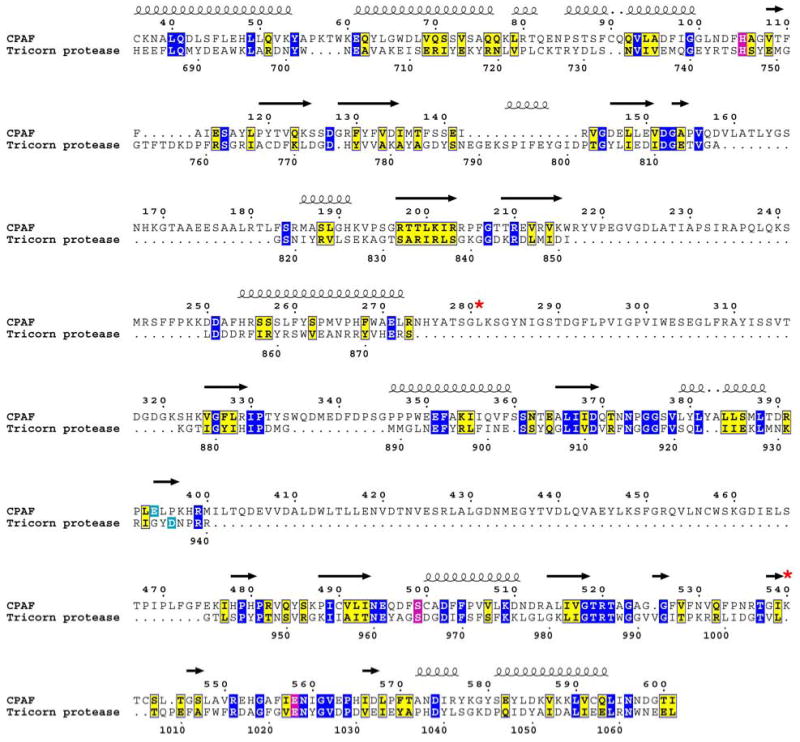
Modified sequence alignment between tricorn protease and CPAF based on pair-wise comparison of profile Hidden Markov Models (HMMs) as implemented in HHPRED [26, 27]. Identical amino acids are boxed in blue and homologous amino acids are boxed in yellow. The residues forming the catalytic triads are boxed in magenta and the substrate specificity residues are boxed in cyan. A red star designates residues in CPAF that were altered by site-directed mutagenesis. The CPAF mutants were tested for proteolytic activity (see text and Figure 4). Secondary structural elements are indicated as spirals for α-helices and arrows for β-strands based on the known structure of the tricorn protease [pdb code 1k32 [25]]. Note the lengthy intervening sequences in CPAF (residues 159-183, 217-249, 274-321, 335-345, 401-474) not found in tricorn protease that presumably form structural elements distinct from the catalytic domains common to tricorn and CPAF (see Figure 2 and Supplementary Figure 1). This figure was prepared in part using the program ESPript [50].
Table 1.
Top-scoring structures that can serve as templates for the CPAF amino acid sequence in the program MODELLER [28] as identified by the program HHPRED [26, 27]. The column labeled “Prob” is the probability that the database match is a true positive. “E-values” are defined in the same way as in PSI-BLAST [46]. The “P-value” is equal to the “E-value” divided by the number of HMMs in the protein data bank. The “Score” column gives the total score that includes the score from the secondary structure comparison, which is listed in the column labeled “SS”. “Cols” contains the total number of matched columns in the query–template alignment and the remaining columns describe the range of aligned residues in the query and template [26, 27].
| No. Hit |
PDB Code |
Protein |
Prob |
E-val |
P-val |
Score |
SS |
Cols |
Query HMM |
Template HMM |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1k32 [25] | Tricorn Protease Thermatoga maritima |
100 | 0 | 0 | 335.6 | 32.0 | 363 | 36-601 | 657-1040 (1045) |
| 2 | 1fc6 [45] | PS II D1 Protease Scenedesmus obliquus |
100 | 0 | 0 | 299.5 | 34.8 | 363 | 34-593 | 2-381 (388) |
| 3 | 1j7x [47] | IRBP Interphotoreceptor Xenopus laevis |
100 | 8.9E-38 | 7E-42 | 241.1 | 17.4 | 180 | 321-569 | 104-283 (302) |
| 4 | 2ejy [48] | Erythrocyte membrane protein Homo sapiens |
92.7 | 0.096 | 7.5E-6 | 28.0 | 5.2 | 49 | 108-156 | 16-69 (97)) |
| 5 | 2ev8 [49] | Erythrocyte membrane protein Homo sapiens |
92.1 | 0.093 | 7.3E-6 | 28.0 | 4.6 | 49 | 108-156 | 16-69 (97) |
Figure 2.
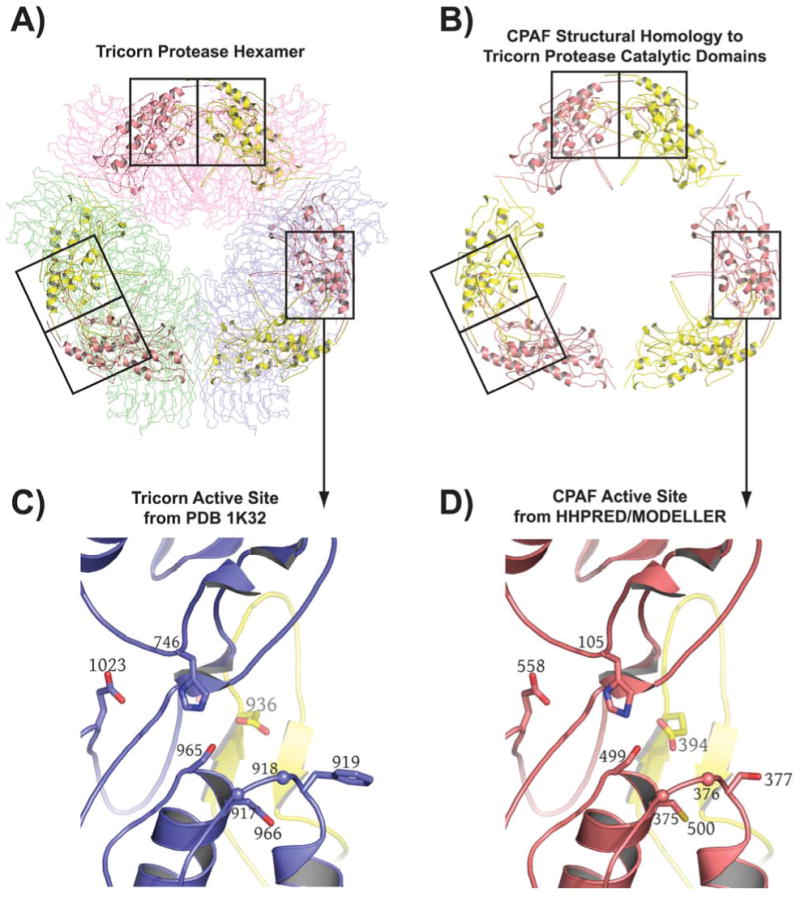
Computational prediction of CPAF fold and catalytic residues. A) The highest scoring hit returned for the CPAF amino acid sequence by HHPRED [26, 27] is the structure of the tricorn protease [25], a 720 kDa proteolytic system from Thermoplasma acidophilum that degrades cytosolic proteins analogously to the proteasome [43] (see text). The tricorn protease structure is a hexamer formed by a trimer of dimmers that has the shape of a distorted hexagon. The dimers forming the long sides of the hexagon are shown as Cα-traces and are colored in light pink, light green, and blue, respectively. The amino acid residues of CPAF that map onto the catalytic domain of the tricorn protease are shown in cartoon format. B) The amino acid residues of CPAF that map onto the catalytic domains of the tricorn protease taken out of the context of the tricorn protease. The functional active site requires contributions from both a yellow and a salmon subunit, corresponding to the catalytic domains of two-fold related tricorn protease subunits, suggesting that for CPAF, the minimal functional proteolytic unit is a CPAFn:CPAFc dimer (see text). C) The tricorn protease active site. Note that the specificity-determining residue, E936 shown in yellow, comes from a two-fold related catalytic domain subunit of the tricorn protease. D) CPAF active site identified by HHPRED [26, 27] and modeled by the program MODELLER [28]. The CPAF H105, S499, E558 catalytic triad and the E394 specificity-determining residue of CPAF corresponds to the H746, S945, D1023 catalytic triad and E936 specificity-determining residue of tricorn protease. This figure was prepared using the program PyMol (Delano Scientific, www.pymol.org).
Substitution of H105, S499 or E558 with Alanine blocks CPAF proteolytic activity
CPAF mutants with alanine substitution of the putative catalytic residues (H105A, S499A, E558A) together with the wild type (Wt), a cleavage-defective control mutant [24] and an unrelated alanine substitution mutant (K540A) were expressed as GST fusion proteins. The fusion proteins purified onto the glutathione-conjugated agarose beads were checked for both quantity and quality on a SDS-polyacrylamide gel (Figure 3). Each clone expressed an equivalent amount of full-length GST-CPAF fusion proteins migrating at the predicted molecular weight position (96 kDa). Free GST molecules were always present in all fusion preps. We then used the bead-bound fusion proteins as the source of enzyme to digest cellular proteins extracted from normal HeLa in a cell-free assay. The residual substrate proteins were monitored with corresponding antibodies on a Western blot (Figure 4). The transcription factors RFX5 (panel a) and USF-1 (b) were detected in the nuclear extract (lane 1) but completely degraded by the endogenous CPAF in L2S100 (lane 2). Both the Wt CPAF and the CPAF carrying a substitutional mutation of K540, a residue predicted not to participate in catalysis, with alanine, also completely degraded the transcription factors. However, the three CPAF mutants each with a putative catalytic residue replaced by an alanine (H105A, S449A & E558A) failed to degrade either RFX5 or USF-1. The processing-deficient mutant L281G also failed to degrade these substrates, which is consistent with our previous findings [24]. We further compared these CPAF preps for their ability to degrade cytosolic substrates including the BH3-only domain protein Puma (panel c) and the cytokeratin 8 (d). Similarly, the CPAF mutants with the putative catalytic residue replacements were not able to efficiently degrade these two cytosolic substrates compared to the Wt and the unrelated mutant. These results together have demonstrated that the three residues H105, S499 & E558 are each required for optimal proteolytic activity of CPAF.
Figure 3.

The CPAF mutants used in this study. A) The various CPAF mutants were generated as GST fusion proteins. Cleavage of full-length CPAF into CPAFn and CPAFc occurs at residue 283. B) The GST-CPAF fusion proteins were purified on beads and were loaded into a SDS PAGE gel in varying amounts as indicated by the bead volume at the top of the figure. After the proteins are separated via electrophoresis, the gels were stained with the Coommassie blue dye. After destaining, the quality and quantity of the fusion proteins were inspected and compared between different samples. The protein band image was acquired using a dried gel.
Figure 4.
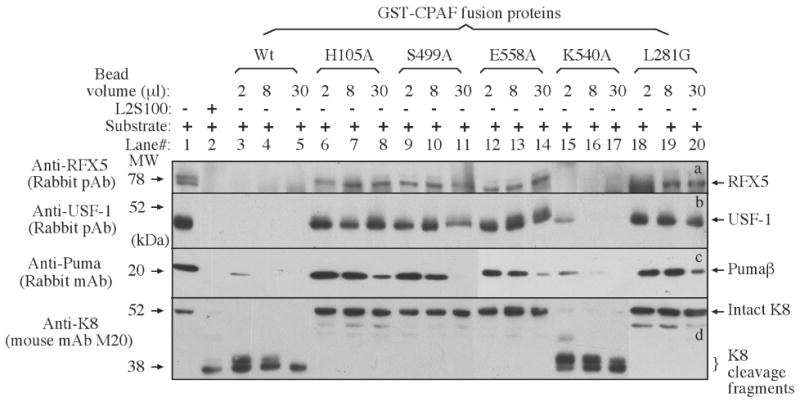
Degradation of target proteins by CPAF mutants in a cell-free assay. Either nuclear extract (NE) containing RFX5 (panel a) & USF-1 (b) or cytosolic extract (CE) containing Puma (c) and keratin 8 (d) were used as substrates to mix with the enzyme source (either L2S100 or various GST-CPAF fusion proteins) and the mixtures were incubated for 1 h at 37 °C. The entire mixture from each reaction was loaded into the corresponding lanes as indicated on top of the figure. After electrophoresis, the resolved protein bands were blotted onto nitrocellulose membrane for corresponding antibody detection as indicated along the left side of the figure. One independent set of enzyme/substrate reactions was used for detecting each substrate. The intact full-length keratin 8 has a molecular weight of 52 kDa and CPAF is known to cleave keratin 8 into 38 kDa fragments [17].
Substitution of H105, S499 or E558 also blocks CPAF processing
We have previously shown that CPAF activity is dependent on CPAF processing into two fragments [22, 24]. A substitutional mutation of L281 with glycine at the predicted cleavage site blocked both CPAF processing and proteolytic activity [24]. CPAF processing has been used to assess CPAF activity [21, 24]. We then tested whether mutation of the catalytic residues can also affect CPAF processing. As expected, both the Wt and the unrelated control mutant CPAF preps generated the free CPAFc fragments, indicating that these preps were at least partially processed (Figure 5), which is consistent with their ability to degrade substrate proteins as described above. Interestingly, CPAF mutants with replacements of either the catalytic residues (H105A, S499A & E558A) or a cleavage site residue (L281G) were not processed at all since no free CPAFc fragments were detected from these preps. These observations suggest that the three putative catalytic residues also play critical roles in CPAF processing.
Figure 5.
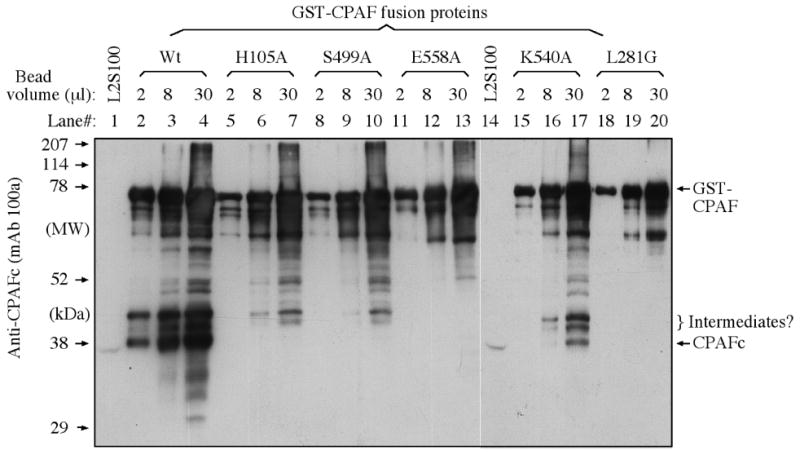
Detection of CPAF processing using a Western blot. The various GST-CPAF fusion proteins were loaded onto a SDS PAGE gel as described in the legend to Figure 3. After electrophoresis, the proteins bands were transferred onto nitrocellulose membrane for detection with the anti-CPAFc mAb 100a. All CPAF preps displayed the full-length GST-CPAF fusion protein along with fragments or varying length. However, only the Wt (lanes 2-4) and the unrelated CPAF mutant K540A (lane 17) displayed a protein band migrating at the position similar to band of CPAFc from the L2S100 sample (lanes 1 & 14).
Discussion
Since its discovery, much progress has been made toward understanding the biochemical properties of CPAF and the role of CPAF in chlamydial pathogenesis and vaccine development [21, 32-41]. However, the catalytic nature of CPAF had remained elusive. Because the relationship between CPAF and other proteases were not obvious using sequence alignment methods alone (see RESULTS section), we used computational methods in an effort to provide insight into possible CPAF mechanism(s). In traditional “protein threading” analyses, the amino acid sequence of an unknown structure is scanned against a database of known structures [42]. For each known structure, which serves as a potential scaffold for the unknown protein's amino acid sequence, a scoring function assesses the compatibility of the sequence to the structural template. High scores yield possible 3-D models. Such methods have great utility because amino acid sequences diverge much more rapidly than 3-D structures, so although a given protein sequence may not possess significant amino acid identify with other proteins in the sequence database, it may still be quite compatible with 3-D scaffolding present in the structural database. In other words, two proteins may have very similar 3-D folds even though they possess very little sequence identity.
In the current study, we used the programs HHPRED [26, 27] and MODELLER [28] to predict putative catalytic residues of CPAF and found an excellent alignment of CPAF residues with the structure of the catalytic domains and the catalytic triads of tricon protease (Figure 1 and Table 1), a large serine protease from Thermoplasma acidophilum [25] (Figure 2). The results strongly suggest that CPAF is also a serine protease with a catalytic triad consisting of amino acid residues H105, S499, and E558. Thus, S499 likely serves as the nucleophile attacking the carbonyl carbon atom of the residue to be cleaved in the substrate, while H105 serves to polarize the hydroxyl group of S499, and E558 orients H105 via a bound water molecule and makes it a better proton acceptor via electrostatic effects. Tricorn has been shown to exhibit both tryptic and chymotryptic specificities [43]. The X-ray structure reveals that specificity for basic P1 residues (preceding the cleavable bond) is conferred by D936, which is provided by the dyad-related subunit [25] (Figure 2C). As shown in Figure 2D, this specificity determinant (E394) also appears to be present in CPAF. Site-directed mutagenesis analyses have allowed us to confirm that these three residues are indeed critical for CPAF enzymatic activity in degrading four known CPAF substrate proteins. This conclusion is consistent with the observation that none of the point mutations affected the CPAF overall structure since the solubility of all CPAF preps remained essentially the same. Furthermore, this conclusion is apparently supported by a recent crystal structure of CPAF [44], although at this writing the coordinates are not available to permit a comparison to our model.
Intriguingly, as shown in Figure 2D, a cysteine residue, C500, sits immediately adjacent to the S499 of the predicted catalytic triad of CPAF and it is tempting to speculate that CPAF may also function as a cysteine protease, endowing it with broader substrate specificity than that demonstrated by tricorn protease. It is also interesting to note that, as shown in Figure 1 and Supplementary Figure 1, the CPAF sequences that map to the 3-D structure of the catalytic domains of tricorn protease are interspersed with sequences that do not seem to map with any known structure in the protein data bank. This suggests that these interspersing residues are likely involved in the higher order assembly of CPAF and not in catalysis, and that the overall oligomeric composition of CPAF is different than the hexameric arrangement of subunits in tricorn protease. In this context, it should be noted that the second-highest scoring molecule in the protein data bank for the CPAF query sequence is for the structure of the catalytic domains of the photosystem II D1 serine protease from Scenedesmus obliquus (pdb code 1FC6 [45]) which functions as a homodimer and not a homohexamer.
An additional intriguing finding is that mutation of the three putative catalytic residues also affects CPAF processing. Since CPAFc starts with the residue G284, the cleavage site has to be upstream of and close to this residue. Clearly, none of the three catalytic residues could be located in the cleavage site. Therefore, we hypothesize that CPAF processing might depend on its own enzymatic activity. This hypothesis is consistent with the recent finding that artificially induced oligmerization of CPAF can lead to CPAF fusion protein processing and activation [21].
Besides its role in pathogenesis, CPAF has been found to induce protective immunity against chlamydial infection. CPAF is one of the most immunodominant antigens during C. trachomatis infection in humans in terms of antibody production [29, 33, 35]. Although the human serum antibodies can neutralize CPAF enzymatic activity in test tubes, antibodies may play a very limited role in regulating CPAF activity inside the Chlamydia-infected cells. Indeed, the CPAF-induced protective immunity in mice was found to depend on a Th1 dominant and IFNγ-mediated response [39-41]. The characterization of CPAF proteolytic activity may enable us to design small molecule inhibitors that are cell-permeable for blocking CPAF activity in the Chlamydia-infected cells so that the infected cells can be efficiently detected and attacked by T lymphocytes.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the US National Institutes of Health (to G. Zhong) and from the Robert A. Welch Foundation AQ-1399 (to P.J. Hart). Support for the X-ray Crystallography Core Laboratory by the Office of the Vice President for Research at the University of Texas Health Science Center is also gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hackstadt T, Fischer ER, Scidmore MA, Rockey DD, Heinzen RA. Trends Microbiol. 1997;5:288–293. doi: 10.1016/S0966-842X(97)01061-5. [DOI] [PubMed] [Google Scholar]
- 2.Grayston JT, Wang S. J Infect Dis. 1975;132:87–105. doi: 10.1093/infdis/132.1.87. [DOI] [PubMed] [Google Scholar]
- 3.Wright HR, Turner A, Taylor HR. Lancet. 2008;371:1945–1954. doi: 10.1016/S0140-6736(08)60836-3. [DOI] [PubMed] [Google Scholar]
- 4.Mabey DC, Solomon AW, Foster A. Lancet. 2003;362:223–229. doi: 10.1016/S0140-6736(03)13914-1. [DOI] [PubMed] [Google Scholar]
- 5.Rekart ML, Brunham RC. Sex Transm Infect. 2008;84:87–91. doi: 10.1136/sti.2007.027938. [DOI] [PubMed] [Google Scholar]
- 6.Vanrompay D, Harkinezhad T, van de Walle M, Beeckman D, van Droogenbroeck C, Verminnen K, Leten R, Martel A, Cauwerts K. Emerg Infect Dis. 2007;13:1108–1110. doi: 10.3201/eid1307.070074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theegarten D, Sachse K, Mentrup B, Fey K, Hotzel H, Anhenn O. Respir Res. 2008;9:14. doi: 10.1186/1465-9921-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kauffold J, Melzer F, Berndt A, Hoffmann G, Hotzel H, Sachse K. Theriogenology. 2006;66:1816–1823. doi: 10.1016/j.theriogenology.2006.04.042. [DOI] [PubMed] [Google Scholar]
- 9.Bobo LD, Novak N, Munoz B, Hsieh YH, Quinn TC, West S. J Infect Dis. 1997;176:1524–1530. doi: 10.1086/514151. [DOI] [PubMed] [Google Scholar]
- 10.Stephens RS. Trends Microbiol. 2003;11:44–51. doi: 10.1016/s0966-842x(02)00011-2. [DOI] [PubMed] [Google Scholar]
- 11.Cheng W, Shivshankar P, Li Z, Chen L, Yeh IT, Zhong G. Infect Immun. 2008;76:515–522. doi: 10.1128/IAI.01064-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng W, Shivshankar P, Zhong Y, Chen D, Li Z, Zhong G. Infect Immun. 2008;76:942–951. doi: 10.1128/IAI.01313-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong G, Fan T, Liu L. J Exp Med. 1999;189:1931–1938. doi: 10.1084/jem.189.12.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong G, Liu L, Fan T, Fan P, Ji H. J Exp Med. 2000;191:1525–1534. doi: 10.1084/jem.191.9.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong G, Fan P, Ji H, Dong F, Huang Y. J Exp Med. 2001;193:935–942. doi: 10.1084/jem.193.8.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pirbhai M, Dong F, Zhong Y, Pan KZ, Zhong G. J Biol Chem. 2006;281:31495–31501. doi: 10.1074/jbc.M602796200. [DOI] [PubMed] [Google Scholar]
- 17.Dong F, Su H, Huang Y, Zhong Y, Zhong G. Infect Immun. 2004;72:3863–3868. doi: 10.1128/IAI.72.7.3863-3868.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. J Exp Med. 1998;187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong F, Pirbhai M, Xiao Y, Zhong Y, Wu Y, Zhong G. Infect Immun. 2005;73:1861–1864. doi: 10.1128/IAI.73.3.1861-1864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer SF, Vier J, Kirschnek S, Klos A, Hess S, Ying S, Hacker G. J Exp Med. 2004;200:905–916. doi: 10.1084/jem.20040402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paschen SA, Christian JG, Vier J, Schmidt F, Walch A, Ojcius DM, Hacker G. J Cell Biol. 2008;182:117–127. doi: 10.1083/jcb.200804023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong F, Sharma J, Xiao Y, Zhong Y, Zhong G. Infect Immun. 2004;72:3869–3875. doi: 10.1128/IAI.72.7.3869-3875.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heuer D, Brinkmann V, Meyer TF, Szczepek AJ. Cell Microbiol. 2003;5:315–322. doi: 10.1046/j.1462-5822.2003.00278.x. [DOI] [PubMed] [Google Scholar]
- 24.Dong F, Pirbhai M, Zhong Y, Zhong G. Mol Microbiol. 2004;52:1487–1494. doi: 10.1111/j.1365-2958.2004.04072.x. [DOI] [PubMed] [Google Scholar]
- 25.Brandstetter H, Kim JS, Groll M, Huber R. Nature. 2001;414:466–470. doi: 10.1038/35106609. [DOI] [PubMed] [Google Scholar]
- 26.Soding J. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 27.Soding J, Biegert A, Lupas AN. Nucleic Acids Res. 2005;33:W244–248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sali A, Blundell TL. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 29.Sharma J, Zhong Y, Dong F, Piper JM, Wang G, Zhong G. Infect Immun. 2006;74:1490–1499. doi: 10.1128/IAI.74.3.1490-1499.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong G, Reis e Sousa C, Germain RN. Proc Natl Acad Sci U S A. 1997;94:13856–13861. doi: 10.1073/pnas.94.25.13856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong G, Castellino F, Romagnoli P, Germain RN. J Exp Med. 1996;184:2061–2066. doi: 10.1084/jem.184.5.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaw AC, Vandahl BB, Larsen MR, Roepstorff P, Gevaert K, Vandekerckhove J, Christiansen G, Birkelund S. Cell Microbiol. 2002;4:411–424. doi: 10.1046/j.1462-5822.2002.00200.x. [DOI] [PubMed] [Google Scholar]
- 33.Sharma J, Bosnic AM, Piper JM, Zhong G. Infect Immun. 2004;72:7164–7171. doi: 10.1128/IAI.72.12.7164-7171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong F, Zhong Y, Arulanandam B, Zhong G. Infect Immun. 2005;73:1868–1872. doi: 10.1128/IAI.73.3.1868-1872.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma J, Dong F, Pirbhai M, Zhong G. Infect Immun. 2005;73:4414–4419. doi: 10.1128/IAI.73.7.4414-4419.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cong Y, Jupelli M, Guentzel MN, Zhong G, Murthy AK, Arulanandam BP. Vaccine. 2007;25:3773–3780. doi: 10.1016/j.vaccine.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li W, Guentzel MN, Seshu J, Zhong G, Murthy AK, Arulanandam BP. Clin Vaccine Immunol. 2007;14:1537–1544. doi: 10.1128/CVI.00274-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li W, Murthy AK, Guentzel MN, Seshu J, Forsthuber TG, Zhong G, Arulanandam BP. J Immunol. 2008;180:3375–3382. doi: 10.4049/jimmunol.180.5.3375. [DOI] [PubMed] [Google Scholar]
- 39.Murphey C, Murthy AK, Meier PA, Neal Guentzel M, Zhong G, Arulanandam BP. Cell Immunol. 2006;242:110–117. doi: 10.1016/j.cellimm.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murthy AK, Chambers JP, Meier PA, Zhong G, Arulanandam BP. Infect Immun. 2007;75:666–676. doi: 10.1128/IAI.01280-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murthy AK, Cong Y, Murphey C, Guentzel MN, Forsthuber TG, Zhong G, Arulanandam BP. Infect Immun. 2006;74:6722–6729. doi: 10.1128/IAI.01119-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bowie JU, Luthy R, Eisenberg D. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 43.Tamura T, Tamura N, Cejka Z, Hegerl R, Lottspeich F, Baumeister W. Science. 1996;274:1385–1389. doi: 10.1126/science.274.5291.1385. [DOI] [PubMed] [Google Scholar]
- 44.Huang Z, Feng Y, Chen D, Wu X, Huang S, Wang X, Xiao X, Li W, Huang N, Gu L, Zhong G, Chai J. Cell Host Microbe. 2008;4:529–542. doi: 10.1016/j.chom.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 45.Liao DI, Qian J, Chisholm DA, Jordan DB, Diner BA. Nat Struct Biol. 2000;7:749–753. doi: 10.1038/78973. [DOI] [PubMed] [Google Scholar]
- 46.Bhagwat M, Aravind L. Methods Mol Biol. 2007;395:177–186. doi: 10.1007/978-1-59745-514-5_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loew A, Gonzalez-Fernandez F. Structure. 2002;10:43–49. doi: 10.1016/s0969-2126(01)00698-0. [DOI] [PubMed] [Google Scholar]
- 48.Kusunoki H, Kohno T. Biochem Biophys Res Commun. 2007;359:972–978. doi: 10.1016/j.bbrc.2007.05.215. [DOI] [PubMed] [Google Scholar]
- 49.Kusunoki H, Kohno T. Proteins. 2006;64:804–807. doi: 10.1002/prot.21028. [DOI] [PubMed] [Google Scholar]
- 50.Gouet P, Courcelle E, Stuart DI, Metoz F. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.