

Abstract
Assigned from data sets measured in water at 2, 25, and 60 °C containing 13C=O NMR chemical shifts and [θ]222 ellipticities, helical propensities are reported for the twenty genetically coded amino acids, as well as for norvaline and norleucine. These have been introduced by chemical synthesis at central sites within length-optimized, spaced, solubilized Ala19 hosts. The resulting polyalanine-derived, quantitative propensity sets express for each residue its temperature-dependent but context-independent tendency to forgo a coil state and join a preexisting helical conformation. At 2 °C their rank ordering is: P ⪡ G < H < C, T, N < S < Y, F, W < V, D < K < Q < I < R, M < L < E < A; at 60 °C the rank becomes: H, P < G < C < R, K < T, Y, F < N, V < S < Q < W, D < I, M < E < A < L. The ΔΔ G values, kcal/mol, relative to alanine, for the cluster T, N, S, Y, F, W, V, D, Q, imply that at 2 °C all are strong breakers: ΔΔ Gmean = +0.63 ± 0.11, but at 60 °C their breaking tendencies are dramatically attenuated and converge toward the mean: ΔΔ Gmean = +0.25 ± 0.07. Accurate modeling of helix-rich proteins found in thermophiles, mesophiles, and organisms that flourish near 0 °C thus requires appropriately matched propensity sets. Comparisons are offered between the temperature-dependent propensity assignments of this study and those previously assigned by the Scheraga group; the special problems that attend propensity assignments for charged residues are illustrated by lysine guest data; and comparisons of errors in helicity assignments from shifts and ellipticity data show that the former provide superior precision and accuracy.
Introduction
Much prior effort has been directed toward quantitative prediction of aqueous helix-coil energetics from peptide and protein residue compositions and sequences.1,2,3 These predictions have likely medical relevance.4 During the folding of many biosynthesized protein sequences, a pivotal early event is the formation of solvent-exposed, weakly stabilized, short helical regions,5 and their mutation-induced destabilization with resulting mis-folding may be a primary factor in the onset of particular human degenerative diseases.6 Algorithms that tie helical sequence to energetics can thus provide chemical insight into mutation-based mis-folding, and during the 1990s a widely cited helicity algorithm was introduced.7
In addition to dependencies of helicity on sequence length and end-region structure, this and related algorithms embody parameters assigned from helicity-modifying single and double guest substitutions within medium-length host peptides. Although these characterized a large inventory of energetic effects,8 for practical reasons, temperature dependences for these parameters were usually extrapolated from data measured at the low end of the aqueous temperature range (0 to 10 °C).
Such extrapolations may result in large errors, since the single pertinent host-guest study, carried out over the range of 10 to 70 °C, reveals temperature dependences that are strongly residue-specific.9 A need for new temperature-dependent helicity data is also underlined by a remarkable recent observation. Genomic analyses of thermophilic organisms show that seven amino acid residues appear within their protein helices with unusual frequency,10,11 yet five of these residues are helix breakers at 0 °C, and alanine, the strongest helix former at this temperature,12 is not included among the seven. We here report a new host-guest study, designed to meet stringent accuracy requirements and carried out over a large temperature range.13
A useful host must be experimentally tractable, and it must retain detectable helicity over most of the aqueous temperature range. Among homopeptides, a polyalanine core region Alan 12 ≤ n ≤ 45 meets the first condition if it is N- and C-linked through spacing elements to solubilizing, aggregation-inhibiting polylysine caps.14 In a series of foundation studies, we have previously calibrated host helicity assignments from both circular dichroism ellipticities [θ]222 15 and 13C=O NMR chemical shifts16 and reported a definitive set of length- and temperature-dependent values for the alanine helical propensity w Ala[n,T].13,17 We have also demonstrated that propensity assignment accuracies are optimized by the 19-residue lengths of chemically synthesized mutants 1, helically characterized by chemical shifts, and mutants 2, helically characterized by ellipticities.13
| (1) |
| (2) |
This report consummates these efforts. It assigns temperature-dependent helical propensities for the 20 genetically coded amino acid residues and for selected others. It then correlates these with the classic benchmarks reported by the Scheraga group.9
The properties of polyalanine hosts ensure that new modeling opportunities can follow from these assignments. For any helical sequence, the set of its propensities embodies helicity contributions that result solely from residue composition, and a second set characterizes context effects that embody helicity perturbations attributable to the sequence order. A polyalanine-derived propensity set combined with a compatible context set would uniquely allow chemically based structural modeling of the fundamental energetics of helix formation. In the following section we define basic concepts that are developed in the discussion and also raise a practical issue. How accurate are propensity values that are solely assigned from CD ellipticities measured for mutants 2?
Helical Propensities and Context Effects
Based on the hypothesis that local effects control helical stability, most helicity algorithms are built from two sets of parameters. The first contains the twenty quantitative helical propensities, each reflecting for a particular residue, its temperature-dependent equilibration between coil and α-helical states. A much larger second set consists of temperature-dependent energetic corrections for paired charge, polar, and hydrophobic interactions between a guest side chain and those of its four neighbors, Figure 1a. The contribution to global helicity from helical propensities thus reflects residue composition, and the contribution from the set of context parameters embodies sequence-dependence.2,18,19
Figure 1.
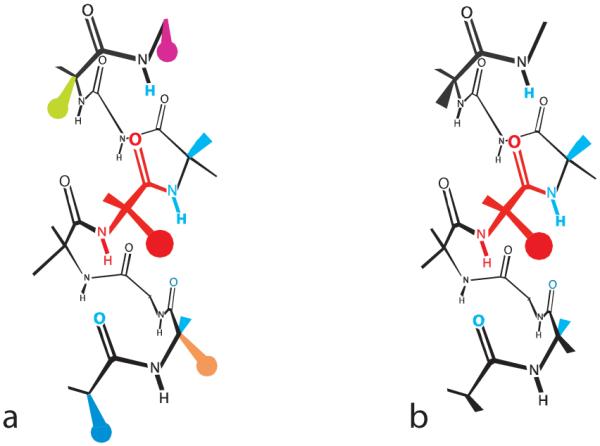
Contacts of a red guest residue at a mutant site i within a typical α-helix (a) and within an idealized α-helix in which local context effects are minimal (b). In (a), the red guest atoms contact the cyan neighboring backbone as well as side chain atoms of residues at (i ± 3) and (i ± 4) separations (green, magenta, orange, and blue circles). Both helical propensities and context effects are required to characterize the energetics of these interactions. For case (b), the contacts between side chains are minimal, and only the cyan contacts perturb helicity significantly. Their sequence-independent energetics are defined by the helical propensities.
Standard modeling tools of molecular chemistry should, at least in principle, be applicable to any helical sequence to predict its temperature-dependent helix-coil energetics in water. In fact, model validation has been plagued by doubts concerning the reliability of available helical propensity sets, many of which have been challenged as incorporating hidden host-dependent biases. The polyalanine regions of mutants 1 and 2 can resolve this imbroglio, since it is generally agreed that a helical context that consists solely of alanine side chains, with their minimal hydrophobic bulk and absence of charge or polarity, provides the closest experimental approximation to the modeler’s context-free α-helix of Figure 1b.20
Background
The polyalanine mutants 1 and 2 have a second essential feature. We have previously tailored their lengths to maximize sensitivity to guest substitution,13 and alone among the medium length uncharged homopeptide hosts that can be constructed from the natural amino acids, they can retain helicity over a maximal temperature range. Only the oligopeptide mutants synthesized and studied by the Scheraga group have been optimized in this manner.9 As an introduction to the data sections that follow, we compare and contrast these two design strategies.
Studies carried out in the 1970s defined the experimental foundations for all subsequent quantitative studies of peptide helicity. The α-helices formed by polylysine or polyglutamate oligopeptides allowed the first experimental characterization of the highly cooperative temperature- and length-dependent helicities that characterize isolated peptides solvated by water. The data were inconsistent with simple all-or-none helix-coil energetic models, but good fits resulted from two-parameter algorithms, based on statistical mechanical ensembles of equilibrated non-helical, completely helical, and partially helical conformations.21 In these algorithms, cooperativity is attributed to an energetically unfavorable helix initiation step that is followed by a series of helix propagation steps. In each of these, the marginally favorable helical weight of a single new residue is added to the weight of the previous helical sequence for each ensemble conformation.
The resulting overall ensemble weighting ensures that if a peptide or oligopeptide that lacks tertiary packing exhibits detectable helicity, it will melt reversibly and progressively over a broad temperature region. A small temperature increment corresponds to a correspondingly small degree of melting, reflecting a proportionate increase in the relative abundance of conformations that contain shorter helices.
In the widely used Lifson-Roig algorithm,22 propagation is modeled by a temperature- and residue-dependent parameter w, the helical propensity, which is an equilibrium constant that defines the incremental helicity change for a sequence if its length is increased by one residue. For helix-breakers, w values lie in the range 0 to 1.0, but for helix-formers, w usually lies between 1.0 and 1.8.
The overall helicity contribution from propagation for any helical region is proportional to the product of the w values of its residues, and their mean magnitude within a sequence defines the minimal length required for it to exhibit detectable helicity. If this mean lies between 1.0 and 1.1, a detectably helical sequence must contain more than a hundred residues, but a peptide of moderate length can be detectably helical if the mean w value exceeds 1.3.
Prior to 1989, when Marqusee, Robbins, and Baldwin reported that short peptides are highly helical if they are alanine-rich,23 oligopeptides were the sole mutant options, and consistent with a prior study,24 in 1971 Scheraga and coworkers reported a pair of helical oligopeptides constructed from covalently modified glutamines that met the stringent needs of a breakthrough host-guest helicity study, designed to be conducted in water over a broad temperature range.25
The large perturbing effects of guests on melting curves posed a design challenge that was solved by use of a pair of hosts, derived from structurally similar helix-forming residues. Substitutions by helix formers within the less helical host, formed from N5-(3-hydroxypropyl-L-glutamine), yielded mutants with a planned melting range 10 to 70 °C, and this range was shared by mutants derived from helix breakers, incorporated within a more helical host formed from N5-(4-hydroxybutyl-L-glutamine). Analysis of the resulting mutant data yielded the first set of temperature dependent helical propensities for the natural amino acids. Ten of these decreased monotonically with increasing temperature, five exhibited curved temperature dependences with maxima within the range of 15 to 40 °C, and the remaining five increased monotonically with increasing temperature. Within the latter group, the β-branched hydrophobic residue valine, a helix breaker near 0 °C, becomes a weak helix former at higher temperatures, a property confirmed from a study of helical polyvalines.26 The helical propensity of alanine ranked it sixth in the list of helix formers, a value that decreased within the range 0 to 60 °C from 1.1 to 1.0, but very different values were reported in the 1990s for alanine residues of alanine-rich peptides of medium length that were solubilized by spaced polar or charged residues and studied at or near 0 °C.27 In these reports, alanine was ranked as the strongest helix former, with a wAla between 1.5 and 1.8. These assignments were initially questioned, and alternatives were offered that attributed the high helicities of these peptides to context-dependent helix-stabilizing effects of glutamine and lysine residues.28 Subsequent guest mutations within other hosts supported the new alanine rank order,29 and later low temperature assignments of wAla within polyalanines of adequate length confirmed it.14g,14h,30
To lay a foundation for the present study, we characterized wAla[n,T] values for central residue contexts of tLeu-Alan-tLeu, n = 9 to 23, in water between 2 and 80 °C, using 13C=O NMR chemical shifts of the alanine backbone amides as helicity monitors. The following assignments for wAla[19,T] resulted: 2 °C, 1.56; 10 °C, 1.52; 25 °C, 1.39; 37 °C, 1.30; 52 °C, 1.22; 60 °C, 1.17; 80 °C, 1.11.13,17 Rather than using a pair of hosts for mutation studies over the temperature range of 2 to 80 °C, we tailored the polyalanine host length to provide an optimal trade-off between the helicity properties of mutants derived from guests that are strong helix-breakers as well as those that are strong helix-formers. For breaking guests, the mutant length must be sufficiently long to retain characterizable helicity within the range 60 to 80 °C; for forming guests, the mutant length must be sufficiently short to allow characterization of the helicity difference between mutant and host within the range 2 to 10 °C. Modeling was initially used to select an optimal mutant length of nineteen residues, and this choice was then validated empirically for mutants 1.13 The strong helix-former norvaline and the pair of strong helix breakers glycine and proline were selected as guests, since their propensities were expected to bracket those for the natural residues. For these mutants 13C=O NMR shift data were measured within 2 to 80 °C, and wGly, wPro, and wNva were assigned with acceptable precision. Values for wNva closely resembled those for wAla, decreasing comparably with increasing temperature, but values for wGly and wPro increased significantly, suggesting that sequences of helical proteins found in mesophiles or thermophiles can include these residues with a reduced folding penalty. Comparisons of the sets of temperature dependent propensities assigned by the Scheraga group and those from the present study are explored in the discussion section.
Precision for Circular Dichroism Ellipticities at 222 nm, [θ]222, Measured for Mutants 2
Nearly universally regarded as the “gold standard” for quantitative helicity asssignments, the experimental per-residue molar ellipticities [θ]222,Exper,n,T allow calculation of fractional helicities FH, defined as the mole fraction of helical residues within a partially helical sequence, Equations (1) & (2). The intensity of the helical CD chromophore at 222 nm, together with the minimal intensity of chromophores at that wavelength for all other peptide backbone conformations, justifies a frequently applied approximation that disregards the coil term [θ]222,C,n,T, as seen in Equation (3). The [θ]222,Exper,n,T data for mutants 2 are corrected by subtraction of the minor contributions of N- and C-caps,14f,14h,16 and FH values are then assigned from host-assigned [θ]222,H,n,T and [θ]222,C,n,T31.
| (3) |
To assign precisions for [θ]222,Exper,n,T, we carried out nineteen (Ala → Nle) single residue replacements within the series 2 host peptide, giving the T-dependent ellipticities of Figure 2a. Two motivations governed selection of the norleucine ≡ Nle residue for this assignment. At the three temperatures of this study, Nle is a slightly stronger helix former than alanine32 (compare the solid lines of Figure 2a with the broken lines of Figure 2b), which ensures that the mutant ellipticity signal at 60 °C remains sufficiently intense to be interpretable. Moreover, lysine and norleucine share a carbon skeleton, providing useful comparisons for lysine data, as discussed in a later section.
Figure 2.

(a) Values of [θ]222 for specifically sited single Nle ≡ norleucine guests within the tLeu-Ala19-tLeu host at 2 °C (blue), 25 °C (orange), and 60 °C (magenta). The solid lines correspond to mean values of [θ]222. (b) Values of [θ]222 for spaced pairs of Nle (open squares) and Nva ≡ norvaline guests (solid triangles) within the series 2 Ala19 host at 2 °C (blue), 25 °C (orange), and 60 °C (magenta).The dotted lines correspond to [θ]222 values for this host.
Convincing site dependences are not detectable within the Figure 2a data set, and treated as randomly distributed, ellipticities at all temperatures have standard deviations of 3 to 4 % of the average value. Padmanabhan et al reported similar values at 0 °C for alanine-rich peptides,20a and in studies of the length dependence of [θ]222,Exper for a series 2 capped Alan series, 12 ≤ n ≤ 20, we assigned respective relative measurement precisions for [θ]222,Exper, n,T at 2, 25, and 60 °C of 4 %, 5 %, and ca. 10 %.14f The higher latter value can be attributed to the weak high-temperature CD intensities typical of short helical peptides (n < 17).
The data of Figure 2b illustrate an important consequence. Two sets of seven [θ]222 measurements are shown for mutants that incorporate pairs of contiguous or spaced norleucine and norvaline guests. A comparison with Figure 1a shows that the residue pairs at (9, 12) and (8, 12) spacings may have (i, i+3) and (i, i+4) side chain contacts, with resulting helicity perturbations. The mutants that contain (i, i+4) guest pairs have slightly larger than average [θ]222 values, and those that contain (i, i+3) guest pairs have slightly smaller values, but both lie within measurement error, and neither can be used to verify or refute the existence of the corresponding helicity changes. Thus [θ]222 measurements lack the precision required for assignments of small context-dependent helicity perturbations. In the next two sections, we explore the key issues of methodological precision and accuracy, preconditions for any rigorous discussion of propensity values.
Precisions of 13C=O NMR Chemical Shift Measurements for Alanine Residues; T-Dependent Shift Scans for the Spaced, Solubilized Series 1 Ala19 Host and for its Single-Site Mutants; Accuracies of Shift-derived FHi
The filled circles of Figure 3a provide complete 13C=O NMR chemical shift scans measured in water at 2, 25, and 60 °C for alanine residues of the host. Also shown are analogous mutant 1 alanine site scans for guests: Leu, Ile, Nle, Nva, and Val. The error calculated from repeated measurements is ± 0.02 ppm; implying that the shift precision, defined relative to the shift range, exceeds that assigned in the preceding section for [θ]222 values.
Figure 3.

Values of 13C=O NMR chemical shifts in water at 2 °C (blue), 25 °C (orange), 60 °C (magenta). Host values are filled circles. Values for mutants 1 tLeu-Ala9XxxAla9-tLeu are coded as follows: a. open triangles: Xxx = Nva; filled triangles: Xxx = Val. b. Filled squares: Xxx = Nle; open squares: Xxx = Leu; filled diamonds: Xxx = Ile. The closed-line fits to host data that appear in a. are repeated in b. Each shift is site-coded within each peptide by a specific 13C/12C ratio.
The NMR chemical shift of a specific backbone 13C=O nucleus reflects a weighted average of contributions from local magnetic anisotropies. These are residue-specific and sensitive to changes in neighboring structure and local solvation, but the helix-coil conformational anisotropy difference almost invariably dominates, allowing accurate identification of helical regions within globular proteins as well as assignment of fractional site helicities FHi for peptides.33,34
Except at end regions, the host shift scans of Figure 3a define smooth envelopes with maximal values near the sequence center. This is the signature of averages over equilibrated ensemble conformations, of which more than 60 % contribute to experimental shifts at central sites, but only 11 % contribute at end sites.13,17 The environment of backbone atoms near end regions cause their shifts to deviate significantly from envelope extrapolations. Pertinent anisotropies include atypical solvent exposure and proximity to the terminal helix dipoles and to spacer side chains.
End region shift issues also arise for partially helical conformations within the ensemble, whose abundance-weighted averages define the experimental shifts. For maximal FHi assignment accuracy, we modify the standard two-state helix-coil model by assigning experimentally derived weights to conformations with helical regions that start or stop at each site i. Both FHi, Equation (4), and modeled chemical shifts δ13C=Oi,Exp, Equation (5), are thus written as sums of weights for conformations that are initiated (I), terminated (T), or continued (C) at that site. The required four temperature-dependent calibration constants of Equation (5) have been previously characterized.17
| (4) |
| (5) |
What perturbations are expected for alanine 13C=O chemical shifts if a guest side chain replaces an alanine methyl at site 10? The mutant data of Figures 3ab show that large fluctuations in shift magnitude appear at sites 6 to 9, which lie N-terminal to the guest. These are plausibly attributable to local shift anisotropies attributable to side chain contacts, guest-induced distortions in backbone angles that maintain helical structure, and local solvent shielding. Perturbations C-terminal to the guest are small and almost invariably lie within measurement error. The 13C=O chemical shifts thus offer a unique advantage as helicity reporting tools, provided their accuracy is maximized by introducing 13C-labels solely within alanine regions characterized by smooth shift envelopes.13 We usually rely on central sites within the Ala9 C-terminal region.
To test reporting fidelity for these sites, we correlated two 21-guest sets of shifts, measured at alanine sites 14 and 15. The filled squares of Figure 4 depict the results. Within measurement error, identical shift rank orders are seen, with the following correlation coefficients and standard deviations, 2 °C, 0.997, 0.03 ppm; 25 °C, 0.997, 0.02 ppm; 60 °C, 0.98, 0.02 ppm. These values confirm the expected FHi precision and accuracy.
Figure 4.

Shift regressions measured in water at neutral pH. a. 2 °C (blue), b. 25 °C (orange), c. 60 °C (magenta). Mutants containing aspartate, glutamate, and histidine were studied at pH 7; [θ]222 values for Cys and Trp mutants were not measured; Pro data are omitted for clarity since they fall far to the left of depicted graph sites. Filled squares correlate 13C=O chemical shifts at sites 15 and 14 for 21 series 1 mutants, and lines depict linear regressions. Open squares correlate [θ]222 data measured in mutants 2 with selected site 14 shift data. Mean rank orders for shift data appear below, with red vertical lines that distinguish helix breakers from formers: 2 °C: P ⪡ G < H < C, T, N < S < Y, F, W < V, D < K < Q < I < R, M <|L < E < Nva, Nle < A 25 °C: P ⪡ H, G < C, T < N < S, Y, F < D, V < K < W < Q < R < I < M <|E < L, Nva < Nle, A 60 °C: H, P < G < C < R, K < T, Y, F < N, V < S < Q < W, D <|I, M < E < Nva < A < L, Nle
An analogous optimization of measurement sites is unavailable if [θ]222 ellipticities replace shifts as quantitative helicity monitors. Both FH and [θ]222 are global averages for entire helical regions, and for series 2 mutants, the [θ]222 values and FH assignments invariably include potentially anomalous contributions from guests and their neighboring alanines. The accuracy of Equation (3), used to calculate FH from [θ]222 data, thus rests on the accuracy of its calibration constant [θ]222,H,n,T, which is necessarily assigned from host helicity data. In the Supporting Information we show that for specific guests, use of host-derived values for [θ]222,H,n,T underestimates mutant FH35.
From modeled data we have previously shown that over the range of possible guests, shifts and [θ]222 values correlate strictly linearly. The open squares of Figure 4 test the quality of this correlation for our experimental data. At each temperature, ellipticity- and shift-derived rank orders correlate quite well, although three to four ellipticities deviate significantly. The correlation coefficients and standard deviations for linear regressions between sets of [θ]222 and site 14 shifts lie within generally acceptable limits: 2 °C, 0.94, 1.4; 25 °C, 0.93, 1.0; 60 °C, 0.95, 0.4 (SD units are deg cm2 dm-1). These values show that this correlation is inferior to that seen for the closed squares of Figure 4; nonetheless, for many purposes, shifts and ellipticities provide nearly equivalent measures of helicity changes induced by a range of helix forming and breaking guests.
Since the above data correlations are not dependent on Lifson-Roig analyses, they might be said to maximize interpretational transparency, but they preclude one essential correlation, which is available if raw data are replaced by modeled propensities wXxx/wAla, assigned using previously reported protocols.13,17, 22c Table I shows the resulting correlations.
Table I.
T-Dependent Linear Regression Slopes and Intercepts with Standard Deviations and Correlation Coefficients for the Pair of wXxx/wAla Sets Assigned from [θ]222 Values of Mutants 2 and 13C=O NMR Chemical Shifts at Site 14 of Mutants 1. Mean Δ are Percentage Differences for Shift-vs. [θ]222-Derived wXxx/wAla: Xxx = Nle, Nva, A, E, L, K, M, I, Q, R, D, V, F, Y, S, N, T, G, P
| T, °C | Slope | Intercept | Fit (SD) | CC | Mean Δ, % |
|---|---|---|---|---|---|
| 2 | 1.06 | - 0.15 | 0.10 | 0.95 | + 30 |
| 25 | 1.07 | - 0.10 | 0.08 | 0.96 | + 18 |
| 60 | 1.00 | + 0.07 | 0.09 | 0.92 | - 11 |
The correlation coefficients CC of column 5 are consistent with the previously cited, data-derived values of Figure 5, and columns 2 and 3 report intercepts and slopes of linear regressions carried out on paired wXxx/wAla sets assigned at 2, 25, and 60 °C. Within assignment error, these are in quantitative agreement with the expected values of 0 and 1.0. These findings show that this pair of wXxx/wAla sets passes normal correlation tests.36 However, as seen in Figure 5, one additional important correlation fails. For a majority of residues at 2 and 25 °C, the ellipticity-derived wXxx/wAla are smaller than those calculated from shifts. This difference is expressed in the last Table I column as an averaged percentage of the shift-derived value. We attribute the consistently lower value of ellipticity-derived wXxx/wAla assignments to guest-dependent decreases in the Equation (3) calibration term [θ]222,H,n,T. In so doing, we are reasoning from evidence that, unlike extinction coefficients of UV-vis spectroscopy, values of [θ]222 are very sensitive to local structural perturbations. For example, the empirically assigned values for the parameter X of Equation 2, used to assign length-dependence to [θ]222,H,n,T, demonstrate this sensitivity. As shown in the Supporting Information, values for X allow calculation of the different contributions of an average end region alanine and a core alanine to the [θ]222,H,n,T mean value. For the host, the end region value is 30 to 70 % less than the core value.
Figure 5.

Squares define pairs of wXxx/wAla, assigned for each site 10 residue from [θ]222 values, vertical axis and from 13C=O NMR chemical shifts, bottom axis. Temperatures: a. T = 2 °C; b. T = 25 °C; c. T = 60 °C. Data were measured in H2O (ellipticities) and in D2O (shifts); for charged residues, shifts were measured in 10 mM Tris buffer, pD = 7.0. Shift-derived wXxx/wAla are averages of the assignments at sites 12, 14, and 15, and squares that lie on dashed lines correspond to wXxx/wAla values that are identical.
A test of the validity of the two-state model that underlies Equation (1) is provided by temperature- and guest-dependent ellipticity values at 203 nm.37 Superimposed plots of CD spectra for the host peptide at 2, 25, and 60 °C exhibit a well-defined isodichroic point at 202.5 ± 1 nm, implying that as the temperature is increased from 2 to 60 °C, the constants [θ]203,H,n,T and [θ]203,C,n,T of Equation (1) are equal and assume the expected literature intensity. If one adds to this plot the corresponding nine CD spectra of series 1 mutants containing the guests Xxx = Nle, Nva, and Abu = α-aminobutyric acid, this isodichroic point remains invariant in both wavelength and intensity.17 However the temperature-dependent CD spectra of mutants containing guests with branched aliphatic side chains, rings, or polar side chains lack well-defined isodichroic points and exhibit intensity deviations at 203 nm of up to 30 %. These elliticities are inconsistent with a two-state helix-coil dichroic model.
In the Supporting Information, we further show that nearly all guest discrepancies of Figure 5 at 2 and 25 °C are consistent with decreases in [θ]222,H,n,T that fall in the relatively narrow range of 5 to 15 %. A larger decrease is seen for the Gly mutant. This is expected if glycine, with its unique conformational freedom, induces an increase in local 310 helical character.38 We conclude that for the natural amino acid residues, CD ellipticity data are valuable as confirmations of the 13C=O chemical shift-derived host-guest helical propensities, but by themselves, they lack the accuracy required for primary propensity assignments.
Helical Propensities wXxxx[T] for Amino Acids at a Central Site within a Polyalanine Context.39
Figure 6 depicts temperature-dependent helical propensities for twenty two amino acid residues, assigned in water within a polyalanine context. At 2 °C the propensity of alanine dominates. Its nearest competitor among the natural uncharged residues is leucine, but wAla[T] is 40 % larger thanwLeu[T], and in turn, this value is substantially larger than either wMet[T] or wIle[T]. At 25 °C, wAla[T] is 30 % larger than wLeu[T], but at 60 °C, within assignment error, these propensities are equal. Over the full temperature range, relatively large spacings between propensity values are characteristics of the 2 °C assignments. With an increase in temperature, convergence occurs, reflecting in part the presence of both negative and positive slopes for key residues.
Figure 6.

Interpolated wXxx[T] values assigned from the chemical shift-derived relative helical propensities wXxx/wAla of Figure 5 by wAla[T] multiplication followed by T-dependent quadratic fits as detailed in the Supporting Information. The depicted curves for wAla[T], wNva[T], wGly[T], and wPro[T] have been reported previously.13 See this report for a discussion of the problems that attend accurate assignment of wPro[T] at low temperatures.
The alanine helical propensity decreases significantly and nearly linearly with temperature, and its values and slope are most closely matched by propensities for the two residues Nva and Nle that bear straight alkyl side chains.32 Values for wArg[T] also decrease with a comparable slope, and perhaps coincidentally, the side chains of Arg and Nva share the same three-carbon backbone sequence. For the remaining fourteen uncharged residues, the slopes of the temperature dependences of their wXxx[T] values are positive. These are largest over the 25 to 60 °C interval, and expressed as percentages over the full range, the increases for the following residues are noteworthy: Trp: 67 %, Ser: 83 %, Thr: 85 %, and Asn, 110 %.
Large increases are also seen for the strong helix breakers Gly and Pro. At 2, 25, and 60 °C, wAla[T]/wGly[T] has respective values 9, 8, and 2.5, and the corresponding values for wAla[T]/wPro[T] are ≥ 100, 30, and 4. Since alanine is the strongest former and proline is the strongest breaker, these ratios define the full temperature-dependent propensity ranges, which decrease at least 25-fold as the temperature is raised from 2 to 60 °C.
What molecularly based conclusions can be drawn from these findings? Substantial prior effort has been directed toward elucidation of aqueous helix-coil energetics,40 and significant variables include residue-dependent coil entropies, coil and helix solvation energetics, and hydrophobic interactions41 of the helical backbone with residue side chains. The residue- and temperature-dependent propensities of Figure 6 are at least qualitatively consistent with currently accepted thermodynamic models, yet none of the latter predict wXxx[T] values quantitatively, and in a definitive recent overview Makhatadze has suggested that computer simulation, rather than experiment, may provide an optimal future strategy.40
Helical propensities that take the form of rank-ordered lists of helix breakers and formers are widely used as heuristic tools for spotting helical and non-helical regions within protein primary sequences. Three such lists, derived from the findings of this report, appear in the legend of Figure 4. We propose that these lists are likely to be most useful if they are supplemented by quantitative information that can be derived from Figure 5. Inspection of its three panels reveals clustered relative helical propensities (horizontal axes) for glutamine, aspartate, valine, tryptophan, phenylalanine, tyrosine, serine, asparagine, and threonine. At 2 °C, the mean propensity value for this cluster is 0.33 ± 0.07; at 25 °C, the mean increases slightly to 0.38 ± 0.07; but at 60 °C, the increase in this mean value is dramatic: 0.68 ± 0.08. Remarkably this effect is observed for both residues with non-polar, hydrophobic side chains and for residues with short, polar side chains. Given appropriate local sequences, this mean magnitude is likely to be increased by stabilizing (i, i ± 3) and (i, i ± 4) context effects,20b suggesting that at high temperatures, a majority of residues may act as strong helix formers.
Charged residues
Lysine data of Figure 7 provide a preliminary picture of helix-perturbing complexities caused by residues with charged side chains. It has long been recognized that host mutants derived from these residues are exceptions to the generalization that local effects dominate helical stability; for these residues, propensities are strongly site dependent.42 Structurally speaking, a lysine residue is a norleucine in which a terminal ε-hydrogen of the long, potentially flexible Nle chain has been replaced by a charged -NH3+ function. A comparison of ellipticities for the site scans of Figures 2a and 7a suggests that this terminal charge strongly interacts with the helix backbone and on average, strongly diminishes the host helicity. In the earlier discussion of Figure 2a data, we noted the absence of detectable wNle[T] site dependences, and in Figure 2b we detected only hints of (i, i ± 4) and (i, i ± 3) context effects for mutants containing Nle pairs. The corresponding Figures 7ab display dramatic site variations and evidence for local Lys-Lys energetic interactions that are helix-destabilizing. However, caution should be used in generalizing applications for these data, since they were measured in the absence of salt. At higher than physiological concentrations, sodium chloride has been shown to diminish the helix destabilizing interactions between alanine-rich and sequences containing Lys-NH3+ functions.23
Figure 7.

(a) Values of [θ]222 for specifically sited single Lys+ guests within the series 2 host at 2 °C (blue), 25 °C (orange), and 60 °C (magenta) at neutral pH in pure water. The solid lines correspond to values of [θ]222 for the host. (b) Values of [θ]222 for spaced pairs of Lys+ guest triplets within this host at 2 °C (blue), 25 °C (orange), and 60 °C (magenta). The following helical propensities are assigned at 2 °C: for (a), single substitutions at sites 4, 10, and 16, wLys[2] = 0.54, 0.70, ≥ 1.1. For the left-to-right triple substitutions of (b), <wLys[2]> = 0.53, 0.56, 0.40, 0.37, 0.43. For these, the helicity rank order is thus: (i, i ± 5) > (i, i ± 1) > (i, i ± 2) ≥ (i, i ± 4) > (i, i ± 3).
As detailed in the Supporting Information, these findings are shared by mutants containing the other residues that bear positively charged side chains. Allowing for the similar site-10 values for wLys[T] and wArg[T] and the much lower values seen in Figure 6 for wHis[T], one finds that the site-dependent variations in ellipticities of Figure 2a are shared with corresponding Arg and His mutants. A dramatic increase in [θ]222 intensity results if these guests are sited at any of the four C-terminal sequence residues, and this increase converges to a maximum at site 19. This is the expected signature of helix stabilization that results from placement of a positive charge near the C-terminal dipole of a helix backbone.43
Owing to these site and context dependences, helical propensities for central site charged residues provide at best a qualitative approximation to their range of helicity contributions at other sites. Histidine provides an extreme example. A recent PDB search of helical protein regions identified the 100 most frequently encountered four-residue sequences. All residues appear in these sequences, with the single exception of histidine.44 Our data suggest an explanation. Over a full temperature range of our study, alone among charged residues, His consistently ranks among the four strongest helix breakers, unless it is sited at a C-terminus of a helical region.
A more general issue is posed by the large asymmetric influence of centrally sited charged residues on the relative weighting of ensemble conformations. Specifically, at 2 °C, the central site wLys[2] values lie above 1.6 for all conformations with helical regions that terminate at that site, but its values for conformations that are initiated or continued at that site are smaller by about a factor of three. As a result, for sequences of medium length in which charge-derived context effects are minor, a central substitution by a charged Lys, Arg, or His residue will substantially skew site helicities to favor the N-terminal half of the sequence. Owing to averaging, an FH is relatively insensitive to this effect, but FHi values, assigned at a pair of sites that are spaced equally from a central charged residue, will be skewed to lower or to higher relative values, depending upon the charge sign, and judiciously sited 13C= O-labeled alanines should allow experimental characterization of this helicity-biasing effect.
Comparison of Two Temperature-Dependent Helical Propensity Sets; Summary
As noted above, the Scheraga group previously reported helical propensities for a large temperature range using mutants of oligopeptide hosts that were constructed from N5-(3-hydroxypropyl and 4-hydroxybutyl)-L-glutamine residues. The linear side chains of the resulting hosts contain five or six methylenes, polar secondary amides and polar hydroxyl functions at their termini. Conformational side chain diversity is thus a likely host feature. In addition to flexibility attributable to the normal gauche-anti conformers of alkyl regions, conformational freedom at the single amide-linked C-C=O and C-NH bonds that appear near the side chain centers should resemble the permissive glycine Ψ and Φ values. Some of the coil state freedom is lost in a coil-helix transition, but side chain motion is likely to be retained in helical conformations. Hydrophobic and/or polar context effects are thus expected for guests introduced within these hosts,45 and conceivably, retained conformational diversity in the helical state may increase with temperature, with a resulting increase in the crowding of bulky guest side chains.
A new test of fit is appropriate if, as seen in the 2 °C plot of Figure 6, propensities for alanine, glycine, and proline are distanced from those of their neighbors. This test excludes the outliers and compares the means of values for the remaining propensities. For the Figure 6 assignments these means are: 2 °C 0.65; 25 °C 0.65; 60 °C 0.84, and the analogous means for the Scheraga propensities are: 2 °C 0.98; 25 °C 0.95; 60 °C 0.86. At 2 and 25 °C, the latter exceed the former by 50 %, suggesting the presence of helix stabilizing context effects within the Scheraga hosts. Also informative are comparisons for pairs of residue values. Inspection of Figure 6 shows that as the temperature is increased from 2 to 60 °C, wXxx[T] for alanine, arginine+, and glutamate decrease by 15 to 25 %; the values for histidine remain constant, and values increase significantly for the set that includes the remaining natural residues. For glycine, proline, asparagine, threonine, valine, and serine at 0 to 40 °C, the Scheraga propensities also increase with temperature. For alanine, leucine, phenylalanine, cysteine, and lysine+, the Scheraga propensities decrease slightly or are constant with a small maximum, but for the remaining nine residues these propensities decrease by 15 to 55 %. Relatively speaking, for these guests, the Scheraga hosts become less hospitable as the temperature is raised.
Figure 8 correlates the 60 °C Scheraga propensities with those assigned in the minimally interactive polyalanine host. A poor correlation is seen for the five residues with charged side chains and is likely attributable to the high context sensitivity of members of this residue class. For valine and phenylalanine the Scheraga propensities lie substantially above the correlation line, but not outside of the plausible range of hydrophobic context effects reported by Creamer and Rose.20b If propensities for these seven residues are excluded, the comparison passes correlation coefficient and slope tests. The overall quality of correlation at 60 °C thus reveals the dominant operation of helical propensity differences, even in the presence of potentially large context effects.
Figure 8.
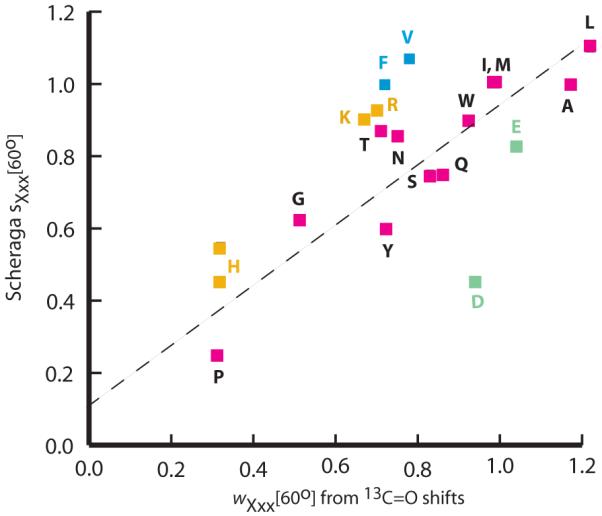
A comparison of 60 °C helical propensity sets. The vertical axis plots Scheraga s values9c versus Figure 6 wXxx[T] values, horizontal axis. Vertical His and His+ values are both depicted; the Figure 6 His value was assigned at pH 7. The correlation coefficient for all residues is 0.68; it increase to 0.91 if data for F, V, and the charged residues are excluded.
At 2 and 25 °C, the interpretive significance of slopes and correlation coefficients are compromised by the outlier properties of wAla[T], wGly[T], and wPro[T].46 Examination of the temperature-dependent values of the ratios wAla[T]/wGly[T] and wAla[T]/wPro[T] reveals the problem. At 2 °C, our wAla[T]/wPro[T] ratio is at least 100, but the Scheraga ratio is ca. 15; at 25 °C the corresponding ratios are 30 and 5; but at 60 °C, they converge to 4 and 3. At 2 °C, our wAla[T]/wGly[T] ratio is 9, but the Scheraga ratio is ca. 2.1; at 25 °C the corresponding ratios are 8 and 1.8; but at 60 °C, they converge to 2.4 and 1.6.
Context corrections that arise in typical helical sequences derived from globular proteins are likely to reduce our high outlier wAla[T] and our low outlier wGly[T] values. In a recent report,13 we used spaced, solubilized, spaced mutants tLeu3-AlaNva4XxxNva4Ala9-tLeu3 to reassign these propensities within a local polynorvaline context, which embodies host-guest hydrophobic β-, γ-, and δ-CH contacts. The resulting context-dependent wGly[T] converge to 0.5 at all temperatures, and in the range 25 to 60 °C, wAla[T] decreases from ca 1.2 to 1.0. These are close to the Scheraga values.
We have here reported the first context-independent, temperature-dependent helical propensity sets for the natural and for selected other amino acid residues. These sets and the methodology used in their assignments jointly provide a rigorous foundation for further studies. Our most important result is the demonstration that in a polyalanine context, the helix-forming tendencies of the large majority of natural amino acid residues increase significantly as the temperature is raised. This result implies a subsantial increase in the percentage of natural residues that can act as helix formers.
As we have noted in the introduction, characterization at a chemical level of the dependencies of peptide helicity on residue composition and sequence requires assignment of temperature-dependences for both propensities and a linked set of context dependences. For the latter assignments, the series 1 polyalanine mutants are ideally suited, owing to a substantial length region that can accommodate pairs of guests. The practical consequences of our Figure 7 findings include a more satisfactory foundation for molecular modeling experiments, a better predictive understanding of the effects of site mutations on early folding intermediates that have been linked to human degenerative diseases, and practical insights into protein stability for living organisms that include mesophiles and thermophiles. Although 80 °C almost certainly defines the practical upper limit for guest mutation studies in hosts of structure 1, the wAla[T] value near 1.1 that we have assigned at 80 °C suggests the feasibility of using hosts with considerably longer lengths. In effect, one should be able to apply lessons from the extensive prior oligopeptide methodology to spaced, solubilized polyalanines. Moreover, the practical aqueous temperature limit of 100 °C may be extendable through use of additives such as ethylene glycol, which we have previously utilized to carry out low temperature CD studies.15
Experimental Section
Synthesis, Purification, and Characterization of Peptides
Peptides 1 and 2 were synthesized on a 0.02-0.03 mole scale using a PE Biosystems Pioneer Peptide Synthesizer with standard 9-fluorenylmethyleneoxy-carbonyl (Fmoc/HBTU) chemistry,14f and peptides were cleaved from the resin as reported previously.14f, 14h, 15 For unlabeled couplings, nine equivalents of Fmoc-AA-OH and HATU were used. For 13C=O labeled sites, HBTU and three equivalents of a mixture of labeled and non-labeled Fmoc-Ala-OH with 13C percentages that varied from 100% to 25 % were used and each labeled site was then double-coupled with nine equivalents of non-labeled Fmoc-Ala-OH. Purification was carried out on a Waters 2690 HPLC with auto-injector and 996 detector using YMC ODS-AQ 200 Å 4.5 × 150 mm columns. Further details concerning preparation and characterization of peptides 2 appear in reference 14h.
NMR Experiments
Peptide 1 samples containing uncharged guest residues were analyzed unbuffered in pure D2O at concentrations of 1 to 2 mM; for peptides containinf charged guests, 10 mM Tris buffer (pD = 7.0) was used. The 13C spectra were collected using VNMR 6.1c software on a Varian Inova-500 MHz NMR spectrometer, equipped with an Oxford Instruments superconducting magnet and a 5 mm dual broadband RF (50-202 MHz), CP/MS 13C-31P{1H} solids probe. Proton-decoupled carbon resonance spectra were measured at temperatures of 2, 25, and 60°C. Probe temperatures were maintained by a Varian variable temperature unit and calibrated using 100 % methanol for the range 1 to 40 °C and ethylene glycol at 60 °C according to instructions provided by Varian. The calibrated values agree within 1 °C with values reported by Van Geet.47 For all experiments, the 13C chemical shifts were referenced at each temperature relative to 0.1 M DSS (sodium 2,2-dimethyl-2-silapentane-5-sulfonate) by setting its 1H methyl shift value to zero.48 The assigned 13C=O chemical shift values of this report appear in the Supporting Information.
Circular Dichroism Experiments
UV spectra of solutions of peptides 2 in 18.2 M Ω water were measured on a double-beam Varian Cary 100 Bio UV-visible spectrophotometer, and their concentrations were assigned using the Trp UV chromophore with molar absorptivity at 280 nm of 5560 cm-1 M-1.14h All peptides exhibited a normal peak shape for the Trp chromophore. CD spectra were measured at ca 12 mM peptide concentrations using a single-beam Aviv 62DS spectropolarimeter equipped with a thermostatic temperature controller that was calibrated using d-10-camphorsulfonic acid. Peptides containing charged guests were studied in pH 1.2 and pH 7 phosphate buffers. CD spectra were measured after twelve-minute temperature equilibrations in 10 mm Hellma QS strain-free suprasil cells. Five successive spectra, 195-255 nm, were measured at 1.0 nm bandwidth and 0.5 nm step size, averaged, and smoothed using Aviv 62DS version 4.0 software. The Supporting Information contains peptide [θ]222 values cited in the text.
Supplementary Material
Acknowledgements
The authors thank the NIH, Grant GM 13453, for generous support.
REFERENCES
- (1).Doig AJ. Biophys. Chem. 2002;101-102:281–285. doi: 10.1016/s0301-4622(02)00170-9. [DOI] [PubMed] [Google Scholar]
- (2).Shi Z, Olson CA, Bell AJ, Jr., Kallenbach NR. Biopolymers. 2001;60:366–380. doi: 10.1002/1097-0282(2001)60:5<366::AID-BIP10177>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- (3).Maison W, Arce E, Renold P, Kennedy RJ, Kemp DS. J. Am. Chem. Soc. 2001;123:10245–10254. doi: 10.1021/ja010812a. [DOI] [PubMed] [Google Scholar]
- (4).Kelly JW. Curr. Opin. Struct. Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- (5).(a) Baldwin RL, Rose GD. Trends Biochem. Sci. 1999;24:77–83. doi: 10.1016/s0968-0004(98)01345-0. [DOI] [PubMed] [Google Scholar]; (b) Werner JH, Dyer HB, Fesinmeyer RM, Andersen NH. J. Phys. Chem. B. 2002;106:487–494. [Google Scholar]
- (6).(a) Viguera AR, Villegas V, Aviles FX, Serrano L. Folding & Design. 1996;2:23–33. doi: 10.1016/S1359-0278(97)00003-5. [DOI] [PubMed] [Google Scholar]; (b) Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Hang Z, Fletterick RJ, Cohen FE, Prusiner SB. Proc. Natl. Acad. Sci. U.S.A. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Lacroix E, Viguera AR, Serrano L. J. Mol. Biol. 1998;284:173–192. doi: 10.1006/jmbi.1998.2145. [DOI] [PubMed] [Google Scholar]; (b) Muñoz V, Serrano L. J. Mol. Biol. 1995;245:297–308. doi: 10.1006/jmbi.1994.0024. [DOI] [PubMed] [Google Scholar]
- (8).Muñoz V, Serrano L. Biopolymers. 1997;41:495–509. doi: 10.1002/(SICI)1097-0282(19970415)41:5<495::AID-BIP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- (9).(a) Wójcik J, Altmann K-H, Scheraga HA. Biopolymers. 1990;30:121–134. doi: 10.1002/bip.360300112. [DOI] [PubMed] [Google Scholar]; (b) Scheraga HA. In: Perspectives in Structural Biology. Vijajan M, Yathindra N, Kolaskar AS, editors. Indian Academy of Sciences; Bangalore: 1999. pp. 275–292. [Google Scholar]; (c) Scheraga HA, Vila JA, Ripoli DR. Biophys. Chem. 2002;101-102:255–265. doi: 10.1016/s0301-4622(02)00175-8. [DOI] [PubMed] [Google Scholar]
- (10).Chakrabartty S, Varadarajan R. Biochemistry. 2002;41:8152–8161. doi: 10.1021/bi025523t. [DOI] [PubMed] [Google Scholar]
- (11).Zeldovich KB, Berezovsky IN, Shakhnovich EI. PLoS Comput. Biol. 2007;3:62–72. doi: 10.1371/journal.pcbi.0030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rohl CA, Chakrabartty A, Baldwin RL. Protein Sci. 1995;5:2623–2637. doi: 10.1002/pro.5560051225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nasr KA, Schubert CR, Török M, Kennedy RJ, Kemp DS. Biopolymers. 2009;91:311–320. doi: 10.1002/bip.21129. [DOI] [PubMed] [Google Scholar]
- (14).(a) Kemp DS, Curran TC, Boyd JG, Allen TA. J. Org. Chem. 1991;56:6683–6697. [Google Scholar]; (b) Kemp DS, Allen TA, Oslock SL. J. Am. Chem. Soc. 1995;117:6641–6657. [Google Scholar]; (c) Kemp DS, Allen TA, Oslock SL, Boyd JG. J. Am. Chem. Soc. 1996;118:4240–4248. [Google Scholar]; (d) Kemp DS, Oslock SL, Allen TA. J. Am. Chem. Soc. 1996;118:4249–4255. [Google Scholar]; (e) Cammers-Goodwin A, Allen TA, Oslock SL, McClure KF, Lee JH, Kemp DS. J. Am. Chem. Soc. 1996;118:3082–3090. [Google Scholar]; (f) Miller JS, Kennedy RJ, Kemp DS. Biochemistry. 2001;40:305–309. doi: 10.1021/bi0019500. [DOI] [PubMed] [Google Scholar]; (g) Kennedy RJ, Tsang K-Y, Kemp DS. J. Am. Chem. Soc. 2002;124:934–944. doi: 10.1021/ja016285c. [DOI] [PubMed] [Google Scholar]; (h) Miller JS, Kennedy RJ, Kemp DS. J. Am. Chem. Soc. 2002;124:945–962. doi: 10.1021/ja011726d. [DOI] [PubMed] [Google Scholar]
- (15).Job GE, Kennedy RS, Heitmann B, Miller JS, Walker SM, Kemp DS. J. Am. Chem. Soc. 2006;128:8227–8233. doi: 10.1021/ja060094y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kennedy RJ, Walker SM, Kemp DS. J. Am. Chem. Soc. 2005;127:16961–16968. doi: 10.1021/ja054645g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Details concerning these assignments appear in the Supporting Information of reference 13.
- (18).Zerkowski JA, Powers ET, Kemp DS. J. Am. Chem. Soc. 1997;119:1153–1154. [Google Scholar]
- (19).Kallenbach NR, Spek EJ. Methods Enzymol. 1998;295:26–41. doi: 10.1016/s0076-6879(98)95033-9. [DOI] [PubMed] [Google Scholar]
- (20).(a) Padmanabhan S, York EJ, Gera L, Stewart JM, Ridgeway T, Baldwin RL. Biochemistry. 1994;33:8604–8609. doi: 10.1021/bi00194a027. [DOI] [PubMed] [Google Scholar]; (b) Creamer TP, Rose GD. Protein Sci. 1995;4:1305–1314. doi: 10.1002/pro.5560040706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Poland D, Scheraga HA. Statistical Mechanical Theory of Order-Disorder Transitions in Biological Macromolecules. Academic Press; New York: 1970. [Google Scholar]
- (22).(a) Lifson S, Roig A. J. Chem. Phys. 1961;34:1963–1974. [Google Scholar]; (b) Qian H, Schellman JA. J. Phys. Chem. 1992;96:3987–3994. [Google Scholar]; (c) Kemp DS. Helv. Chim. Acta. 2002;35:4392–4423. [Google Scholar]
- (23).Marqusee S, Robbins VH, Baldwin RL. Proc. Natl. Acad. Sci. U.S.A. 1989;86:5286–5290. doi: 10.1073/pnas.86.14.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lotan N, Yarov A, Berger A. Biopolymers. 1966;4:365–368. [Google Scholar]
- (25).Von Dreele PH, Lotan N, Ananthanarayanan VS, Andreatta RH, Poland D, Scheraga HA. Macromolecules. 1971;4:408–417. [Google Scholar]
- (26).Gō M, Hesselink FT, Gō N, Scheraga HA. Macromolecules. 1974;7:459–467. doi: 10.1021/ma60040a013. [DOI] [PubMed] [Google Scholar]
- (27).(a) Padmanabhan S, Marqusee S, Ridgeway T, Laue TM, Baldwin RL. Nature. 1990;344:268–270. doi: 10.1038/344268a0. [DOI] [PubMed] [Google Scholar]; (b) Park SH, Shalongo W, Stellwagen E. Biochemistry. 1993;32:7048–7053. doi: 10.1021/bi00078a033. [DOI] [PubMed] [Google Scholar]; c) Chakrabartty A, Korteme T, Baldwin RL. Protein Sci. 1994;3:843–852. doi: 10.1002/pro.5560030514. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rohl CA, Chakrabartty A, Baldwin RL. Protein Sci. 1996;5:2623–2637. doi: 10.1002/pro.5560051225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).(a) Vila J, Williams RL, Grant JA, Wójcik J, Scheraga HA. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7821–7825. doi: 10.1073/pnas.89.16.7821. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Williams L, Kather K, Kemp DS. J. Am. Chem. Soc. 1998;120:11033–11043. [Google Scholar]
- (29).(a) Lyu PC, Liff MI, Marky LA, Kallenbach NR. Science. 1990;250:669–673. doi: 10.1126/science.2237416. [DOI] [PubMed] [Google Scholar]; (b) O’Neil KT, DeGrado WF. Science. 1990;250:646–650. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]; (c) Blaber M, Zhang X-J, Lindstrom JD, Pepiot SD, Baase WA, Matthews BW. J. Mol. Biol. 1994;235:600–624. doi: 10.1006/jmbi.1994.1016. [DOI] [PubMed] [Google Scholar]
- (30).Kennedy RJ. Ph. D. Thesis. Massachusetts Institute of Technology; Cambridge, Massachusetts: 2004. [Google Scholar]
- (31).Heitmann B, Job GE, Kennedy RS, Miller JS, Walker SM, Kemp DS. J. Am. Chem. Soc. 2005;127:1690–1704. doi: 10.1021/ja0457462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).(a) Padmanabhan S, Baldwin RL. J. Mol. Biol. 1991;219:135–137. doi: 10.1016/0022-2836(91)90553-i. [DOI] [PubMed] [Google Scholar]; (b) Lyu PC, Sherman JC, Chen A, Kallenbach NR. Proc. Natl. Acad. Sci. U.S.A. 1991;88:5317–5326. doi: 10.1073/pnas.88.12.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wishart DS, Richards F,M, Sykes BD. J. Mol. Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- (34).Wishart DS, Sykes BD. Methods Enzymol. 1994;239:363–392. doi: 10.1016/s0076-6879(94)39014-2. [DOI] [PubMed] [Google Scholar]
- (35).As detailed in the Supporting Information, evidence includes a 203 nm test as well as quantitative bounds on required corrections to [θ]222,H,n,T.
- (36).Myers JK, Pace CN, Scholtz JM. Biochemistry. 1997;36:10923–10929. doi: 10.1021/bi9707180. [DOI] [PubMed] [Google Scholar]
- (37).(a) Holtzer MH, Holtzer A. Biopolymers. 1993;32:1675–1677. doi: 10.1002/bip.360321209. [DOI] [PubMed] [Google Scholar]; (b) Wallimann P, Kennedy RJ, Miller JS, Shalongo W, Kemp DS. J. Am. Chem. Soc. 2002;125:1203–1220. doi: 10.1021/ja0275360. [DOI] [PubMed] [Google Scholar]
- (38).(a) Long HF, Tycko R. J. Am. Chem. Soc. 1998;120:7039–7048. [Google Scholar]; (b) Toniolo C, Polese A, Formaggio F, Crisma M, Kamphuis J. J. Am. Chem. Soc. 1996;118:2744–2745. [Google Scholar]
- (39).Other workers have reported helical propensities for guest residues within polyalanines or alanine-rich hosts. In addition to references 12, 27b, and 27d, the following should be noted:Zhou NE, Monera OD, Kay CM, Hodges RS. Protein Peptide Lett. 1994;1:114–119.Luo P, Baldwin RL. Proc. Natl. Acad. Sci. U.S.A. 1999;96:4930–4935. doi: 10.1073/pnas.96.9.4930.
- (40).Makhatadze GI. Adv. Protein Chem. 2006;72:199–226. doi: 10.1016/S0065-3233(05)72008-8. [DOI] [PubMed] [Google Scholar]
- (41).Dill KA. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- (42).(a) Armstrong KM, Baldwin RL. Proc. Natl. Acad. Sci. U.S.A. 1993;90:11337–11340. doi: 10.1073/pnas.90.23.11337. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hyghues-Despointes GMP, Scholtz JM, Baldwin RL. Protein Sci. 1993;2:1604–1611. doi: 10.1002/pro.5560021006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Aqvist J, Lueke H, Quiocho F, Warshel A. Proc. Natl. Acad. Sci. U.S.A. 1991;88:2026–2030. doi: 10.1073/pnas.88.5.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Acevedo OE, Lareo LR. OMICS. 2005;9:391–399. doi: 10.1089/omi.2005.9.391. [DOI] [PubMed] [Google Scholar]
- (45).See reference 20a for data derived from peptide host experiments that provide insight into this issue.
- (46).The slope and CC linearity tests are maximally sensitive to deviations in randomly selected data points only if the data sets are nearly evenly spaced. A good approximation to this ideal is seen in Figure 8. If one or both sets must be characterized as a central data cluster with high and low outliers, values for CC and slope yield only a three-point correlation: high value, mean value for cluster, low value.
- (47).Van Geet AL. Anal. Chem. 1968;40:2227–2229. [Google Scholar]
- (48).Markley JL, Bax A, Arata Y, Hilbers CW, Kaptein R, Sykes BD, Wright PE, Wüthrich K. Pure Appl. Chem. 1998;70:117–142. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.