

Abstract
The transcriptome of ataxic muscles from α-tocopherol transfer protein deficient (ATTP-KO), 23-month old, mice was compared with that of their normal littermates. Genes encoding sarcolipin (sln) and ubiquitin carboxyl-terminal hydrolase (uchl1) were over-expressed (≥ 10-fold) in ataxic muscles. SLN is a 3.2 kDa membrane protein that binds to sarcoplasmic reticulum calcium ATPase, regulates Ca++ transport and muscle relaxation–contraction cycles. UCHL1 is a 24.8 kDa member of proteosome proteins; it is over-expressed in myofibrillar myopathy and is associated with neurodegenerative diseases. Furthermore, six additional transcripts, three encoding thin-filament proteins and three encoding Ca++ sensing proteins that participate in contraction-relaxation cycle, and eight transcripts that encode members of lysosomal proteins were also over-expressed in ataxic muscles. These observations suggest that chronic α-tocopherol (AT) deficiency activates critical genes of muscle contractility and protein degradation pathways, simultaneously. The magnitude of induction of sln and uchl1 was lower in asymptomatic, 8-month old, ATTP-KO mice and in 8-month old mice fed an AT-depleted diet. These studies suggest sln and uchl1 genes as novel targets of AT deficiency and may offer molecular correlates of well documented descriptions of neuromuscular dysfunctions in AT-deficient rodents. Since the neuromuscular deficits of ATTP-KO mice appear to be similar to those of patients with ATTP mutations, it is suggested that over-expression of sln and uchl1 may also contribute to AT-sensitive ataxia in humans.
Keywords: Ataxia, AVED, calcium homeostasis, muscle relaxation, myopathy, tocopherol transfer protein, ubiquitin, vitamin E
Introduction
α-Tocopherol transfer protein (ATTP) is a ∼32 kDa protein that selects α-tocopherol (AT) from seven other members of the dietary vitamin E family and preferentially transfers AT to lipoproteins secreted by the liver [1]. ATTP is abundantly expressed in liver [2–4], but its expression is low or undetectable in peripheral organs and various regions of the central nervous system [5]. Therefore, hepatic ATTP is a major determinant of blood and tissue concentrations of AT [6,7]. The physiological importance of ATTP and AT is underscored by the discovery of several mutations in attp gene in patients displaying ataxia with vitamin E deficiency, AVED [8,9]. Although progressive neuromuscular disease associated with vitamin E deficiency in patients with AVED or abetalipoproteinaemia or chronic cholestasis [10,11] is well documented, the identity of the genes that may contribute to the failure of the neuromuscular system remain to be clarified.
The development of transgenic mice that recapitulate the systemic AT-deficiency and neuromuscular phenotypes of AVED patients with attp mutations offer a useful model to investigate mechanisms of actions of AT in vivo [12–17]. Most studies of the genetic model of AT-deficiency (ATTP-KO mice) have focused on defining AT-dependent changes in the central nervous system, which is severely depleted in AT [5,12] in spite of the mice consuming a diet containing sufficient AT to maintain normal fertility. For example, somatosensory evoked potentials in the cerebral cortex of 18–20 month old ATTP-KO mice were shown to be markedly attenuated when compared to those recorded from WT mice [14]. This observation may be attributed to low expression of genes encoding synaptic and calcium regulatory proteins in the cerebral cortex of ATTP-KO mice [12,18].
Muscles and various regions of the brains of ATTP-KO mice are severely AT-depleted, possibly from birth, compared to their WT littermates, in spite of consuming a diet that contains sufficient AT for fertility [5,19]. An earlier study described atrophic muscle fibres in 20-month old ATTP-KO, but not in the muscles of 12-month old ATTP-KO, ataxic mice [14]. These observations suggest that histological changes appear after the initiation of ataxia. More comprehensive descriptions of muscle fibre necrosis, decrease in cytochrome oxidase activity and ‘a waddling ataxic gait’ has also been described in ∼1-year old rats that consumed an AT-depleted diet, post-weaning [20]. However, temporal changes in the activity of muscle genome imposed by chronic AT-deficiency are less well characterized. A previous study addressed temporal changes in muscle gene expression during ∼1-year of AT-deficiency, imposed by an AT-deficient diet, that did not produce obvious neuromuscular deficits [21].
We hypothesized that simultaneous changes in the transcription of distinct sets of multiple, functionally-related, genes are likely to play an important role in the emergence and persistence of the ataxic phenotype that occurs late during chronic AT-deficiency. The present study aimed at identifying the possible contribution of ∼8500 muscle genes in the pathogenesis of ataxia, defined by shorter stride lengths of ‘old’, 23-month, ATTP-KO mice [14,18] compared to their normal littermates. In addition, some of the most responsive AT-sensitive genes identified in ‘old’ ataxic mice were also assayed in asymptomatic, ‘adult’, 8-month mice to define separately the contribution of age and AT-deficiency obtained by either the deletion of the ATTP-gene or by feeding an AT-depleted diet.
Materials and methods
Diets and mice
The AT-depleted and AT-sufficient (35 IU tocopheryl acetate/Kg diet) diets used in this study were prepared as previously described [22,23]. Mice were allowed to access food and water ad libitum.
All the experiments in mice were humanely conducted and approved by Institutional Animal Care and Use Committee, University of California, Davis. Male mice from our colony of WT and ATTP-KO mice were studied [5]. Eight-month old ‘adult’ mice (six WT mice and four ATTP-KO mice) and 22–23 month old, ‘old’, mice (five WT mice and four ATTP-KO) were used for these studies. The group of mice assigned for 8 month study included six WT mice which were randomly sub-grouped to be fed either the AT-depleted (n=3) or the AT-sufficient (n=3) diets.
Skeletal muscle collection and processing
Mice were anaesthetized with intraperitoneal injection of pentobarbital (120 mg/Kg). After collection of blood by cardiac puncture, quadriceps and gastrocnemius muscles were exposed, dissected and immediately frozen in dry-ice. The frozen samples were stored at −80°C.
RNA extraction and sample preparation for GeneChip and qRT-PCR analysis
Frozen quadriceps and gastrocnemius muscles from each mouse were pulverized in pre-frozen porcelain mortar and pestle (in dry ice) to obtain finely powdered frozen muscle which was transferred to a 2.0 ml conical glass-teflon homogenizer. RNA was extracted with PARIS™ protein and RNA isolation kit essentially as described by the manufacturer (Applied Biosystems, Foster City, CA). Total RNA was quantified spectrophotometrically.
GeneChip (Mouse Genome 430 A 2.0) analysis was performed as previously described [24], using four GeneChips. A 5 μg aliquot of each of the four total RNA extracts (two extracts from WT-muscles and two extracts from ATTP-KO muscles) were processed. The scanned GeneChip images were processed to obtain hybridization signal intensities as described in Affymetrix GeneChip Operating System, V 1.4 (Affymetrix, Santa Clara, CA). To obtain the list of genes that were differentially expressed between the ATTP-KO and the WT mice, the ‘.cel’ files were imported into dChip software (1.0.01). The data were normalized and processed to obtain differentially expressed genes with default settings [25]. The data from WT-mice were used as baseline and those from the ATTP-KO mice were used as experimental to obtain fold-changes. The list of genes with fold-changes was then edited to exclude all the genes that showed less than 2-fold change and those that did not have annotations. The data were tabulated to show gene names, their fold-change attributed to ATTP-KO genotype, Affymetrix probe set identifiers and gene number in mouse genome database. The Affymetrix probe set identifiers offer an opportunity to identify, from the Affymetrix Database, the precise deoxynucleotide sequences of probes used in the Mouse genome 430 A 2.0 GeneChips and for designing oligonucleotide primers for quantitative PCR verifications.
Quantitative real-time PCR (qRT-PCR) was performed as previously described [22]. Nineteen aliquots of cDNAs synthesized from muscle RNA extracts from 19 different mice were used for validation of the selected genes identified by the GeneChip assay. The selection criterion was based of their relevance to the novel findings of this study. The oligonucleotide sequences of primer pairs were obtained using Primer Express software (Applied Biosystems) and their sequences are shown in Table I.
Table I.
List of genes selected from GeneChip data and subjected to qRT-PCR validation. The specific primer pairs [deoxynucleotide sequences (5′ to 3′) are shown for each primer] for each gene. The oligonucleotide sequences from Affymetrix probeset identifiers were used to design primers of the respective genes using the primer design software. The primers were custom prepared and used as described in the Materials and methods section.
| Gene name | Forward primer | Reverse primer |
|---|---|---|
| ATPase, Ca++ transporting, cardiac muscle, slow twitch 2 | GTGGCCAGATTGCTCTACAGTG | AGGGCCCATTAGAAAGCATGT |
| B cell leukaemia/lymphoma 6 | GAAGTTTTCAATGATGGACGGG | GCCTACACTTCAAAAAGGGATGG |
| CD36 antigen | GAAAATCAAGCTCCTTGGCATG | ACTCCAATCCCAAGTAAGGCC |
| Cathepsin B | GAACCACTGTGGCATTGAATCA | TCAGTGCGTGGGATTCCAG |
| Cathepsin L | ACCACTGTGGACTTGCCACC | CCCATCAATTCACGACAGGAT |
| Cathepsin S | GACGACCCCTCCTGTACGG | GCCAACCACAAGAACACCATG |
| Glyceraldehyde-3-phosphate dehydrogenase | TTGTGGAAGGGCTCATGACC | TCTTCTGGGTGGCAGTGATG |
| Sarcolipin | TGTGCCCCTGCTCCTCTTC | TGATTGCACACCAAGGCTTG |
| Troponin C, cardiac/slow skeletal | GAAGGACGACAGCAAAGGGA | CGGAAGAGATCCGACAGCTC |
| Troponin I, skeletal, slow 1 | CAGCCTATGCGCACACCTTT | TCCCCTTTGTGTGCCATTTC |
| Ubiquitin carboxy-terminal hydrolase L1 | ACGGCCATCTGTACGAGCTC | CATGGTTCACTGGAAAGGGC |
| α-tocopherol transfer protein | TGGAAACTCAACGCAATGGAG | GCCAGCCTTCCAGGTCAAA |
Statistics
The means, standard error of means (SEMs) and significance of difference (p-values) in fold-change determined by qRT-PCR were calculated by t-test using GraphPad Prizm software 4.0 (La Jolla, CA).
Results
The analytical strategy included confirmation of genotype of the mice (Figure 1), obtaining the primary list of AT-sensitive genes (and their functional assignments) from muscles of 23-month old ataxic mice (Table II), confirmation of the selected genes by qRT-PCR (Table III) and assaying the AT-sensitive, qRT-PCR confirmed genes in the AT-deficient muscles of 8-month old mice (Table IV).
Figure 1.
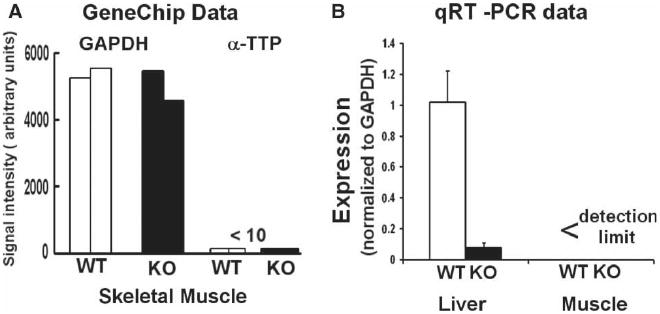
α-TTP mRNA (ATTP) expression in muscles and livers of 2-year old wild type (WT) and α-tocopherol transfer protein deficient (KO) mice. ATTP and GAPDH mRNA expressions were assayed by GeneChips and qRT-PCR. The GeneChip data are from muscles of WT (n=2) and ATTP-KO (n=2) mice. The qRT-PCR data were normalized to GAPDH expression in each sample. ATTP expression in KO-liver was very low (p <0.00001) and it was undetectable in muscles. The qRT-PCR data are mean±SEM, for five WTand four KO tissues.
Table II.
α-tocopherol sensitive genes in muscles of 23-month old ataxic, male mice. Gene expression data from WT muscles (n=2 mice) were used as baseline and those from ATTP-KO muscles (n=2 mice) were used as experimental to calculate fold-change by dChip data analysis software for Affymetrix arrays. The data were grouped according to function from the list of 57 genes that were differentially expressed.
| Functional group and gene name | Fold change | probe set ID | Accession # |
|---|---|---|---|
| Cytoskeletal and Ca sensitive | |||
| sarcolipin | 14.7 | 1420884_at | AK008863 |
| periostin, osteoblast-specific factor | 6.5 | 1423606_at | BI110565 |
| PDZ and LIM domain 5 | 3.7 | 1421413_a_at | NM_022554 |
| troponin I, skeletal, slow 1 | 3.6 | 1450813_a_at | NM_021467 |
| ATPase, Ca++ transporting, cardiac muscle, slow twitch 2 | 3.5 | 1452363_a_at | AA245637 |
| tropomyosin 3, gamma | 3.4 | 1449996_a_at | NM_022314 |
| troponin C, cardiac/slow skeletal | 2.8 | 1418370_at | NM_009393 |
| myosin, heavy polypeptide 7, cardiac muscle, beta | 2.7 | 1448553_at | NM_080728 |
| capping protein (actin filament) muscle Z-line, beta | 2.6 | 1453960_a_at | AK007209 |
| vimentin | 2.3 | 1450641_at | M24849 |
| matrix Gla protein | 2.2 | 1448416_at | NM_008597 |
| calsequestrin 2 | 2.0 | 1422529_s_at | NM_009814 |
| Protein degradation | |||
| ubiquitin carboxy-terminal hydrolase L1 | 8.4 | 1448260_at | NM_011670 |
| cathepsin S | 6.2 | 1448591_at | NM_021281 |
| lysozyme | 4.1 | 1423547_at | AW208566 |
| P lysozyme structural | 3.6 | 1436996_x_at | AV066625 |
| cathepsin B | 3.3 | 1448732_at | M14222 |
| proteasome (prosome, macropain) 26S sub-unit, non-ATPase | 2.4 | 1451056_at | BB034143 |
| ubiquitin-conjugating enzyme E2D 3 (UBC4/5 homologue, yeast) | 2.3 | 1455480_s_at | BG070759 |
| cathepsin L | 2.1 | 1451310_a_at | J02583 |
| lysosomal-associated protein transmembrane 4A | 2.0 | 1423368_at | BI695636 |
| Immune-inflammation | |||
| immunoglobulin kappa chain, constant region | 5.7 | 1452417_x_at | AV057155 |
| lectin, galactose binding, soluble 3 | 4.4 | 1426808_at | X16834 |
| apolipoprotein E | 2.9 | 1432466_a_at | AK019319 |
| beta-2 microglobulin | 2.7 | 1452428_a_at | AI099111 |
| CD36 antigen | 2.3 | 1450883_a_at | BB534670 |
| apolipoprotein D | 2.3 | 1416371_at | NM_007470 |
| histocompatibility 2, D region | 2.2 | 1451784_x_at | L36068 |
| histocompatibility 2, class II antigen A, alpha | 2.1 | 1435290_x_at | BE688749 |
| Thiol oxidation and metabolic stress | |||
| metallothionein 2 | 3.3 | 1428942_at | AA796766 |
| cytochrome P450, family 2, sub-family e, polypeptide 1 | 3.2 | 1415994_at | NM_021282 |
| thioredoxin interacting protein | 3.2 | 1415997_at | AF173681 |
| heat shock protein family, member 7 (cardiovascular) | 2.8 | 1421290_at | BG968304 |
| esterase D/formylglutathione hydrolase | 2.1 | 1417825_at | NM_016903 |
| cysteine and glycine-rich protein 3 | 2.1 | 1460318_at | NM_013808 |
| spermine oxidase | −2.1 | 1424268_at | BC004831 |
| Vesicular and secretory | |||
| synaptophysin-like protein | 3.0 | 1422879_at | BE333485 |
| synaptopodin 2 | 2.9 | 1450828_at | NM_080451 |
| reticulon 4 | 2.4 | 1421116_a_at | NM_024226 |
| vesicle-associated membrane protein 3 | −2.8 | 1437708_x_at | BB552111 |
| Mitochondrial | |||
| diaphorase 1 (NADH) | 3.2 | 1422185_a_at | NM_029787 |
| pyruvate dehydrogenase E1 alpha 1 | 2.6 | 1449137_at | NM_008810 |
| indolethylamine N-methyltransferase | 2.4 | 1418697_at | NM_009349 |
| glutamate oxaloacetate transaminase 2, mitochondrial | 2.3 | 1417716_at | U82470 |
| Miscellaneous | |||
| carbonic anhydrase 3 | 3.4 | 1460256_at | NM_007606 |
| tumour differentially expressed 2 | 3.2 | 1454811_a_at | AV026664 |
| ADP-ribosylation factor-like 6 interacting protein 2 | 3.2 | 1416794_at | NM_019717 |
| neural precursor cell expressed, developmentally down-regulated gene 4 | 2.9 | 1451109_a_at | BG073415 |
| glucan (1,4-alpha-), branching enzyme 1 | 2.2 | 1420654_a_at | NM_028803 |
| protein phosphatase 3, catalytic sub-unit, beta isoform | 2.2 | 1427468_at | M81483 |
| integral membrane protein 2A | 2.1 | 1451047_at | BI966443 |
| musculoskeletal, embryonic nuclear protein 1 | 2.1 | 1427201_at | AJ277212 |
| cell division cycle 42 homologue (S. cerevisiae) | 2.1 | 1449574_a_at | BF143638 |
| HESB like domain containing 2 | 2.1 | 1423652_at | AV209097 |
| protein kinase inhibitor, alpha | 2.0 | 1420858_at | AK010212 |
| DEAD (Asp-Glu-Ala-Asp) box polypeptide 51 | −2.0 | 1428728_at | AK010015 |
| B-cell leukaemia/lymphoma 6 | −2.3 | 1421818_at | U41465 |
Table III.
Validation of GeneChip data by qRT-PCR assay. The genes selected from the list of 57 differentially expressed genes were analysed by qRT-PCR. Muscle RNA extracts from ‘old’, 23-month mice were used for validation. Four samples (two from WT mice and two from ATTP-KO mice) used for GeneChip assay and additional muscle RNA extracts from five mice (three WT mice and two ATTP-KO mice) were processed. The expression data from WT muscles were used as baseline and those from ATTP-KO muscles were used as experimental to calculate fold-changes as described in the Materials and methods section.
| Fold change (ATTP-KO/WT) | |||
|---|---|---|---|
| Gene name | symbol | GeneChip (n=2) |
qRT-PCR Mean±SEM (n=4-5) |
| sarcolipin | Sln | 14.7 | 33.0±1.1 |
| troponin C1 | Tnnc1 | 2.8 | 4.8±0.4 |
| troponin I | Tnni | 3.6 | 2.3±0.4 |
| calcium transporting ATPase | Atp2a2 | 2.0 | 3.4±0.5 |
| protein kinase inhibitor, alpha | Pkia | 2.0 | 1.7±0.4 |
| ubiquitin caroxy-terminal hydroxylase L1 | Uchl1 | 8.4 | 10.2±1.5 |
| CD 36 | CD36 | 2.3 | 3.3±0.8 |
| cathepsin S | Ctss | 6.2 | 3.0±0.3 |
| cathepsin B | Ctsb | 3.3 | 4.3±1.6 |
| cathepsin L | Ctsl | 2.1 | 1.4±0.4 |
| B-cell leukemia/lymphoma 6 | Bcl6 | −2.3 | −1.9 ± 0.3 |
Table IV.
Changes in the expression of the sarcolipin and ubiquitin carboxy-terminal hydrolase 1 gene clusters in skeletal muscles from ‘adult’, 8-month old, pre-ataxic and ‘old’, 23-month, ataxic mice. The expression of the seven genes of sln gene cluster and four genes of uchl1 gene cluster was assayed by qRT-PCR of cDNA samples from skeletal muscles of ‘adult’, 8-month, mice fed either an AT-sufficient or an AT-depleted diet. Fold-change in the expression of the genes in the AT-depleted muscles was obtained by comparing its expression with that in the muscles of mice fed the AT-sufficient diet. Similarly, the effect of AT-deficiency imposed by the deletion of ATTP gene were obtained by comparing the expression of the genes in ‘adult’, 8-month, AT-depleted muscles of ATTP-KO mice with their WT-littermates.
| Fold change determined by qRT-PCR | ||||
|---|---|---|---|---|
| Genotype | WT | KO | KO | |
| Age | 8-month | 8-month | 23-month | |
| Diet | DEFICIENT | NORMAL | NORMAL | |
| Gene name | symbol | Mean±SEM (n=3-5) | Mean±SEM (n=3-5) | Mean±SEM (n=4-5) |
| sarcolipin | Sln | 11.3±2.3 | 6.7±1.8 | 33.0±1.1 |
| troponin C1 | Tnnc1 | 1.3±0.3 | 1.4±0.4 | 4.8±0.4 |
| troponin I1 | Tnni1 | 3.6±0.6 | 1.5±0.2 | 2.3±0.4 |
| calcium transporting ATPase | Atp2a2 | 3.1±0.5 | −1.2±0.7 | 3.4±0.5 |
| calsequestrin 2 | Casq2 | 1.1±0.2 | 1.5±0.3 | 1.7±0.4 |
| tropomyosin 3, gamma | Tpm3 | 1.1±0.2 | 1.8±0.4 | 3.3±0.9 |
| myosin, heavy polypeptide 7 | Myh7 | −1.5 ± 0.6 | 1.1±0.2 | 1.5±0.2 |
| ubiquitin carboxy-terminal hydroxylase L1 | Uchl1 | 4.2±1.0 | 7.6±0.5 | 10.2±1.5 |
| cathepsin S | Ctss | 4.1±0.8 | 1.9±0.2 | 3.0±0.3 |
| cathepsin B | Ctsb | 1.6±0.2 | 1.3±0.1 | 4.3±1.6 |
| cathepsin L | Ctsl | 1.0±0.2 | −1.5±0.8 | 1.4±0.4 |
| pre-ataxic | ataxic | |||
Confirmations of ATTP-KO and WT genotypes
Figure 1 shows that the expression of ATTP mRNA in muscles of 2-year old ataxic mice, assayed by either GeneChips or qRT-PCR, is below the limits of detection. In contrast, ATTP mRNA expression is robust (similar to that of GAPDH as indicated by the ratio of 1) in WT, 2-year old, male mice and is very low in ATTP-KO mice, as expected. A previous study has shown that the muscles and other tissues of ATTP-KO mice are deficient in AT [19].
AT-sensitive muscle transcriptomes
To determine the genome-wide signature of mRNA changes associated with chronic AT-deficiency and the resulting ataxic phenotype, muscle RNA extracts from ‘old’ WT and ATTP-KO mice were processed. The GeneChip assay detected ∼12 000 mRNAs in muscles. Comparison of ATTP-KO- with that of WT-muscle transcriptomes identified 57 genes that were differentially expressed (≥ 2-fold and the difference in signal intensity for each gene between the two data sets was ≥ 100 units) (Table II). Fifty-three genes were over-expressed in ATTP-KO compared with WT-muscles. These genes were then clustered according to their functions and literature descriptions. Six groups of genes each including 4–7 genes were obtained (Table II). Thirteen genes could not be assigned to any of the six clusters and are listed as miscellaneous in Table II. The functional groups included genes that encode: cytoskeletal and calcium sensing-regulated proteins, those that participate in protein degradation, immune-inflammation, thiol-oxidation and metabolic stress, vesicular and secretory functions and those associated with mitochondria.
Over-expression of genes that affect Ca++ mediated muscle contraction-relaxation cycles
A cluster of four-genes (sarcolipin (sln), troponin I (tnni), troponin C (tnnc) and calsequestrin 2 (casq2)), whose encoded proteins are known to participate in Ca++ dependent muscle contraction and relaxation cycles, were over-expressed in muscles of ATTP-KO compared with WT littermate mice (Table II). There was a 14-fold induction of sln, the gene encoding SLN, a 3.6 kDa protein [26] that inhibits re-uptake of sarcoplasmic calcium by inhibiting the activity of ATPase, Ca++ transporting 2, also known as SER-CA2A2 [27]. Gene encoding a target of SLN, SERCA2A2 was also induced. Table II also shows a 2-fold induction of the gene that encodes calsequestrin 2, the main Ca++ buffer of sarcoplasmic reticulum [28]. At least three additional genes that regulate calcium-dependent contraction-relaxation cycles of skeletal muscles were also co-activated with sln (Table II). Coordinated inductions of tnni, tnnci and tpm3 are particularly remarkable because the encoded proteins form the thin filaments of striated muscles and they have been suggested to form an inhibitory unit that prevents interactions between actin and myosin [29] and, thereby, may modulate muscle relaxation and contraction cycles. This cluster of seven genes is designated hereafter as ‘the sarcolipin cluster’.
Activation of proteolytic and inflammatory responses by chronic AT-deficiency
Genes associated with proteolysis were over-expressed in ataxic, AT-deficient muscles (Table II). Many of the genes in this cluster (e.g. cathepsins B, L and S) are associated with lysosomes and phagosomes [30]. Three genes encoding members of the ubiquitin-dependent proteosomal pathway (some of which are associated with processing by phagosomes [31]) are also included in this cluster.
Ataxic muscles of ATTP-KO mice also displayed markers of overactive inflammatory response (Table II). Inductions of genes encoding members of major histocompatibilty class II and beta-2 microglobulin are included in one group for their roles in immune functions that depend on ubiquitin-proteosomal pathways [31,32]. Co-inductions of CD36 and apolipoproteins D and E genes are noteworthy because they participate in lipoprotein metabolism and are frequently implicated in inflammatory responses [33,34]; CD36 is a receptor for oxidized low density lipoproteins and apolipoproteins are integral components of these macromolecular complexes. A 3-fold induction of gene encoding cytochrome P4502e1 in the ATTP-KO muscles is further suggestive of increased oxidation of lipids and of proteins which may be targeted to proteosome complexes [35,36] in AT-deficient muscles.
QRT-PCR assay of selected genes validates GeneChip data
To address the possible relation between the ataxic phenotype and mRNA expression, we focused on genes that have previously been associated with muscle contraction-relaxation cycles and muscle protein degradation (Table III). The induction and repression of all the genes selected from the list of 57 differentially expressed genes, identified by the GeneChip assay, were confirmed by qRT-PCR assay of the same RNA samples used for the GeneChip assay (Table III). The precise magnitude of the change obtained by the two methods differed; for example, the GeneChip assay under estimated sln induction, compared to that obtained by qRT-PCR assay (Table III). Furthermore, muscle RNA extracts from additional ‘old’ mice (n=5, three WT mice and two ATTP-KO mice) also showed the predicted change in the expression of the same genes (Table III). Hence, qRT-PCR validation assays for the expression of genes identified by the GeneChip assay attest to the reliability and reproducibility of the analytical procedures used for the genome-wide analysis of muscle mRNAs and the initial analytical step in the search for AT-sensitive genes in ataxic muscles.
Effect of age and dietary AT-deficiency on the sarcolipin gene cluster
The contribution of age on the expression of the sarcolipin gene cluster, implicated in the regulation of muscle contraction-relaxation cycle, was assayed in muscles from ‘adult’ WTand ATTP-KO mice and in adult mice fed the AT-depleted diet. A 6-fold-induction of sln was detected in the ATTP-KO mice when compared to the expression of sln in muscles from WT mice (Table IV). Furthermore, when WT mice were fed the AT-depleted diet, muscle sln was also induced (Table IV). Thus, data from three different groups of mice of two different genotypes demonstrate that muscle sln expression is a sensitive indicator of AT-deficiency. Moreover, sln is the most responsive AT-sensitive gene within the sln cluster, in muscles of both ‘adult’ and ‘old’ mice (Table IV).
Two genes, one encoding TROPONIN I and the other encoding calcium transporting ATPase2a2 (also known as SERCA2a2), appear to be more sensitive to diet-induced AT-deficiency compared to deficiency resulting from the deletion of the ATTP-gene. Data in Table IV also suggest differential sensitivity of each gene within the sln cluster to AT-deficiency. For example, the large induction of troponin C1 (tnnc1) and that of tropomyosion 3, gamma (tpm3) is seen only in the muscles of ‘old’ ATTP-KO mice.
Effect of age and dietary AT-deficiency on the uchl1-gene cluster
Four of the nine genes in uchl1 gene cluster were selected for qRT-PCR verification in ‘adult’ and ‘old’muscles (Table IV). Expression of three of the four genes in this cluster was lower in muscles of ‘adult’ ATTP-KO compared with ‘old’ ATTP-KO mice. The expression of uchl1 was activated, 4.2- and 7.6-fold, by AT deficiency obtained by AT-deficient diet and by the deletion of ATTP gene, respectively; both these inductions were lower than that detected in muscles from ‘old’ ATTP-KO mice. Similarly, the fold-inductions of two of the three cathepsins were also lower in ‘adult’ AT-deficient mice, as indicated by ≤ 1.6-fold change, when compared to their inductions in ‘old’ mice (Table IV).
Collectively, these results show that AT-deficiency caused by either dietary depletion or by the deletion of ATTP-gene activates sln- and uchl1-gene clusters. The magnitude of these inductions was higher in ‘old’ mice compared to that in ‘adult’ mice.
Discussion
Transcriptomic programme for ataxic phenotype in skeletal muscles of ATTP-KO mice
The most significant and novel result of this study is the identification, by two independent analytical procedures, a robust induction of seven muscle genes, the sln cluster (Table IV), whose simultaneous activation may contribute to age-related ataxia, defined by shorter stride-lengths of ATTP-KO mice compared to their WT-littermates [14,18]. The sln cluster of seven genes consisted of sln, tnni, tnnc, tpm3, ATP2a2 (also known as serca2a2), myh7 and casq2. Remarkably, five of these seven genes, sln, tnni, tnnc, serca2a2 and casq2, encode proteins that are known to play an essential role in calcium mediated contraction–relaxation cycles of muscles [26,29,37]. The lack of co-ordinated induction of all these genes in younger, AT-deficient, mice (Table IV) which were asymptomatic prompt the interpretation that the sarcolipin cluster of ‘old’ ATTP-KO mice may be associated with the neuromuscular deficits observed in mice with severe AT-deficiency [14,18].
Of the seven genes in the sarcolipin gene cluster, sln showed a > 14-fold induction; the highest induction in this group, and it was independently confirmed by qRT-PCR of RNA extracts from nine different muscle samples (Table IV). Additional confirmation of sln induction by AT-deficiency was also obtained by qRT-PCR data from muscles of younger ATTP-KO mice and from muscles of WT mice fed an AT-depleted diet (Table IV). Previously mice displaying myopathy showed a > 70-fold induction of sln in response to the deletion of nebulin-gene [38]. sln encodes a 3.1 kDa protein that inhibits the activity of SERCA2, a Ca++ pump that actively removes Ca++ from sarcoplasm and participates in the recovery of muscle from contraction [27]. Hence, over-expression of sln detected in muscles of AT-deficient mice may delay muscle recovery from contraction. The muscles of ∼10-month old, AT-deficient rats also displayed abnormal electromyographic properties [39], possibly due to over-expression of sln. This postulate is further reinforced by the observations that the forced over-expression of sln in rat soleus muscle inhibits SERCA, decreases luminal Ca++ and impairs contractility [40]. Our preliminary electrophysiological experiments that assayed isometric contractions of gastrocnemius muscle group via sciatic nerve stimulation showed that muscles from a 1-year old ATTP-KO mouse failed to maintain tetanic force and failed to recover after titanic stimulation when compared to the muscle of a 1-year old WT mouse (Carlsen and Bodine, unpublished). Further electrophysiological experiments are necessary to validate these preliminary observations and to further distinguish between the contribution of motor units and muscle fibre functions.
Two previous studies have suggested co-regulation of SLN and SERCA [26,41]. The data in Tables II–IV suggest that the expressions of the two mRNAs are also co-regulated. Hence, the robust induction of sln and other members of sarcolipin gene cluster is a novel and unanticipated action of AT-deficiency in skeletal muscle and may contribute to the ataxic phenotype of the ATTP-KO mice. Further studies are necessary to assay the expression of SLN protein and its fibre-type distribution in ataxic muscles of ATTP-KO mice. Our preliminary studies with commercially available anti-SLN polyclonal antibody have been unsuccessful both in cardiac atria (positive control where SLN is abundantly expressed [26]) and in skeletal muscles of ATTP-KO mice. We note with interest that SLN was first detected as a protein that co-purified with SERCA1 from fast twitch skeletal muscle [42] and was identified as a proteolipid protein and designated ‘sarcolipin’ [43]. A lack of SLN protein induction in the presence of a large induction of mRNA would implicate a role for micro RNA(s) which are known to block protein synthesis [44,45]. Experimental evaluations of these possibilities remain major challenges in addressing the molecular basis of AT-sensitive neuromuscular functions.
The large induction of sln was accompanied by the co-inductions of tnni, tnnc, tpm3 and casq2 (Tables II, III – IV), all of which participate in regulating Ca++ dependent muscle contraction and relaxation cycles (Figure 2). Co-inductions of tnni, tnnc and tpm3 may be consistent with the observation that TNNI, TNNC and TPM3 form an inhibitory unit that prevents the interactions between actin and myosin [29]. Tables II–IV also show induction of the gene that encodes casq2, the main Ca++ buffer of sarcoplasmin reticulum [28]. A previous study has shown that over-expression of casq2 in cardiac muscle decreases contractility [46]. Therefore, over-expression of these four genes may further add to the inhibitory actions of sln over-expression on skeletal muscle contractility of ATTP-KO mice. Future studies are necessary to determine if the changes in mRNAs detected here are translated into the respective proteins and if all the members of the sarcolipin gene cluster are necessary to display short-stride lengths of the old, ATTP-KO mice. Our data (Table IV) from ‘adult’, 8-month old, asymptomatic, AT-deficient mice suggest that the co-inductions of all the genes within the sarcolipin gene cluster may be required for the display of shorter stride lengths of ATTP-KO mice compared to those of their WT-littermates.
Figure 2.
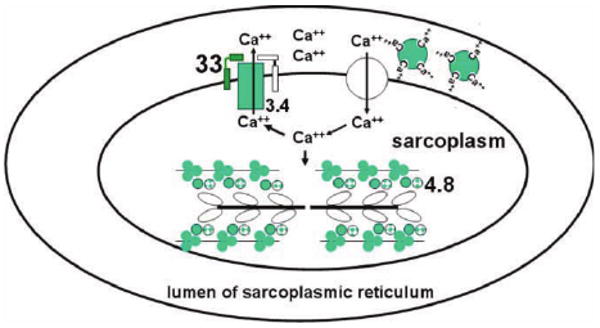
Schematic representation of proteins that participate in calcium-dependent regulation of muscle contraction-relaxation cycles. Calcium is stored, bound to calsequestrin, in the lumen of sarcoplasmic reticulum. It is released, through ryanodine receptors, into the sarcoplasm in response to a nerve stimulus, causing muscle contraction. To initiate relaxation, Ca++ is pumped out by SERCA2. Activity of SERCA2 is inhibited by SLN and PLN. Genes encoding proteins (shades of green or grey, darker=higher expression) were over-expressed in ataxic muscles of 2-year old male, ATTP-KO mice compared to those in age-matched, male WT-mice. The numbers indicate fold-induction of the mRNA, obtained by qRT-PCR, in the ATTP-KO muscles.  sarcolipin (SLN),
sarcolipin (SLN),  troponin I and troponin c,
troponin I and troponin c,  tropomyosin,
tropomyosin,  phospholamban (PLN),
phospholamban (PLN),  sarco-endoplasmic reticulum calcium ATPase2 (SERCA2),
sarco-endoplasmic reticulum calcium ATPase2 (SERCA2),  calsequestrin,
calsequestrin,  myosin of thick filament,
myosin of thick filament, 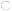 ryanodine receptor.
ryanodine receptor.
The only previous skeletal muscle study of chronic (up to ∼13 months) vitamin E deficiency in rats that used GeneChips identified 56 AT-sensitive genes, all of which were induced by dietary vitamin E deficiency [21]. Hence, two studies that used different in vivo models to produce AT-deficient muscles and different data analysis protocols arrived at very similar numbers of AT-sensitive genes. There is also a considerable overlap in the functional groups of genes that are modulated by severe AT-deficiency. The induction of the cluster of genes that encode vesicular and secretory function (Table II) is also noteworthy because two previous studies have documented a role for AT in the modulation of genes that regulate vesicle assembly and function [12,47]. Comparison of the muscle AT-sensitive genes in the two studies gives important clues about the genes that may contribute to the ataxic phenotype of vitamin E deficient rodents and, possibly, humans. The data from dietary AT-deficient rats [21] were remarkable for the lack of change in the sarcolipin gene cluster described in this study (Tables II, III and IV). It is also noted that the rats were asymptomatic. This observation underscores the possible relevance of the sarcolipin gene cluster in the delayed display of ataxia in the mouse model of AVED [14,18].
AT-sensitive changes in the cerebral cortex may also contribute to the ataxia phenotype of AT-deficient mice [12,14] and neurological deficits of AT-deficient rats [48]. Previous studies in rodents [49] and humans [50] have suggested that chronic AT-deficiency results in initial loss of neural functions. Hence, the transcriptomic responses of skeletal muscles of ATTP-KO mice described here may be in addition to neural deficits. Further studies that address the changes in gene expression programmes of the various regions of the central nervous system, the peripheral nervous system and the muscular system are needed to characterize the chronology of molecular changes that are associated with the emergence of neuromuscular phenotypes associated with chronic AT-deficiency.
AT-deficiency activates protein degradation and inflammation pathways
Muscles of ATTP-KO mice had increased expression of genes that encode proteolytic enzymes associated with lysosomes and phagosomes and members of proteosomal pathway (Tables II, III and IV). Presence of increased levels of lysosomes in muscles of an AT-deficient patient have also been described [11]. The induction of cathepsins (Tables II, III and IV) was also reported in the previous study [21]. Unlike the previous study, the present study detected over-expression of CD36 gene (Tables II, III and III), which encodes the receptor for oxidized lipoproteins [51,52], in ‘old’ but not in ‘adult’ AT-deficient mice. Since the induction of CD36 was detected only in the ‘old’ ataxic mice we infer that the inflammatory process is also a late event which may be triggered by increased accumulation of oxidized phospholipids [53] in AT-deficient muscles. Genes encoding apolipoproteins D and E genes were also activated by AT-deficiency (Table II). Products of these genes are often used as markers of increased inflammation [33,34] and have been reported to be induced in myopathies [54,55] that are not associated with AT-deficient muscles. Comparative analysis of muscle inflammatory markers associated with various myopathies may enable identification genes that are specifically associated with chronic AT-deficiency.
Temporal disconnect between the onset of AT-deficiency and the appearance of neuromuscular phenotypes
The molecular mechanisms that account for the long lag-phase (months) between the initiation of AT-deficiency and the display of neuromuscular phenotype remain unexplained. The phenomenon is extensively documented. For example, the maximum length of deficiency in the asymptomatic rats was 1-year post-weaning [21], which contrasts with almost 2 years of AT-deficiency, including the pre-weaning period, in ataxic, ATTP-KO mice.
It can be hypothesized that ‘pro-ataxic’ metabolites, yet to be identified, are generated and accumulate in terminally differentiated muscle fibres during chronic AT-deficiency. Since tocopherols are primarily sequestered in biological membranes [56], it is reasonable to propose that AT-depleted biological membranes, including nuclear membranes, are a significant source of these ‘pro-ataxic’ metabolites. Furthermore, phospholipids, a ubiquitous class of membrane lipids, are also present in the nucleoplasm [57] and have been suggested to be associated with, and modulate the activity of, chromatin [58]. Hence, AT-depleted muscle membranes may have altered phospholipid metabolism. The present GeneChip data may be interpreted to support this hypothesis. The assay identified a 2-fold induction of cyp2e1 (Table II, Thiol oxidation and metabolic stress group). A previous study has suggested that AT inhibits CYP2E1 activity [59]. CYP2E1 has been suggested to increase reactive oxygen species [35,36] which may affect activities of redox-sensitive transcription factors such as nuclear factor (erythroid-derived 2)-like 2 (NRF2), nuclear factor of kappa light polypeptide gene enhancer in B-cells, (NF-κB) and hypoxia-inducible factor [60–62]. A previous transcriptomic analysis of lungs from ATTP-KO mice suggested modulation of AhR-NRF2-NF-κB transcription factor networks [22]. A search of the muscle transcriptomic data (Table II) for genes that may be induced by the activation of these transcription factors identified genes such as diaphorase 1 (also known as NAD(P)H:menadione oxidoreductase), metallothionein 2, thioredoxin interacting protein and heat shock protein family, member 7 (Table II). The previous GeneChip analysis of AT-deficient muscles from rats fed AT-deficient diet for ∼1-year also suggested increased expression of seven genes related to oxidative stress [21]. Collectively, these data offer mRNA correlates of metabolic pathways which may contribute to temporal changes in metabolites, possibly derived from lipids of membranes and nucleoplasm, which may result in an altered transcriptomic programme, precipitating failure of the neuromuscular functions with chronic AT-deficiency.
Conclusions
Comparative analysis of skeletal muscle transcriptomes from ‘old’, 23-month, ataxic, ATTP-KO mice and their WT-littermates identified two genes, sln and uchl1, as sensitive responders of AT-deficiency. Sln encodes a ∼3.2 kDa proteolipid protein that is an inhibitor of sarcoplasmic reticulum calcium transporting ATPase and it regulates relaxation-contraction cycles. Furthermore, six additional genes whose encoded proteins participate in relaxation-contraction cycle were also over-expressed in ataxic muscles. Hence, these seven co-regulated genes appear to form a functional cluster, ‘the sarcolipin gene cluster’, which may contribute to delayed ataxia in α-tocopherol-transfer protein deficient mice and possibly in AVED patients. These transcriptomic data suggest a role for α-tocopherol in the transcriptional regulation of genes that affect calcium dynamics across sarcoplasmic reticulum.
Acknowledgments
We thank Professor Maret Traber (Linus Pauling Institute, Orgeon State University) for her valuable comments and suggestions in the preparation of this manuscript. We also thank Professor Jerry Last for his generous gift of Mouse Genome Affymetrix arrays. The research was funded by the following agencies: NIH Grant #ES011985, USDA grant # 35200-13456, University of California Clinical Nutrition Research Unit and University of California, Davis, Center for Human Nutrition Research Pilot Research Grant.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Morley S, Cacchini M, Zhang W, Virgulti A, Noy N, Atkinson J, Manor D. Mechanisms of ligand transfer by the hepatic tocopherol transfer protein. J Biol Chem. 2008;283:17797–17804. doi: 10.1074/jbc.M800121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sato Y, Arai H, Miyata A, Tokita S, Yamamoto K, Tanabe T, Inoue K. Primary structure of alpha-tocopherol transfer protein from rat liver. Homology with cellular retinaldehyde-binding protein. J Biol Chem. 1993;268:17705–17710. [PubMed] [Google Scholar]
- 3.Sato Y, Hagiwara K, Arai H, Inoue K. Purification and characterization of the alpha-tocopherol transfer protein from rat liver. FEBS Lett. 1991;288:41–45. doi: 10.1016/0014-5793(91)80999-j. [DOI] [PubMed] [Google Scholar]
- 4.Arita M, Sato Y, Miyata A, Tanabe T, Takahashi E, Kayden HJ, Arai H, Inoue K. Human alpha-tocopherol transfer protein: cDNA cloning, expression and chromosomal localization. Biochem J. 1995;306:437–443. doi: 10.1042/bj3060437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gohil K, Oommen S, Quach HT, Vasu VT, Aung HH, Schock B, Cross CE, Vatassery GT. Mice lacking alpha-tocopherol transfer protein gene have severe alpha-tocopherol deficiency in multiple regions of the central nervous system. Brain Res. 2008;1201:167–176. doi: 10.1016/j.brainres.2008.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manor D, Morley S. The alpha-tocopherol transfer protein. Vitam Horm. 2007;76:45–65. doi: 10.1016/S0083-6729(07)76003-X. [DOI] [PubMed] [Google Scholar]
- 7.Traber MG. Vitamin E regulatory mechanisms. Annu Rev Nutr. 2007;27:347–362. doi: 10.1146/annurev.nutr.27.061406.093819. [DOI] [PubMed] [Google Scholar]
- 8.Gotoda T, Arita M, Arai H, Inoue K, Yokota T, Fukuo Y, Yazaki Y, Yamada N. Adult-onset spinocerebellar dysfunction caused by a mutation in the gene for the alpha-tocopherol-transfer protein. N Engl J Med. 1995;333:1313–1318. doi: 10.1056/NEJM199511163332003. [DOI] [PubMed] [Google Scholar]
- 9.Ouahchi K, Arita M, Kayden H, Hentati F, Ben Hamida M, Sokol R, Arai H, Inoue K, Mandel JL, Koenig M. Ataxia with isolated vitamin E deficiency is caused by mutations in the alpha-tocopherol transfer protein. Nat Genet. 1995;9:141–145. doi: 10.1038/ng0295-141. [DOI] [PubMed] [Google Scholar]
- 10.Kayden HJ. Tocopherol content of adipose tissue from vitamin E-deficient humans. Ciba Found Symp. 1983;101:70–91. doi: 10.1002/9780470720820.ch6. [DOI] [PubMed] [Google Scholar]
- 11.Guggenheim MA, Ringel SP, Silverman A, Grabert BE, Neville HE. Progressive neuromuscular disease in children with chronic cholestasis and vitamin E deficiency: clinical and muscle biopsy findings and treatment with alpha-tocopherol. Ann NY Acad Sci. 1982;393:84–95. doi: 10.1111/j.1749-6632.1982.tb31235.x. [DOI] [PubMed] [Google Scholar]
- 12.Gohil K, Schock BC, Chakraborty AA, Terasawa Y, Raber J, Farese RV, Jr, Packer L, Cross CE, Traber MG. Gene expression profile of oxidant stress and neurodegeneration in transgenic mice deficient in alpha-tocopherol transfer protein. Free Radic Biol Med. 2003;35:1343–1354. doi: 10.1016/s0891-5849(03)00509-4. [DOI] [PubMed] [Google Scholar]
- 13.Terasawa Y, Ladha Z, Leonard SW, Morrow JD, Newland D, Sanan D, Packer L, Traber MG, Farese RV., Jr Increased atherosclerosis in hyperlipidemic mice deficient in alpha-tocopherol transfer protein and vitamin E. Proc Natl Acad Sci USA. 2000;97:13830–13834. doi: 10.1073/pnas.240462697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yokota T, Igarashi K, Uchihara T, Jishage K, Tomita H, Inaba A, Li Y, Arita M, Suzuki H, Mizusawa H, Arai H. Delayed-onset ataxia in mice lacking alpha-tocopherol transfer protein: model for neuronal degeneration caused by chronic oxidative stress. Proc Natl Acad Sci USA. 2001;98:15185–15190. doi: 10.1073/pnas.261456098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuddihy SL, Ali SS, Musiek ES, Lucero J, Kopp SJ, Morrow JD, Dugan LL. Prolonged alpha-tocopherol deficiency decreases oxidative stress and unmasks alpha-tocopherol-dependent regulation of mitochondrial function in the brain. J Biol Chem. 2008;283:6915–6924. doi: 10.1074/jbc.M702572200. [DOI] [PubMed] [Google Scholar]
- 16.Nishida Y, Yokota T, Takahashi T, Uchihara T, Jishage K, Mizusawa H. Deletion of vitamin E enhances phenotype of Alzheimer disease model mouse. Biochem Biophys Res Commun. 2006;350:530–536. doi: 10.1016/j.bbrc.2006.09.083. [DOI] [PubMed] [Google Scholar]
- 17.Ren YR, Nishida Y, Yoshimi K, Yasuda T, Jishage K, Uchihara T, Yokota T, Mizuno Y, Mochizuki H. Genetic vitamin E deficiency does not affect MPTP susceptibility in the mouse brain. J Neurochem. 2006;98:1810–1816. doi: 10.1111/j.1471-4159.2006.03994.x. [DOI] [PubMed] [Google Scholar]
- 18.Gohil K, Godzdanker R, O'Roark E, Schock BC, Kaini RR, Packer L, Cross CE, Traber MG. Alpha-tocopherol transfer protein deficiency in mice causes multi-organ deregulation of gene networks and behavioral deficits with age. Ann NY Acad Sci. 2004;1031:109–126. doi: 10.1196/annals.1331.012. [DOI] [PubMed] [Google Scholar]
- 19.Leonard SW, Terasawa Y, Farese RV, Jr, Traber MG. Incorporation of deuterated RRR- or all-rac-alpha-tocopherol in plasma and tissues of alpha-tocopherol transfer protein–null mice. Am J Clin Nutr. 2002;75:555–560. doi: 10.1093/ajcn/75.3.555. [DOI] [PubMed] [Google Scholar]
- 20.Thomas PK, Cooper JM, King RH, Workman JM, Schapira AH, Goss-Sampson MA, Muller DP. Myopathy in vitamin E deficient rats: muscle fibre necrosis associated with disturbances of mitochondrial function. J Anat. 1993;183:451–461. [PMC free article] [PubMed] [Google Scholar]
- 21.Nier B, Weinberg PD, Rimbach G, Stocklin E, Barella L. Differential gene expression in skeletal muscle of rats with vitamin E deficiency. IUBMB Life. 2006;58:540–548. doi: 10.1080/15216540600871100. [DOI] [PubMed] [Google Scholar]
- 22.Gohil K, Oommen S, Vasu VT, Aung HH, Cross CE. Tocopherol transfer protein deficiency modifies nuclear receptor transcriptional networks in lungs: modulation by cigarette smoke in vivo. Mol Aspects Med. 2007;28:453–480. doi: 10.1016/j.mam.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 23.Oommen S, Vasu VT, Leonard SW, Traber MG, Cross CE, Gohil K. Genome wide responses of murine lungs to dietary alpha-tocopherol. Free Radic Res. 2007;41:98–109. doi: 10.1080/10715760600935567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vasu VT, Hobson B, Gohil K, Cross CE. Genome-wide screening of alpha-tocopherol sensitive genes in heart tissue from alpha-tocopherol transfer protein null mice (ATTP(-/-)) FEBS Lett. 2007;581:1572–1578. doi: 10.1016/j.febslet.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Babu GJ, Bhupathy P, Carnes CA, Billman GE, Periasamy M. Differential expression of sarcolipin protein during muscle development and cardiac pathophysiology. J Mol Cell Cardiol. 2007;43:215–222. doi: 10.1016/j.yjmcc.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhupathy P, Babu GJ, Periasamy M. Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J Mol Cell Cardiol. 2007;42:903–911. doi: 10.1016/j.yjmcc.2007.03.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reyes-Juarez JL, Juarez-Rubi R, Rodriguez G, Zarain-Herzberg A. Transcriptional analysis of the human cardiac calsequestrin gene in cardiac and skeletal myocytes. J Biol Chem. 2007;282:35554–35563. doi: 10.1074/jbc.M707788200. [DOI] [PubMed] [Google Scholar]
- 29.Galinska-Rakoczy A, Engel P, Xu C, Jung H, Craig R, Tobacman LS, Lehman W. Structural basis for the regulation of muscle contraction by troponin and tropomyosin. J Mol Biol. 2008;379:929–935. doi: 10.1016/j.jmb.2008.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bechet D, Tassa A, Taillandier D, Combaret L, Attaix D. Lysosomal proteolysis in skeletal muscle. Int J Biochem Cell Biol. 2005;37:2098–2114. doi: 10.1016/j.biocel.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 31.Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, Laplante A, Princiotta MF, Thibault P, Sacks D, Desjardins M. Phagosomes are competent organelles for antigen cross-presentation. Nature. 2003;425:402–406. doi: 10.1038/nature01912. [DOI] [PubMed] [Google Scholar]
- 32.Loureiro J, Ploegh HL. Antigen presentation and the ubiquitin-proteasome system in host-pathogen interactions. Adv Immunol. 2006;92:225–305. doi: 10.1016/S0065-2776(06)92006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim E, Tolhurst AT, Qin LY, Chen XY, Febbraio M, Cho S. CD36/fatty acid translocase, an inflammatory mediator, is involved in hyperlipidemia-induced exacerbation in ischemic brain injury. J Neurosci. 2008;28:4661–4670. doi: 10.1523/JNEUROSCI.0982-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teupser D, Mueller MA, Koglin J, Wilfert W, Ernst J, von Scheidt W, Steinbeck G, Seidel D, Thiery J. CD36 mRNA expression is increased in CD14+ monocytes of patients with coronary heart disease. Clin Exp Pharmacol Physiol. 2008;35:552–556. doi: 10.1111/j.1440-1681.2007.04836.x. [DOI] [PubMed] [Google Scholar]
- 35.Goasduff T, Cederbaum AI. NADPH-dependent microsomal electron transfer increases degradation of CYP2E1 by the proteasome complex: role of reactive oxygen species. Arch Biochem Biophys. 1999;370:258–270. doi: 10.1006/abbi.1999.1399. [DOI] [PubMed] [Google Scholar]
- 36.Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beard NA, Wei L, Cheung SN, Kimura T, Varsanyi M, Dulhunty AF. Phosphorylation of skeletal muscle calsequestrin enhances its Ca(2+) binding capacity and promotes its association with junctin. Cell Calcium. 2008;44:363–373. doi: 10.1016/j.ceca.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 38.Ottenheijm CA, Fong C, Vangheluwe P, Wuytack F, Babu GJ, Periasamy M, Witt CC, Labeit S, Granzier H. Sarcoplasmic reticulum calcium uptake and speed of relaxation are depressed in nebulin-free skeletal muscle. Faseb J. 2008;22:2912–2919. doi: 10.1096/fj.07-104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goss-Sampson MA, Kriss A, Muddle JR, Thomas PK, Muller DP. Lumbar and cortical somatosensory evoked potentials in rats with vitamin E deficiency. J Neurol Neurosurg Psychiatry. 1988;51:432–435. doi: 10.1136/jnnp.51.3.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tupling AR, Asahi M, MacLennan DH. Sarcolipin over-expression in rat slow twitch muscle inhibits sarcoplasmic reticulum Ca2+ uptake and impairs contractile function. J Biol Chem. 2002;277:44740–44746. doi: 10.1074/jbc.M206171200. [DOI] [PubMed] [Google Scholar]
- 41.Vangheluwe P, Schuermans M, Zador E, Waelkens E, Raeymaekers L, Wuytack F. Sarcolipin and phospholamban mRNA and protein expression in cardiac and skeletal muscle of different species. Biochem J. 2005;389:151–159. doi: 10.1042/BJ20050068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacLennan DH, Yip CC, Iles GH, Seeman P. Isolation of sarcoplasmic reticulum proteins. Cold Spring Harbor Symp Quant Biol. 1972;37:469–478. [Google Scholar]
- 43.Wawrzynow A, Theibert JL, Murphy C, Jona I, Martonosi A, Collins JH. Sarcolipin, the ‘proteolipid’ of skeletal muscle sarcoplasmic reticulum, is a unique, amphipathic, 31-residue peptide. Arch Biochem Biophys. 1992;298:620–623. doi: 10.1016/0003-9861(92)90457-8. [DOI] [PubMed] [Google Scholar]
- 44.Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 46.Sato Y, Ferguson DG, Sako H, Dorn GW, 2nd, Kadambi VJ, Yatani A, Hoit BD, Walsh RA, Kranias EG. Cardiac-specific overexpression of mouse cardiac calsequestrin is associated with depressed cardiovascular function and hypertrophy in transgenic mice. J Biol Chem. 1998;273:28470–28477. doi: 10.1074/jbc.273.43.28470. [DOI] [PubMed] [Google Scholar]
- 47.Nell S, Bahtz R, Bossecker A, Kipp A, Landes N, Bumke-Vogt C, Halligan E, Lunec J, Brigelius-Flohe R. PCR-verified microarray analysis and functional in vitro studies indicate a role of alpha-tocopherol in vesicular transport. Free Radic Res. 2007;41:930–942. doi: 10.1080/10715760701416988. [DOI] [PubMed] [Google Scholar]
- 48.Hyland S, Muller D, Hayton S, Stoecklin E, Barella L. Cortical gene expression in the vitamin E-deficient rat: possible mechanisms for the electrophysiological abnormalities of visual and neural function. Ann Nutr Metab. 2006;50:433–441. doi: 10.1159/000094635. [DOI] [PubMed] [Google Scholar]
- 49.Hayton SM, Kriss T, Wade A, Muller DP. Effects on neural function of repleting vitamin E-deficient rats with alphatocopherol. J Neurophysiol. 2006;95:2553–2559. doi: 10.1152/jn.00842.2005. [DOI] [PubMed] [Google Scholar]
- 50.Sokol RJ, Bove KE, Heubi JE, Iannaccone ST. Vitamin E deficiency during chronic childhood cholestasis: presence of sural nerve lesion prior to 2 1/2 years of age. J Pediatr. 1983;103:197–204. doi: 10.1016/s0022-3476(83)80344-8. [DOI] [PubMed] [Google Scholar]
- 51.Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA. CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem. 1993;268:11811–11816. [PubMed] [Google Scholar]
- 52.Luangrath V, Brodeur MR, Rhainds D, Brissette L. Mouse CD36 has opposite effects on LDL and oxidized LDL metabolism in vivo. Arterioscler Thromb Vasc Biol. 2008;28:1290–1295. doi: 10.1161/ATVBAHA.107.161653. [DOI] [PubMed] [Google Scholar]
- 53.Hazen SL. Oxidized phospholipids as endogenous pattern recognition ligands in innate immunity. J Biol Chem. 2008;283:15527–15531. doi: 10.1074/jbc.R700054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haslett JN, Kunkel LM. Microarray analysis of normal and dystrophic skeletal muscle. Int J Dev Neurosci. 2002;20:359–365. doi: 10.1016/s0736-5748(02)00041-2. [DOI] [PubMed] [Google Scholar]
- 55.Zhou X, Dimachkie MM, Xiong M, Tan FK, Arnett FC. cDNA microarrays reveal distinct gene expression clusters in idiopathic inflammatory myopathies. Med Sci Monit. 2004;10:BR191–BR197. [PubMed] [Google Scholar]
- 56.Atkinson J, Epand RF, Epand RM. Tocopherols and tocotrienols in membranes: a critical review. Free Radic Biol Med. 2008;44:739–764. doi: 10.1016/j.freeradbiomed.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 57.Hunt AN. Completing the cycles; the dynamics of endonuclear lipidomics. Biochim Biophys Acta. 2006;1761:577–587. doi: 10.1016/j.bbalip.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 58.Albi E, Lazzarini R, Viola Magni M. Phosphatidylcholine/sphingomyelin metabolism crosstalk inside the nucleus. Biochem J. 2008;410:381–389. doi: 10.1042/BJ20070758. [DOI] [PubMed] [Google Scholar]
- 59.Caro AA, Cederbaum AI. Role of intracellular calcium and phospholipase A2 in arachidonic acid-induced toxicity in liver cells overexpressing CYP2E1. Arch Biochem Biophys. 2007;457:252–263. doi: 10.1016/j.abb.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gorlach A, Bonello S. The cross-talk between NF-kappaB and HIF-1: further evidence for a significant liaison. Biochem J. 2008;412:e17–e19. doi: 10.1042/BJ20080920. [DOI] [PubMed] [Google Scholar]
- 61.Ji LL. Modulation of skeletal muscle antioxidant defense by exercise: role of redox signaling. Free Radic Biol Med. 2008;44:142–152. doi: 10.1016/j.freeradbiomed.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 62.Chen XL, Dodd G, Thomas S, Zhang X, Wasserman MA, Rovin BH, Kunsch C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am J Physiol Heart Circ Physiol. 2006;290:H1862–H1870. doi: 10.1152/ajpheart.00651.2005. [DOI] [PubMed] [Google Scholar]