

Abstract
Recent studies indicate that chromatin regulatory complexes produce biological specificity in the way that letters produce meanings by combinations into words. Combinatorial assembly of chromatin regulatory complexes may be critical for maximizing the information content provided by arrays of histone modifications.
The DNA within each mammalian cell is compacted about 5000-fold into chromatin with hierarchical levels of complexity, the simplest being a nucleosome with about 7-fold compaction. This compaction is controlled by at least three mechanisms: DNA methylation, histone modifications, and the ATP-dependent chromatin remodeling complexes. The latter contain SWI2/SNF2-like ATPases and regulate the presence and position of nucleosomes on DNA, enabling the binding of transcription factors to nucleosomes and the facilitation of DNA recombination, repair, and viral integration (Cairns, 2007).
The vertebrate genome contains ∼30 genes encoding proteins similar to the yeast SWI2/SNF2 ATPases, which are essential for mating type switching and nutrient responses in yeast (Neigeborn and Carlson, 1984; Stern et al., 1984). The diversity of this ATPase family in vertebrates suggests that the function of this class of proteins is both broader and yet more specific than anticipated. Compounding this potential diversity is the observation that these ATPases are generally present in complexes with 4 to 12 other protein subunits. In vertebrates, these subunits are often encoded by gene families, indicating that the diversity of these complexes might be further enhanced by combinatorial assembly. Characterized complexes include those containing the SNF2L, SNF2H, Mi-2α, Mi-2β, Brg, and Brm ATPases (Figure 1).
Figure 1. Combinatorial Assembly of Chromatin Regulatory Complexes.
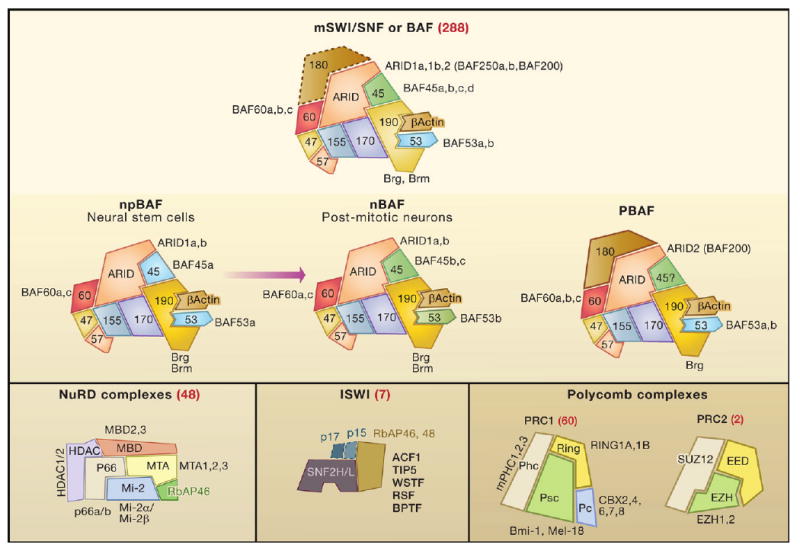
Shown is the predicted combinatorial diversity for the mammalian chromatin regulatory complexes: BAF (mSWI/SNF), NuRD, ISWI, and Polycomb (the number of possible combinations is shown in parentheses in red).
(Top) Three examples of BAF complexes illustrate respelling of the chromatin remodeling word by switching subunit composition. The subunits are depicted as interlocking pieces in which a similar shape of the subunit denotes homology and thereby a specific position in the complex. Subunits shown in dashed outline are inconstant components of the complexes. The positions of the proteins in the complexes are arbitrary 2D projections, except for actin and BAF53, which contact the catalytic domain of Brg (Zhao et al., 1998). The depicted area of each subunit is roughly proportional to its mass.
The mSWI/SNF or BAF complexes of vertebrates illustrate the features of combinatorial assembly and prompt an analogy: the subunits of these complexes are like letters and can be assembled in different combinations to form new complexes (words). Although initially thought to be analogous to yeast SWI/SNF complexes, recent genome-wide studies have found that vertebrate mSWI/SNF or BAF complexes most commonly repress their target genes, whereas the yeast SWI/SNF complex has only been found to activate its targets. These mechanistic differences likely derive from differences in subunit composition (Lessard et al., 2007: Cairns, 2007; Zhao et al., 1998) (Table S1 and Figure S1 available online). The subunits appear to have been shuffled between complexes, with some subunits lost and new ones incorporated, at the evolutionary origin of multicellular organisms. This remodeling may have occurred in response to the appearance of linking histones and the need for extensive cell type specialization.
Biochemical studies have defined certain rules for combinatorial assembly of the vertebrate mSWI/SNF or BAF family of complexes. First, 20 genes encode the 11 subunits of these complexes (Table 1) giving a total of 288 predicted assemblies. Second, studies using antibodies specific for the products of genes encoding different subunit family members revealed that these subunits are freely combined with other subunits in cultured cells (Wang et al., 1996; Zhao et al., 1998). Third, the 11 subunits resist dissociation even under near-denaturing conditions. Importantly, challenge with in vitro synthesized subunits indicated that these subunits are not exchangeable (Zhao et al., 1998), suggesting that the complexes may form stable, conformationally specific assemblies, which would be critical for diversifying their functions. Thus, BAF complexes can be viewed as an 11-letter chromatin remodeling word with 288 possible spellings (Figure 1). During evolution, expansion of the gene families encoding the different subunits first appeared in the genomes of organisms that shared a common ancestor about 500 million years ago, the dawn of vertebrate life (Figure S1). Thus, combinatorial assembly of these complexes, along with combinatorial use of transcription factors (Levine and Tjian, 2003), may have contributed to the diversification of gene function in vertebrates by endowing a common set of genes with different expression patterns.
Table 1. Subunits of Combinatorially Assembled Vertebrate Chromatin Remodeling Complexes.
| Complex | Subunits | Domains | |
|---|---|---|---|
| mSWI/SNF (BAF) | Brg, Brm | ATPase, Bromodomain, HSA, BRK | |
| BAF170 | Chromo-related domain, SWIRM, SANT, Leu-zipper | ||
| BAF155 | Chromo-related domain, SWIRM, SANT, Leu-zipper | ||
| BAF57 | HMG, coiled-coil | ||
| BAF47/SNF5/Ini | SNF5 domain | ||
| BAF60a, b, c | SWIB/MDM2 domain | ||
| β-actin | actin | ||
| BAF45d | PHD, Krüppel, N-terminal | ||
| npBAF-specific | BAF53a | actin-related protein | |
| npBAF-specific | BAF45a,d | PHD, Krüppel | |
| nBAF-specific | BAF53b | actin-related protein | |
| nBAF-specific | BAF45b, c | PHD, Krüppel, N-terminal | |
| BAF-specific | BAF250a, b | ARID | |
| PBAF-specific | BAF200/ARID2 | ARID, RFX, Zn finger | |
| PBAF-specific | BAF180 | Bromodomain (6), BAH, HMG | |
| NuRD/Mi-2 | Mi-2α, β | ATPase, Chromodomain, PHD | |
| MTA1, 2, 3 | BAH, ELM, SANT, Zn finger | ||
| HDAC1, HDAC2 | HDAC domain | ||
| RbAP46, 48 | WD40 | ||
| MBD2, MBD3 | MBD | ||
| P66a, p66b | Zn finger | ||
| ISWI | NURF | SNF2L | ATPase, HAND, SANT, SLIDE |
| BPTF | PHD, Bromodomain, HMG, WAC, WAKZ | ||
| RbAP46/48 | WD40 | ||
| ACF/WCRF | SNF2H | ATPase, HAND, SANT, SLIDE | |
| ACF1/WCRF180 | PHD, Bromodomain, WAC, LH, BAZ1, BAZ2, WAKZ | ||
| CHRAC | SNF2H | ATPase, HAND, SANT, SLIDE | |
| ACF1/WCRF180 | PHD, Bromodomain, WAC, LH, BAZ1, BAZ2, WAKZ | ||
| CHRAC-15, 17 | Histone-fold domain | ||
| WICH | SNF2H | ATPase, HAND, SANT, SLIDE | |
| WSTF | PHD, Bromodomain, WAC, LH, BAZ1, BAZ2, WAKZ | ||
| RSF | SNF2H | ATPase, HAND, SANT, SLIDE | |
| RSF-1/p325 | PHD | ||
| NoRC | SNF2H | ATPase, HAND, SANT, SLIDE | |
| TIP5 | PHD, Bromodomain, MBD, HMG, LH, BAZ1, BAZ2, WAKZ | ||
| SNF2H/cohesin | SNF2H | ATPase, HAND, SANT, SLIDE | |
| NuRD (Mi-2, HDAC1/2, MBD2/3, MTA1/2, RbAp46/48) | |||
| Cohesin (hRAD21, hSMC1/2, SA1/SA2) | |||
| PRC1 | CBX2/HPC1, CBX4/HPC2, CBX6, CBX7, CBX8/HPC3 | Chromodomain, AT-hook | |
| PHC1, 2, 3 | FCS Zn finger, SAM domain | ||
| RING1A, RING1B | RING finger | ||
| Bmi1, Mel18, MBLR, NSPc1 | RING finger | ||
| PRC2 | EZH1, EZH2 | SET, SANT | |
| EED | WD40 | ||
| SUZ12 | Zn finger | ||
Domains with the ability to interact with histones are shown in bold.
Chromatin Remodeling Words: Misspelling and Respelling
An appropriate test of the analogy to the assembly of words is to examine the biological consequences of different “spellings” or “misspellings” (i.e., substitution of one letter or subunit for another by genetic or biochemical manipulation) that might create new meanings. Vertebrate SWI/SNF complexes contain two alternative ATPases, Brg or Brm, but not both (Figure 1). A growth suppressor or tumor suppressor role for Brg and Brm was first proposed from studies of cell lines that lack both the Brg and Brm subunits (Dunaief et al., 1994). Introduction of Brg slowed growth of the cell lines and induced differentiation, whereas Brm had little effect. Brg mutant mice die on embryonic day 3 due to growth arrest of the inner cell mass and trophoblast (Bultman et al., 2000), whereas Brm-deficient mice are slightly larger than normal but otherwise viable (Reyes et al., 1998). Brg is one of the few known genes required for zygotic gene activation, a process that is not affected by the absence of Brm (Bultman et al., 2006). In addition, Brg but not Brm plays an essential and specific role in the self-renewal of embryonic stem (ES) cells (Bultman et al., 2000) and neural stem cells (Lessard et al., 2007), lymphocyte development (Chi et al., 2002), and limb morphogenesis (Indra et al., 2005). These genetic studies suggested that the functions of these two ATPases in BAF complexes are different but left open the possibility that this was simply due to patterns of expression.
In vitro biochemical studies also suggested that the Brg- and Brm-based ATPases have different activities. Brg and Brm preferentially interact with different classes of transcription factors through their unique N-terminal domains (Kadam and Emerson, 2003). Brg binds to zinc finger proteins such as KLF and GATA family transcription regulators, whereas Brm interacts with two ankyrin repeat proteins involved in Notch signaling (however, Brm mutants have no defect in Notch signaling). Thus, these studies also implicate distinct roles for complexes containing Brg and Brm. However, a rigorous test of the biological specificity of the ATPase subunit by expressing Brg or Brm in a mouse with the opposite mutant background has not been reported.
The 60 kDa (BAF60 family) subunit is encoded by three genes, but only one subunit type is present per complex (Figure 1) (Wang et al., 1996). BAF60c is expressed selectively in the embryonic heart, and downregulation of BAF60c by RNA interference in the early mouse embryo resulted in defective heart development and death at embryonic days 10–11 (Lickert et al., 2004). Transgenic expression of the BAF60b protein repressed the heart defect suggesting that “misspelling” of the chromatin remodeling word did not change the original biological meaning. However, the small amount of tissue available and lack of subunit-specific antibodies made it impossible to examine the degree of expression of the protein in the proper site or its incorporation into complexes, leaving the authors to speculate that the functions of the BAF60b and BAF60c subunits are interchangeable.
A good example of how nature respells chromatin remodeling words is provided by the PBAF complex (Figure 1). Using a nuclear receptor-dependent in vitro transcription assay, Tjian and colleagues purified chromatin remodeling cofactors from cultured cells that supported transcription in response to ligand-activated nuclear receptors. Remarkably, the responsible complex contained Brg selectively paired with BAF180/Polybromo (Lemon et al., 2001). In these PBAF complexes, BAF200 (ARID2) replaces BAF250a (ARID1a) and BAF250b (ARID1b), which also contain ARID domains (Figures 1 and S1) (Yan et al., 2005). PBAF complexes activated vitamin D receptor-dependent transcription in response to vitamin D, whereas BAF complexes containing BAF250 but not BAF200 or BAF180 failed to do so (Lemon et al., 2001). BAF180 contains six Bromodomains that bind to acetylated histone, and so this subunit could help to target the complex. Mice lacking BAF180 have defects in heart development that are consistent with a role in responses to retinoic acid (Wang et al., 2004b), but earlier retinoic acid-dependent processes do not appear to be affected. Most BAF180 in nontransformed cells resides in larger complexes lacking Brg (Lessard et al., 2007), making interpretation of the genetic data less clear. The combinatorial compositions of the complexes in the affected heart field remain undefined. PBAP, the Drosophila homolog of PBAF, shares most subunits with BAF complexes but also contains Polybromo, BAP170 (an ARID2 homolog), and a protein similar to BAF45a (SAYP) (Figure S1) (Chalkley et al., 2008). Although flies lacking either Polybromo or BAP170 are viable, double mutant flies lacking both PBAP subunits have defects in metamorphosis and fail to activate genes involved in innate immunity but surprisingly have normal responses to the insect nuclear hormone ecdysone (Carrera et al., 2008). As this phenotype is distinct from those caused by mutations in BAP complex subunits, it is likely that the PBAP combination of subunits has a distinct function.
Recent studies suggest that combinatorial assembly may underlie different aspects of pluripotency. Mutation of three subunits of BAF complexes–Brg, BAF155, and BAF47–severely compromises the survival of the inner cell mass of the mammalian embryo (Bultman et al., 2000; Guidi et al., 2001; Kim et al., 2001; Klochendler-Yeivin et al., 2000), suggesting an essential role of BAF complexes in the self-renewal and differentiation of pluripotent ES cells. This phenotype appears specific to pluripotent cells, as Brg is not essential for proliferation of fibroblasts or glia but is essential for the multipotency of neural stem cells (Bultman et al., 2000; Lessard et al., 2007). Remarkably, mouse embryos lacking BAF250a (ARID1a) form the inner cell mass but do not gastrulate or form mesoderm. ES cells derived from these embryos have severely compromised pluripotency and defective self-renewal (Gao et al., 2008). In contrast, ES cells lacking BAF250b show a mild reduction in proliferation, decreased expression of pluripotency genes, and more rapid differentiation (Yan et al., 2008). Thus, different aspects of ES cell function may require complexes with a different “letter” at the BAF250 position, although rescue of null mutations with the homologous proteins will be necessary to justify this conclusion.
In studies of growth regulatory chromatin remodeling complexes, it is often forgotten that mutation of complex components may result in a growth advantage leading to the predominance of clones lacking one or more subunits of a specific complex. For example, multiple human tumor cell lines such as SW13 cells lack Brg and/or Brm (Dunaief et al., 1994; Wong et al., 2000), malignant rhabdoid tumors lack BAF47 (Versteege et al., 1998), and other cell lines lack BAF57 and BAF155. HeLa tumor cells lack the BAF45 family and have only about 30,000 complexes per cell, whereas nontransformed cells have about 10 times this number (Lemon et al., 2001; Lessard et al., 2007). Thus, the study of nontransformed cells is essential to understanding the biochemical and functional diversity of this family of chromatin remodeling complexes.
Studies of BAF complexes in the developing nervous system have defined an essential switch in subunits at mitotic exit that constitutes a natural respelling of the chromatin remodeling word. Mitotic exit in the nervous system marks the end of multipotency. The transition from neural stem cells to neurons occurs with anatomical precision in the embryo making this a convenient system to study the switch between multipotency and committed fate. The first purification and proteomic analysis of complexes from nontransformed vertebrate cells showed that multipotent neural stem cells and progenitors have complexes containing BAF53a and BAF45a but not BAF60b (npBAF in Figure 1). Complexes in postmitotic neurons (nBAF) have BAF53b, BAF45b but not BAF60b (Lessard et al., 2007; Wu et al., 2007). Both complexes appear to be polymorphic at positions occupied by other subunits. Subunit exchange occurs at mitotic exit probably within one cell division, and the complexes of progenitor cells and neurons appear to be mutually exclusive indicating that respelling is precise (Lessard et al., 2007). These findings set the stage for a definitive test of whether different complexes have different functions.
The npBAF complexes appear to be both necessary and sufficient for neural stem cell self-renewal (Lessard et al., 2007), whereas nBAF complexes are essential for dendritic and axonal morphogenesis (Wu et al., 2007). These studies suggest that these two families of complexes function differently, but could the functional specificity be due to their different expression patterns? A more rigorous test was provided by mice lacking BAF53b, a dedicated subunit of postmitotic neuron-specific nBAF complexes (Wu et al., 2007). BAF53b is exclusively expressed in neurons, and null mice die perinatally with a failure to nurse, reduced neurite outgrowth, and reduced synapse formation. The latter defect is probably due to the fact that nBAF complexes directly bind and regulate cytoskeleton genes necessary for dendritic morphogenesis. The lethal defects in BAF53b null mice were rescued by BAF53b but not by BAF53a (Wu et al., 2007). These studies demonstrate that complexes containing BAF53a and BAF53b have distinct functions that are not simply a product of their pattern of expression. Thus, combinatorial assembly, at least at this position in BAF complexes, produces functional diversity.
Words of Other Chromatin Regulatory Complexes
The gene families encoding the subunits of other chromatin remodeling and modifying complexes suggest that combinatorial assembly might also dictate specificity in these complexes (Table 1; Figure 1). The NuRD/Mi-2 complexes can be viewed as a chromatin remodeling word with six letters containing both a SNF2-like ATPase and a histone deacetylase (Xue et al., 1998). All six subunits are encoded by gene families in vertebrates giving 48 possible complexes (Bowen et al., 2004). Although genetic experiments in which a null mutation in one family member is substituted with another (misspelling) have not been reported, substantial biochemical evidence indicates that at least some of the complexes have different functions.
The ISWI family of chromatin remodelers appear to be two- to four-letter chromatin remodeling words based on the alternative ATPases, SNF2L and SNF2H (Figure 1), the mammalian homologs of the Drosophila ISWI ATPase (Eberharter and Becker, 2004). Not only do they differ in their expression pattern, but they assemble into at least seven distinct complexes. SNF2L is a component of the NURF complex, interacting with BPTF and RbpAp46/48 (Figure 1; Table 1). BPTF contains two PHD domains, which likely bring the chromatin remodeling activity of these complexes to genetic loci with specific histone modifications (Wysocka et al., 2006). The closely related protein, SNF2H, is found in at least six complexes (hACF, hCHRAC, hWICH, RSF, NoRC, and SNF2H/cohesin; Figure 1; Table 1) (Eberharter and Becker, 2004). Interestingly, three protein partners of SNF2H in these complexes belong to the BAZ/WAL family (ACF1 in hACF and hCHRAC, WSTF in WICH, TIP5 in NoRC) (Jones et al., 2000). In addition, the NURF component BPTF also shares WAC, WAKZ, PHD, and Bromodomains with these BAZ/WAL family proteins. Thus, combinatorial assembly of SNF2L/2H proteins with a family of homologous subunits appears to diversify ISWI complexes structurally (Eberharter et al., 2001; Hamiche et al., 1999; Ito et al., 1999; Langst et al., 1999). Genetic and biochemical analyses indicate that ISWI complexes play important roles in transcriptional regulation, heterochromatin replication, chromatin assembly, and chromatin higher-order structure. For example, TIP5 in the NoRC complex localizes in the nucleolus and mediates transcriptional silencing of ribosomal RNA (rRNA) (Strohner et al., 2001). WSTF, but not the closely related ACF1, associates with mitotic chromosomes and maintains chromatin structures during DNA replication (Bozhenok et al., 2002; Poot et al., 2004). In contrast, ACF1 containing complexes function in chromatin formation and assembly (Eberharter et al., 2001; Fyodorov et al., 2004).
Polycomb Group (PcG) complexes modify histones and are key players in development (Schuettengruber et al., 2007). Two types of PcG complexes (PRC1 and PRC2) with different histone modification activities maintain developmental regulatory genes in a silenced state. Vertebrate PRC1-like complexes can be considered as four-letter words with at least 60 predicted combinations (Whitcomb et al., 2007). The four core subunits (PHC, CBX, Bmi1, and RING1) (Table 1) are homologs of Drosophila Ph, Pc, Psc, and dRing, respectively (Figure 1). For example, in mammals, there are at least four paralogs of the Drosophila RING finger protein Psc–Bmi1, Mel18, MBLR, and NSPc1–that interact with other PcG proteins to form a set of distinct but related complexes. For example, NSPc1 together with Ring1B and other PRC1 subunits is a component of a BCOR corepressor complex (Gearhart et al., 2006), whereas MBLR together with Ring1B is implicated in the E2F6 complex (Ogawa et al., 2002).
Genetic studies suggest that the mammalian orthologs of Psc may have overlapping and distinct functions. For example, Bmi1 and Mel18 share 63% amino acid identity, but only Bmi1 is required for maintaining hematopoietic and neural stem cells postnatally (Lessard and Sauvageau, 2003; Molofsky et al., 2003). Disruption of Bmi1 or Mel18 in mice results in posterior transformation of the axial skeleton, but their homeotic phenotypes are clearly distinct (Akasaka et al., 1996; van der Lugt et al., 1994). The nonoverlapping functions of Mel18 and Bmi1 may reflect the fact that they participate in distinct PcG complexes (Elderkin et al., 2007).
How many functions do PRC1 family members have and do specific complexes carry them out? One of the functions of PRC1-like complexes is to monoubiquitinate chromatin on histone H2A at lysine residue 119. Although there are four Ring finger domains in PRC1, only the Ring fingers of the Ring1B and Ring1A subunits perform this function (Buchwald et al., 2006; Wang et al., 2004a). Biochemical data also suggest that the Chromodomain-containing CBX family proteins (homologs of Drosophila Pc) bind differentially to trimethylated H3K27 and H3K9 and may target PRC1 complexes to different loci (Bernstein et al., 2006). But whether histone ubiquitination follows differential targeting of PRC1-like complexes to distinct loci by different Pc or Psc paralogs remains to be shown.
PRC2 complexes contain EED, SUZ12, and either EZH1 or EZH2. Hence, they can be viewed as three-letter words with only two possible combinations. The catalytic subunit EZH2 places the dimethyl and trimethyl mark on H3K27 and is thought to be required for PRC1 action. Recent studies suggest that EZH1- and EZH2-based complexes have distinct and overlapping functions (Shen et al., 2008). Although EED is encoded by one gene, four proteins are produced by alternative translational starts possibly adding to the diversity. (Alternative splice forms have not been included in the calculations of diversity in Figure 1, but they could contribute to diversity as indicated with different translated EED gene products.) These complexes appear to have different in vitro specificity toward H3K27 and H1K26 (Kuzmichev et al., 2004). Although both EED and EZH2 are essential for di- and trimethylation of H3K27 in ES cells, they are surprisingly not required to generate pluripotent ES cells or for their self-renewal (Shen et al., 2008). The lack of an effect of EZH2 mutation on pluripotency is not due to compensation by EZH1 because removal of EED, which produces an even more severe defect in H3K27 methylation, does not compromise pluripotency but does produce severe defects in later stages of embryonic development (Chamberlain et al., 2008). Hence, conditional alleles will be essential to understanding the unique contributions of EZH1- and EZH2-based complexes.
Chromatin Remodeling Words and Histone Codes
Most of these findings support the contention that combinatorial assembly of chromatin regulatory complexes underlies at least part of their biological specificity. The distinct complexes are not distinguished purely by a single subunit but rather by a combination of subunits, some of which, like BAF45b and BAF53b, occur together. Thus, similar to words such as “quiescence” and “queasiness,” different meanings are produced by combinations, yet some combinations, like “qu,” seem invariant. How might specificity arise at a molecular level?
The current model for the function of ATP-dependent remodeling complexes is that a core ATPase is necessary for the mobility of nucleosomes (Narlikar et al., 2002) and the other subunits interact with transcription factors that recruit the complexes to their sites of action. Thus, one possibility is that the different composite surfaces produced by the different combinations of subunits contact specific transcription factors (Figure 2A). This mechanism may apply to PBAF complexes and their interaction with nuclear receptors (Lemon et al., 2001). The composite surfaces produced by the combination of BAF180, ARID2, and Brg with other subunits might provide a docking site for specific transcription factors (nuclear receptors in this case).
Figure 2. Biological Specificity by Combinatorial Assembly.
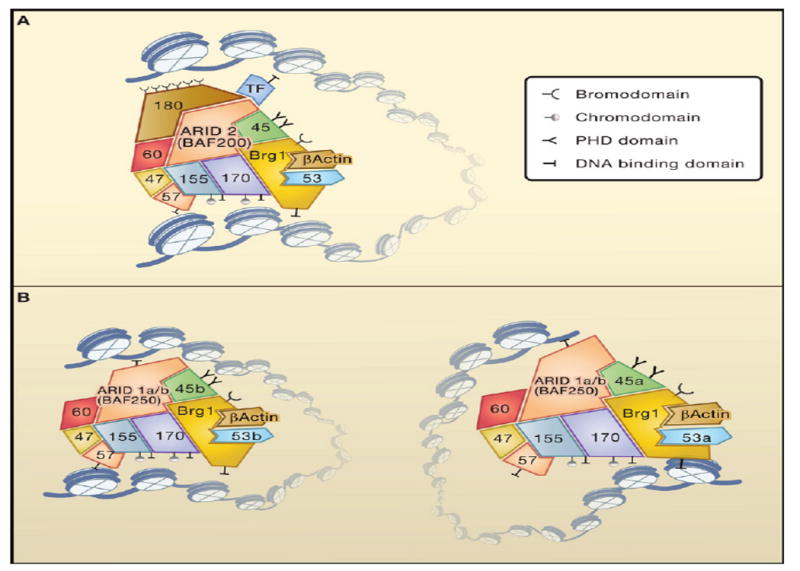
Alternative, but not mutually exclusive, models for generating biological specificity by combinatorial assembly of ATP-dependent chromatin remodeling complexes. (A) Composite surface model in which transcription factors (TF) bind at interfaces of the subunits. (B) Conformational model in which the arrangement of histone modification recognition domains and DNA-binding domains within the complex leads to recognition of specific loci. Brg, BAF155, BAF170, BAF45, and BAF57 have either DNA-binding domains or domains that bind to modified histones and hence could target the complex to specific developmental loci independent of, or in cooperation with, transcription factors. (Not all of the domains are shown, but an exhaustive list appears in Table 1.) BAF-mediated looping of chromatin (suggested by in vitro studies) illustrates the size of the BAF complex and its potential multivalent interactions with chromatin.
A more complex mechanism is suggested by recent studies showing that BAF complexes might be polymorphic readers of histone modifications. These complexes contain two PHD domains in BAF45 (Lessard et al., 2007), seven acetylated histone-binding Bromodomains (six on BAF180 and one on Brg/Brm), and two Chromo-related domains (one in BAF155 and one in BAF170), which might bind to methylated histones (Brehm et al., 2004). In addition, the complexes contain seven DNA-binding domains with relatively little sequence specificity (at least individually). Thus, different combinations of subunits within an essentially nonexchangeable complex could produce conformationally specific combinations allowing recognition of complementary arrays of modified histones (Figure 2B). The need to generate specific three-dimensional arrays of targeting moieties might explain why chromatin remodeling activities are nearly always part of large multisubunit complexes. This line of reasoning also predicts different targets for npBAF and nBAF complexes, which contain different double PHD domain proteins (BAF45a or BAF45b). Indeed, npBAF complexes containing the BAF45a subunit regulate about 400 target genes in neural stem cells with almost no overlap among the 200 target genes regulated by nBAF complexes containing BAF45b in neurons (Lessard et al., 2007; Wu et al., 2007).
The likely possibility that BAF complexes are polymorphic readers of histone modifications predicts that combinatorial assembly of chromatin remodeling complexes operates synergistically with the proposed histone code to produce biological specificity. Reading arrays of histone modifications with combinatorially generated assemblies of Bromodomains, Chromodomains, and PHD domains would allow highly specific targeting of chromatin regulators to matching three-dimensionally specific arrays of modified histones. The large size of BAF complexes (about 12-fold bigger than a nucleosome) should enable reading of multiple histone modifications on adjacent nucleosomes (Figure 2B). Biological specificity generated by this mechanism may be the product of the number of arrays of histone modifications on gene regulatory regions and the number of conformationally specific arrays of recognition domains on the surface of the chromatin regulatory complexes. Future studies on the interdependence of the two mechanisms will be required to test this speculation. However, present evidence strongly supports a combinatorial strategy for generating and reading diverse chromatin landscapes.
Supplementary Material
Footnotes
Supplemental Data: Supplemental Data include one table and one figure and can be found with this article online at http://www.cell.com/supplemental/S0092-8674(09)00015-4.
References
- Akasaka T, Kanno M, Balling R, Mieza MA, Taniguchi M, Koseki H. Development. 1996;122:1513–1522. doi: 10.1242/dev.122.5.1513. [DOI] [PubMed] [Google Scholar]
- Bernstein E, Duncan EM, Masui O, Gil J, Heard E, Allis CD. Mol Cell Biol. 2006;26:2560–2569. doi: 10.1128/MCB.26.7.2560-2569.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen NJ, Fujita N, Kajita M, Wade PA. Biochim Biophys Acta. 2004;1677:52–57. doi: 10.1016/j.bbaexp.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Bozhenok L, Wade PA, Varga-Weisz P. EMBO J. 2002;21:2231–2241. doi: 10.1093/emboj/21.9.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm A, Tufteland KR, Aasland R, Becker PB. Bioessays. 2004;26:133–140. doi: 10.1002/bies.10392. [DOI] [PubMed] [Google Scholar]
- Buchwald G, van der Stoop P, Weichenrieder O, Perrakis A, van Lohuizen M, Sixma TK. EMBO J. 2006;25:2465–2474. doi: 10.1038/sj.emboj.7601144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G, Magnuson T. Mol Cell. 2000;6:1287–1295. doi: 10.1016/s1097-2765(00)00127-1. [DOI] [PubMed] [Google Scholar]
- Bultman SJ, Gebuhr TC, Pan H, Svoboda P, Schultz RM, Magnuson T. Genes Dev. 2006;20:1744–1754. doi: 10.1101/gad.1435106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns BR. Nat Struct Mol Biol. 2007;14:989–996. doi: 10.1038/nsmb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrera I, Zavadil J, Treisman JE. Mol Cell Biol. 2008;17:5238–5250. doi: 10.1128/MCB.00747-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley GE, Moshkin YM, Langenberg K, Bezstarosti K, Blastyak A, Gyurkovics H, Demmers JA, Verrijzer CP. Mol Cell Biol. 2008;28:2920–2929. doi: 10.1128/MCB.02217-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SJ, Yee D, Magnuson T. Stem Cells. 2008;26:1496–1505. doi: 10.1634/stemcells.2008-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi TH, Wan M, Zhao K, Taniuchi I, Chen L, Littman DR, Crabtree GR. Nature. 2002;418:195–199. doi: 10.1038/nature00876. [DOI] [PubMed] [Google Scholar]
- Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K, Luban J, Begemann M, Crabtree GR, Goff SP. Cell. 1994;79:119–130. doi: 10.1016/0092-8674(94)90405-7. [DOI] [PubMed] [Google Scholar]
- Eberharter A, Becker PB. J Cell Sci. 2004;117:3707–3711. doi: 10.1242/jcs.01175. [DOI] [PubMed] [Google Scholar]
- Eberharter A, Ferrari S, Langst G, Straub T, Imhof A, Varga-Weisz P, Wilm M, Becker PB. EMBO J. 2001;20:3781–3788. doi: 10.1093/emboj/20.14.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elderkin S, Maertens GN, Endoh M, Mallery DL, Morrice N, Koseki H, Peters G, Brockdorff N, Hiom K. Mol Cell. 2007;28:107–120. doi: 10.1016/j.molcel.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Fyodorov DV, Blower MD, Karpen GH, Kadonaga JT. Genes Dev. 2004;18:170–183. doi: 10.1101/gad.1139604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Tate P, Hu P, Tjian R, Skarnes WC, Wang Z. Proc Natl Acad Sci USA. 2008;105:6656–6661. doi: 10.1073/pnas.0801802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearhart MD, Corcoran CM, Wamstad JA, Bardwell VJ. Mol Cell Biol. 2006;26:6880–6889. doi: 10.1128/MCB.00630-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi CJ, Sands AT, Zambrowicz BP, Turner TK, Demers DA, Webster W, Smith TW, Imbalzano AN, Jones SN. Mol Cell Biol. 2001;21:3598–3603. doi: 10.1128/MCB.21.10.3598-3603.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamiche A, Sandaltzopoulos R, Gdula DA, Wu C. Cell. 1999;97:833–842. doi: 10.1016/s0092-8674(00)80796-5. [DOI] [PubMed] [Google Scholar]
- Indra AK, Dupe V, Bornert JM, Messaddeq N, Yaniv M, Mark M, Chambon P, Metzger D. Development. 2005;132:4533–4544. doi: 10.1242/dev.02019. [DOI] [PubMed] [Google Scholar]
- Ito T, Levenstein ME, Fyodorov DV, Kutach AK, Kobayashi R, Kadonaga JT. Genes Dev. 1999;13:1529–1539. doi: 10.1101/gad.13.12.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MH, Hamana N, Nezu J, Shimane M. Genomics. 2000;63:40–45. doi: 10.1006/geno.1999.6071. [DOI] [PubMed] [Google Scholar]
- Kadam S, Emerson BM. Mol Cell. 2003;11:377–389. doi: 10.1016/s1097-2765(03)00034-0. [DOI] [PubMed] [Google Scholar]
- Kim JK, Huh SO, Choi H, Lee KS, Shin D, Lee C, Nam JS, Kim H, Chung H, Lee HW, et al. Mol Cell Biol. 2001;21:7787–7795. doi: 10.1128/MCB.21.22.7787-7795.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klochendler-Yeivin A, Fiette L, Barra J, Muchardt C, Babinet C, Yaniv M. EMBO Rep. 2000;1:500–506. doi: 10.1093/embo-reports/kvd129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmichev A, Jenuwein T, Tempst P, Reinberg D. Mol Cell. 2004;14:183–193. doi: 10.1016/s1097-2765(04)00185-6. [DOI] [PubMed] [Google Scholar]
- Langst G, Bonte EJ, Corona DF, Becker PB. Cell. 1999;97:843–852. doi: 10.1016/s0092-8674(00)80797-7. [DOI] [PubMed] [Google Scholar]
- Lemon B, Inouye C, King DS, Tjian R. Nature. 2001;414:924–928. doi: 10.1038/414924a. [DOI] [PubMed] [Google Scholar]
- Lessard J, Sauvageau G. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR. Neuron. 2007;55:201–215. doi: 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Tjian R. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- Lickert H, Takeuchi JK, Von Both I, Walls JR, McAuliffe F, Adamson SL, Henkelman RM, Wrana JL, Rossant J, Bruneau BG. Nature. 2004;432:107–112. doi: 10.1038/nature03071. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Nature. 2003;425:962–967. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar GJ, Fan HY, Kingston RE. Cell. 2002;108:475–487. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Neigeborn L, Carlson M. Genetics. 1984;108:845–858. doi: 10.1093/genetics/108.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. Science. 2002;296:1132–1136. doi: 10.1126/science.1069861. [DOI] [PubMed] [Google Scholar]
- Poot RA, Bozhenok L, van den Berg DL, Steffensen S, Ferreira F, Grimaldi M, Gilbert N, Ferreira J, Varga-Weisz PD. Nat Cell Biol. 2004;6:1236–1244. doi: 10.1038/ncb1196. [DOI] [PubMed] [Google Scholar]
- Reyes JC, Barra J, Muchardt C, Camus A, Babinet C, Yaniv M. EMBO J. 1998;17:6979–6991. doi: 10.1093/emboj/17.23.6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Cell. 2007;128:735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, Orkin SH. Mol Cell. 2008;32:491–502. doi: 10.1016/j.molcel.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern M, Jensen R, Herskowitz I. J Mol Biol. 1984;178:853–868. doi: 10.1016/0022-2836(84)90315-2. [DOI] [PubMed] [Google Scholar]
- Strohner R, Nemeth A, Jansa P, Hofmann-Rohrer U, Santoro R, Langst G, Grummt I. EMBO J. 2001;20:4892–4900. doi: 10.1093/emboj/20.17.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lugt NM, Domen J, Linders K, van Roon M, Robanus-Maandag E, te Riele H, van der Valk M, Deschamps J, Sofroniew M, van Lohuizen M, et al. Genes Dev. 1994;8:757–769. doi: 10.1101/gad.8.7.757. [DOI] [PubMed] [Google Scholar]
- Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. Nature. 2004a;431:873–878. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- Wang W, Cote J, Xue Y, Zhou S, Khavari PA, Biggar SR, Muchardt C, Kalpana GV, Goff SP, Yaniv M, et al. EMBO J. 1996;15:5370–5382. [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhai W, Richardson JA, Olson EN, Meneses JJ, Firpo MT, Kang C, Skarnes WC, Tjian R. Genes Dev. 2004b;18:3106–3116. doi: 10.1101/gad.1238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitcomb SJ, Basu A, Allis CD, Bernstein E. Trends Genet. 2007;23:494–502. doi: 10.1016/j.tig.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Wong AK, Shanahan F, Chen Y, Lian L, Ha P, Hendricks K, Ghaffari S, Iliev D, Penn B, Woodland AM, et al. Cancer Res. 2000;60:6171–6177. [PubMed] [Google Scholar]
- Wu JI, Lessard J, Olave IA, Qiu Z, Ghosh A, Graef IA, Crabtree GR. Neuron. 2007;56:94–108. doi: 10.1016/j.neuron.2007.08.021. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, et al. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. Mol Cell. 1998;2:851–861. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- Yan Z, Cui K, Murray DM, Ling C, Xue Y, Gerstein A, Parsons R, Zhao K, Wang W. Genes Dev. 2005;19:1662–1667. doi: 10.1101/gad.1323805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Wang Z, Sharova L, Sharov AA, Ling C, Piao Y, Aiba K, Matoba R, Wang W, Ko MS. Stem Cells. 2008;26:1155–1165. doi: 10.1634/stemcells.2007-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Wang W, Rando OJ, Xue Y, Swiderek K, Kuo A, Crabtree GR. Cell. 1998;95:625–636. doi: 10.1016/s0092-8674(00)81633-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.