


Abstract
With the development of high-throughput experimental techniques such as microarray, mass spectrometry and large-scale mutagenesis, there is an increasing need to automatically annotate gene sets and identify the involved pathways. Although many pathway analysis tools are developed, new tools are still needed to meet the requirements for flexible or advanced analysis purpose. Here, we developed an R-based software package (SubpathwayMiner) for flexible pathway identification. SubpathwayMiner facilitates sub-pathway identification of metabolic pathways by using pathway structure information. Additionally, SubpathwayMiner also provides more flexibility in annotating gene sets and identifying the involved pathways (entire pathways and sub-pathways): (i) SubpathwayMiner is able to provide the most up-to-date pathway analysis results for users; (ii) SubpathwayMiner supports multiple species (∼100 eukaryotes, 714 bacteria and 52 Archaea) and different gene identifiers (Entrez Gene IDs, NCBI-gi IDs, UniProt IDs, PDB IDs, etc.) in the KEGG GENE database; (iii) the system is quite efficient in cooperating with other R-based tools in biology. SubpathwayMiner is freely available at http://cran.r-project.org/web/packages/SubpathwayMiner/.
INTRODUCTION
In recent years, high-throughput experimental techniques such as microarray, mass spectrometry, and large-scale mutagenesis identified hundreds of interesting genes and gene products. For interpreting these high-throughput experimental data, biologists often study the functional relationships among these genes or gene products. One commonly used approach is to annotate these genes to biological pathways, such as Kyoto Encyclopedia of Genes and Genomes (KEGG) (1), and identify the statistically significantly enriched pathways. Many groups have developed pathway analysis tools relative to annotation and identification. These tools include PathwayExplorer (2), KOBAS (3,4), PathExpress (5), WebGestalt (6), KAAS (7), PathMAPA (8) and ArrayXPath II (9) and have become the commonly used tools.
Biological pathways contain complex pathway structure information. For example, a metabolic pathway in KEGG can be naturally modeled as a network or graph with compounds (substrates and products) as nodes and chemical reactions (enzymes) as edges. Studies showed that pathway structure information can provide more delicate biological insights and help us understand higher-order functions of the biological system (10–12). In this article, we developed a new pathway analysis tool relative to pathway annotation and identification, which applies pathway structure information to pathway identification. According to pathway structure information provided by KEGG, our system can detect distance similarity among enzymes in each pathway and mine each sub-pathway in which distance among all enzymes is no greater than the parameter k (a user-defined distance). Gene sets can then be annotated to these sub-pathways through assigning EC numbers for them and matching them to these sub-pathways. Furthermore, the significantly enriched sub-pathways can be identified using statistical method such as hypergeometric test. With different setting of the distance parameter k, the identification of sub-pathways is able to become more flexible. For evaluating our method, our system was applied to differentially expressed gene sets of lung cancer. We found that some pathways associated with lung cancer but not significant in entire pathway identification were highly significant in our sub-pathway identification. The results indicate that there is a positive effect on the flexible identification of metabolic pathways in our system.
As a new pathway analysis tool, SubpathwayMiner overcomes some limitations of the existing tools through some effective ways. First, the system applies pathway simplification technique and sub-pathway mining method to metabolic pathways, and then facilitates sub-pathway identification of metabolic pathways. Second, storage and update of data relative to pathway analysis can easily be operated by users themselves. Consequently, users will always receive the most up-to-date pathway analysis results. Third, the system can support multiple species (about 100 eukaryotes, 714 bacteria and 52 archaea) and different gene identifiers (Entrez Gene IDs, NCBI-gi IDs, UniProt IDs, PDB IDs, etc.) in the KEGG GENE database through an effective way to automatically store and update data. Fourth, it is quite efficient in cooperating with other R-based tools in computational biology and bioinformatics because the system is an R-based system (13).
MATERIALS AND METHODS
SubpathwayMiner is implemented in R, an open source programming environment (13), and adopts a module design to provide more flexibility. Figure 1 depicts the schematic overview of the system. The system is composed of four modules: storage and update of data, sub-pathway mining, annotation and identification of pathways, visualization of results. Storage and update module can get and update data relative to analysis of pathways from the KEGG GENE database. Sub-pathway mining module is used to mine sub-pathways for flexible identification of metabolic pathways. Annotation and identification module helps users to annotate and identify pathways or sub-pathways. Visualization module provides three methods for displaying analysis results.
Figure 1.

Schematic overview of SubpathwayMiner.
Storage and update of data
A new method (the function updateOrgAndIdType in R) is presented here, which enables users to store and update data automatically for pathway analysis. These data can be automatically downloaded from KEGG, converted, and stored directly in the SubpathwayMiner environment variable as a database rather than in an external DBMS (database management system). These data can be updated automatically on request by the user. By this method, the system can synchronize data with the KEGG GENE database and can support most organisms and cross-reference identifiers in the KEGG GENE database. We have also considered that this method may be time consuming for several organisms in which many genes may be in common (e.g. Homo sapiens and Mus musculus). We thus present two methods to solve the problem and to provide more flexibility. On the one hand, SubpathwayMiner uses two functions (loadKE2G and saveKE2G) to save and load the SubpathwayMiner environment variable easily. Through the functions users can update data relative to a certain organism one time only and use repeatedly them in the future. On the other hand, the environment variables of organisms with well-annotated genomes have been provided in the SubpathwayMiner package.
Sub-pathway mining
Sub-pathway mining module is used to mine sub-pathways for flexible identification of metabolic pathways. However, sub-pathway mining has become a general problem in view of the complex structures of metabolic pathways. Fortunately, whether or not a gene can be annotated to a pathway is completely dependent on enzymes rather than compounds in the pathway. We thus convert each metabolic pathway to an undirected graph with enzymes as nodes. Two nodes in an undirected graph are connected by an edge if there is a common compound in the enzymes corresponding reactions. As a result, the metabolic pathway is simplified when chemical compounds are omitted from the graph. According to this pathway simplification method, the sub-pathway mining problem can be considered as a sub-graph mining problem. Many sub-graph mining methods are theoretically available. In the current system, we mine sub-pathways based on distance similarity among enzymes. Some studies suggest that the functional similarity between two enzymes increases as their distance in pathways decreases (12,14). Our sub-pathway mining strategy thus tends to find the sub-pathways in which all enzymes have highly similar functions. To do it, we adopt the k-clique concept in social network analysis (15) to define sub-pathways based on distance similarity among enzymes. In social network analysis, a k-clique in a graph is considered as a sub-graph where the distance between any two nodes is no greater than k. When we consider each k-clique as a sub-pathway of metabolic pathways, sub-pathways can be mined by using the special k-clique algorithm provided by RGBL package (16). SubpathwayMiner provides users with the default value of parameter k (k = 4). Users can also choose the parameter according to their needs. The distance among all enzymes in mined sub-pathways decreases as the value of the parameter k reduces. If we set a smaller value of parameter k, more compact sub-pathways based on distance will be produced. For example, if we set k = 3, the distance between all enzymes in the mined sub-pathway from citrate cycle pathway is no >3 (Figure 2c).
Figure 2.

A visualized example of sub-pathway mining. (a) A metabolic pathway in KEGG, citrate cycle (TCA cycle). (b) The metabolic pathway is converted to the undirected graph using our pathway simplification programming (the function updateGraphs in SubpathwayMiner). (c) A 3-clique sub-pathway in which distance between any two enzymes is no >3. It is mined from the undirected graph that the pathway corresponds to (surrounded by a black line in Figure 2a and b).
The following describes the step-by-step method for mining sub-pathways:
Downloading automatically the corresponding XML file of metabolic pathways from the KEGG KGML (The current version is at ftp://ftp.genome.jp/pub/kegg/xml/map/).
Taking out the relationship of enzymes from each XML file. Two enzymes are connected by an edge if their corresponding reactions have a common compound.
Simplifying metabolic pathways. Each metabolic pathway is converted to an undirected graph with enzymes as nodes (Figure 2b). Two are connected by an edge if there is a common compound in the enzymes corresponding reactions.
Saving the simplification version of each metabolic pathway to the environment variable KE2G that is used as the core database of SubpathwayMiner.
Setting up the distance parameter k (k = 1, 2, 3, … , n). The setting of parameter k is flexible. Users can choose an appropriate parameter according to their needs. The distance among all enzymes in sub-pathways decreases as the value of parameter k reduces, which will product more delicate identification of pathways.
For the simplification version of each metabolic pathway, mine k-cliques of this pathway according to distance parameter k. Each k-clique is treated as a sub-pathway (Figure 2c).
Collecting all sub-pathways (k-cliques) in metabolic pathways. The identifier of each sub-pathway is given with its pathway identifier plus a sub-pathway number (e.g. ‘path: 00010_1’).
Annotation and identification of pathways
Annotation and identification module can provide annotation and identification of sub-pathways or entire pathways. When users select annotation of entire pathways, the function getAnn will assign pathway numbers for a set of genes submitted by users according to gene-pathway relationship saved in the environment variable. When users select sub-pathway annotation of metabolic pathways, the function will assign genes to EC numbers and match them to sub-pathways. To identify the statistically significantly enriched pathways, p-values are calculated using the hypergeometric distribution. The default background distribution is considered to be the whole genome (the system also permits users to choose their own background distribution). For each pathway (an entire pathway or a sub-pathway) that occurs in the set of genes submitted for analysis, the system counts the total number of genes in the set that are involved in the pathway. If the whole genome has a total of m genes, of which t are involved in the pathway under investigation, and the set of genes submitted for analysis has a total of n genes, of which r are involved in the same pathway, then the p-value can be calculated to evaluate enrichment significance for that pathway as follows:
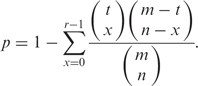 |
When many correlated pathways (entire pathways or sub-pathways) are considered, a high false positive discovery rate is likely to result. For this reason, the system also provides the FDR-corrected q-values (if applicable) for reducing the false positive discovery rate (17,18).
Annotation and identification module provides the function cutoffAnn for identifying the statistically significantly enriched pathways or sub-pathways. As our system adopts a module design where annotation and identification module is relatively independent, the module can be used to annotate and identify user-defined sub-pathways. Users can also annotate and identify their own sub-pathways through mining sub-pathways based on the simplification version of metabolic pathways.
Visualization of results
Visualization module provides three methods for displaying results. As illustrated in Figure 3a, the first method (the function printAnn) converts a list of results to a data frame in R that can be easily saved as a tab-delimited text file by using the function write in R. The second method (the function gotoKEGG) visualizes pathways through linking to the KEGG website (Figure 3c). On the pathway map, enzymes are colored red if the according enzyme is identified in the submitted set of genes. If users choose sub-pathways annotation of metabolic pathways, the third method (the function plotAnn) is available. It visualizes sub-pathways as an undirected graph (Figure 3b). Enzymes are colored red if the according enzyme is identified in the submitted gene sets.
Figure 3.

Screenshots of visualization provided in SubpathwayMiner. (a) Display results using a data frame in R. Each row corresponds to information of the pathway that genes are annotated to. The first column contains pathway identifiers. Relevant pathways are listed in ascending order of p-values and multiple-comparison corrected q-values. (b) Visualize a sub-pathway as the undirected graph. Enzymes are colored red if the according enzyme is identified in the submitted sets of genes. (c) Visualize a pathway through linking to the KEGG website. On the pathway map, enzymes are colored red if the according enzyme is identified in the submitted set of genes.
RESULTS
SubpathwayMiner is available for pathway annotation and identification of any interesting gene/protein sets with identifiers supported by the system (Entrez Gene IDs, NCBI-gi IDs, UniProt IDs, PDB IDs, etc.). For example, the system is not limited to pathway analysis of gene expression data. It can also receive interesting gene sets from certain other approaches, such as the ensemble decision approach by the authors (19).
A key function of SubpathwayMiner is sub-pathway identification of metabolic pathways. For comparison of entire pathway and sub-pathway identification, we showed an example application of SubpathwayMiner to a gene expression data, analyzed initially by Landi et al. (20). The data was publicly available at the GEO database (accession number GSE10072). The pathway data got from KGML_v0.6.1 (ftp://ftp.genome.jp/pub/kegg/release/archive/kgml/KGML_v0.6.1/map).
We first identified a total of 1313 differentially expressed genes using the significance analysis of microarray (SAM) method (21) (FDR <0.01) and Fold-change (FD >1.5 or <0.667). We then used SubpathwayMiner to annotate these differentially expressed genes to entire pathways and sub-pathways (k = 4) of metabolic pathways. The results showed that these genes were annotated to 87 entire pathways and 307 sub-pathways of metabolic pathways. With the strict cutoff of p-values <0.01, our system identified seven statistically significantly enriched entire pathways of metabolic pathways and 36 enriched sub-pathways corresponding to 10 entire pathways of metabolic pathways. The average overlap between the significant sub-pathways found within each single pathway was also calculated according to the Sokal and Sneath coefficient (22) (Table 1). We have found that three entire pathways, which were included in 10 entire pathways that 36 sub-pathways correspond to, were not statistically significant (p > 0.01). They were respectively path:00350 (tyrosine metabolism), path:00260 (glycine, serine and threonine metabolism), and path:00564 (glycerophospholipid metabolism). When we only adopt entire pathway identification method, these pathways may be ignored because of their high p-values. However, some sub-pathways of these pathways were statistically significant in our system. The result indicates that these significant sub-pathways included in pathways of high p-values may be associated with cancer initiation or progression. For looking for knowledge support, we searched PUBMED database. The results showed that gene macrophage migration inhibitory factor (MIF), which was differentially expressed and annotated in 5 sub-pathways (path:00350_5, path:00350_6, path:00350_7, path:00350_8 and path:00350_12) of the pathway path:00350, was associated with risk of recurrernce after resection of lung cancer (23). MIF was also associated with beast cancer (24), colorectal cancer (25) and prostate cancer (26), etc. Gene alcohol dehydrogenase 1B (ADH2), a differentially expressed gene annotated to these sub-pathways, was reported to be associated with esophageal cancer, aerodigestive cancer, breast cancer and colorectal cancer (27–30). One differentially expressed gene annotated in a sub-pathway (path:00260_9) of the pathway path:00260, aldo-keto reductase family 1, member B10 (AKR1B10), was found to be useful as a new marker for identification of high lung cancer risk patients in usual interstitial pneumonia (31). Mashkova et al. showed that AKR1B10 was a potential oncogene and elevated transcription level is important for squamous cell lung cancer tumorogenesis (32). Genes annotated in two sub-pathways (path:00564_1 and path:00564_2) of the pathway path:0000564 were found not to be obviously associated with lung cancer. However, two of them, Gene CHPT1 (choline phosphotransferase 1) and PLA2G4A (phospholipase A2, group IVA), were associated with breast cancer (33) and colon cancer (34). Moreover, some evidences were found in the literature for the biological significance of the highly enriched sub-pathways. Studies showed that some enzymes in sub-pathways of the ‘tyrosine metabolism’ pathway, including monoamine oxidase (MAO), aldehyde reductase (AR), catechol-Omethyltransferase (COMT), alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (AD), were found to be highly associated with cancer (35–37). Moreover, norepinephrine and its metabolism catalyzed by these enzymes were also found to be associated with cancer initiation and progression (37–41). In the process of norepinephrine metabolism, norepinephrine is deaminated by MAO to 3,4-dihydroxyphenylglycolaldehyde (DOPEGAL). DOPEGAL is then converted by the sequential actions of AR, COMT, ADH and AD to 3,4-dihydroxyphenylglycol (DHPG), 3-methoxy-4-hydroxyphenylglycol (MHPG), 3-methoxy-4-hydroxyphenylglycolaldehyde (MOPEGAL) and formation of vanillylmandelic acid (VMA), respectively (37). These evidences indicate that the sequential actions of enzymes (MAO, AR, COMT, ADH and AD), which are in the sub-pathways identified by our method, may play an important role in cancer initiation and progression. The above biological knowledge mining highly supports our analysis. We thus propose that pathways, which are statistically significant in sub-pathways but not in entire pathways, may be highly associated with cancer initiation and progression.
Table 1.
The statistically significantly enriched sub-pathways identified by SubpathwayMiner for differentially expressed genes from lung cancer
| Entire pathway ID (p-values/overlapa) | Entire pathway name | Sub-pathway ID | Sub-pathway p-values |
|---|---|---|---|
| Path:00350 (0.1037/49%) | Tyrosine metabolism | Path:00350_12 | 0.003248 |
| Path:00350_3 | 0.002156 | ||
| Path:00350_5 | 0.004418 | ||
| Path:00350_6 | 0.003799 | ||
| Path:00350_7 | 0.006378 | ||
| Path:00350_8 | 0.003975 | ||
| Path:00260 (0.01109/0) | Glycine, serine and threonine metabolism | Path:00260_9 | 0.002947 |
| Path:00564 (0.01057/88%) | Glycerophospholipid metabolism | Path:00564_1 | 0.006880 |
| Path:00564_2 | 0.007549 | ||
| Path:00010 (0.00012/27%) | Glycolysis/gluconeogenesis | Path:00010_2 | 0.004475 |
| Path:00010_3 | 0.0008654 | ||
| Path:00010_4 | 0.001629 | ||
| Path:00010_5 | 0.002797 | ||
| Path:00010_6 | 0.0004917 | ||
| Path:00010_7 | 0.003566 | ||
| Path:00220 (0.004746/59%) | Urea cycle and metabolism of amino groups | Path:00220_3 | 0.006607 |
| Path:00220_5 | 0.003975 | ||
| Path:00220_6 | 0.006607 | ||
| Path:00220_7 | 0.004745 | ||
| Path:00230 (0.000128/35%) | Purine metabolism | Path:00230_1 | 0.001154 |
| Path:00230_10 | 0.006450 | ||
| Path:00230_11 | 0.0008266 | ||
| Path:00230_2 | 0.009967 | ||
| Path:00230_4 | 0.001097 | ||
| Path:00230_6 | 0.001063 | ||
| Path:00230_8 | 0.006450 | ||
| Path:00230_9 | 0.0005034 | ||
| Path:00565 (0.0011/64%) | Ether lipid metabolism | Path:00565_2 | 0.004406 |
| Path:00565_3 | 0.005799 | ||
| Path:00565_4 | 0.003269 | ||
| Path:00590 (0.002233/37%) | Arachidonic acid metabolism | Path:00590_1 | 0.0009760 |
| Path:00590_2 | 0.002773 | ||
| Path:00590_3 | 0.002411 | ||
| Path:00590_4 | 0.003772 | ||
| Path:00480 (0.005111/0) | Glutathione metabolism | Path:00480_1 | 0.005110 |
| Path:00670 (0.009117/0) | One carbon pool by folate | Path:00670_1 | 0.009117 |
aThe average overlap between the significant sub-pathways found within each single pathway.
DISCUSSION
In this article, we apply pathway structure information to pathway identification. We use a pathway simplification method to convert each metabolic pathway to an undirected graph, and then implement sub-pathway identification by mining sub-pathways based on k-clique concept in social network analysis. In fact, methods to mine sub-pathways are presented by some studies in recent years. For instance, Ogata et al. found conserved pathway motifs in metabolic pathways (12). Koyutürk et al. (11) found frequently occurring patterns and modules in the KEGG pathways. However, these methods are not fit for implementing sub-pathway annotation and identification in term of different purpose of research. Therefore, we present a new sub-pathway mining method fit for sub-pathway identification of metabolic pathways. For evaluating our method, our system was applied to differentially expressed gene sets of lung cancer. We find that although some pathways are not significant in entire pathway identification, they are highly significant in our sub-pathway identification. Interestingly, these differentially expressed genes annotated to these sub-pathways are found to be highly associated with cancer initiation and progression. This indicates that our sub-pathway identification method is able to recall some pathways that are associated with cancer initiation and progression; however, those pathways are ignored by the entire pathway identification method.
The sub-pathway identification method provided by SubpathwayMiner tends to identify certain local areas of pathways because the method is based on k-clique concept in social network analysis. For example, in the ‘Results’ section, some of differentially expressed genes of lung cancer were annotated to the ‘Tyrosine metabolism’ pathway. As illustrated in Figure 4, these differentially expressed genes (red enzymes) annotated to the ‘Tyrosine metabolism’ pathway are mostly concentrated in local areas of the pathway. Thus, some sub-pathways corresponding to local areas of the pathway are statistically significant although the entire pathway is not statistically significant (Table 1). These identified sub-pathways usually perform certain type-specific functions compared with their entire pathways. For example, we have found that the sub-pathways (path:00350_5, path:00350_6, path:00350_7, path:00350_8) can efficiently contain the ‘norepinephrine metabolism’ pathway which is highly associated with cancer initiation and progression, and which belongs to a minor pathway (or sub-pathway) of the ‘Tyrosine metabolism’ pathway (in the right-bottom part of Figure 4). This indicates that certain cancer may be more associated with these genes concentrated in local areas of pathways. It may be a common biological phenomenon that some genes tend to perform certain type-specific functions (e.g. norepinephrine metabolism), which may cause the certain results (e.g. cancer). These type-specific functions tend to distribute in local areas of the pathway instead of entire pathway.
Figure 4.

The tyrosine metabolism pathway where the differentially expressed genes of lung cancer were annotated. The enzymes identified in all genes of Homo sapiens were colored green. The enzymes identified in the submitted genes were colored red. The results show that these genes were mostly concentrated in local areas of the pathway such as the right-bottom part of the figure.
SubpathwayMiner provides much flexibility in annotation and identification of pathways. It uses a new method to automatically store data relative to pathway annotation and identification. This enables our system to support most of organisms in the KEGG GENE database. Data can also be automatically updated on demand by the user. Therefore, users are able to receive the most up-to-date pathway analysis results. Our system is developed in R programming environment, which has proved to be a powerful tool for computational biology and bioinformatics. More and more computational biology and bioinformatics studies are carried out in R environment (42,43). The functions provided by SubpathwayMiner can easily be applied to these R-based studies. For example, the system developed here can efficiently support pathway analysis of probe sets of microarrays by cooperating with bioconductor (http://www.bioconductor.org). Currently, the system supports pathway analysis of probe sets from about 40 kinds of Affymetrix chips and from some other kinds of microarrays (e.g. Illumina chips) using the probe-gene relationship provided by bioconductor. SubpathwayMiner's definition of sub-pathways is based on distance similarity among enzymes because of adopting k-clique concept in social network analysis. The sub-pathway identification method can thus efficiently identify local areas of pathways. Moreover, some studies suggest that the functional similarity between two enzymes increases as their distance in pathways decreases (12,14). This indicates that the sub-pathway mining strategy presented here tends to find the sub-pathways in which enzymes have highly similar functions. In addition, the present method for mining sub-pathways can provide great flexibility in identification of sub-pathways, especially in the highly connected pathways which commonly occur in some well-annotated genomes (e.g. Homo sapiens and Saccharomyces cerevisiae). For example, the system is able to divide the ‘tyrosine metabolism’ pathway (Figure 4) into 12 sub-pathways (when k = 4) and then to identify significantly enriched sub-pathways within these sub-pathways. The sub-pathway identification can also be made more flexible by using different values of the distance parameter k. It can be expected that expect that SubpathwayMiner will be a beneficial pathway annotation and identification tool.
FUTURE DEVELOPMENT
We plan to adopt two strategies to improve our current system in the future. First, the current system uses the k-clique concept to mine sub-pathways. However, some other methods based on mining sub-graphs may be available. Therefore, we will add more sub-pathway mining methods to mine sub-pathways. This will provide more sub-pathway identification strategies for users. Second, the current system supports sub-pathway identification of metabolic pathways. Furthermore, we will extend sub-pathway identification to more KEGG pathways. These strategies will no doubt increase abilities of sub-pathway identification in our system. Because our system adopts the module design, the extension of the system will become more available.
FUNDING
The National Natural Science Foundation of China (grant nos. 30871394, 30370798 and 30571034), the National High Tech Development Project of China, the 863 Program (grant nos. 2007AA02Z329), the National Basic Research Program of China, the 973 Program (grant nos. 2008CB517302) and the National Science Foundation of Heilongjiang Province (grant nos. ZJG0501, 1055HG009, GB03C602-4, and BMFH060044). Funding for open access charge: National High Tech Development Project of China, the 863 Program (grant nos. 2007AA02Z329).
Conflict of interest statement. None declared.
REFERENCES
- 1.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mlecnik B, Scheideler M, Hackl H, Hartler J, Sanchez-Cabo F, Trajanoski Z. PathwayExplorer: web service for visualizing high-throughput expression data on biological pathways. Nucleic Acids Res. 2005;33:W633–W637. doi: 10.1093/nar/gki391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu J, Mao X, Cai T, Luo J, Wei L. KOBAS server: a web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006;34:W720–W724. doi: 10.1093/nar/gkl167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21:3787–3793. doi: 10.1093/bioinformatics/bti430. [DOI] [PubMed] [Google Scholar]
- 5.Goffard N, Weiller G. PathExpress: a web-based tool to identify relevant pathways in gene expression data. Nucleic Acids Res. 2007;35:W176–W181. doi: 10.1093/nar/gkm261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–W748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan D, Sun N, Cheung KH, Guan Z, Ma L, Holford M, Deng X, Zhao H. PathMAPA: a tool for displaying gene expression and performing statistical tests on metabolic pathways at multiple levels for Arabidopsis. BMC Bioinformatics. 2003;4:56. doi: 10.1186/1471-2105-4-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung HJ, Park CH, Han MR, Lee S, Ohn JH, Kim J, Kim J, Kim JH. ArrayXPath II: mapping and visualizing microarray gene-expression data with biomedical ontologies and integrated biological pathway resources using Scalable Vector Graphics. Nucleic Acids Res. 2005;33:W621–W626. doi: 10.1093/nar/gki450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antonov AV, Dietmann S, Mewes HW. KEGG spider: interpretation of genomics data in the context of the global gene metabolic network. Genome Biol. 2008;9:R179. doi: 10.1186/gb-2008-9-12-r179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koyuturk M, Grama A, Szpankowski W. An efficient algorithm for detecting frequent subgraphs in biological networks. Bioinformatics. 2004;20(Suppl. 1):i200–i207. doi: 10.1093/bioinformatics/bth919. [DOI] [PubMed] [Google Scholar]
- 12.Ogata H, Fujibuchi W, Goto S, Kanehisa M. A heuristic graph comparison algorithm and its application to detect functionally related enzyme clusters. Nucleic Acids Res. 2000;28:4021–4028. doi: 10.1093/nar/28.20.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Team RDC. R: a language and environment for statistical computing. R Foundation Statistical Computing. 2008 [Google Scholar]
- 14.Guo X, Liu R, Shriver CD, Hu H, Liebman MN. Assessing semantic similarity measures for the characterization of human regulatory pathways. Bioinformatics. 2006;22:967–973. doi: 10.1093/bioinformatics/btl042. [DOI] [PubMed] [Google Scholar]
- 15.Wasserman S, Faust K. Social Network Analysis: Methods and Applications. New York: Cambridge University Press; 1994. [Google Scholar]
- 16.Huber W, Carey VJ, Long L, Falcon S, Gentleman R. Graphs in molecular biology. BMC Bioinformatics. 2007;8(Suppl. 6):S8. doi: 10.1186/1471-2105-8-S6-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strimmer K. fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics. 2008;24:1461–1462. doi: 10.1093/bioinformatics/btn209. [DOI] [PubMed] [Google Scholar]
- 18.Strimmer K. A unified approach to false discovery rate estimation. BMC Bioinformatics. 2008;9:303. doi: 10.1186/1471-2105-9-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Rao S, Wang Y, Gong B. Gene mining: a novel and powerful ensemble decision approach to hunting for disease genes using microarray expression profiling. Nucleic Acids Res. 2004;32:2685–2694. doi: 10.1093/nar/gkh563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS ONE. 2008;3:e1651. doi: 10.1371/journal.pone.0001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sokal R, Sneath PHA. Principles of Numerical Taxonomy. San Francisco, CA: W.H. Freeman and Co.; 1963. [Google Scholar]
- 23.White ES, Flaherty KR, Carskadon S, Brant A, Iannettoni MD, Yee J, Orringer MB, Arenberg DA. Macrophage migration inhibitory factor and CXC chemokine expression in non-small cell lung cancer: role in angiogenesis and prognosis. Clin. Cancer Res. 2003;9:853–860. [PubMed] [Google Scholar]
- 24.Xu X, Wang B, Ye C, Yao C, Lin Y, Huang X, Zhang Y, Wang S. Overexpression of macrophage migration inhibitory factor induces angiogenesis in human breast cancer. Cancer Lett. 2008;261:147–157. doi: 10.1016/j.canlet.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 25.Yao K, Shida S, Selvakumaran M, Zimmerman R, Simon E, Schick J, Haas NB, Balke M, Ross H, Johnson SW, et al. Macrophage migration inhibitory factor is a determinant of hypoxia-induced apoptosis in colon cancer cell lines. Clin. Cancer Res. 2005;11:7264–7272. doi: 10.1158/1078-0432.CCR-05-0135. [DOI] [PubMed] [Google Scholar]
- 26.Meyer-Siegler KL, Iczkowski KA, Vera PL. Further evidence for increased macrophage migration inhibitory factor expression in prostate cancer. BMC Cancer. 2005;5:73. doi: 10.1186/1471-2407-5-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen YJ, Chen C, Wu DC, Lee CH, Wu CI, Lee JM, Goan YG, Huang SP, Lin CC, Li TC, et al. Interactive effects of lifetime alcohol consumption and alcohol and aldehyde dehydrogenase polymorphisms on esophageal cancer risks. Int. J. Cancer. 2006;119:2827–2831. doi: 10.1002/ijc.22199. [DOI] [PubMed] [Google Scholar]
- 28.Hashibe M, McKay JD, Curado MP, Oliveira JC, Koifman S, Koifman R, Zaridze D, Shangina O, Wunsch-Filho V, Eluf-Neto J, et al. Multiple ADH genes are associated with upper aerodigestive cancers. Nat. Genet. 2008;40:707–709. doi: 10.1038/ng.151. [DOI] [PubMed] [Google Scholar]
- 29.Terry MB, Knight JA, Zablotska L, Wang Q, John EM, Andrulis IL, Senie RT, Daly M, Ozcelik H, Briollais L, et al. Alcohol metabolism, alcohol intake, and breast cancer risk: a sister-set analysis using the Breast Cancer Family Registry. Breast Cancer Res. Treat. 2007;106:281–288. doi: 10.1007/s10549-007-9498-7. [DOI] [PubMed] [Google Scholar]
- 30.Matsuo K, Wakai K, Hirose K, Ito H, Saito T, Suzuki T, Kato T, Hirai T, Kanemitsu Y, Hamajima H, et al. A gene-gene interaction between ALDH2 Glu487Lys and ADH2 His47Arg polymorphisms regarding the risk of colorectal cancer in Japan. Carcinogenesis. 2006;27:1018–1023. doi: 10.1093/carcin/bgi282. [DOI] [PubMed] [Google Scholar]
- 31.Li CP, Goto A, Watanabe A, Murata K, Ota S, Niki T, Aburatani H, Fukayama M. AKR1B10 in usual interstitial pneumonia: expression in squamous metaplasia in association with smoking and lung cancer. Pathol. Res. Pract. 2008;204:295–304. doi: 10.1016/j.prp.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 32.Mashkova TD, Oparina N, Zinov'eva OL, Kropotova ES, Dubovaia VI, Poltaraus AB, Fridman MV, Kopantsev EP, Vinogradova TV, Zinov'eva MV, et al. Transcription TIMP3, DAPk1 and AKR1B10 genes in squamous cell lung cancer. Mol. Biol. (Mosk) 2006;40:1047–1054. [PubMed] [Google Scholar]
- 33.Akech J, Sinha Roy S, Das SK. Modulation of cholinephosphotransferase activity in breast cancer cell lines by Ro5-4864, a peripheral benzodiazepine receptor agonist. Biochem. Biophys. Res. Commun. 2005;333:35–41. doi: 10.1016/j.bbrc.2005.05.071. [DOI] [PubMed] [Google Scholar]
- 34.Parhamifar L, Jeppsson B, Sjolander A. Activation of cPLA2 is required for leukotriene D4-induced proliferation in colon cancer cells. Carcinogenesis. 2005;26:1988–1998. doi: 10.1093/carcin/bgi159. [DOI] [PubMed] [Google Scholar]
- 35.Fowler JS, Logan J, Wang GJ, Volkow ND, Telang F, Zhu W, Franceschi D, Shea C, Garza V, Xu Y, et al. Comparison of monoamine oxidase a in peripheral organs in nonsmokers and smokers. J. Nucleic Med. 2005;46:1414–1420. [PubMed] [Google Scholar]
- 36.Eisenhofer G, Huynh TT, Hiroi M, Pacak K. Understanding catecholamine metabolism as a guide to the biochemical diagnosis of pheochromocytoma. Rev. Endocr. Metab. Disord. 2001;2:297–311. doi: 10.1023/a:1011572617314. [DOI] [PubMed] [Google Scholar]
- 37.Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol. Rev. 2004;56:331–349. doi: 10.1124/pr.56.3.1. [DOI] [PubMed] [Google Scholar]
- 38.Sood AK, Bhatty R, Kamat AA, Landen CN, Han L, Thaker PH, Li Y, Gershenson DM, Lutgendorf S, Cole SW. Stress hormone-mediated invasion of ovarian cancer cells. Clin. Cancer Res. 2006;12:369–375. doi: 10.1158/1078-0432.CCR-05-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang EV, Donovan EL, Benson DM, Glaser R. VEGF is differentially regulated in multiple myeloma-derived cell lines by norepinephrine. Brain Behav. Immun. 2008;22:318–323. doi: 10.1016/j.bbi.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landen CN, Jr, Lin YG, Armaiz Pena GN, Das PD, Arevalo JM, Kamat AA, Han LY, Jennings NB, Spannuth WA, Thaker PH, et al. Neuroendocrine modulation of signal transducer and activator of transcription-3 in ovarian cancer. Cancer Res. 2007;67:10389–10396. doi: 10.1158/0008-5472.CAN-07-0858. [DOI] [PubMed] [Google Scholar]
- 41.Yang EV, Sood AK, Chen M, Li Y, Eubank TD, Marsh CB, Jewell S, Flavahan NA, Morrison C, Yeh PE, et al. Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res. 2006;66:10357–10364. doi: 10.1158/0008-5472.CAN-06-2496. [DOI] [PubMed] [Google Scholar]
- 42.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc. Natl Acad. Sci. USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]