


Abstract
Genome-scale microarray experiments for comparative analysis of gene expressions produce massive amounts of information. Traditional statistical approaches fail to achieve the required accuracy in sensitivity and specificity of the analysis. Since the problem can be resolved neither by increasing the number of replicates nor by manipulating thresholds, one needs a novel approach to the analysis. This article describes methods to improve the power of microarray analyses by defining internal standards to characterize features of the biological system being studied and the technological processes underlying the microarray experiments. Applying these methods, internal standards are identified and then the obtained parameters are used to define (i) genes that are distinct in their expression from background; (ii) genes that are differentially expressed; and finally (iii) genes that have similar dynamical behavior.
INTRODUCTION
Microarray technology provides a genome-wide screening and monitoring of expression levels for thousands of genes simultaneously, and has been extensively applied to a broad range of biological and medical problems in order to identify changes in expression between different biological states. The immense amount of information that can be obtained from microarray studies enables us to address a variety of different research aims but still presents a challenge for data analysis, especially in terms of mutually exclusive parameters such as sensitivity and specificity. Many excellent reviews have been written on this subject (1–4). Our intention is, rather than providing an overview of available approaches, to offer a presentation of our methodological approaches with the main emphasis of using internal standards as means of robust evaluation strategy. Some of the methods have been published at least in part, others are completely new.
Methods based on conventional t-tests estimate the probability (P) that a difference in gene expression occurred by chance. If the threshold for probability chosen as significant in the context of a small sized experiment is applied in another microarray experiment, it can have a high false positive rate. For example, if the P threshold is 0.01, then even a set of random data satisfying the null hypothesis will result in one false positive per every 100 genes tested. A microarray containing tens of thousands of genes will generate hundreds of false positive results.
Two of the most popular approaches to address this problem are to make adjustment of thresholds or to use various combinatory calculations in order to improve the power (sensitivity) and specificity of the statistical conclusions. Due to its simplicity, the Bonferroni adjustment was used frequently despite its well-known conservativeness. The correction of P threshold by dividing the desired significance by the total number of statistical tests performed, ensures the achievement of a desired false positive rate over the entire set of genes, but conversely sets a criterion that can be too strict for each individual gene. Specificity is gained at the expense of sensitivity. Thus, the method does not reject hypotheses as often as it should and therefore it lacks power. This is of course a paradoxical situation, since the statistical significance for each individual measurement apparently depends on the total number of unrelated measurements.
None of the various attempts to improve Bonferroni adjustments has helped to resolve the problem. The most popular of such adjustments, the so called false discovery rate (FDR) control (5,6) that has been introduced into microarray analysis by Benjamini and Hochberg (7) enables to estimate the measure of the proportion of rejected null hypotheses. All genes are ranked according to their P-values and tested against individualized thresholds: the smallest observed P-value is tested against the strictest threshold, and the remaining P-values against successively more relaxed thresholds. In other tests, e.g. in the popular significance analysis of microarrays (SAM) method (8,9), the use of individualized thresholds improves the conservativeness of the Bonferroni test, though the improvement is only partial and often minor.
The relative difference in gene expressions computed from replicated hybridizations provides a control for random fluctuations, the power of which depends essentially on the number of replicates. To improve statistical significance of biological variation without increasing the number of replicates, additional controls are needed. In the aforementioned methods, like SAM, ‘instead of performing more experiments’, which are expensive and labor intensive, Tusher et al. (8) generated a large number of controls using re-sampling methods such as bootstrap or permutation to estimate the underlying distribution from the observed data. However, generation of larger number of controls by using combinatory approaches instead of performing more experiments is somewhat illusory in that it does not truly increase the amount of information being analyzed.
Fortunately, there exists an adequate resource to increase the power of statistical tests by using the massive quantity of information inherently obtainable in each microarray experiment. We introduce here an approach in which the paired comparison of gene expression in two different situations is accompanied by the associative test—checking the hypothesis that each given gene in the experimental group has common features and can be associated with an internal standard. Internal standard in this context is considered as a large family of genes sharing some useful features for analysis, which in turn are neither dependent on the particular gene sequence nor on the level of expression, and are also not dependent on the coordinate position in the chip.
The methodology of the evaluation described in this communication will serve us as a stepping stone to our further effort of using internal standards for analysis in a statistically robust manner, functional associations through clustering and networking genes having similar dynamical behavior. These methods are equally applicable to time course dynamics initiated by various treatments and to natural variations of genes involved in essential dynamical processes in biological systems as well. This we intend to describe in the follow-up article (in preparation).
Early variants of some procedures described here were first included in the Matlab toolbox for microarray data analysis MDAT described in Knowlton et al. (10), while the improved and modified version exists now and is available on request.
MATERIALS AND METHODS
Gene expression datasets
This work uses a wide spectrum of experimental data that were only partially published.
The expression datasets were obtained with the use different sources of mRNA and different microarray technologies. They include Mouse Atlas 1.2 membranes and Mouse plastic 5K arrays Human Cancer Atlas 1.2 membranes (Clontech, Palo Alto, CA). Most data were obtained with the use of high-density microarrays.
Custom microarrays were prepared at the Oklahoma Medical Research Foundation Microarray Core Facility using commercially available libraries of oligonucleotides: Human Genome Oligo Ser Version 2.0 and mouse genome set, version 2.0 (Qiagen, Valencia, CA).
All data of recent years were obtained with the use of Affimetrix U133 Plus 2.0 and U95 GeneChips (Human) and Mouse genome 430 2.0 arrays, and the BedArray technology—Illumina Sentrix® Expression BeadChip microarrays.
Microarray data analysis
Our methods of data normalization and analysis are based on the use of internal standards that characterize some aspects of system behavior such as technical variability. In general, an internal standard is constructed by identifying a large family of similarly behaving genes. These internal standards are used to estimate in a robust manner those parameters that describe some state of the experimental system such as the identification of genes expressed distinctly from background, differentially expressed genes and genes having similar dynamical behavior. This will be elaborated in detail in the Results section.
Résumé of calculations steps
Upon providing in the Result section, detailed explanations and arguments about the chosen path of calculations, procedures summarizing the calculation steps are presented in six sequential step-by-step résumés.
Step-by-step Résumé 1: individual normalization of the microarray data to background.
Step-by-step Résumé 2: determination of parameters and adjustment of the normalized profiles.
Step-by-step Résumé 3: two-sample data adjustment.
Step-by-step Résumé 4: multi- sample data adjustment.
Step-by-step Résumé 5: reference group of equally expressed genes.
Step-by-step Résumé 6: gene expression analysis.
RESULTS
Statistical monitoring of weak spots
Among the most controversial aspects of the treatment of data that are related to low-intensity signals, is the procedure that enables to distinguish between true (specific) hybridization signals and technological noise. In this context, we consider the genes either as ‘expressed’ or ‘non-expressed’ though this discrimination is not based on biological but rather on technological difference. Depending on the sensitivity of the used technology and on technical quality of experiments, the same low-expression level genes could be treated in high-quality experiments as being expressed (distinctively from nonspecific noise), while in ‘soiled’ experiments (with high level of non-specific hybridizations and/or background noise) they would fall in the category of non-expressed genes. The importance of discrimination of these genes is related to their different information content for subsequent analytical procedures.
Ratio of expressed to non-expressed genes is not a meaningful term
Ratio analysis is commonly employed to determine expression differences between two samples. However, any procedure that uses raw intensities to infer relative expression is imperfect due to the fact that accuracy is signal-level-dependent, with variations increasing dramatically for low intensity signals (9,11,12). Besides, only those ratios that are based on expressed genes are meaningful. The best demonstration of this statement could be obtained with array consisting of duplicated spots for each gene (13). Figure 1 presents results of such an analysis with the use of data from Clontech membrane array (analogous results were obtained also with Perkin-Elmer Micromax cDNA arrays of 2400 human genes spotted in duplicates—not shown). The histogram for the distribution of all spots on the array is presented in Figure 1A. Ratios of duplicated spots that should be equal to 1 with some systematic variations are depicted in Figure 1B. However, this appeared to be the case only for genes expressed above certain threshold level (in this particular set, the threshold being 3). Below this threshold, the ratios are highly variable, demonstrating the absence of any agreement with the duplicate expressions. It follows that the removal of the background level spots should precede any microarray data analysis based on the use of expression ratios.
Figure 1.

Ratio of the duplicated spots in the area of background noise is meaningless. (A) Localization of the normally distributed background noise in the histogram of all microarray gene expressions using iterative exclusion procedure (see Figure 2 and explanations in text). (B) Ratio of the expression levels of the duplicated spots demonstrates increased variability in the area of low-intensity expressions. Fragment of array with duplicated spotting is shown in the right-upper corner. (C) Lack of correlation between the intensities of duplicated spots of low intensities. The axes present intensities of the duplicated spots.
Technologically non-expressed genes represent non-correlated noise
The distribution of the ratios similar to that presented in Figure 1B could also be obtained with expression profiles of samples from a homogenous group, where one expects equal expression of the vast majority of genes. Drastically distorted ratios below a certain level of expression suggest that low levels of gene expression lack any correlation (Figure 1C). A sharp border that discriminates correlated expressions from non-correlated noise is obtained when ‘sliding window’ approach for comparison of the ratio variations (Figure 2) is used. In the presented comparison one set is sorted, while keeping gene association with the second set. Thereafter, an F-test is performed for the standard deviation (SD) of ratios of genes in the ‘window’ (the 10th lowest one is sample one) compared with the SD of ratios of all remaining genes with highest expression. When the window moves like a stencil along the data stream, one obtains comparative characteristics of ratio variability depending upon expression level. There is a sharp border for the P-value (probability for identity of SD in F-test) in this dependence as shown in Figure 2B. Above this threshold, there are all possible levels of P-values from 0 to 1 (10 sequential genes could have very similar levels of expression when the majority of genes in homogenous group of samples are equally expressed), however there are no exclusions for low-expression levels, i.e. all P-values here are close to zero indicating absence of any correlation in the noise level expressions. The border obtained for background noise appears to be in good agreement with the method for obtaining the zone of normally distributed background noise through iterative procedure described below.
Figure 2.

Selection of the normally distributed background noise in the presence of low expressed genes. (A) Histogram of the low-spot distribution after iterative cutting off the expressed genes (see details in text). The presence of low-expressed genes causes in some instances skewing the right side of the background distribution even in high-quality microarrays. For this case, only the left-portion residual after trimming is not distorted by the presence of expressed genes. For estimation of the parameters of the noise distribution, a new histogram is created by substituting the right portion of the background distribution with the mirror image of the left portion. The parameters of the noise distribution are estimated by non-linear fitting of a normal distribution function to this histogram. (B) The sliding window method for estimation of the changes in correlation between gene expressions depending on the level of expression. The F-test is performed for SD of ratios of genes in the ‘window’ (for 10 genes with lowest expression in sample one) compared with SD of ratios of all remaining genes with highest expression. The appearance of the sharp decrease of the P-values (probability for identity of SD in F-test) evidences about the existance of the area of low expression whose variations exceeded significantly the variations of the majority of the rest gene expressions. The position of the sharp decrease of the P-values shows the border for the non-correlated background noise.
Normally distributed additive noise is a convenient internal standard for ‘non-expressed genes’
Several methods have been developed to select ‘non-expressed genes’ and hence to diminish the influence of background noise. One such solution is to ignore genes that yield low total abundance transcripts, another one is to exclude weak spots arbitrarily [in the work of Kooperberg et al. (11)] an intensity cutoff was chosen such that the relative error in ratios was <25%) and still other one is to compare spot expressions with local background level (see Dozmorov et al., 2004 (13) for review). Those procedures for flagging and excluding weak spots that are not based on robust statistical criterion remain problematic since potentially valuable data might be discarded. This issue is compounded by the fact that in biological systems a number of key regulators might be expressed at low levels presumably to ensure a tight control of the expression of regulatory entities (14,15).
The work of Churchill et al. (14) is the first example of solving the problem efficiently with the use of an internal standard. The two main sources of heterogeneity in gene expression variations are indicated in Rocke and Durbin (16) by including the ‘additive component’, prominent at low-expression levels, and the ‘multiplicative component’, prominent at high-expression levels. The intensity measurement yi, j for gene I ∈ I = {i1, … ,in} in sample j ∈ J = { j1, … , jm} is modeled by the equation yi,j = ai,j + (mi,j eh + ei, j), where ai, j is the normal background, mij is the expression level in arbitrary units, ei, j, is the additive error term within a spot, and h is the second error term, which represents the multiplicative component. Gene expression data obtained with the standard procedure of local background subtraction will not exclude spot intensities ei, j, which present additive noise above background levels. The distribution of the spots with ei, j, as predominant member of intensity depends on the array technology used and on the quality of data. Atlas arrays (Clontech) are a good example of high-quality membrane-based arrays exemplifying high specificity and low levels of background. Background spots comprise up to 50% of all spots on the array. The nearly normal distribution of this noise can be seen in a histogram of all intensity values (Figure 3A and B). Parameters of this distribution were estimated with the use of the multi-step iterative procedure.
Figure 3.

Procedure of normalization of the gene expression profile. (A) The histogram of overall gene expressions fits poorly to a normal distribution, with noticeably extended left and right tails. Values at the left tail results from the background correction procedure, while values at the right tail correspond to genes expressed above background. (B) Normal probability plot demonstrates deviations from normality in the tails of the A-distribution. (C) The results of iterative removal of residual background spots demonstrate a good fit to normal distribution. This histogram is used for the estimation of the parameter of normal distribution through the non-linear least-squares curve fitting procedure. Once the parameters of the normally distributed background noise are determined, all expression data are transformed, yielding mean = 0, SD = 1 for background distribution. All gene expressions are presented now in the SD units of the background distribution.
First—the expressed genes are excluded one by one as their values exceed the mean ± 2SD of the core of non-discarded genes. This procedure is repeated in an iterative manner until no additional spot is excluded and the resulting non-discarded values represent the set of non-expressed genes (Figure 3C).
Second—the parameters of the additive noise are estimated by non-linear fitting of a normal distribution function to the core of non-expressed values. The parameters of this distribution [average (Av), SD and the number of members] completely characterize this internal standard of ‘absence of expression’. After that data normalization proceeds by assigning to each experimental value, a normalized score S using the formula S’ = (S – Av)/SD. As a result, the internal standard of the ‘absence of expression’ has a mean of zero and SD = 1 and all gene expressions on array are presented in the SD units of this internal standard.
The iterative procedure described above for discarding the gene expression that alters the normality of the background noise is efficient only with array technology that yields a major gap between the value range of this noise distribution and the set of values of the expressed genes. This was the case with the data obtained with high-quality Clontech membrane array using very sensitive radioactive probes and ensuring that for the probe synthesis only gene-specific primers are used. With these measures, the distance between normally distributed additive noise and majority of low-expressed genes in the histogram is promoted (Figure 3A).
This is not always the case when oligo or random primers are employed. Even in high-quality fluorescently labeled oligonucleotide microarrays (Affimetrix), the distribution of low-intensity noise spots might turn out to be unsatisfactory. The right side of the distribution is often skewed by the abundance of low-expressed genes. This skewness of the distribution can be present even in the histogram obtained upon application of the iterative procedure as shown in Figure 2A. For this case, only the left side of the histogram is used for the estimation of the parameters of the noise distribution. A new histogram is created substituting the right portion of the background distribution with the mirror image of the left portion. Curve fitting is then applied to the new histogram in order to obtain parameters of the noise distribution for subsequent normalization of the array data. This approach to the characterization of the noise distribution seems to be more adequate than attempts to approximate the distorted distribution with artificial combination of overlapping distributions (17,18).
Microarray profiles with relatively low content of non-expressed genes generate another type of problem for localization of the background distribution. The background level spots represent only a relatively small portion of all spots (<30%) in these arrays, thus their distribution is not as prominent as in the previous examples when viewed in a histogram of all spots. The automated iterative procedure for selection of background described above will not locate the background distribution. Therefore, it is necessary to perform a special preliminary step intended to increase the area of the background distribution and focus the iteration procedure onto this area—initial selection of the lowest 30th percentile of data. Then, the new sub-set is trimmed and subsequently curve fitted (see above).
Statistical significance of gene expression—signal/noise discrimination
As we demonstrated earlier (13) the additive noise distribution is quite homogenous over the whole chip after the background correction procedure that makes it possible to use weak spots from the entire chip for estimation parameters of its distribution and use them as a united internal standard for non-expressed genes. Discrimination of ‘expressed’ from ‘non-expressed genes’ is based upon the use of recognized statistical criteria instead of subjective cutoff rules. The power of this statistical criterion is determined by the content of the internal standard—normally several thousand members—and this enables to use relatively high-statistical thresholds without loss of the sensitivity of the selection.
In a replicated experiment, genes that are expressed distinctively above the background noise are readily identified by paired analysis. As it is demonstrated below, data from a replicated experiment upon proper normalization can be used for statistical discrimination of even very weakly expressed genes from the normally distributed noise. Genes with low-level signals—even within background area—could also be identified distinctively from the background due to their higher stability (low SD in replicate measurements).
Step-by-step Résumé 1: individual normalization of the microarray data to background.
The mean and SD are calculated. Using these as a starting point, data beyond +2SD above the mean are cut and discarded. The mean and SD are recalculated and data beyond −2SD below the mean are cut and discarded. This trimming of outlier values above and below is continued, further refining the SD estimate, until no additional cuts can be made.
The rest of data are used for creation of the 10 bar histogram of expression distribution.
Interactive curve fitting for the trimmed data is performed. Using final trimmed data mean and SD are estimated. Theoretical normal distribution is established with estimated mean and SD. Using the theoretical estimate, a non-linear least square curve fitting procedure is performed in order to improve the SD estimate. The quality of fitting is determined visually. If there is some visual distortion of the right tail (proposed presence of weak gene expression) the procedure is repeated using a new user-defined mean (Histogram bars 1–5) and estimating the new distribution on the bars to the left of the chosen one.
In case of low-quality arrays with the abundance of weak expressions distributed too close to background noise the initial choice of the lowest 30th percentile of data is selected to eliminate highly expressed values. Then, the new sub-set is trimmed and subsequently curve is fitted as described above.
Once an appropriate fit is achieved and parameters of the normally distributed background noise is determined as m and s then all the data is Z-transformed 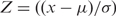 yielding Mean = 0, SD = 1 for background distribution. All gene expressions are presented now in the SD units of the background distribution.
yielding Mean = 0, SD = 1 for background distribution. All gene expressions are presented now in the SD units of the background distribution.
Finally, the data are log-transformed in such a manner that the negative values are substituted with the log of the minimum positive value.
The follow-up is given in the Step-by-step Résumé 2.
Data adjustment
Individual normalization of data from each chip to their background is not sufficient for making their profiles comparable, because first—backgrounds are often different in different experiments, and second—there might be several additional reasons for systemic differences in the expression profiles that can be compensated only by two-parametric regression procedure. This procedure is described in details in the next section and the important feature of it is that this procedure is based on the comparison of potentially equivalent gene expression correlated in compared profiles. The background non-correlated noise could be a serious obstacle for such procedure as it is shown in Figure 1. Knowledge of the background distribution parameters enables to remove the non-correlated noise from correlation adjustments. The threshold 3SD above the mean of background excluded the noise with excess before the final adjustment is made.
The observed variations of the intensity of spots result from biological changes in gene expressions and also due to stochastic and systemic variations that occur in every microarray experiment. In order to accurately and precisely measure gene expression changes, it is important to minimize systemic variations and to estimate the contribution of stochastic variations. Systemic variations appear due to differences in experimental conditions and come from many sources such as procedures of sample handling, methods of cell cultures, methods for mRNA isolation, extraction and amplification, hybridization conditions and labeling efficiencies, as well as due to contamination by genomic DNA [major sources of fluctuations in microarray experiments were listed and discussed in several publications (19)]. The purpose of normalization is to minimize systematic variations in the measured gene expression levels of replicative experiment. Once this is achieved, estimation of the parameters of the stochastic variations the biological differences can be more readily accomplished.
In several excellent reviews, there were proposed different methods of normalization that relieve us from the necessity to discuss them in details (1–3,20). We will note only that the two independent sources of systemic variability in microarray data (additive and multiplicative) need normalization procedures.
Two sample data adjustment
The regression analysis of duplicates from the same array (Figure 1C) presents an excellent example of data having only stochastic variations. Neither multiplicative variations due to differences in hybridization or due to labeling conditions nor additive variations due to non-compensated background noise occur. Both these sources of systemic variations are equal for duplicated spots at the same chip. After exclusion of the area of common non-correlated noise and log-transformation of the data, gene pairs are presented in the scatter plot as dots close to the straight line intercepting the origin with slope 1. The log-transformation is the simplest one making individual gene spots deviating from regression line independently on the level of gene expression and which is normally distributed. The normality can be proven graphically (normal probability plot) or analytically—applying Kolmogorov–Smirnov criteria.
A scatter plot of data from two independent arrays will demonstrate additional systemic variations: ‘additive’—due to differences in background (leading to the deviation of the regression line from the coordinate origin; position of the initial regression line is shown as blue straight line in Figure 4C) and ‘multiplicative’—due to the overall difference in the spot densities (leading to the change of the slope of regression line)—Figure 4C. Transformation of one of these datasets will minimize this differences and make the scatter plot similar to the one obtained for duplicated spots where additional multiplicative and additive differences are absent (compare Figures 1C and 4C).
Figure 4.

Two-sample data adjustment. (A) Histograms for the spots in two arrays. (B) Histograms for normalized to background and log-transformed data. Normal distribution curves fitted to the truncated histograms (as in Figure 3) are shown in green. (C) Profiles of control pool (y-axis) and patient pool (x-axis) adjusted to each other through linear regression with excluded background noise (black spots) and potential outliers. Blue line, position of the regression line before adjustment; green line, position after adjustment. (D) Data of the plot presented in the transformed coordinates. Right side shows the nearly normal distribution of the deviations from equity of expression. The use of the majority of equally expressed genes as an internal standard presents opportunity to select differentially expressed genes as outliers from this standard beyond of some statistical thresholds. These genes are shown as open circles.
An array of gene expression profiles may be conceptualized as a vector of outcomes in the scatter plot of data. Let Yk = (Y1k, Y1k, … , YJk) denote the array, where Yjk denotes the expression of the j-th gene in the k-th sample (j = 1, 2, … , J; k = 1, 2, … , K).
in which (aj, bj) are gene-specific additive and multiplicative factors, 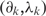 are the sample-specific regression coefficients, and
are the sample-specific regression coefficients, and 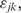 is used to depict variations due to all unknown sources. Estimated regression factors are used for overall adjustment of the expression levels in one sample to another as
is used to depict variations due to all unknown sources. Estimated regression factors are used for overall adjustment of the expression levels in one sample to another as 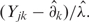 After these adjustment relations of the expressions in two samples presented as Yjk = aj + bjxk are obtained where aj presents the difference in local background and bj—multiplicative factor. For data acquisition with local background subtraction the aj are minimized or even disappear and a log-transformation produces expressions differing by the additive close to normal distribution noise log(bj) that is an unified measure variation in gene expression essentially unrelated to the influence of level of expression.
After these adjustment relations of the expressions in two samples presented as Yjk = aj + bjxk are obtained where aj presents the difference in local background and bj—multiplicative factor. For data acquisition with local background subtraction the aj are minimized or even disappear and a log-transformation produces expressions differing by the additive close to normal distribution noise log(bj) that is an unified measure variation in gene expression essentially unrelated to the influence of level of expression.
The described adjustment leads to maximal similarity of expression of all genes in two arrays. This procedure, however, will be incorrect in the presence of differentially expressed genes, because it will aspire to make them equally expressed also. It means that the presence of differentially expressed genes can seriously impede the adjustment procedure. Generally, their influence could be minimal if they are distributed more or less symmetrically around regression line. However, the presence of not compensated outliers might influence the bias adjustment drastically, especially when such unbalanced outliers are present in the area of very high expressions—usually area less populated with spots. These outliers violate the assumption of normally distributed residuals in least squares regression. They tend to pull the least squares fit too much in their direction by getting considerably more ‘weight’ than they deserve.
Various methods were proposed to diminish the distorting influence of differentially expressed genes. They were based mainly on arbitrary estimations of permissible distances from equity line. The procedure of revealing and down weighting could be produced on the strong statistical basis using another internal standard—family of equally expressed genes. Fortunately, in any normal experiment, the majority of genes are equally expressed, and their variations around regression line have prominent distribution that can be elicited by the iteration procedure described earlier for background data analysis. Such stochastic distribution of the deviations of gene positions looks very similar to the distribution obtained for duplicated spots in Figure 1C. A histogram of these deviations (Figure 4D) includes the normal distribution with tails distorted by the presence of differentially expressed genes that could be selected and excluded once the parameters of the normal distribution are determined.
The stochastic distribution of the random variations is typically unknown. In our practice of making hundreds of analyses using different technological platforms, we were never confronted with a violation of the normality assumption, nevertheless, if hypothetically the assumptions of normality are violated, some non-parametric criteria will be more reliable for making statistical inferences—as. For example, Thomas et al. (21) proposed to use Z-scores that is closely connected with Wilcoxon rank sum statistics (22). Z-scores do not require any distributional assumptions or homogeneity of deviations. In practice, Z-scores are expected to be similar to t-test statistics, when the distribution of expression levels can be approximated by the normal distribution. When these assumptions are violated, Z-scores will differ from t-statistics and will be more reliable for making statistical inferences.
Step-by-step Résumé 2: determination of parameters and adjustment of the normalized profiles.
The first step is the determination of the parameters of the background of the array—Av and SD of normally distributed low-level expressions in array with subsequent normalization of all expressions in array. A normalized score, ‘S’, is obtained [S = (PV – Av)/SD], where PV is the original pixel value for the spot, and Av and SD are the mean and SD of the set of background spots. The distribution of S has mean of zero and SD = 1 over the set of background genes in the normalized array. We accept S = 3SD above the mean background level as the preliminary criterion for distinguishing expressed from non-expressed genes. Only genes expressed above background are used for the second step ‘adjustment’ as described below.
The second step is the adjustment of the normalized profiles to each other by robust regression analysis of genes expressed above the background. This procedure is based on the selection of equally expressed genes as a homogenous family of genes with normally distributed residuals defined as deviations from regression line. The parameters of this distribution are obtained by iterative procedures similar to the one used before for the selection of the kernel part of normally distributed background noise. Outliers are thereafter determined as having deviations not associated with this internal standard of equity in expression including thousands of members (Figure 4D).
The follow-up is given in the Step-by-step Résumé 3.
Nonlinear regression
Linear regression analysis will be valid only if (i) the hybridization signal is linearly related to target concentration and (ii) the majority of the genes expressed in both samples are expressed equally. Bias adjustment transforms the dependence between two samples into a simple multiplicative model (see above). Sometimes, however such a model is inadequate. Such cases can be identified on the scatter plot when a straight line fits the data poorly and instead a curved shape results. The use of straight line for normalization can lead to a high rate of false positive results. A variety of approaches to normalize such gene expression data have been proposed, including a cubic spline transformation (23,24), and locally weighted linear regression [Lowess; see for review Do et al. (2006) (2), Bolstad et al. (2003) (20) and Wu (2001) (25)].
Remarkably, the assumption that non-linear transformation is always beneficial for tests for differentially expressed genes has never been properly tested. Making the choice in favor of the non-linear normalization procedures, it is necessary to keep in mind that serious problems might occur in cases where the non-linearity is the result of non-homogenous distribution of differentially expressed genes of opposite directions. From this perspective, the non-linear transformation can be beneficial for the adjustment of profiles of samples from a homogenous group. However in a comparative analysis, this method bears a definite danger of losing sensitivity of discrimination of the differences in gene expression.
The examination of examples of non-linear distribution of gene expression in the regression plot indicates that in most cases essential non-linearity is present in the area of low-gene expressions. The exclusion of the background area and of the closely associated low-expressed genes is able to diminish considerably the influence of such non-linearity.
The residual essential non-linearity is an evidence of the low quality of the technological procedure and the best way to correct it is to avoid it in the first place. Examination of the quality of the data from high-throughput platforms ‘prior to interpretative analysis’ is a critical step that will help researchers to avoid contaminating their otherwise well-conducted study with samples harmful to overall analysis and interpretation.
Step-by-step Résumé 3: two-sample data adjustment.
Regression analysis of two-sample data gives residuals (deviations from regression line) for each gene expressed >3 (=0.477 after log-transformation) in both samples.
The mean and SD of all residuals are calculated. Using these values as a starting point for data trimming as described above, the parameters of the normal distribution of the majority of residuals are obtained.
The probability of belonging to the normal distribution of the majority of residuals (for equally expressed genes) is estimated for each gene (each residual).
Genes having probability less than 1/N (N—number of all genes expressed >3 in both samples) are excluded and the regression analysis for the rest of them is used for estimation and exclusion of additive and multiplicative factors.
The result of adjustment can be presented in transformed coordinates with indicated borders ± (1/N) for differentially expressed genes (Figure 4D).
The follow-up is given in the Step-by-step Résumé 4.
Multiple-sample data adjustment
Many of the issues that we discussed in the two-sample case, such as bias correction, remain important for replicate experiments, although we will not discuss them further. Often the two-sample methods can be generalized to handle replicate experiments. For example, we can extend the methods for bias correction by normalizing across a series of N samples, rather than one sample against another. In this case, the solution involves fitting a normalization curve in an N-dimensional space. However, in practice, we successfully use different iterative procedures of normalization to common averaged profile as detailed in Figure 5. In this multi-step procedure, we use averaged profile for bias adjustment of each individual profile with subsequent recalculation of the averaged profile and repetitive adjustment.
Figure 5.

Multi-sample data adjustment to the averaged profile using robust regression procedure. Data first normalized to background with procedure described above (A). The averaged profile (B) is created and data at each sample adjusted to this average profile with robust regression procedure (C). After that, a new averaged profile from transformed data is created. Several subsequent cycles may be necessary for the best result, however, for the data initially normalized to background, two-steps adjustment is usually enough.
Step-by-step Résumé 4: Multi-sample data adjustment.
Averaged profile is calculated and each sample is adjusted to the averaged profile using robust regression procedure described earlier for two-sample adjustment.
New averaged profile is calculated from transformed profiles of the samples and the adjustment procedure is repeated.
Several subsequent adjustment may be necessary for the best result, however for the data initially normalized to background two steps of adjustment are usually enough.
The result of the adjustment can be presented in transformed coordinates in form of Mean + SD of multiplicated residuals for each gene (Figure 6A).
Figure 6.

Reference group—the main internal standard for Associative Analysis of differentially expressed genes. The reference group (B) is created from initial distribution of the residuals (A) after trimming of hyper-variably expressed genes (HVE genes) with use F-test. (C and D) Reference group as an internal standard for equity in expression [normally distributed deviations in the left part (E)] and for stability of expression [F-distributed SDs in the right side (F)].
The follow-up is given in the Step-by-step Résumé 5.
Reference group—an internal standard for replicate experiment
One of the problems in performing a reliable t-test from microarray data is to obtain accurate estimates of the SDs of individual gene measurements based on only a few measurements. It has been, however, observed that an overall reciprocal relationship exists between variance and gene expression levels, and that genes expressed at similar levels exhibit similar variance (26). Beside that, there were obtained transformations depriving variance dependence on the gene expression levels (27). Log-transformation is one of the simplest examples of such transformation. Therefore, it is possible to use this prior knowledge to obtain more robust estimates of variance for any gene by examining the expression levels of other genes within a single experiment.
After normalization, the residuals from the calibration data are used to provide prior information on variance components in the analysis of comparative experiments. After adjustment of the each array profile to the averaged profile for the control group, we obtain two new standards joined by the common name ‘reference group’.
First, all genes are represented here by their residuals (relatively averaged profile) that after normalization and log-transformation loose their sample dependent and expression level dependent individualities (Figure 6A and C). As soon as absolute majority of genes in homogenous group are equally expressed, their residuals demonstrate very similar to normal distribution (Figure 6E).
Second, the residuals of these genes in the replicated experiment could be presented as mean ± SD. For the majority of genes, their replicate variations are relatively small and homogenous following to the standard F-distribution. The small portion of genes having enormously high (statistically distinctive from the rest) variation present so called hypervariable genes (HV-genes), whose nature was discussed elsewhere (28,29). To get the internal standard for gene variability, HV-genes should be excluded by iterative procedure similar to described above (for normally distributed background events and for normally distributed residuals of equally expressed genes). The only difference is that in this procedure, the F-test is used as a criterion for the exclusion of outliers. To perform the F-test, we compare two estimates of variance, one from the variability of expression levels of the entire group, and the other from the variability of the expression level of every given gene. If the gene variability estimate is much higher than the total-group estimate, we have evidence that the given gene does not share the same stability as a majority of genes and should be excluded from the reference group. The procedure continues until no more genes could be excluded in this manner. The result of all these exclusions is a new internal standard—the reference group, composed of genes expressed above background in control samples with normal low variability of expression (as determined by an F-test) and whose residuals approximate a normal distribution.
Very similar standards for equity of expression and stable variability were introduced earlier by Rocke and Durbin (16). However, none of them were cleaned from HV-gene contamination, with the consequence that the standards were biased, thus decreasing significantly the sensitivity of the criteria.
Step-by-step Résumé 5: reference group of equally expressed genes.
In course of normalization with bias adjustment
residuals as differences between final normalized expression and the average before last adjustment are calculated;
SD of all residuals taken together are calculated;
SD for all genes individually are calculated;
F-test is performed on every gene to determine if the variability is higher than that of all genes;
all genes whose SD is higher than in step (ii) and/or fail F-test are excluded;
SD for all remaining genes are recalculated;
steps (iv)–(vi) are repeated until no further genes can be excluded
The follow-up is given in the Step-by-step Résumé 6.
Associative analysis—identification of differentially expressed genes
The use of the reference group created in the previous section, as an internal standard enables to carry out differential gene expression analysis, and what is of utmost importance, it solves the problem of mutually exclusive characteristics of sensitivity and specificity. For this purpose, we use an associative t-test (30) developed as a modification of the ‘General Error Model’ (16) in which the replicated residuals for each gene of the experimental group are compared with the entire set of residuals from the reference group. The null hypothesis is checked to determine if gene expression in the experimental group is associated with the reference group. The significance threshold is corrected to make the appearance of false positive determinations improbable.
Selecting differentially expressed genes relies on five statistical steps.
Assume Group 1 has n samples and k genes and Group 2 has m samples and k genes. A Student’s t-test is performed, with (n + m − 2) degrees of freedom, in order to determine if the genes are equally expressed.
Then an associative t-test is performed, with (m + k − 2) degrees of freedom to see if the gene belongs to the group of equally expressed genes with stabile variability. Selections passing through both tests have high sensitivity (Student’s t-test with normal low threshold P < 0.05) and high specificity (subsequent associative t-test with corrected threshold P < 1/k excludes all false positive determinations).
Another two Student’s t-tests are used to establish the distinction from technical noise—discrimination of ‘expressed’ from ‘non-expressed’ genes.
Finally, the ratio of gene expressions in Groups 2 and 1 is used to help exclude statistically significant but not biologically significant changes.
Clearly, simple discriminations based on ‘fold changes’ or ratios are insufficient for drawing proper conclusions. But, we use foldness restrictions as an addition to the statistical analysis of differentially expressed genes to concentrate attention on the most prominent differences first of all.
The t-test assumes that the replicate data have an underlying normal distribution. This assumption is reasonable, especially if the replicate samples are relatively homogeneous. Note that the assumption of normality is different in these two subsequent steps of the analysis. In the first step—paired comparison—in most cases, we have relatively few replicate samples and it is difficult to test for normality having only a few data points. Therefore, we often adopt the assumption of normality because it is hard to prove otherwise. In the second step—associative analysis—we use the reference group as an internal standard and proved that after log-transformation and exclusion of outliers with iterative procedure the rest of residuals has a distribution whose normality is confirmed by statistical and graphical criterions.
The two step procedure allows the use of traditional low-level significance cutoffs (P < 0.05) at the first step without the risk of including false positive selections. These false positives are excluded in subsequent second step—associative analysis having extreme statistical power enabling to use the significance cutoff corrected to the number of comparisons without risk to loose sensitivity. The use of the reference group enables to receive all benefits of the thousands replicates of technical variations—deviations from equity—to increase statistical power of the comparative analysis. This analysis is based on an idea, which is opposite to the commonly held view that large-scale array experiments suffer from compensatory tradeoffs in sensitivity and specificity. In fact, the procedures presented herein demonstrate that large scale datasets are extraordinary information-rich and provide means for discrimination of common technical variation from individual biological variability. More evidence of this is presented in a power analysis (Figure 7).
Figure 7.

Power analysis. Estimation of the number of microarray experiments required to obtain reliable results from a comparison of data from patients and controls. The sample size was estimated using PASS 2005 (Keysville, Utah). Our experience with different array technologies (including ‘Illumina’, which is used here) indicates that a coefficient of variation between 0.25 and 0.5 is typical among expressed genes. The left portion of graph demonstrates the dependence of the power of analysis on the number of replicates for a paired T-test with a statistical threshold of α = 0.05. On the right portion of the graph, power analysis results from an associative analysis are estimated. An associative analysis with threshold of α = 0.0001 has power comparable with a paired T-test using a threshold of α = 0.05. Results of this analysis will be used for estimating the number of replicate experiments required for selection of differentially expressed genes. For example 2- to 3-fold difference can be observed with power 1 − β = 0.8 with a 6-replicate experiment.
Step-by-step Résumé 6: in this step, gene expression analysis is described.
Selection with a Student’s t-test for replicates using the commonly accepted significance threshold of P < 0.05. It keeps the commonly accepted sensitivity level, however a significant proportion of genes identified at this threshold level as differentially expressed will be false positive determinations.
An associative t-test in which the replicated residuals for each gene of the experimental group are compared with the entire set of residuals from the reference group defined above. Ho hypothesis is checked if gene expression in experimental group presented as replicated residuals (deviations from averaged control group profile) is associated with highly representative (several hundreds members) normally distributed set of residuals of gene expressions in the reference group. The significance threshold is corrected to make the appearance of false positive determinations improbable. Only genes that passed through both tests were presented in the result tables.
Genes expressed distinctively from background were determined by analysis of the association of each replicated gene expression with normally distributed background having Av = 0 and SD = 1. Genes expressed distinctively from background in one group and not distinctive from background in another group are given as further example of differentially expressed genes.
Data filtration and error exclusion procedures
Selection of ‘bad’ samples
The local errors in the data acquisitions will be able to produce significant increase of the SD for given gene in replicated experiment. It is possible to use the F-test for selection of such errors, however the problem of the sensitivity/specificity alternative will prevent from accurate estimation of outliers. At the same time, the summary estimation of such outliers in every given sample will enable to characterize overall quality of the array data in every chip. We propose a simple program for the chip quality estimation. In a homogenous group of samples for each gene in the array, we estimate the changes in its variability by comparing the SD of the total set of expressions with the SD obtained after exclusion of one replicate after another. If the F-test results in probability for no difference being <0.05 then this gene expression in the given sample is considered as an outlier. Finally, the resulting outliers estimated for every sample are considered as of bad quality and are excluded from analysis. The use of non-corrected low threshold P < 0.05 produces massive presence of false positive selections. The sum of these false selections should be comparable for all good quality samples presenting internal standard of good quality sample that can be used for statistical selection of bad samples with significantly elevated number of outliers. The program EFILTER produces the histogram of the numbers of outliers in samples for the visual inspection of the group quality.
Ranking of selections
Another method of data filtration is based on the comparison of the results of many differential expression analyses produced with sequential exclusion of samples one by one to determine the dependence of the conclusion about any selection on the exact group’s content. This method determines the robustness of the differential gene expression selections and deliberates them from the influence of singular experimental errors.
The analysis uses standard Associative Analysis algorithms (30). The ‘leave-one-out’ approach excludes one sample from the group—one by one until all possible singular exclusions are produced—with estimation of the frequency of positive selection for every gene. This approach produces accurate ranking estimation of the robustness for most selections. However, it is not safe from the effect of singular errors of measurements, because the presence of one such outlier within any of the replicate is able to mask its difference of expression and diminishes the rank of otherwise ideal selection. The next modification of the procedure makes it defended from this effect of singular errors. Note that the exclusion of the ‘bad’ replicate and re-estimation of the robustness of the given selection will produce results devoid of the outlier influence. The new algorithm can be named as ‘leave-two-out’, because includes preliminary step—exclusion of one sample—with subsequent application ‘leave-one-out’ procedure for the rest of the samples in consideration. For an experiment having total number of samples equal n (sum of samples in both compared groups), this algorithm will produce a set of n ranks for each excluded sample and highest of them will be the one most independent on the worse replicate. Compared with previous EFILTER procedure, the TWOEX algorithm provides the opportunity to benefit even from a relatively bad sample, incorporating only expressions and excluding erroneous measurements. Based on the use of standard program for associative analysis, this algorithm enables to produce ranking estimation with selected restriction on the minimal expression and foldness being an adequate addition to the standard associative analysis.
Estimation of the quality of differential expression analyses
For the estimation of quality, we use ‘artificial’ data with controlled differences in gene expressions. The presumably homogenous group of samples was divided into two sub-groups. One of them was used as a control, whereas in another sub-group (experimental) artificial changes in gene expressions were introduced. Towards this aim, all data were sorted according to the averaged gene expression in experimental group. The entire data set was split into 1000 gene blocks, and thereafter controlled balanced (±) changes were introduced into 20% of data of experimental group. Within each block (1000 genes) 100 genes received positive changes—multiplied by ‘foldness’, and 100 genes received negative changes—divided by ‘foldness’). One such block is presented in Figure 8. After applying the analysis procedure, the resulting number of selections is compared with true selections for determination of the ‘Sensitivity’ and ‘Specificity’ of the given analysis as it is shown in Figure 8.
Figure 8.
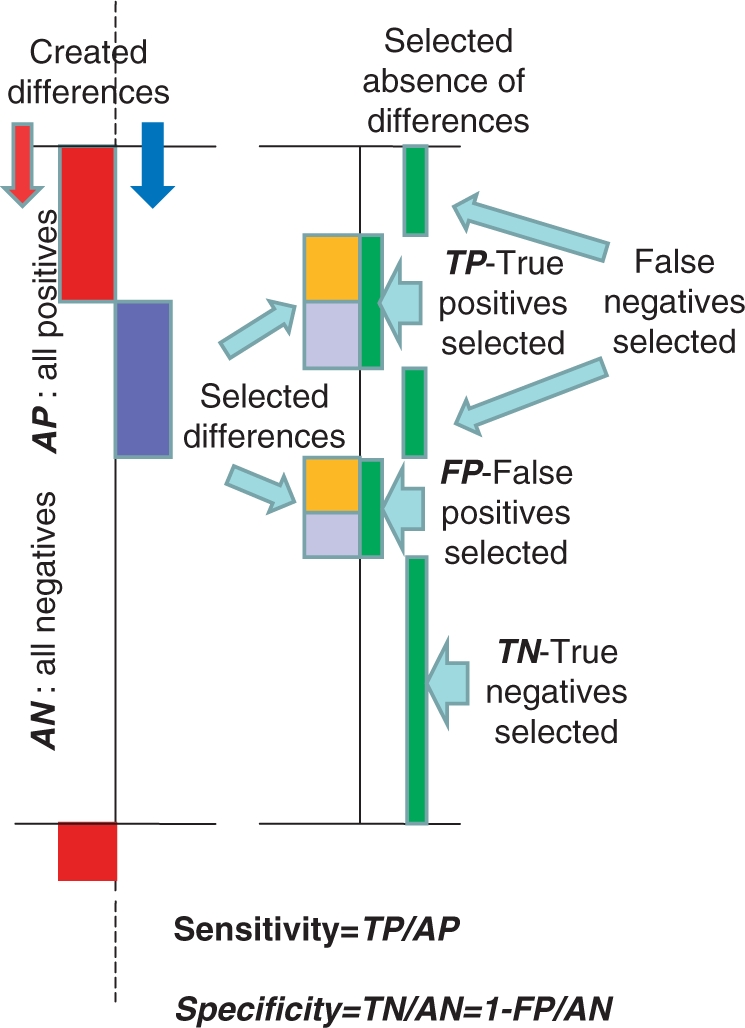
Test system for determination of the sensitivity and specificity of the differential gene expression analyses. The presumingly homogeneous group of samples was divided to two equal sub-groups one of which not changed used as a control and another one used as a experimental group with introduced changes. Here is shown a fragment of these experimental dataset with introduced positive (red) and negative (blue) changes in the 20% portion of gene expression—left part. Right part presents differences selected by the differential gene expression analysis (left of the vertical axis) with indication on the right side from the axis which of this selection is true (co-incidenting with the artificially made selections) and which are false. The sensitivity of selections is determined here as a proportion of true positive selections within all produced changes, whereas a specificity determined as a proportion of true negative selections. The fragments with artificial changes presented here are evenly distributed along all experimental group.
The presented system enables to compare different methods of data normalization, and it enables also to estimate the role of restrictions made in course of differential gene expression analysis. The following designations were used in this analysis.
Fd—‘foldness’ of controlled changes in the data;
Fa—minimal ‘foldness’ of Associative Analysis;
Em—minimal expression for genes selected as being expressed distinctly from background in Associative Analysis.
Results using data obtained with mRNA collected from peripheral blood mononuclear cells from healthy donors with the use of ‘Illumina microarray’ technology are presented in Figure 9. Quality of analysis is estimated here by the two parameters: sensitivity is determined as a proportion of true positive selections within all introduced changes, and specificity determined as 1—portion of false positive selections among all not changed expressions (31).
Figure 9.

Sensitivity/specificity (Sn/Sp) characteristics of the normalization and analyses procedures. (A) Dependence of the analysis quality of the foldness of changes in gene expression: along ordinates Fd-foldness of controlled changes of data/Fa-minimal foldness accepted for results of differential gene expression analysis/Em = 20-minimal expression above background. (B) Dependence of the analysis quality of the number of replicates. Fd/Fa/Em = 2/1.5/20. (C) Comparison of normalization methods: two-step analysis (presented above) versus Quantile normalization versus Lowess normalization. (D) Comparison of analysis methods: associative analysis—SAM with Quantile normalization—SAM with Loess normalization. Abscise—sensitivity and specificity of the analysis as described in text.
Figure 9A demonstrates the dependence of sensitivity and specificity in terms of the relationship between Fa and Fd. When Fa < Fd, the Associative Analysis of normalized data selects more that 80% of changes. Sensitivity drops down sharp when the Fa becomes comparable or even higher than the ‘foldness’ of introduced changes Fd.
The number of replicates is the most essential parameter form the output quality. Figure 9B shows a sharp decrease of the sensitivity of analysis for the number of replicates <4. Five to six replicates could be recommended as minimal size of the groups, whereas the usually used four replicated experiments might loose up to 20% of true differences. These numbers could vary in different microarray technologies and with the use samples from different sources. This result could be used as an alternative of the standard methods for the estimation of the power of analysis and the number of replicates necessary to achieve desired quality of analysis. The advantage of this approach is in the use of real data with practically not distorted infrastructure (variations and their distributions over expression levels) for estimation of the quality of the future analysis.
The method presented here enables the comparison of the quality of different types of analysis and influence of different normalization methods. In Figure 9C, we compared results of associative analysis with use different methods of normalization. It appeared that the use of our two-step normalization procedure and two popular methods Quantile (Q) and Lowess (L) (32) produced very comparable results except the area of highly expressed genes (first, thousand genes with highest expression) where quality of analysis based on the use Q- and L-normalizations significantly worse compared with two-step normalization presented here as it is shown in Figure 9C. Quite obvious that the same difference in quality was presented in comparison of our Associative Analysis based on the use two-step normalization and SAM analysis that used Quantile and Loess normalizations (32; Figure 9D).
To estimate the stability of the obtained estimations, we modified the quality analysis by using several variants of arbitrary splitting of the total dataset to two equal sub-groups (column permutation with subsequent splitting). The averaged result of five permutations presented in Figures 9A and B demonstrates relative stability of these estimations.
DISCUSSION
Current statistical methods do not adequately address mutually exclusive characteristics of sensitivity and specificity in microarray experiments monitoring the expression levels of thousands genes simultaneously. The common practice to use low-significance thresholds (P < 0.05) will result in a large number of false positive selections. Attempts to increase stringency by raising the threshold of significance above this value will cause a compensatory decrease in sensitivity and a resultant increase in false negative selections.
In measurements of gene expressions, the biological component is accompanied with variations of non-biological origin coming from a number of different sources. Normalization reduces systemic variations, while not affecting random variations. Common practice is to obtain information about random variation from replicated measurements. The number of replicates is critical for the accuracy of estimation of random variation and biological component as well. The use of large numbers of replicates is able to improve the situation in microarray experiments as well (33,34), although it can be rather expensive and labor intensive. Fortunately, there is a real resource to increase the power of statistical tests by using the enormous mass of information coming from each microarray experiments. We introduce here an approach based on the use of internal standards—large families of genes sharing some important features, while not being dependent on any particular gene sequence, level of expression, or coordinate position on the chip. Here were discussed standards for equity in gene expression, stability, standard for expressions below the sensitivity of the system (standard for ‘non-expressed’ genes). Deprived with dependence on the level of expression elements such standards bear information about experimental variation replicated thousands times by the count of the elements in the standard. This is an alternative to replications for increasing the power of statistical criterions. The increase of the power from such huge ‘replication’ should be tremendous.
The two main problems should be resolved before using this approach. Is the distribution of the elements of the internal standard normal and how to determine parameters of this distribution? Usually, each internal standard is contaminated with outliers. For example, majority of genes are equally expressed in any homogenous group and have a relatively small variability, however there are always some genes that does not share these features. Reduction of the influence of outliers is a critical step in the analyses based on the use of internal standards. Fortunately, this contamination with outliers is always relatively small and can be selected and removed with simple procedures.
The problem of normality is solved for this standard in several different ways. The selection of the normally distributed additive noise (background) is solved by using only the left portion of the non-distorted part of distribution for fitting to normal distribution. Standard of equity of expression and standard of the stability (reference group) appeared to have normal distribution after exclusion of outliers in the simple iterative procedure. It means that the rest of the distribution obtained after sequential truncation steps was always satisfactory fitted to the normal distribution. Even if there is some contamination with not normally distributed members, it is not essential and does not interfere with the normality of the rest.
Procedures similar to associative analysis have been previously proposed by Newton et al. (35); Rocke and Durbin (16); Tseng et al. (36). However, there are critical differences between these methods and ours. For example, in Rocke and Durbin (16), all genes were used as a reference group without exclusion of HV-genes. The presence of HV-genes increases the SD of the residuals in the reference group, thus reducing the power of the associative analysis.
There were versatile assumptions about the distribution of the background level signals and additive error term in the literature. Rocke and Durbin (16) were the first to suggest the use of iterative procedure for estimation of background parameters similar to the procedure presented here. Our approach goes one step further and demonstrates that the apparent deviation of the additive noise distribution from normality is produced by the presence of the weak signals overlapping with the noise. These results enable the skewed distribution presented in Figure 2 to be treated as a normally distributed additive noise distorted on its right side by the presence of low gene expressions.
The estimation of the performance of microarray data analysis demonstrated an advantage of the proposed here normalization and analysis methods over the popular normalization (Quantile, Loess) and analysis (SAM) procedures. The application of the methods presented here to various biological and clinical problems demonstrated their ability to reveal essential features of the systems under investigations [see for example (28–30,37–43)], confirmed by the subsequent analysis of signaling pathways involved, transcription factor analysis and comparison with other publications. In some applications, the parallel use of different approaches to the analysis of the same data demonstrated advantage of the internal standard based methods over others in the selection of the gene sets reasonably associated with the studied phenomenon [see for example Dozmorov and Centola (2003) (30)].
Internal standard-based analysis enables to improve the power of microarray analysis at several levels. In the next part, we will demonstrate that the knowledge of the parameters governed by internal standards can be used for analysis in a statistically robust manner also for functional associations through clustering and networking genes having similar dynamical behavior.
FUNDING
National Institutes of Health (grants P20 RR020143, R01 AI045050 and P30 AR053483); National Institutes of Health/National Center for Research Resources – Centers of Biomedical Research Excellence (grant IRG-05-066-04); American Cancer Society (grant IRG-05-066-04). Funding for open access charge: American Cancer Society (grant IRG-05-066-04).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
Authors thank Michael Centola, Richard Miller, Edward Wakeland and Nicholas Chiorazzi for fruitful discussions, Nicholas Knowlton and Shengguang Qian for help with programming and Jonathan Wren and Teodor Ene for the help in the preparation of this article.
REFERENCES
- 1.Lee NH, Saeed AI. Microarrays: an overview. Methods Mol. Biol. 2007;353:265–300. doi: 10.1385/1-59745-229-7:265. [DOI] [PubMed] [Google Scholar]
- 2.Do JH, Choi DK. Normalization of microarray data: single-labeled and dual-labeled arrays. Mol. Cells. 2006;31:254–261. [PubMed] [Google Scholar]
- 3.Hua J, Balagurunathan Y, Chen Y, Lowey J, Bittner ML, Xiong Z, Suh E, Dougherty ER. Normalization benefits microarray-based classification. J. Bioinform. Syst. Biol. 2006:43056. doi: 10.1155/BSB/2006/43056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saviozzi S, Calogero RA. Microarray probe expression measures, data normalization and statistical validation.Comp. Funct. Genomics. 2003;4:442–446. doi: 10.1002/cfg.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holm S. A simple sequentially rejective multiple test procedure. Scand. J. Statistics. 1979;6:65–67. [Google Scholar]
- 6.Cheng C, Pounds S. False discovery rate paradigms for statistical analyses of microarray gene expression data. Bioinformation. 2007;1:436–446. doi: 10.6026/97320630001436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B. 1995;57:289–300. [Google Scholar]
- 8.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin D, Shkedy Z, Burzykowski T, Ion R, Göhlmann HW, Bondt AD, Perer T, Geerts T, Van den Wyngaert I, Bijnens L. An investigation on performance of Significance Analysis of Microarray (SAM) for the comparisons of several treatments with one control in the presence of small-variance genes. Biom. J. 2008;50:801–823. doi: 10.1002/bimj.200710467. [DOI] [PubMed] [Google Scholar]
- 10.Knowlton N, Dozmorov I, Centola M. Microarray data analysis toolbox (MDAT) for normalization, adjustment and analysis of gene expression data. Bioinformatics. 2004;20:3687–3690. doi: 10.1093/bioinformatics/bth424. [DOI] [PubMed] [Google Scholar]
- 11.Kooperberg C, Fazzio TG, Delrow JJ, Tsukiyama T. Improved background correction for spotted DNA microarrays. J. Comput. Biol. 2002;9:55–66. doi: 10.1089/10665270252833190. [DOI] [PubMed] [Google Scholar]
- 12.Attoor S, Dougherty ER, Chen Y, Bittner ML, Trent JM. Which is better for cDNA-microarray-based classification: ratios or direct intensities. Bioinformatics. 2004;20:2513–2520. doi: 10.1093/bioinformatics/bth272. [DOI] [PubMed] [Google Scholar]
- 13.Dozmorov I, Knowlton N, Tang Y, Centola M. Statistical monitoring of weak spots for improvement of normalization and ratio estimates in microarrays. BMC Bioinformatics. 2004;5:53. doi: 10.1186/1471-2105-5-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Churchill GA, Oliver B. Sex, flies and microarrays. Nat. Genet. 2001;29:355–356. doi: 10.1038/ng1201-355. [DOI] [PubMed] [Google Scholar]
- 15.Wei J, Riley RM, Wolfinger RD, White KP, Passador-Gurgel G, Gibson C. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat. Gen. 2001;2:389–395. doi: 10.1038/ng766. [DOI] [PubMed] [Google Scholar]
- 16.Rocke DM, Durbin BA. A model for measurement error for genhe expression analysis. J. Comput. Biol. 2001;8:557–569. doi: 10.1089/106652701753307485. [DOI] [PubMed] [Google Scholar]
- 17.Takeya M, Matsuda T, Iwamoto M, Tsumura M, Nakaguchi T, Miyake Y. Noise analysis of duplicated data on microarrays using mixture distribution modeling. Opt. Rev. 2007;14:97–104. [Google Scholar]
- 18.Sidorov IA, Hosack DA, Gee D, Yang J, Cam MC, Lempicki RA, Dimitrov DS. Oligonucleotide microarray data distribution and normalization. Inform. Sci. 2002;146:67–73. [Google Scholar]
- 19.Schuchhardt J, Beule D, Malik A, Wolski E, Eickhoff H, Lehrach H, Herzel H. Normalization strategies for cDNA microarrays. Nucleic Acids Res. 2000;28:E47. doi: 10.1093/nar/28.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 21.Thomas JG, Olson JM, Tapscott SJ, Zhao LP. An efficient and robust statistical modeling approach to discover differentially expressed genes using genomic expression profiles. Genome Res. 2001;11:1227–1236. doi: 10.1101/gr.165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheadle C, Vawter MP, Freed WJ, Becker KG. Analysis of microarray data using Z score transformation. J. Mol. Diagn. 2003;5:73–81. doi: 10.1016/S1525-1578(10)60455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Workman C, Jensen LJ, Armer H, Berka R, Gautier L, Nielser HB, Saxild HH, Nielsen C, Brunak S, Knudsen S. A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-9-research0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujita A, Sato JR, Rodrigues LO, Ferreira CE, Sogayar MC. Regulatory dendritic cells protect against cutaneous chronic graft-versus-host disease mediated through CD4+CD25+Foxp3+ regulatory T cells. BMC Bioinformatics. 2006;7:469. doi: 10.1182/blood-2007-04-086470. [DOI] [PubMed] [Google Scholar]
- 25.Wu TD. Analysing gene expression data from DNA microarrays to identify candidate genes. J. Pathol. 2001;195:53–65. doi: 10.1002/1096-9896(200109)195:1<53::AID-PATH891>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 26.Hatfield GW, Hung SP, Baldi P. Differential analysis of DNA microarray gene expression data. Mol. Microbiol. 2003;47:871–877. doi: 10.1046/j.1365-2958.2003.03298.x. [DOI] [PubMed] [Google Scholar]
- 27.Durbin BP, Rocke DM. Variance-stabilizing transformations for two-color microarrays. Bioinformatics. 2004;20:660–667. doi: 10.1093/bioinformatics/btg464. [DOI] [PubMed] [Google Scholar]
- 28.Dozmorov I, Knowlton N, Tang Y, Shields A, Pathipvanich P, Jarvis J, Centola M. Hypervariable genes – experimental error or hidden dynamics. Nucleic Acids Res. 2004;32:e147. doi: 10.1093/nar/gnh146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dozmorov IM, Centola M, Knowlton N, Tang Y-H. Mobile-classification in microarray experiments. Scand J. Immunol. 2005;62(Suppl. 1):84–91. doi: 10.1111/j.1365-3083.2005.01614.x. [DOI] [PubMed] [Google Scholar]
- 30.Dozmorov IM, Centola M. An associative analysis of gene expression array data. Bioinformatics. 2003;19:204–211. doi: 10.1093/bioinformatics/19.2.204. [DOI] [PubMed] [Google Scholar]
- 31.Khodarev NN, Park J, Kataoka Y, et al. Receiver operating characteristic analysis: a general tool for DNA array data filtration and performance estimation. Genomics. 2003;81:202–209. doi: 10.1016/s0888-7543(02)00042-3. [DOI] [PubMed] [Google Scholar]
- 32.Chiogna M, Massa MS, Risso D, Romualdi C. A comparison on effects of normalisations in the detection of differentially expressed genes. BMC Bioinformatics. 2009;13:61. doi: 10.1186/1471-2105-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pavlidis P, Li Q, Noble WS. The effect of replication on gene expression microarray experiments. Bioinformatics. 2003;19:1620–1627. doi: 10.1093/bioinformatics/btg227. [DOI] [PubMed] [Google Scholar]
- 34.Glynne RJ, Ghandour G, Goodnow CC. Genomic-scale gene expression analysis of lymphocyte growth, tolerance and malignancy. Curr. Opin. Immunol. 2000;12:210–214. doi: 10.1016/s0952-7915(99)00076-x. [DOI] [PubMed] [Google Scholar]
- 35.Newton MA, Kendziorski CM, Richmond CS, Blattner FR, Tsui KW. On differential variability of expression ratios: improving statistical inference about gene expression changes from microarray data. J. Comput. Biol. 2001;8:37–52. doi: 10.1089/106652701300099074. [DOI] [PubMed] [Google Scholar]
- 36.Tseng GC, Oh MK, Rohlin L, Liao JC, Wong WH. Issues in cDNA microarray analysis: quality filtering, channel normalization, models of variations and assessment of gene effects. Nucleic Acids Res. 2001;29:2549–2557. doi: 10.1093/nar/29.12.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torgerson TR, Genin A, Chen C, Zhang M, Zhou B, Anover S, Frank MB, Dozmorov I, Ocheltree E, Kulmala P, et al. FOXP3 Inhibits Activation-Induced NFAT2 Expression in Human T Cells. J. Immunol. 2009;183:907–915. doi: 10.4049/jimmunol.0800216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saban MR, O’Donnell MA, Hurst RE, Wu X-R, Simpson C, Dozmorov I, Davis C, Anant S, Vadigepalli R, Saban R. Molecular networks discriminating mouse bladder responses to intravesical bacillus calmette-guerin (BCG), LPS, and TNF-α. BMC Immunol. 2008;9:4. doi: 10.1186/1471-2172-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jorgensen ED, Dozmorov I, Frank MB, Centola M, Albino AP. Global gene expression analysis of human bronchial epithelial cells treated with tobacco condensates. Cell Cycle. 2004;3:1154–1168. [PubMed] [Google Scholar]
- 40.Dozmorov IM, Saban MR, Gerard NP, Lu B, Nguyen NB, Centola M, Saban R. Neurokinin 1 receptors and neprilysin modulation of mouse bladder gene-regulation. Physiol. Genomics. 2003;12:239–250. doi: 10.1152/physiolgenomics.00141.2002. [DOI] [PubMed] [Google Scholar]
- 41.Dozmorov IM, Saban MR, Knowlton N, Centola M, Saban R. Connective molecular pathways of experimental bladder inflammation. Physiol. Genomics. 2003;15:209–222. doi: 10.1152/physiolgenomics.00130.2003. [DOI] [PubMed] [Google Scholar]
- 42.Jarvis J, Dozmorov I, Jiang K, Frank MB, Szodoray P, Alex P, Centola M. Novel approaches to gene expression analysis of active polyarticular juvenile rheumatoid arthritis. Arth. Res. Therapy. 2003;6:R15–R31. doi: 10.1186/ar1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurella S, Yaciuk JC, Dozmorov I, Frank MB, Centola M, Farris AD. Transcriptional modulation of TCR, Notch and Wnt signaling pathways in SEB anergized CD4+ T cells. Genes Immunity. 2005;6:596–608. doi: 10.1038/sj.gene.6364245. [DOI] [PMC free article] [PubMed] [Google Scholar]