


Abstract
Apurinic/apyrimidinic (AP) sites are the most frequently found mutagenic lesions in DNA, and they arise mainly from spontaneous base loss or modified base removal by damage-specific DNA glycosylases. AP sites are cleaved by AP endonucleases, and the resultant gaps in the DNA are repaired by DNA polymerase/DNA ligase reactions. We identified the gene product that is responsible for the AP endonuclease activity in the hyperthermophilic euryarchaeon, Pyrococcus furiosus. Furthermore, we detected the physical interaction between P. furiosus AP endonuclease (PfuAPE) and proliferating cell nuclear antigen (PCNA; PfuPCNA) by a pull-down assay and a surface plasmon resonance analysis. Interestingly, the associated 3′–5′ exonuclease activity, but not the AP endonuclease activity, of PfuAPE was stimulated by PfuPCNA. Immunoprecipitation experiments using the P. furiosus cell extracts supported the interaction between PfuAPE and PfuPCNA in the cells. This is the first report describing the physical and functional interactions between an archaeal AP endonuclease and PCNA. We also detected the ternary complex of PfuPCNA, PfuAPE and Pfu uracil-DNA glycosylase. This complex probably functions to enhance the repair of uracil-containing DNA in P. furiosus cells.
INTRODUCTION
Abasic or apurinic/apyrimidinic (AP) sites in DNA are generated by ionizing radiation, reactive oxygen species and spontaneous base loss (1–3). As many as 10 000 purine bases are reportedly lost spontaneously in a mammalian cell each day (4). AP sites are potentially mutagenic and cytotoxic, because of the inhibition of DNA replication and transcription. AP sites are also formed by damage-specific DNA glycosylases as repair intermediates during base excision repair (BER) (5,6). In the next step, DNA strand is cleaved at the AP sites by two different types of enzyme, AP endonucleases and AP lyases, which attack different phosphodiester bonds at the AP site (5). AP endonuclease is more ubiquitous and two families, represented by Escherichia coli exonuclease III (EcoExoIII) and endonuclease IV (EcoEndoIV), are now widely recognized based on the amino-acid sequence similarity (7). These are also called XthA (ExoIII) and Nfo (EndoIV) families. XthA is expressed constitutively and accounts for 90% of the total AP endonuclease activity in E. coli cells, whereas Nfo accounts for 10% of the activity (8,9). In Saccharomyces cerevisiae, the Apn1 and Apn2 proteins were identified as Nfo and XthA homologs, respectively (10,11). In contrast to Nfo in E. coli, S. cerevisiae Apn1 is the major AP endonuclease, representing >97% of the AP endonuclease activity in the yeast cell. S. cerevisiae Apn2 is thought to act as a backup enzyme for Apn1 (11). Interestingly, the Nfo homologs have not been identified in mammalian cells, but two XthA-homologous proteins, Ape1 and Ape2, were identified in human cells (12,13). Human Ape1 reportedly has strong AP endonuclease activity, but Ape2 exhibits relatively weak activity. The main catalytic property distinguishing Nfo from XthA is the lack of 3′ exonuclease activity as described in the recent review article (5) and also in the textbook (14), although a report in the year 2003 showed that Nfo and its homolog from the hyperthermophilic eubacteria, Thermotoga maritime and Thermus thermophilus, have the exonuclease activity (15,16). The physiological function of the AP endonuclease-associated 3′-exonuclease activity is not fully understood. Based on biochemical studies of human Ape1 and Ape2, this enzymatic activity is supposed to play an important role in the processing of a 3′-mismatched nucleotide, to compensate for the lack of proofreading activity by DNA polymerase β (Polβ)(17,18). Interestingly, the 3′–5′ exonuclease activity of S. cerevisiae Apn2 is stimulated by interacting with proliferating cell nuclear antigen (PCNA), which is known as a processivity factor for the replicative DNA polymerases (19). Many protein factors involved in replication-linked processes also interact with PCNA. Basically, these PCNA-binding proteins interact with PCNA via a conserved motif called the PCNA-interacting protein (PIP) box (20,21). The PIP box consists of the sequence QXXhXXaa, where X represents any amino acid, h represents hydrophobic residues (e.g. Leu, Ile or Met) and a represents aromatic residues (e.g. Phe, Tyr or Trp). It was reported that S. cerevisiae Apn2 physically interacts with PCNA via a PIP box-like motif (427NKSLDSFF434) in the middle of the C-terminal region of the protein (19). During the preparation of this manuscript, a report describing the human Ape2–PCNA interactions was published (22). The report also showed that PCNA strongly stimulates the 3′–5′ exonuclease and 3′-phosphodiesterase activities of Ape2, but has no effect on its AP-endonuclease, as the case of S. cerevisiae Ape2.
We have been studying the PCNA-interacting proteins from the hyperthermophilic euryarchaeon Pyrococcus furiosus (23–27). This organism grows at high temperature (optimal growth temperature of 100°C), where the rate of cytosine deamination to uracil would be accelerated (28). Therefore, it is assumed that an efficient repair mechanism for uracil excision exists in hyperthermophilic Archaea. Basically, uracil bases in DNA are recognized and removed by uracil-DNA glycosylases (UDGs). The UDGs have been classified into five families, based on their substrate specificity and amino-acid sequence motifs in the active site (29,30). Among them, family 4 UDG (UDGa) and family 5 UDG (UDGb) are found in thermophilic bacteria and Archaea. We previously reported that P. furiosus UDG (PfuUDG), which belongs to family 4, based on its sequence, interacts with PCNA (26). Since the first step of the uracil excision repair pathway is coupled with PCNA, there would be a PCNA-initiated BER pathway.
To date, a number of archaeal AP endonucleases, which are homologous to XthA (31–33) and Nfo (34,35), have been reported. However, the physical interaction between archaeal AP endonuclease and PCNA has not been described. After the AP sites are cleaved by AP endonucleases, the resultant gaps will be repaired, either by short-patch BER (SP-BER) or long-patch BER (LP-BER), according to the molecular model of the BER process. In the former pathway, a one nucleotide gap is filled by Polβ and then sealed by the XRCC1-DNA ligase III (LigIII) complex. In LP-BER, 2–10 nucleotides are replaced by strand displacement synthesis, catalyzed by DNA polymerase δ/ε, flap endonuclease and DNA ligase I. Similar to the maturation of Okazaki fragments during DNA replication, PCNA is thought to be involved in LP-BER. Since the functional homologs of eukaryotic Polβ and LigIII have not been identified in Archaea, the archaeal BER process is thought to be accomplished by the LP-BER pathway (26,34). However, relatively little is known about the molecular mechanisms underlying archaeal LP-BER.
In this study, we report the physical and functional interactions between P. furiosus PCNA (PfuPCNA) and AP endonuclease. By using purified recombinant proteins, we identified the gene product bearing the AP endonuclease and 3′–5′ exonuclease activities for synthetic DNA substrates, and designated it as PfuAPE. The physical interaction between PfuAPE and PfuPCNA was confirmed by a pull-down assay and a surface plasmon resonance (SPR) analysis. Intriguingly, we observed a stimulatory effect of PfuPCNA on the 3′–5′ exonuclease activity, but not on the AP endonuclease activity, of PfuAPE. This stimulatory effect was also identified in the BER reconstitution system, using the PfuUDG, PfuAPE and PfuPCNA proteins. Taken together, we propose that PfuAPE and PfuPCNA form a functional complex to generate a gap of several nucleotides for efficient DNA repair synthesis.
MATERIALS AND METHODS
Cloning of the genes encoding Nfo homologs and their mutant proteins
The genes encoding the two Nfo homologs, PF0258 and PF1383, were amplified by PCR directly from P. furiosus genomic DNA, using the oligonucleotides 5′-dGCGCCATATGTTCAAAATTGATAGGCTAAG-3′ and 5′-dGCGCGGATCCTTAAATTTTTAGCTCCTCCC-3′ as the forward and reverse primers for PF0258 and 5′-dGCGCCATATGAAAGTTGGAGTTAGCATATA-3′ and 5′-dGCGCGGATCCTCACCTCATCAATCTCTGAA-3′ as the forward and reverse primers for PF1383. The amplified genes were cloned into the pGEM-T Easy vector (Promega), and their nucleotide sequences were confirmed. The cloned genes were excised by NdeI-BamHI digestion and inserted into the corresponding sites of pET21a or pET28a (Novagen). The resultant plasmids were designated as pET28a-PF0258 and pET21a-PF1383. Amino-acid substitutions were introduced into the PF0258 gene on the pET28a-PF0258 plasmid by PCR-mediated mutagenesis (QuikChange site-directed mutagenesis kit; STRATAGENE), using the appropriate primers. Their sequences are available upon request.
Overproduction and purification of PF0258 proteins
To obtain the recombinant PF0258 protein, E. coli Rosetta (DE3) pLysS cells (Novagen) carrying pET28a-PF0258 were grown in a liter of LB medium, containing 50 µg/ml ampicillin and 34 µg/ml chloramphenicol, at 37°C. The cells were cultured to an A600 = 0.4, and expression of the PF0258 gene was induced by adding isopropyl β-d-thiogalactopyranoside to a final concentration of 1 mM and continuing the culture for 4 h at 45°C. The PF0258 gene was expressed as a soluble protein by cultivating the cells at a high temperature. Some genes from thermophilic microorganisms are reportedly expressed efficiently by high-temperature cultivation (36). After cultivation, the cells were harvested and disrupted by sonication in buffer A (20 mM phosphate buffer, pH 7.4, 0.5 M NaCl and 20 mM imidazole). The soluble cell extract obtained by centrifugation at 12 000g for 15 min was heated at 80°C for 20 min. The heat-resistant fraction, obtained by centrifugation, was subjected to chromatography on a HisTrap HP column (GE Healthcare). The proteins that eluted at 0.19–0.28 M imidazole were dialyzed against buffer B (50 mM Tris–HCl, pH 8.0, 0.1 mM EDTA, 0.5 mM DTT and 10% glycerol). The dialyzate was loaded onto a Hitrap Q HP column (GE Healthcare), which was developed with a 0–1 M NaCl gradient in buffer B. The proteins were eluted at 0.09 M NaCl, pooled and stored at 4°C. The mutated PF0258 proteins prepared in this study were purified by the same procedures. The purification of PfuPCNA and its mutants was performed as described previously (37). PfuUDG was prepared as described previously (26). The purity of each protein used in this study was evaluated by SDS-PAGE. The protein concentrations were calculated by measuring the absorbance at 280 nm. The theoretical molar extinction coefficients of the molecules were calculated based on the numbers of tryptophan and tyrosine residues. The calculated molar extinction coefficients of the PF0258, PfuPCNA and PfuUDG proteins are 39420, 7450 and 24410 M−1cm−1, respectively.
Overproduction and purification of the PF1383 protein
To obtain the recombinant PF1383 protein, E. coli BL21-CodonPlus (DE3)-RIL cells (STRATAGENE) carrying pET21a-PF1383 were grown in a liter of LB medium, containing 50 µg/ml ampicillin and 34 µg/ml chloramphenicol, at 37°C. The cells were cultured to an A600 = 0.4, and expression of the PF1383 gene was induced by adding isopropyl β-d-thiogalactopyranoside to a final concentration of 1 mM and continuing the culture for 3 h at 37°C. After cultivation, the cells were harvested and disrupted by sonication in buffer B. The soluble cell extract, obtained by centrifugation at 12 000g for 15 min, was heated at 80°C for 20 min. The heat-resistant fraction obtained by centrifugation was treated with 0.15% polyethyleneimine, to remove the nucleic acids. The soluble proteins were precipitated by 80% saturated ammonium sulfate. The precipitate was resuspended in buffer C (50 mM Tris–HCl, pH 8.0, 1 M (NH4)2SO4, 0.1 mM EDTA, 0.5 mM DTT and 10% glycerol) and was subjected to chromatography on a Hitrap Phenyl HP column (GE Healthcare), which was developed with a 1–0 M ammonium sulfate gradient in buffer B. The proteins that eluted at 0.48–0.17 M ammonium sulfate were dialyzed against buffer D (50 mM Tris–HCl, pH 8.0, 0.1 M NaCl, 0.1 mM EDTA, 0.5 mM DTT and 10% glycerol). The dialyzate was loaded onto a Hitrap Q HP column (GE Healthcare), which was developed with a 0.1–1 M NaCl gradient in buffer B. The proteins were eluted at 0.16 M NaCl, pooled and stored at 4°C. The purity of the protein was evaluated by SDS-PAGE. The protein concentration was calculated by measuring the absorbance at 280 nm. The theoretical molar extinction coefficient of the PF1383 protein is 10 430 M−1cm−1.
AP endonuclease assay
The substrate used in the AP endonuclease assay was a 49-bp DNA duplex containing a synthetic AP site analog, tetrahydrofuran (F). The F-containing oligonucleotide was synthesized as described earlier (38) on an Applied Biosystems 394 DNA synthesizer, using a phosphoramidite of 3-hydroxy-2-(hydroxymethyl)tetrahydrofuran (dSpacer) purchased from Glen Research, and was purified by HPLC, using a Waters µBondasphere C18 15 µ 300 Å column (7.8 × 300 mm) at a flow rate of 2.0 ml/min, with a linear gradient of acetonitrile in 0.1 M triethylammonium acetate (pH 7.0). The 49-mer deoxynucleotide was labeled with 32P at the 5′ terminus, and was annealed to the 49-mer deoxynucleotide (5′-dAGCTATGACCATGATTACGAATTGCTTAATTCGTGCAGGCATGGTAGCT-3′) in TAM buffer (40 mM Tris–acetate, pH 7.8 and 0.5 mM magnesium acetate). The purified P. furiosus AP endonuclease proteins (various concentrations) were incubated with 5 nM DNA substrate, prepared as described above, in 20 µl of assay buffer (50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 10 mM MgCl2, 1 mM DTT and 0.1 mg/ml BSA) at 60°C for 15 min. Various amounts (units) of EcoEndoIV (New England Biolabs Inc.) were used as a positive control with 5 nM DNA substrate, in 20 µl of commercial buffer (NEBuffer 3; New England Biolabs), containing 50 mM Tris–HCl (pH 7.9), 100 mM NaCl, 10 mM MgCl2 and 1 mM DTT, at 37°C for 15 min. Reactions were terminated with 5 µl of stop solution (98% formamide, 10 mM EDTA, 0.1% bromophenol blue and 0.1% xylene cyanol). Samples were heated at 100°C for 5 min and chilled rapidly on ice prior to loading onto a denaturing 12% polyacrylamide gel containing 7 M urea. After electrophoresis, the gels were dried and were scanned with an FLA-5000 image analyzer (FUJIFILM) to detect the 32P-labeled DNA.
DNA-binding assay
Various concentrations (0–200 nM) of the purified PF0258 and PF1383 proteins were incubated with a 32P-labeled 49-bp DNA duplex (5 nM) containing a synthetic AP site analog, tetrahydrofuran, in 20 µl of binding buffer (50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 1 mM MgCl2, 1 mM DTT and 0.1 mg/ml BSA) at 37°C for 10 min. After this incubation, a 5 µl aliquot of loading buffer (30% glycerol, 0.1% xylene cyanol and 0.1% bromophenol blue) was added, and then the complexes were analyzed by 6% PAGE. After electrophoresis, the gels were dried and were scanned with an FLA-5000 image analyzer (FUJIFILM) to detect the 32P-labeled DNA.
3′–5′ exonuclease assay
The substrate used in the 3′–5′ exonuclease assay was a 49-bp DNA duplex containing a one nucleotide gap. The 21-mer deoxynucleotide (5′-dAGCTACCATGCCTGCACGAAT-3′), labeled with 32P at the 5′ terminus, and the 27-mer deoxynucleotide (5′-dAAGCAATTCGTAATCATGGTCATAGCT-3′) were annealed to the 49-mer deoxynucleotide (5′-dAGCTATGACCATGATTACGAATTGCTTAATTCGTGCAGGCATGGTAGCT-3′) in TAM buffer (40 mM Tris–acetate, pH 7.8 and 0.5 mM magnesium acetate). Various concentrations of the purified PfuAPE (PF0258 product) were incubated with 5 nM DNA substrate, prepared as described above, in 20 µl of assay buffer (50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 10 mM MgCl2, 1 mM DTT and 0.1 mg/ml BSA) at 60°C for 15 min. Reactions were terminated with 5 µl of stop solution (98% formamide, 10 mM EDTA, 0.1% bromophenol blue and 0.1% xylene cyanol). Samples were heated at 100°C for 5 min and chilled rapidly on ice prior to loading onto a denaturing 12% polyacrylamide gel containing 7 M urea. After electrophoresis, the gels were dried and were scanned with an FLA-5000 image analyzer (FUJIFILM) to detect the 32P-labeled DNA.
3′ phosphatase assay
The substrate used in the 3′ phosphatase assay was a 49-bp DNA duplex containing a one nucleotide gap. The 21-mer deoxynucleotide (5′-dAGCTACCATGCCTGCACGAATp-3′) labeled with 32P at the 5′ terminus, and the 27-mer deoxynucleotide (5′-dAAGCAATTCGTAATCATGGTCATAGCT-3′) were annealed to the 49-mer deoxynucleotide (5′-dAGCTATGACCATGATTACGAATTGCTTAATTCGTGCAGGCATGGTAGCT-3′) in TAM buffer (40 mM Tris–acetate, pH 7.8 and 0.5 mM magnesium acetate). The purified PfuAPE (200 fmol) was incubated with 5 nM DNA substrate, prepared as described above, in 20 µl of assay buffer (50 mM Tris–HCl, pH 8.0, 50 mM NaCl, 10 mM MgCl2, 1 mM DTT and 0.1 mg/ml BSA) at 60°C for 0, 30, 60 and 120 s. Reactions were terminated with 5 µl of stop solution (98% formamide, 10 mM EDTA, 0.1% bromophenol blue and 0.1% xylene cyanol). Samples were heated at 100°C for 5 min and chilled rapidly on ice prior to loading onto a denaturing 12% polyacrylamide gel containing 7 M urea. After electrophoresis, the gels were dried and were scanned with a FLA-5000 image analyzer (FUJIFILM) to detect the 32P-labeled DNA.
In-vitro pull-down assay
The purified PCNA (10 µM) was incubated with His-tagged PfuAPE (10 µM) in 90 µl of assay buffer (50 mM Tris–HCl, pH 8.0, 0.5 mM DTT and 10% glycerol) at 60°C for 10 min. After this incubation, the protein solution was mixed with 50 µl of Ni-NTA-Superflow beads (QIAGEN) and 630 µl of assay buffer containing 20 mM imidazole with constant rotation at 4°C for 30 min. The beads were subsequently washed with assay buffer containing 20 mM imidazole. The bead-bound proteins were eluted by 200 µl of assay buffer containing 0.5 M imidazole and then were analyzed by 12.5% SDS-PAGE. After electrophoresis, the gels were stained by Coomassie Brilliant Blue R-250.
SPR analysis
A BIACORE (GE Healthcare Bioscience) system was used to study the physical interaction between PfuAPE and PfuPCNA. Purified recombinant PfuAPE was bound to a CM5 sensor chip (research grade) according to the manufacturer’s recommendations. To measure the kinetic parameters, various concentrations of PfuPCNA (0.25, 0.5, 0.75, 1.0 µM) were applied to the immobilized PfuAPE. The physical interaction between PfuAPE and PfuUDG was evaluated in the same manner. All measurements were conducted at 25°C, in buffer containing 10 mM HEPES (pH 7.4), 150 mM NaCl and 0.005% Tween 20. At the end of each cycle, the bound proteins were removed by washing with 2 M NaCl. The equilibrium constant (KD) for PfuPCNA binding to PfuAPE was determined from the association and dissociation curves of the sensorgrams, using the BIAevaluation program (GE Healthcare Bioscience).
Immunoprecipitation experiment
A 20 µl portion of rProtein A Sepharose Fast Flow (GE Healthcare Biosciences) was washed twice with PBS-T (10 mM sodium phosphate, pH 7.5, 150 mM NaCl, 0.1% Tween 20), mixed with 400 μl of PBS-T containing 10 µl of each antiserum, and incubated at room temperature for 1 h on a rotary shaker. Each mixture was washed twice with PBS-T, followed by twice with 0.2 M triethanolamine, pH 8.0. The antibody was cross-linked to Protein A with dimethyl suberimidate 2 HCl (DMS, PIERCE), according to the manufacturer’s protocol. After equilibration of the antibody-conjugated rProtein A Sepharose with PBS-T, a 400 µl aliquot of P. furiosus cell extract (70 mg/ml) was added, and the mixture was incubated for 30 min on a rotary shaker. The precipitates were washed thrice with PBS-T, and the immunoprecipitated proteins were eluted with 40 µl of gel loading solution (50 mM Tris–HCl, pH 6.8, 1% glycerol, 5% β-mercaptoethanol, 0.2% bromophenol blue, 2% SDS). Three microliters of the elution solutions were subjected to western blot analysis. The protein samples were separated by SDS-12% PAGE, and were electroblotted onto a polyvinylidene difluoride membrane. Proteins were visualized by an enhanced chemiluminescence system (Millipore) and an LAS-3000 mini image analyzer (FUJIFILM).
Uracil-initiated BER reconstitution assay
The substrate used in the reconstitution assay was a 49-bp DNA duplex containing a U:G mispair at the center. The 49-mer deoxynucleotide (5′-dAGCTATGACCATGATTACGAATTGUTTAATTCGTGCAGGCATGGTAGCT-3′), labeled with 32P at the 5′ terminus, was annealed to the 49-mer deoxynucleotide (5′-dAGCTACCATGCCTGCACGAATTAAGCAATTCGTAATCATGGTCATAGCT-3′) in TAM buffer (40 mM Tris–acetate, pH 7.8 and 0.5 mM magnesium acetate). The purified PfuUDG (5 nM), PfuAPE (5 nM) and PfuPCNA (100 nM as a trimer) were incubated with 5 nM DNA substrate, prepared as described above, in 20 µl of assay buffer (50 mM Tris–HCl, pH 8.0, 200 mM NaCl, 10 mM MgCl2, 1 mM DTT and 0.1 mg/ml BSA) at 60°C for 30 min. Reactions were initiated by the addition of enzyme and terminated with 5 µl of stop solution (98% formamide, 10 mM EDTA, 0.1% bromophenol blue and 0.1% xylene cyanol). Samples were heated at 100°C for 5 min and chilled rapidly on ice prior to loading onto a denaturing 12% polyacrylamide gel containing 7 M urea. After electrophoresis, the gels were dried and were scanned with an FLA-5000 image analyzer (FUJIFILM) to detect the 32P-labeled DNA.
Model building of the PfuAPE–PfuUDG–PfuPCNA–DNA complex
The molecular model of the PfuAPE–UDG–PCNA–DNA complex was constructed by reference to the crystal structure of EcoEndoIV (PDB code 22nqj) and the previously constructed PfuUDG–PCNA–DNA complex model (26). The modeling was executed with the in-house program CMP (Shirai et al., forthcoming), and the MOE program (Ryoka Systems Inc.) by the procedure shown in the Figure S1. First, the substrate (dsDNA) complex of PfuAPE was created with MOE, based on the EcoEndoIV crystal structure (Figure S1a). Then, the PfuAPE–DNA model (Figure S1c) was loaded on the PCNA by superposing the dsDNAs of the PfuAPE–DNA complex and the PfuUDG–PCNA–DNA models (Figure S1b), which was presented in our previous report (26). PfuUDG was removed from the PfuUDG–PCNA–DNA model to build the PfuAPE–PCNA–DNA model. The structural model of the PfuUDG–PfuPCNA–DNA complex is probably reliable, since UDG interacts with PCNA through an experimentally confirmed PCNA binding site (-152AKTLF156-) as shown previously (26). PfuAPE could not be positioned in the same manner, because no canonical PIP motif was observed in the sequence, and the interaction sites have not been experimentally identified in this study. Therefore, the PfuAPE was positioned according to the substrate DNA orientation. All possible superpositions were examined, and the obtained models were ranked by using CMP. The most appropriate model, which had interactions between PfuAPE and the PIP-binding site of PfuPCNA, and had the PfuAPE-bound ssDNA in a 5′–3′ orientation, consistent with that of PfuUDG, was manually selected from the highly ranked models (Figure S1d). The PfuAPE–PCNA–DNA model (Figure S1b) and PfuUDG–PCNA–DNA model (Figure S1d) were assembled by superposing one subunit of the PCNA ring in the former model onto the other subunit of the latter model, and removing the atomic clashes within the model by energy minimization (Figure S1e).
RESULTS
Search for the gene encoding homologous sequences of AP endonuclease in the P. furiosus genome
To date, there is no report describing an AP endonuclease from Thermococcales (Pyrococcus and Thermococcus species). We therefore performed a PSI-BLAST search for a sequence homologous to E. coli XthA (Exonuclease III) or E. coli Nfo (Endonuclease IV) in the P. furiosus genome. As a result, two genes, PF0258 and PF1383, were found as the Nfo homologs, sharing 13.5% and 13.1% amino-acid sequence identity, respectively (Figure 1A). On the other hand, we were not able to find a gene that has significant amino-acid sequence similarity to XthA in the P. furiosus genome. The PF0258 gene product is predicted to be a putative endonuclease IV, while the PF1383 protein is annotated as a hypothetical protein in the database. However, we noticed that the PF1383 gene forms a putative operon with the PF1384 and PF1385 genes (Figure 1B). In our previous study, we demonstrated that the PF1385 gene product is a UDG belonging to family 4 (26). The AP endonucleases cleave the AP sites generated by damage-specific DNA glycosylases in the early steps of the BER pathway, and therefore, it was interesting to examine whether the PF1385 gene forms a functional operon with PF1383, a putative endonuclease IV gene. Therefore, we cloned the two genes, PF0258 and PF1385, and expressed them in E. coli cells. The recombinant proteins were purified to apparent homogeneity (Figure 2A).
Figure 1.

Escherichia coli endonuclease IV homologs from P. furiosus. (A) Amino-acid sequence alignment of E. coli endonuclease IV (EcoEndoIV) with its homologs from P. furiosus. Asterisks, identical residues between the three proteins; colons and periods, residues with conserved and semiconserved substitutions, respectively. The dashes indicate gaps. The nine residues that play important roles in catalysis by EcoEndoIV are shown in black boxes, and their positions in EcoEndoIV are shown above the corresponding residues. The putative catalytic residues in P. furiosus endonuclease IV-like proteins are shown in grey boxes. (B) The orientations of two putative endonuclease IV genes in the P. furiosus genome are schematically shown. The open reading frame numbers and their predicted functions are indicated.
Figure 2.

Identification of AP endonuclease activity. (A) Purified WT putative endonuclease IV proteins from P. furiosus and their mutant proteins (2 µg) were analyzed by 12.5% SDS–PAGE. The gel was stained with Coomassie Brilliant Blue. (B) An AP site-containing DNA substrate (100 fmol) was incubated with increasing amounts of the putative endonuclease IV proteins from P. furiosus (0, 5, 25 and 125 nM), as described in the ‘Materials and Methods’ section. Escherichia coli endonuclease IV (New England Biolabs Inc.) (0, 5 and 25 units) was used as a positive control of the reaction. The reaction mixtures were analyzed by denaturing PAGE, and the DNA was visualized by autoradiography. (C) An AP site-containing DNA substrate (100 fmol) was incubated with increasing amounts of the putative endonuclease IV proteins from P. furiosus (0, 50, 100, 150 and 200 nM), as described in the ‘Materials and Methods’ section. The protein–DNA complexes were analyzed by 6% PAGE, followed by autoradiography. (D) and (E) An AP site-containing DNA substrate (100 fmol) was incubated with increasing amounts of WT PF0258 protein and its mutants (0, 5, 25 and 125 nM), as described in the ‘Materials and Methods’ section.
Identification of an AP endonuclease in P. furiosus
We then examined the AP endonuclease activity of the two proteins, using a ds DNA substrate containing the AP site analog, tetrahydrofuran (F). As shown in Figure 2B, only the PF0258 protein exhibited AP endonuclease activity similar to that of Nfo (EcoEndoIV). We used EcoEndoIV from a commercial source as a control enzyme. To evaluate the specific activity of PF0258 for the AP site cleavage, an enzymatic activity of 143 pg/units for EcoEndoIV was obtained from the manufacture’s information. The calculated concentrations of EcoEndoIV in lanes 2 and 3 in Figure 2B are 1.1 and 5.7 µM, respectively. The cleavage efficiency in lane 3 is comparable to that of lane 5, which included 5 µM of the PF0258 protein and therefore, these two enzymes seem to have specific activity similar to that of the AP endonucleases. The 3′–5′ exonuclease activity of the PF0258 protein was also observed at a high enzyme concentration (Figure 2B). The PF0258 gene product tended to be insoluble in E. coli cells, and small amount of soluble enzyme sample obtained by cultivation at 45°C was processed for further purification as described in the ‘Materials and Methods’ section. Therefore, the purified enzyme sample may not be stable enough to keep a complete conformation. Further quantitative enzymology is necessary to understand the basal ability, including specific activity, of PF0258 as an AP endonuclease, as well as comparison of the activities of the samples from several different purification procedures. Refolding of the insoluble fraction will be helpful to investigate how the endonuclease activity depends on the preparation procedure. In contrast to PF0258, no enzymatic activity of the PF1383 protein was detected under the same assay conditions, even though the PF1383 product was completely recovered from the soluble fraction of E. coli cell extract without difficulty. We next examined the substrate binding activities of the two proteins. As shown in Figure 2C, the PF0258 protein can bind to DNA, but no binding of the PF1383 protein was detected under the same conditions. Furthermore, we were not able to detect the physical interaction between the PF1383 protein and PfuUDG by using an SPR analysis (data not shown). Since the PF1383 protein was not precipitated after the heat treatment at 80°C for 20 min during the purification procedure, the protein, like other P. furiosus proteins, should be heat-stable. A sequence similarity search revealed that PF1384 gene homologs are also conserved especially in Thermococcales, but the biological function of this endonuclease IV-like PF1383 protein in the cells cannot be predicted at this stage.
The crystal structure of EcoEndoIV complexed with a DNA substrate was previously solved, and its catalytic mechanisms were well studied (39–41). Several amino-acid residues that play important roles in the catalysis have been identified. An amino-acid sequence alignment showed that most of the important residues corresponding to those in EcoEndoIV are conserved in the PF0258 protein (Figure 1A). On the other hand, some important residues are not present in the PF1383 protein. Among them, we examined the importance of two histidine residues, which are not conserved in the PF1383 protein. We therefore prepared the PF0258 mutant proteins, H70A, H110A and H70A/H110A. These mutant proteins were purified by the same procedure as used for the WT PF0258 protein. The mutant proteins were purified to apparent homogeneity (Figure 2A). As shown in Figure 2D, relatively weak AP endonuclease activities were observed from the H70A and H110A mutants. Interestingly, the H70A/H110A mutant retains residual enzyme activity. These observations suggested that these two histidine residues are important for catalysis. However, there are other important residues for the AP endonuclease activity of the PF0258 protein. The structural and mutational analyses of EcoEndoIV revealed that Glu261 plays a central role in catalysis (41). As shown in Figure 1A, the acidic amino-acid residue corresponding to Glu261 in EcoEndoIV is present in the PF0258 protein, but not in PF1383, and therefore, we made two more mutants, E261A and H70A/H110A/E261A (Figure S2), to confirm that the activities detected here are intrinsically derived from PF0258. The mutant PF0258 at E261A retained some activity, however, the protein with the triple mutations of H70A/H110A/E261A completely lost the activity (Figure 2E). Based on these results, we concluded that PF0258 is the sole endonuclease IV homologous gene that encodes the functional AP endonuclease in P. furiosus, and designated the gene product as PfuAPE.
Biochemical characterization of PfuAPE
The AP endonuclease activity of PfuAPE was detected by using a commercial buffer that was optimized for EcoEndoIV. We therefore investigated the optimum reaction conditions for PfuAPE, using a 49-bp DNA duplex containing a synthetic AP site analog, tetrahydrofuran (F). The AP endonuclease activity was optimal in Tris–HCl buffer at pH 7.5–8.0 (Figure 3A). The highest AP endonuclease activity was observed at NaCl concentrations of 100–150 mM (Figure 3B). The residual enzyme activity was detected at relatively high salt concentrations. The enzyme activity was stimulated by the presence of Mg2+ or Mn2+, with an optimum at 10 mM (data not shown). We then tested the effect of increasing salt concentrations on the 3′–5′ exonuclease activity of PfuAPE, using a 49-bp DNA duplex containing a one nucleotide gap, which mimics a cleaved AP site. The 3′–5′ exonuclease activity was optimal at 50–100 mM NaCl. In contrast to the AP endonuclease activity, the 3′–5′ exonuclease activity was completely inhibited at high salt concentrations (>200 mM NaCl) (Figure 3C). To analyze the substrate specificity of the 3′–5′ exonuclease of PfuAPE, we performed the cleavage reactions using a nicked duplex DNA and a 3′-recessed duplex DNA, in parallel with the gapped duplex DNA described above. The exonuclease activity was observed equally for the three types of DNA substrates (Figure S3). We also investigated the preference of PfuAPE for the 3′-terminal structure of the DNA substrate. As shown in Figure 3D, we detected the 3′-phosphatase activity, which is followed by the 3′–5′ exonuclease, in PfuAPE. It would also be important to examine whether PfuAPE has 3′-phosphodiesterase activity, to understand how PfuAPE can contribute to the BER process in the cells. Interestingly, human Ape1 and the Chlamydia pneumoniae AP endonuclease IV homolog are reportedly able to cleave a single-stranded oligonucleotide at the AP site (42,43). As shown in Figure 3E, the endonuclease activity of PfuAPE for the single-stranded oligonucleotide (49-mer) containing a tetrahydrofuran was also observed. This enzyme activity was detected in the reaction buffer either with or without NaCl. A weak AP endonuclease activity of EcoEndoIV for the ssDNA substrate was observed at a high concentration of the enzyme and without NaCl.
Figure 3.

Biochemical properties of PfuAPE. (A) An AP site-containing DNA substrate (100 fmol) was incubated with PfuAPE (20 fmol), as described in the ‘Materials and Methods’ section, at 60°C for 10 min. Bis-Tris–HCl and Tris–HCl were used in the pH ranges of 6.0–7.0 and 7.5–8.8, respectively. (B) An AP site-containing DNA substrate (100 fmol) was incubated with PfuAPE (20 fmol), as described in the ‘Materials and Methods’ section. The NaCl concentration was varied as indicated. (C) The one-nucleotide gap-containing DNA substrate (100 fmol) was incubated with PfuAPE (200 fmol) in a reaction mixture containing various concentrations of NaCl (0, 50, 100, 150, 200, 300, 400 and 500 mM), as described in the ‘Materials and Methods’ section. (D) PfuAPE (200 fmol) was incubated with a DNA substrate (100 fmol) containing a 3′-phosphate at a one-nucleotide gap for 0, 30, 60 and 120 seconds, as described in the ‘Materials and Methods’ section. (E) An AP site-containing 49-mer oligonucleotide (100 fmol) was incubated with increasing amounts of PfuAPE (0, 5, 25 and 125 nM) in a reaction mixture containing either 0 or 100 mM NaCl, as described in the ‘Materials and Methods’ section. EcoEndoIV (0, 5 and 25 units) was also tested in parallel.
Physical interaction between PfuAPE and PfuPCNA
Although the protein–protein interactions between eukaryotic PCNA and AP endonuclease have been reported—e.g. human Ape1 (44), human Ape2 (22,45) and S. cerevisiae Apn2 (19)—there is no direct evidence for archaeal PCNA–AP endonuclease complex formation. To determine the physical interaction between PfuAPE and PfuPCNA, we performed a His-tag pull-down assay. As shown in Figure 4A, the recombinant PfuPCNA protein lacks non-specific binding affinity to the Ni-NTA beads (lanes 2–6). Under these conditions without non-specific binding, we found that a distinct amount of PfuPCNA protein co-eluted with the His-tagged PfuAPE protein (lane 12). Since more than half of the input PfuPCNA remained in the unbound fraction (lane 8), the PfuPCNA–PfuAPE complex seems to be unstable under our experimental conditions. We then performed SPR experiments to determine the equilibrium constant (KD) for the quantitative analysis of the protein–protein interaction. Purified PfuAPE was covalently immobilized on the Biacore CM5 sensor chip, and subsequently, PfuPCNA was injected at different concentrations. The SPR sensorgram showed the physical interaction between PfuPCNA and immobilized PfuAPE (Figure 4B). The calculated KD for WT PCNA was 1.0 × 10−6 M, which is higher than the values for other PCNA-binding proteins in P. furiosus (1.1 × 10−7 M for Lig, 9.9 × 10−8 M for PolBI, 2.2 × 10−7 M for UDG), as determined by our SPR analyses (25–27). The physical interaction between PfuAPE and PfuPCNA appears to be weak, from these results. In the next step, we examined the physical interaction between PfuUDG and PfuAPE, by using the PfuAPE-immobilized sensor chip. As shown in Figure 4C, little response was detected in the sensorgram (<50 RU) for PfuUDG–PfuAPE. Then, PfuPCNA and PfuUDG were sequentially loaded onto the PfuAPE-bound sensorchip. The obtained sensorgram showed that the decreasing resonance units increased again after the injection of PfuUDG, and the resonance units reached a higher point than that from the injection of only PfuPCNA, indicating that the PfuAPE–PfuPCNA–PfuUDG ternary complex is formed on the sensor chip. To confirm the complex formation in the cells, immunoprecipitation was performed, using a P. furiosus cell extract and antibodies against PfuAPE and PfuPCNA. As shown in Figure 4D, the PfuAPE band was clearly detected in the fraction precipitated with the anti-PfuPCNA antibody. Conversely, PfuPCNA was coprecipitated with PfuAPE by an anti-PfuAPE antibody. We previously showed that PfuUDG and PfuPCNA were coprecipitated with each antibody (46).
Figure 4.

Physical interaction between PfuAPE and PfuPCNA. (A) A His-tag pull-down assay was performed using nickel-charged beads to detect the physical interaction between His-tagged PfuAPE and PfuPCNA, as described in the ‘Materials and Methods’ section. (B) An SPR analysis was performed using a BIACORE system to analyze the PfuAPE–PfuPCNA interaction. Purified PfuAPE was immobilized on a sensor chip, and various concentrations of PfuPCNA (0.25, 0.5, 0.75 and 1 µM) were injected for 240 s. The equilibrium constant (KD) was calculated from the obtained sensorgrams. (C) Purified PfuPCNA (1 µM) and PfuUDG (1 µM) were loaded onto the PfuAPE-immobilized sensor chip independently, and their sensorgrams are shown in the left and the middle panels, respectively. PfuPCNA and PfuAPE were sequentially loaded onto the PfuAPE-immobilized sensor chip to investigate the PfuAPE–PfuPCNA–PfuUDG ternary complex formation. The sensorgram is shown in the right panel. (D) Immunoprecipitation analysis of PfuAPE and PfuPCNA. The whole cell extracts were precipitated with anti-PfuAPE and anti-PfuPCNA antibodies, respectively and the precipitates were detected by western blot analyses with each antiserum, as shown on the left side. The whole cell extracts without immunoprecipitation or precipitated with the protein A-sepharose treated with preimmune serum were also loaded as positive and negative controls, respectively.
PfuPCNA enhances the 3′–5′ exonuclease activity of PfuAPE
Like other DNA replicative and DNA repair enzymes, PfuAPE was shown to form a complex with PCNA. This observation prompted us to examine the effect of PfuPCNA on the enzymatic activity of PfuAPE. Therefore, we performed the enzyme assay of PfuAPE in the presence of 0–100 nM PCNA (as a trimer concentration). Unexpectedly, the AP endonuclease activity of PfuAPE was not affected by the presence of PfuPCNA (Figure 5A). Although we tried to find experimental conditions to observe the functional interaction between PfuAPE and PfuPCNA, by varying the relative amounts of PfuAPE, PfuPCNA and substrate DNA under several ionic strength conditions, the stimulatory effect of the PfuPCNA on the AP endonuclease activity was not observed. On the contrary, the 3′–5′ exonuclease activity of PfuAPE was increased in a PfuPCNA concentration-dependent manner (Figure 5B). Since this stimulatory effect was not observed when the monomeric mutant PCNA D143A/D147A (37) was added to the reactions, the toroidal structure of PCNA is surely required for the functional interaction with PfuAPE.
Figure 5.

Functional interaction between PfuAPE and PfuPCNA. (A) PfuAPE (20 fmol) was incubated with an AP site-containing DNA substrate (100 fmol) and increasing amounts of PfuPCNA (0, 10, 25, 50, 75 and 100 nM as a trimer) in a reaction mixture containing 0.2 M NaCl, as described in the ‘Materials and Methods’ section. (B) PfuAPE (200 fmol) was incubated with a one-nucleotide gap-containing DNA substrate (100 fmol) and increasing amounts of WT or a monomeric mutant (D143A/D147A) PfuPCNA (0, 10, 25, 50, 75 and 100 nM as a trimer) in a reaction containing 0.2 M NaCl, as described in the ‘Materials and Methods’ section.
Reconstitution of the early steps in the base excision repair pathway
In this study, we demonstrated that PfuPCNA can stimulate the 3′–5′ exonuclease activity of PfuAPE, using a substrate containing a one nucleotide gap (Figure 5B). Likewise, the UDG activity of PfuUDG is stimulated by PfuPCNA (26). Moreover, the SPR analyses and immunoprecipitation experiments suggested the formation of the PfuAPE–PfuPCNA–PfuUDG ternary complex. Based on these findings, we supposed that PCNA plays a role in the early steps in the uracil excision repair pathway. We therefore examined the effect of PfuPCNA on the PfuUDG and PfuAPE activities by using a dsDNA substrate containing a U:G mispair at the center. As shown in Figure 6, the uracil-containing substrate was unaffected after incubation with PfuAPE (cf. lanes 1 and 2). This result indicates that PfuAPE lacks DNA uridine endonuclease activity, as observed in the Methanothermobacter thermautotrophicus exonuclease III homolog (47). A small amount of product was observed when the substrate was incubated with PfuUDG (lane 3). Since the AP sites generated by the enzymatic activity of UDG are sensitive to spontaneous β-elimination at higher reaction temperatures (34), the product was observed without the heat and alkali treatment, which is used to cleave the AP sites artificially. The amount of the 24-mer product was slightly increased by adding both PfuUDG and PfuAPE (lane 4). The products generated by the AP lyases are known to migrate slightly faster than those resulting from the spontaneous cleavage (34). Since the AP endonuclease activity of PfuAPE is effective at the salt concentration we used in this experiment (Figure 3B), most of the abasic sites produced by PfuUDG would be processed by PfuAPE. We next examined the effect of PfuPCNA on the reconstituted system. Consistent with our previous report on the stimulatory effect of PfuPCNA on the PfuUDG activity, the product formation according to the spontaneous cleavage at the abasic site was increased by adding the WT PCNA (cf. lanes 3 and 7). Since a relatively large amount of product was observed without a hot alkali treatment, the rate of spontaneous cleavage at the abasic sites during the incubation is rather high (lane 7). Interestingly, the uracil-excising activity of an endonuclease IV homolog from the thermophilic bacterium Thermus thermophilus has been reported (16). However, PfuAPE would not have uracil-excising activity, because the product formation was not observed in the presence and absence of PfuPCNA (lanes 2 and 6). Finally, we found that the PfuPCNA can stimulate the 3′–5′ exonuclease activity of PfuAPE, followed by the cleavage of abasic sites (lane 8). The stimulatory effect of PfuPCNA on PfuAPE was not impaired in the presence of the PfuUDG protein, despite the fact that PfuUDG can form a complex with PfuPCNA and PfuAPE (Figure 4C). We then performed the same enzyme assay using the PCNA mutant, D143A/D147A, but the stimulatory effect on the PfuUDG and PfuAPE activities was not observed (lanes 9–12). To show the difference between the spontaneous β-elimination at the AP-site (a partly degraded sugar molecule linked to its 3′-terminus via an additional phosphodiester group, as an AP lyase product) and the additional elimination by NaOH (with a 3′-phosphomonoester group) more clearly, an additional results indicating PfuUDG reactions with and without following NaOH treatment in the absence of PfuAPE are shown in Figure S4. The effect of PCNA on these reactions is also shown in parallel in the figure.
Figure 6.

Reconstitution of the early steps in the BER pathway. The U:G mispair-containing DNA substrate (100 fmol) was incubated with PfuUDG (100 fmol), PfuAPE (100 fmol) and WT or a monomeric mutant (D143A/D147A) PfuPCNA (100 nM as a trimer), as described in the ‘Materials and Methods’ section.
DISCUSSION
We found two genes encoding sequences homologous to E. coli Nfo (EndoIV) in the P. furiosus genome by a PSI-BLAST database search. At the same time, we performed the search on the completely sequenced archaeal genomes, and found that E. coli Nfo (EndoIV) and XthA (ExoIII) homologous genes are conserved in other archaeal genomes. Among them, we selected 13 archaeal species from the different archaeal orders, and the putative AP endonuclease genes in Archaea are summarized in Table 1. The Nfo homologs are completely conserved in all of the archaeal genomes we examined, whereas the XthA homologs are found in only some species. The conservation of AP endonuclease genes in Archaea seems to be different among the species, and it is therefore important to test which AP endonuclease family is responsible for the major AP endonuclease activity in each archaeal species. Interestingly, a number of Nfo homologous genes are conserved in some euryarchaeal genomes. For example, besides the MTH1010 gene, which shows the highest similarity to Nfo, two more candidate genes, MTH1489 and MTH247, are present in the M. thermautotrophicus genome. This multiple gene conservation for the Nfo-like proteins is not observed in crenarchaeal genomes (data not shown). Similar to the M. thermautotrophicus Nfo homologs, two putative Nfo homologs, PF0258 and PF1383, were found in the P. furiosus genome. Our biochemical analyses revealed that the PF0258 protein is the sole AP endonuclease. On the other hand, neither AP endonuclease nor DNA-binding activity with a synthetic DNA substrate was detected with the PF1383 protein (Figure 2B and C). Amino-acid sequence alignments indicated that the PF1383 protein lacks some important amino-acid residues that are critical for the AP endonuclease activity, as shown in this study for PF0258, although the total sequence similarities with E. coli Nfo are 13.5% and 13.1% for PF0258 and PF1383, respectively (Figure 1A). Further analyses are required to predict the function of the PF1383 protein in P. furiosus cells. Biochemical characterizations presented in this study were performed at various temperatures, which differed from the optimal growth temperature of P. furiosus, and therefore, each assay may not provide quantitatively precise results. However, all of the data obtained from the nuclease, DNA binding, SPR, IP and pull-down assays were consistent, and support the conclusions of this study.
Table 1.
Predicted EcoExoIII (XthA) and EcoEndoIV (Nfo) homologs in Archaea
| ExoIII | EndoIV | |
|---|---|---|
| Euryarchaeota | ||
| Archaeoglobus fulgidus | AF0580* | AF2428 |
| Halobacterium salinarum | OE1304F | |
| Methanocaldococcus jannaschii | MJ1311 | |
| Methanopyrus kandleri | MK1021 | |
| Methanosarcina acetivorans | MA3548 | |
| Methanospirillum hungatei | Mhun_1782 | |
| Methanothermobactor thermautotrophicus | MTH212* | MTH1010* |
| Pyrococcus furiosus | PF0258 | |
| Thermoplasma volcanium | TVN0046* | TVN0971 |
| Crenarchaeota | ||
| Aeropyrum pernix | APE_2104.1 | |
| Nitrosopumilus maritimus | Nmar_0064 | |
| Pyrobaculum aerophilum | PAE3257* | |
| Sulfolobus solfataricus | SSO2290 | SSO2156 |
The open reading frames in selected archaeal genomes with homologous sequences to those of EcoExoIII and EcoEndoIV are shown. Asterisks show the genes that were previously reported.
The eukaryotic AP endonucleases belonging to the XthA family can reportedly interact with PCNA (19,22,44,45). While the AP endonuclease activity of Apn2 from S. cerevisiae and human is not stimulated by PCNA, their 3′–5′ exonuclease and 3′-phosphodiesterase activities are clearly stimulated by interacting with PCNA (19,22). In this study, we identified both physical and functional interactions between PfuAPE and PfuPCNA. Hence, this is the first report describing the interaction of an Nfo homolog with PCNA. As observed in the eukaryotic Apn2–PCNA interaction, the 3′–5′ exonuclease activity of PfuAPE alone is stimulated by the PCNA trimer (Figure 5B). This observation strongly indicates that AP endonuclease–PCNA complex formation is a common feature between the eukaryotic and archaeal DNA repair systems. The eukaryotic Ape2 proteins have a PIP box sequence in their C-terminal regions, and bind PCNA at these sites (19,22). Though we were able to detect the physical interaction between PfuAPE and PfuPCNA, a typical PIP box sequence was not found in PfuAPE. To date, we have identified pentapeptide PCNA-binding motifs in P. furiosus DNA ligase (103QKSFF107) (25) and UDG (152AKTLF156) (26). As discussed in our previous report (26), these atypical PCNA-binding motifs appear to be shorter versions of the canonical PIP box sequence, where the successive hydrophobic amino-acid residues play a critical role in binding to PCNA. Moreover, the cluster of basic amino acids adjacent to the binding motif seemed to be important for the interaction. Based on these findings, we tried to identify the PCNA-binding motif in PfuAPE, but failed to find an amino-acid sequence similar to the short PIP box motif. It should be difficult to predict the PCNA-interacting region because a single hydrophobic amino-acid residue in the weakly conserved motif sequence can be critical for the interaction, as shown in our previous study on Aeropyrum pernix DNA ligase (48). In the case of human Ape2–PCNA interaction, the PCNA-dependent stimulation of the exonuclease was only reduced, but was not completely impaired, by the alanine substitutions for Y396 and F397 in the PIP box sequence, indicating that another site, as well as the conserved PIP box, in human Ape2 contributes to the interaction with PCNA. Our next goal is to identify the PCNA-interaction site in PfuAPE precisely, as described below.
It is known that human cells have a repair complex containing UDG, PCNA and APE1, in addition to DNA polymerases α, β, δ and ε, DNA ligase I and XRCC1 (49). We previously reported the physical and functional interactions between PfuPCNA and PfuUDG (26). In this study, we identified the direct interaction between PfuPCNA and PfuAPE. Our SPR analysis detected little binding between UDG and APE, and therefore, it would be most probable that UDG and APE can load onto the same PCNA ring, and this complex should enhance the efficiency of repair of uracil-containing DNA. Using the currently available information, we attempted to build a 3D structure model of the PfuUDG–PfuPCNA–PfuAPE–DNA complex. The detailed procedure of the model building is described in the ‘Materials and Methods’ section, and is shown in Figure S1. The atomic structural model of the PfuAPE–DNA complex can be built based on the EcoEndoIV–DNA cocrystal structure (39), although we have not solved the crystal structure of PfuAPE. By the procedure described in the ‘Materials and Methods’ section, we succeeded in building a reasonable APE–PCNA–UDG–DNA complex without steric hindrance (Figure 7). The DNA strand passing through the PCNA ring is tilted by 30°, to contact both enzymes on the same PCNA ring. A model to switch from PolIII, a normal replication enzyme, to PolIV, a translesion enzyme, on the same β-clamp ring by a DNA tilting mechanism in the E. coli DNA replication process has been proposed recently (50). The PCNA interacting site of PfuAPE is predicted to be the loop containing -23TIAGI27- from this model, as shown in Figure 7. However, this sequence is far from the consensus PCNA-binding motif. Experimental studies are required to confirm the function of this PCNA binding site candidate.
Figure 7.

Molecular model of the PfuAPE–PfuPCNA–PfuUDG–DNA complex. (Left) Top view of the model. PfuAPE and PfuUDG are shown in orange and cyan, respectively. The three subunits of PCNA are colored blue, green and gray. The dsDNA bound to PfuAPE is shown in magenta, and that bound to PfuUDG is shown in yellow. The angle between these dsDNAs is around 30°. (Right) Side view of the model. The PIP-like motif (-152AKTLF156-) of PfuUDG, and the sequence of PfuAP (-23TIAGI27-), which putatively occupies the binding site on another PCNA subunit, are shown in red. The red arrows indicate the 5′–3′ orientation of the ssDNAs used as the substrates of PfuAPE and PfuUDG.
In our previous report, we noted that there are no homologs of eukaryotic Polβ and LigIII, which are important components of the SP-BER pathway, in the P. furiosus genome (26). Pyrobaculum aerophilum reportedly has three family B DNA polymerases, denoted as PaePolB1, PaePolB2 and PaePolB3 (51). Among them, PaePolB2 has been proposed to be a functional homolog of the mammalian Polβ, based on its enzymatic properties (34). The authors hypothesized that PaePolB2 plays a role in SP-BER in P. aerophilum. Meanwhile, it is known that P. furiosus has only one family B DNA polymerase (PfuPolBI) and one family D DNA polymerase (PfuPolD), which are supposed to be replicative DNA polymerases (52). Our previous in-vitro study showed that PolBI prefers gap-filling type substrates to primer-extension type substrates, but the substrate preference of PolD is the opposite (53). In addition, we confirmed that PolBI has strand displacement activity with and without PCNA, although PolD also has the same activity in vitro (Kimizu et al., unpublished results). A comparative study of PfuPolBI and PfuPolD in gap-filling synthesis using a more BER-like substrate would be interesting to identify the DNA polymerase responsible for BER in P. furiosus.
After strand displacement DNA synthesis by the replicative DNA polymerase, the resultant flap DNA structure will be processed by flap endonuclease. At the last step in the BER pathway, the resultant nick will be sealed by an ATP-dependent DNA ligase, which is conserved in all sequenced archaeal genomes. In contrast to the DNA polymerases, only one homolog of eukaryotic DNA ligase I is conserved in Archaea. Interestingly, the halophilic euryarchaeon Haloferax volcanii was shown to have both ATP-dependent and NAD+-dependent DNA ligases (54). However, a genetic analysis revealed that the H. volcanii NAD+-dependent DNA ligase does not play an essential role in the DNA repair pathway.
Taken together, P. furiosus may have only the LP pathway, and the PCNA-dependent 3′-exonuclease activity of PfuAPE at the high salt concentration in the P. furiosus cells is especially important to expand the gap from the AP site suitable for the following DNA polymerase reaction. We propose a basic model of the BER mechanisms in P. furiosus: (i) UDG and APE, recruited onto the PCNA ring, search for the lesion site on the DNA strand, and recognize the uracil site (step 1), (ii) UDG cleaves the glycoside bond, and then APE cleaves the phosphodiester bond at the abasic site (step 2), (iii) the exonuclease of APE expands the nick to make an appropriate gap for loading the DNA polymerase (step 3) and (iv) DNA polymerase (probably PolBI)-PCNA fills in the gap (Figure 8). We observed that the exonuclease activity of the PfuAPE is not processive, and only 5–6 nucleotides were cleaved from the nick site. The gap formation with 5–6 nucleotides may be regulated by a structural change of the DNA. The crystal structure of EcoEndoIV-DNA revealed that the DNA strand bends by about 90° at the AP site (39). This bending may return as the PCNA–APE complex moves in the 5′ direction from the AP site. Notably, the size of the gap and the DNA structure may have a direct relation. The formed gap may be suitable for the DNA polymerase that is in charge of BER to load and start strand synthesis. To further understand the molecular mechanisms underlying the archaeal BER, we are planning to investigate the late steps in the BER pathway by using a series of repair enzymes from P. furiosus, which is one of the simplest model organisms for the BER pathway.
Figure 8.
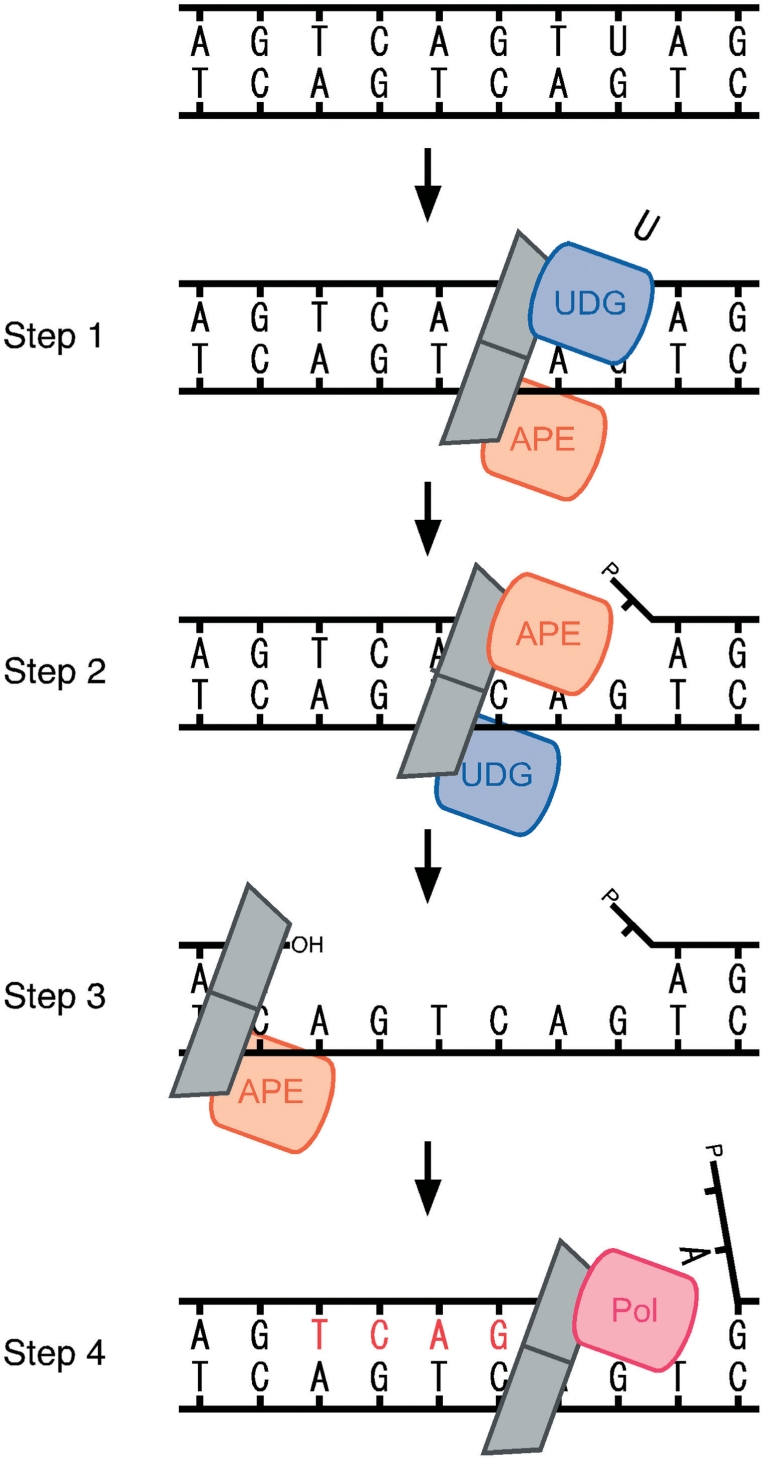
Long patch gap-filling DNA synthesis associated with enhanced 3′–5′ exonuclease activity of PfuAPE. The proposed model of the BER process in P. furiosus is shown. The UDG–PCNA–APE complex searches for the uracil site on the DNA strand and works to remove the uracil and cleave the DNA strand, respectively (steps 1 and 2). Then, the produced nick is expanded by the exonuclease of APE with the help of PCNA, to make a gap suitable for the appropriate binding of DNA polymerase (step 3) and then DNA polymerase fills in the gap to repair the double strand DNA (step 4).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan, 20310134 and EAG7080006 to Y.I. Funding for open access charge: BIRD project from Japan Science and Technology Agency.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Mol CD, Hosfield DJ, Tainer JA. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3′ ends justify the means. Mutat. Res. 2000;460:211–229. doi: 10.1016/s0921-8777(00)00028-8. [DOI] [PubMed] [Google Scholar]
- 2.Boiteux S, Guillet M. Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair. 2004;3:1–12. doi: 10.1016/j.dnarep.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 3.David SS, O'S;hea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 5.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baute J, Depicker A. Base excision repair and its role in maintaining genome stability. Crit. Rev. Biochem. Mol. Biol. 2008;43:239–276. doi: 10.1080/10409230802309905. [DOI] [PubMed] [Google Scholar]
- 7.Robertson AB, Klungland A, Rognes T, Leiros I. Base excision repair: the long and short of it. Cell Mol. Life Sci. 2009;66:981–993. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ljungquist S, Lindahl T, Howard-Flanders P. Methyl methane sulfonate-sensitive mutant of Escherichia coli deficient in an endonuclease specific for apurinic sites in deoxyribonucleic acid. J. Bacteriol. 1976;126:646–653. doi: 10.1128/jb.126.2.646-653.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ljungquist S. A new endonuclease from Escherichia coli acting at apurinic sites in DNA. J. Biol. Chem. 1976;252:2808–2814. [PubMed] [Google Scholar]
- 10.Popoff SC, Spira AI, Johnson AW, Demple B. Yeast structural gene (APN1) for the major apurinic endonuclease: homology to Escherichia coli endonuclease IV. Proc. Natl Acad. Sci. USA. 1990;87:4193–4197. doi: 10.1073/pnas.87.11.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson RE, Torres-Ramos CA, Izumi T, Mitra S, Prakash S, Prakash L. Identification of APN2, the Saccharomyces cerevisiae homolog of the major human AP endonuclease HAP1, and its role in the repair of abasic sites. Gene Dev. 1998;12:3137–3143. doi: 10.1101/gad.12.19.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demple B, Herman T, Chen DS. Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc. Natl Acad. Sci. USA. 1991;88:11450–11454. doi: 10.1073/pnas.88.24.11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hadi MZ, Wilson DM., III Second human protein with homology to the Escherichia coli abasic endonuclease exonuclease III. Environ. Mol. Mutagen. 2000;36:312–324. [PubMed] [Google Scholar]
- 14.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. Washington, DC: ASM Press; 2006. [Google Scholar]
- 15.Kerins SM, Collins R, McCarthy TV. Characterization of an endonuclease IV 3′–5′ exonuclease activity. J. Biol. Chem. 2003;278:3048–3054. doi: 10.1074/jbc.M210750200. [DOI] [PubMed] [Google Scholar]
- 16.Back JH, Chung JH, Park JH, Han YS. A versatile endonuclease IV from Thermus thermophilus has uracil-excising and 3′–5′ exonuclease activity. Biochem. Biophys. Res. Commun. 2006;346:889–895. doi: 10.1016/j.bbrc.2006.05.187. [DOI] [PubMed] [Google Scholar]
- 17.Burkovics P, Szukacsov V, Unk I, Haracska L. Human Ape2 protein has a 3′–5′ exonuclease activity that acts preferentially on mismatched base pairs. Nucleic Acids Res. 2006;34:2508–2515. doi: 10.1093/nar/gkl259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong D, DeMott MS, Demple B. Modulation of the 3′–>5′-exonuclease activity of human apurinic endonuclease (Ape1) by its 5′-incised abasic DNA product. J. Biol. Chem. 2003;278:36242–36249. doi: 10.1074/jbc.M306065200. [DOI] [PubMed] [Google Scholar]
- 19.Unk I, Haracska L, Gomes XV, Burgers PM, Prakash L, Prakash S. Stimulation of 3′–>5′ exonuclease and 3′-phosphodiesterase activities of yeast apn2 by proliferating cell nuclear antigen. Mol. Cell Biol. 2002;22:6480–6486. doi: 10.1128/MCB.22.18.6480-6486.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naryzhny SN. Proliferating cell nuclear antigen: a proteomics view. Cell Mol. Life Sci. 2008;65:3789–3808. doi: 10.1007/s00018-008-8305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 22.Burkovics P, Hajdu I, Szukacsov V, Unk I, Haracska L. Role of PCNA-dependent stimulation of 3′-phosphodiesterase and 3′–5′ exonuclease activities of human Ape2 in repair of oxidative DNA damage. Nucleic Acids Res. 2009;37:4247–4255. doi: 10.1093/nar/gkp357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsumiya S, Ishino S, Ishino Y, Morikawa K. Physical interaction between proliferating cell nuclear antigen and replication factor C from Pyrococcus furiosus. Genes Cells. 2002;7:911–922. doi: 10.1046/j.1365-2443.2002.00572.x. [DOI] [PubMed] [Google Scholar]
- 24.Fujikane R, Shinagawa H, Ishino Y. The archaeal Hjm helicase has recQ-like functions, and may be involved in repair of stalled replication fork. Genes Cells. 2006;11:99–110. doi: 10.1111/j.1365-2443.2006.00925.x. [DOI] [PubMed] [Google Scholar]
- 25.Kiyonari S, Takayama K, Nishida H, Ishino Y. Identification of a novel binding motif in Pyrococcus furiosus DNA ligase for the functional interaction with proliferating cell nuclear antigen. J. Biol. Chem. 2006;281:28023–28032. doi: 10.1074/jbc.M603403200. [DOI] [PubMed] [Google Scholar]
- 26.Kiyonari S, Uchimura M, Shirai T, Ishino Y. Physical and functional interactions between uracil-DNA glycosylase and proliferating cell nuclear antigen from the euryarchaeon Pyrococcus furiosus. J. Biol. Chem. 2008;283:24185–24193. doi: 10.1074/jbc.M802837200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tori K, Kimizu M, Ishino S, Ishino Y. DNA polymerases BI and D from the hyperthermophilic archaeon Pyrococcus furiosus both bind to proliferating cell nuclear antigen with their C-terminal PIP-box motifs. J. Bacteriol. 2007;189:5652–5657. doi: 10.1128/JB.00073-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindahl T, Nyberg B. Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry. 1974;13:3405–3410. doi: 10.1021/bi00713a035. [DOI] [PubMed] [Google Scholar]
- 29.Aravind L, Koonin EV. The alpha/beta fold uracil DNA glycosylases: a common origin with diverse fates. Genome Biol. 2000;1:0007.1–0007.8. doi: 10.1186/gb-2000-1-4-research0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pearl LH. Structure and function in the uracil-DNA glycosylase superfamily. Mutat. Res. 2000;460:165–181. doi: 10.1016/s0921-8777(00)00025-2. [DOI] [PubMed] [Google Scholar]
- 31.Kaneda K, Ohishi K, Sekiguchi J, Shida T. Characterization of the AP endonucleases from Thermoplasma volcanium and Lactobacillus plantarum: contributions of two important tryptophan residues to AP site recognition. Biosci. Biotechnol. Biochem. 2006;70:2213–2221. doi: 10.1271/bbb.60153. [DOI] [PubMed] [Google Scholar]
- 32.Pfeifer S, Greiner-Stöffele T. A recombinant exonuclease III homologue of the thermophilic archaeon Methanothermobacter thermautotrophicus. DNA Repair. 2005;4:433–444. doi: 10.1016/j.dnarep.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 33.Miertzschke M, Greiner-Stöffele T. The xthA gene product of Archaeoglobus fulgidus is an unspecific DNase. Eur. J. Biochem. 2003;270:1838–1849. doi: 10.1046/j.1432-1033.2003.03548.x. [DOI] [PubMed] [Google Scholar]
- 34.Sartori AA, Jiricny J. Enzymology of base excision repair in the hyperthermophilic archaeon Pyrobaculum aerophilum. J. Biol. Chem. 2003;278:24563–24576. doi: 10.1074/jbc.M302397200. [DOI] [PubMed] [Google Scholar]
- 35.Back JH, Chung JH, Park YI, Kim KS, Han YS. Endonuclease IV enhances base excision repair of endonuclease III from Methanobacterium thermoautotrophicum. DNA Repair. 2003;2:455–470. doi: 10.1016/s1568-7864(02)00243-4. [DOI] [PubMed] [Google Scholar]
- 36.Koma D, Sawai T, Harayama S, Kino K. Overexpression of the genes from thermophiles in Escherichia coli by high-temperature cultivation. Appl. Microbiol. Biotechnol. 2006;73:172–180. doi: 10.1007/s00253-006-0448-9. [DOI] [PubMed] [Google Scholar]
- 37.Matsumiya S, Ishino S, Ishino Y, Morikawa K. Intermolecular ion pairs maintain the toroidal structure of Pyrococcus furiosus PCNA. Protein Sci. 2003;12:823–831. doi: 10.1110/ps.0234503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iwai S. Chemical synthesis of oligonucleotides containing damaged bases for biological studies. Nucleosides Nucleotides Nucleic Acids. 2006;25:561–582. doi: 10.1080/15257770600685826. [DOI] [PubMed] [Google Scholar]
- 39.Hosfield DJ, Guan Y, Haas BJ, Cunningham RP, Tainer JA. Structure of the DNA repair enzyme endonuclease IV and its DNA complex: double-nucleotide flipping at abasic sites and three-metal-ion catalysis. Cell. 1999;98:397–408. doi: 10.1016/s0092-8674(00)81968-6. [DOI] [PubMed] [Google Scholar]
- 40.Ivanov I, Tainer JA, McCammon JA. Unraveling the three-metal-ion catalytic mechanism of the DNA repair enzyme endonuclease IV. Proc. Natl Acad. Sci. USA. 2007;104:1465–1470. doi: 10.1073/pnas.0603468104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcin ED, Hosfield DJ, Desai SA, Haas BJ, Björas M, Cunningham RP, Tainer JA. DNA apurinic-apyrimidinic site binding and excision by endonuclease IV. Nat. Struct. Mol. Biol. 2008;15:515–522. doi: 10.1038/nsmb.1414. [DOI] [PubMed] [Google Scholar]
- 42.Marenstein DR, Wilson DM, III, Teebor GW. Human AP endonuclease (APE1) demonstrates endonucleolytic activity against AP sites in single-stranded DNA. DNA Repair. 2004;3:527–533. doi: 10.1016/j.dnarep.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 43.Liu X, Liu J. Chlamydia pneumoniae AP endonuclease IV could cleave AP sites of double- and single-stranded DNA. Biochim. Biophys. Acta. 2005;1753:217–225. doi: 10.1016/j.bbapap.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 44.Dianova II, Bohr VA, Dianov GL. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry. 2001;40:12639–12644. doi: 10.1021/bi011117i. [DOI] [PubMed] [Google Scholar]
- 45.Tsuchimoto D, Sakai Y, Sakumi K, Nishioka K, Sasaki M, Fujiwara T, Nakabeppu Y. Human APE2 protein is mostly localized in the nuclei and to some extent in the mitochondria, while nuclear APE2 is partly associated with proliferating cell nuclear antigen. Nucleic Acids Res. 2001;29:2349–2360. doi: 10.1093/nar/29.11.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kiyonari S, Tahara S, Uchimura M, Shirai T, Ishino S, Ishino Y. Studies on the base excision repair (BER) complex in Pyrococcus furiosus. Biochem. Soc. Trans. 2009;37:79–82. doi: 10.1042/BST0370079. [DOI] [PubMed] [Google Scholar]
- 47.Georg J, Schomacher L, Chong JP, Majerník AI, Raabe M, Urlaub H, Müller S, Ciirdaeva E, Kramer W, Fritz HJ. The Methanothermobacter thermautotrophicus ExoIII homologue Mth212 is a DNA uridine endonuclease. Nucleic Acids Res. 2006;34:5325–5336. doi: 10.1093/nar/gkl604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kiyonari S, Kamigochi T, Ishino Y. A single amino acid substitution in the DNA-binding domain of Aeropyrum pernix DNA ligase impairs its interaction with proliferating cell nuclear antigen. Extremophiles. 2007;11:675–684. doi: 10.1007/s00792-007-0083-0. [DOI] [PubMed] [Google Scholar]
- 49.Parlanti E, Locatelli G, Maga G, Dogliotti E. Human base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteins. Nucleic Acids Res. 2007;35:1569–1577. doi: 10.1093/nar/gkl1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Georgescu RE, Kim SS, Yurieva O, Kuriyan J, Kong XP, O'D;onnell M. Structure of a sliding clamp on DNA. Cell. 2008;132:43–54. doi: 10.1016/j.cell.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fitz-Gibbon ST, Ladner H, Kim UJ, Stetter KO, Simon MI, Miller JH. Genome sequence of the hyperthermophilic crenarchaeon Pyrobaculum aerophilum. Proc. Natl Acad. Sci. USA. 2002;99:984–989. doi: 10.1073/pnas.241636498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ishino S, Ishino Y. Comprehensive search for DNA polymerase in the hyperthermophilic archaeon, Pyrococcus furiosus. Nucleosides Nucleotides Nucleic Acids. 2006;25:681–691. doi: 10.1080/15257770600686485. [DOI] [PubMed] [Google Scholar]
- 53.Ishino Y, Ishino S. DNA polymerases from euryarchaeota. Methods Enzymol. 2001;334:249–260. doi: 10.1016/s0076-6879(01)34473-7. [DOI] [PubMed] [Google Scholar]
- 54.Zhao A, Gray FC, MacNeill SA. ATP- and NAD+-dependent DNA ligases share an essential function in the halophilic archaeon Haloferax volcanii. Mol. Microbiol. 2006;59:743–752. doi: 10.1111/j.1365-2958.2005.04975.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.