

Abstract
Metalloproteins catalyze some of the most difficult and yet important functions in Nature, such as photosynthesis and water oxidation. An ultimate test of our knowledge of how metalloproteins work is by designing novel metalloproteins. Such design can not only reveal hidden structural features that may be missing from studies of native metalloproteins and their variants, but also result in new metalloenzymes for biotechnological and pharmaceutical applications. While it is much more challenging to design metalloproteins than non-metalloproteins, much progress has been made in this area, particularly toward functional design, thanks to recent progress in areas such as computational and structural biology.
Metalloproteins account for nearly half of all proteins in biology. Protein metal-binding sites are responsible for catalyzing some of the most difficult and yet important functions, including photosynthesis, respiration, water oxidation, molecular oxygen reduction, and nitrogen fixation. Much effort has been devoted toward understanding the structure and function of metalloproteins, as summarized in other reviews in this Nature Insight series. The ultimate test is to utilize this knowledge to design new metalloproteins that reproduce structures and functions of native metalloproteins1-3. Metalloprotein design is not an intellectual exercise that simply duplicates biochemical and biophysical studies of native metalloproteins. This bottom-up approach can elucidate structural features that may remain hidden in those studies; while the biochemical and biophysical studies can reveal mostly individual features that result in a loss of function, design requires incorporation of all the structural features needed to attain a function. Armed with insights obtained from the process, metalloprotein design may lead to the design of novel metalloproteins with improved properties such as higher stability, greater efficiency, or even unprecedented functions not found in Nature, for use in an even wider range of biotechnological and pharmaceutical applications.
Despite the promise, metalloprotein design has proven to be more challenging than its non-metal containing counterpart. Both fields require addressing the same issue of polypeptide design, but metalloprotein design involves metal ions that are much larger in number than amino acids, and geometries that are much more variable. On the other hand, most metal-binding sites are highly chromatic and display distinctive magnetic properties, making it easier to characterize the metalloprotein design using metal-based spectroscopic techniques rather than x-ray crystallography or NMR, thus shortening design cycles. Therefore, the field of metalloprotein design has enjoyed much success recently, thanks to advances in biophysical, computational, and structural biology. The field is also continually progressing from pure structural to more functional designs, and from primary coordination sphere to secondary coordination sphere design and beyond. These recent progresses will be summarized here, with focus on creating new and functional metal-binding sites in either de novo designed or native protein scaffolds. In addition, an emerging area of designing novel metalloproteins with expanded functions beyond those available in Nature through the introduction of unnatural amino acids or non-native metal cofactors will be covered. The methodologies used in achieving the goals will be summarized.
Metalloprotein design in de novo designed scaffolds
The purest and most challenging form of metalloprotein design is de novo design or design “from scratch”. De novo design of a metalloprotein involves constructing a polypeptide sequence not directly related any natural protein, to precisely fold into a defined three-dimensional structure that binds a metal ion4. Most work in this area has focused on introducing metal-binding sites into designed α-helical bundles (Fig. 1), which are among the first de novo designed proteins. These α-helical bundles are a common scaffold for a number of heme proteins. Therefore, not surprisingly, heme centers have been among the first and most common metal centers incorporated into de novo α-helical bundles5, from mono heme binding (Fig. 1a)6 to four heme maquettes (Fig. 1b)7 mimicking long-range electron transfer proteins. These proteins were rationally designed by direct comparison to native proteins, such as cytochrome bc1, for clues as to the placement of essential heme binding and structure-stabilizing residues. In addition to electron transfer8 and proton-coupled exchange9 functions, a functional aniline hydroxylase10, peroxidase11, and oxygenase12 have also been engineered into de novo proteins. An exciting development in this field is the design of a di-heme containing four-helix bundle with oxygen transport properties, whose O2 affinities and exchange timescales match those of natural globins13. Remarkably, this designed protein binds O2 even tighter than CO. A key to the success is reduction of helical-interface mobility to exclude water from the heme binding site, thereby reducing heme oxidation and increasing O2 binding stability.
Figure 1. Designed metalloproteins using de novo designed scaffolds.

a, Computer model of a bis-His ligated mono-heme α-helical bundle. Adapted from ref6 courtesy of William DeGrado. b, Computer model of a bis-His ligated multi-heme four α-helical bundle. Reproduced from ref5. c, Computer model of Zn(II) bound His2Cys2 motif of a Zn-finger mimic. Reproduced from ref14. d, X-ray crystal structure of As(III) bound three-stranded coiled-coil (PDB 2JGO)16. e, X-ray crystal structure of di-Zn(II) Due Ferro 1; PDB 1EC5).17
In addition to heme, other metal ions/cofactors have been engineered into de novo designed helical bundles by introducing metal-binding ligands at specific locations to mimic those in native proteins. Examples include the His2Cys2 ligand set found in Zn-finger proteins (Fig. 1c)14 and the His3 set in carbonic anhydrase15 The x-ray crystal structure of an arsenic bound three-stranded coiled-coil protein (Fig. 1d), resembling the putative tris Cys metal-binding environment of the natural arsenic binding protein, ArsR, has been reported, affording the first biologically relevant small molecule mimic of an AsS3 coordination16. Metal-binding ligands Glu and His have also been introduced by retrostructural analysis, whereby the active site of a protein can be separated into idealized secondary structural components (e.g., an α-helix and a β-hairpin), and mathematically described as a root-mean-square deviation from the idealized structure. This strategy led to the construction of dinuclear metalloproteins in α-helical bundles that can bind two Zn(II), Co(II), or Fe(II), with the Zn(II) crystal structure (Fig. 1e) shown to be structurally analogous to the diiron site of ribonuclease reductase and ferritin17. One of these diiron proteins was reported to catalyze the two electron oxidation of 4-aminophenol, similar to the reaction catalyzed by diiron phenol oxidases18,19.
Taking a cue from naturally occurring Cys rich coordination sites in proteins, the introduction of Cys ligands led to the development of α-helical coiled-coil metalloproteins capable of binding Cu(II), Zn(II), Cd(II), Hg(II), and As(III)20-22. One exciting aspect of de novo design is the demonstration that these de novo proteins can stabilize unusual metal coordination states, such as three-coordinate Hg(II) by a tri-helical bundle over the bis-coordination often preferred by Hg(II)23,24. In other cases, metal-binding can actually direct the folding and assembly of an α-helical bundle from either a random coil, less folded, or misfolded state, as was the case of Cd(II)25 and Hg(II) binding26 to Cys containing peptides predisposed to form helices. Interestingly, geometry-selective binding of two different Cd(II) in one designed protein has been achieved and confirmed by the correlation of 113Cd NMR and 111mCd Perturbed Angular Correlation spectroscopy27,28.
In stark contrast to the design of α-helical proteins, de novo design of β-structure proteins is still in its infancy, because there were few model systems with which to study β-sheets in isolation from other protein structures. The de novo design of a redox active rubredoxin mimic, RM1, is a rare example of a structural and functional metallo-β-sheet protein29. RM1 was shown to bind iron and reversibly cycle between the Fe(II/III) oxidation states, mimicking native rubredoxin. The RM1 β-hairpin structure was designed computationally by constraining the positions of the peptide that were strategic in mimicking and/or stabilizing the β-structure and subsequent use of another computer program, SCADS30 to select the most probable amino acids for the remaining positions.
Metalloprotein design in native protein scaffolds
While designing metalloproteins using de novo designed scaffolds offers the prospect of complete control over a protein’s structure and metal-binding properties, our current knowledge of protein folding limits the number of de novo designed scaffolds to only a few types, such as the α-helical bundles discussed above. It has been observed that β-sheet containing proteins tend to support more rigid and more pre-organized metal-binding sites than α-helical proteins, which are inherently more flexible31. Unlike de novo designed scaffolds, the Protein Data Bank contains ~1000 natural protein scaffold types (http://scop.mrc-lmb.cam.ac.uk/scop/count.html#scop-1.73), most of which maintain the same fold and similar stability even after numerous mutations. Therefore, metalloprotein design using native protein scaffolds provides many more scaffold choices that are more tolerant to mutations, allowing for the incorporation of metal-binding sites into proteins with less concern over decreased protein stability. When chosen carefully, native proteins are also relatively easier to crystallize than de novo designed scaffolds, making 3D characterization possible, an objective that is critical to success. Perhaps the strongest argument for designing metalloproteins using native scaffolds is that Nature employs the same approach since the same scaffold is often seen in numerous proteins with diverse metal-binding motifs and functions, suggesting that these native scaffolds are robust and modifiable. For example, the Greek key β-barrel fold has been shown to be used by ~600 different types of proteins with diverse functions, ranging from oxidase, reductase, amylase, and dismutase activities, for instance. Unearthing the secret of how Nature is able to use the same scaffold to design a variety of metal-binding sites is an important goal of metalloprotein design1.
Biochemical techniques, such as site-directed mutagenesis, have been used extensively to study the function of metalloproteins. The loss of function accompanied by certain mutations (usually to highly conserved residues) allows identification of residues essential for function. Although serving a different purpose, the same mutagenesis techniques can be used for metalloprotein design to impart new function into a protein scaffold by the introduction of residues that bind metal ions. This approach can be in the form of redesigning existing metal-binding sites to introduce novel functionality, introducing mononuclear metal-binding sites to proteins that do not natively bind metal ions, or by introducing homonuclear or heteronuclear metal-binding sites into proteins. The designs are often aided by empirical approaches through knowledge/experience, rational design using computer programs, or combinatorial selections.
The redesign of an existing metal-binding site to introduce novel function or metal specificity is the simplest form of metalloprotein design, as this approach relies on the structural differences between template and target proteins, and is amenable to empirical designs through knowledge/experience. Despite its simple form, this approach is still quite powerful in elucidating structural features important for the change, or gain of function by redesign. Heme proteins have been extensively redesigned in this way. They are one of the most diverse classes of proteins, with functions ranging from electron transfer, small molecule (e.g., oxygen and NO) transport, sensing, and activation5,32,33. By systematically changing these characteristics of heme proteins, researchers have been able to convert one type of heme protein into another, as well as introduce novel function or substrate specificity into a heme protein32,34.
The selectivity of designed metalloproteins for specific metals has also been utilized for the purpose of metal-sensing applications. Ratiometric Zn(II) sensors have been created by designing Zn(II) binding sites into green fluorescent proteins and by taking advantage of the resulting fluorescent signal variations due to Zn(II)-dependent conformational changes35-37. The metal-binding site of NikR, a DNA binding protein, has been redesigned to bind UO22+ rather than its native Ni2+ cofactor (Fig. 2a)38, resulting in an artificial protein that binds DNA only in the presence of UO22+. In addition to sensors, designed metal-binding sites have allowed modulation of protein-protein interactions39,40, which are at the heart of many diverse biological functions. Because of complicated non-covalent interactions, it has been difficult to control the interactions well. These designed metal-binding sites allowed specific control of these interactions.
Figure 2. Designed metalloproteins using native scaffolds.

a, Native nickel binding site of NikR (left) and the re-engineered UO22+ binding site of the mutated NikR (right). b, Loop directed mutagenesis of blue copper azurin to yield the dinuclear, purple CuA azurin construct. c, Catalytic heme-copper center in CcO (left) and the designed heme-cooper model in sperm whale Mb (right).
More complex metal-binding site redesign may require rational design using computational biology, such as the simultaneous incorporation and adjustment of functional elements to convert the GlyII enzyme from a glyoxalase, which hydrolyzes thioester bonds, to a β-lactamase41. In order to impart the new functionality, the metal-binding site had to be significantly redesigned in order to change the metal-binding geometry, as well as the substrate binding pocket.
While redesign of metal-binding sites can offer insight into the different structural features between the template and target proteins, those structural features common to the metal-binding sites of both proteins will remain hidden. Introducing metal-binding sites into a protein location where no native metal-binding site is found, can overcome this limitation, although this approach requires a higher level of complexity. A common strategy is to base the design on structural homology between the template protein which contains no metal ions and the target metalloprotein, to introduce metal-binding sites at the corresponding positions. Using this strategy, new Zn(II) binding sites have been introduced into charybdotoxin42 and retinal binding protein43 to mimic carbonic anhydrase (CA).
When there is no structural homology, computer aided design using programs such as Metal-Search44 and Dezymer45, has become indispensable. A Ca(II)46, Pb(II)47, and [Fe(Cys)4] rubredoxin center45 have all been designed into scaffold proteins using computer aided design. In the case of an artificial rubredoxin designed into the non-metalloprotein thioredoxin, it was found to undergo several rounds of air oxidation and reduction by β-mercaptoethanol, similar to native rubredoxins45.
An even higher level of difficulty in metalloenzyme design is the introduction of dinuclear metal-binding sites or metal clusters into proteins. An example is the binuclear CuA center, consisting of two copper ions bridged by two cysteine thiolates, with each copper ion also coordinated to a histidine nitrogen (Fig. 2b). New CuA centers were designed into the cupredoxins azurin (Az)48,49 and amicyanin50,51 using a technique called loop directed mutagenesis (LDM), in which the copper binding loop of cupredoxins is replaced by the corresponding loop in CcO that share similar structural homology. LDM has also been used to introduce lanthanide binding sites into a helix-turn-helix motif by drafting a calcium binding EF-hand loop, and such a designed protein has been shown to promote DNA and phosphate hydrolysis52.
Heteronuclear bimetallic centers are also found in many natural proteins and they perform important biological functions such as oxygen reduction, lignin degradation and nitric oxide reduction. Designing metalloproteins to mimic the enzymes that carry out these complex reactions is even more challenging than designing homonuclear metal-binding sites because of the selective placement and binding of two or more different metal centers. An example is the heme-CuB center in CcO where three histidines bind to a Cu(II) ion that is ~4.5 Å away from the heme iron. Computer-aided design was used to introduce the histidines above the heme of myoglobin (Mb) at positions corresponding to CcO (Fig. 2c)53. Copper binding was confirmed by multiple spectroscopic techniques. Further studies showed the importance of the CuB site in O2 binding and reduction, as well as the significance of a proton network54. This designed protein also displays interesting functional properties, such as nitric oxide reduction55, similar to certain members of CcOs. A Mn(II) binding site has also been designed into cytochrome c peroxidase, resulting in a heme-Mn(II) protein with Mn(II) oxidation activity, as in native manganese peroxidase (MnP)56, a protein involved in lignin biodegradation, which is a critical step in biomass conversions from plant materials to biofuels.
In addition to discrete metal-binding sites, metal nanoparticles have been introduced into proteins57, including Pt nanoparticles in ferritin58 and a small heat shock protein59. The former catalyzes size-selective olefin hydrogenation while the latter catalyzes hydrogen production.
Even though empirical approaches through knowledge/experience and rational design using computer programs have been quite successful in designing metalloproteins, it is still difficult to design certain metalloproteins, especially functional ones, due to the large number of potential interactions within the active site of a metalloenzyme. In this case, combinatorial design and directed evolution of novel metalloenzymes is another powerful choice. For example, directed evolution has been extensively applied to antibodies to develop novel metalloenzymes. Utilizing phage display, the 7G12 antibody protein was evolved to bind heme, with which the oxidation of tyramine could be catalyzed60. A similar technique has recently been expanded to find antibodies for inorganic ligands, such as BINAP and BINOL, with the goal to develop completely novel metalloenzymes61. A striking example of this approach is the use of directed evolution to obtain a new cyt P450 with the ability to selectively hydroxylate propane, a function that has not yet been achieved by small molecule catalysts62.
Designing novel metalloproteins containing unnatural amino acids or non-native metal cofactors
Metalloprotein design using de novo designed or native protein scaffolds has succeeded in providing valuable insights into the fundamental rules that govern protein structure and function. Replicating native protein structure and function, although quite challenging, is not the only goal of protein designers; they want build proteins with properties beyond those found in Nature63. In recent years, the repertoire of functional groups such as amino acids or cofactors available to protein designers has been expanded beyond those available in biological systems.
A myriad of techniques have been developed for the incorporation of unnatural amino acids into a protein scaffold, each with its own advantages and limitations63. Total synthesis of proteins using solid phase peptide synthesis enables incorporation of any synthetic amino acid for which successful protection/deprotection techniques can be applied64, but cost and size constraints limits the use of total synthesis to smaller proteins (~60–100 amino acids). Native chemical ligation (NCL), involving covalent attachment of two synthetic peptides, has expanded this size regime65, but cost is still an issue, at least for large proteins. Expressed protein ligation (EPL), the coupling of a bacterially expressed peptide fragment with a synthetic peptide fragment containing unnatural amino acids, has reduced cost and extended the protein size even further, but it often requires a cysteine residue at the point of ligation to maximize ligation efficiency66, although methods have been developed to use other residues at the point of ligation, with less efficiency. Methods not restricted by protein size include auxotroph generation67, chemical residue modification68, and cavity complementation69, but these methods have limited versatility in terms of the variety of unnatural amino acids that can be incorporated. A promising and versatile technique is the use of custom evolved tRNA synthetase molecules for in vitro and in vivo translation of new residues through recognition of the amber stop codon70. A seminal contribution from this approach was the design of a DNA cleaving protein through site-specific genetic incorporation of the DNA cleaving agent (2,2′-bipyridin-5-yl)alanine into a DNA binding protein71. Upon addition of Cu(II) or Fe(II) and reducing agent to the protein, a double stranded DNA substrate was cleaved at its consensus sequence. The variety of unnatural amino acids this technique can incorporate is higher than those of auxotroph generation, chemical residue modification, or cavity complementation, but lower than those of total synthesis, NCL or EPL.
The expansion of the amino acid regime has made significant contributions toward determining systematically the role of each amino acid in tuning the activity of the metal center toward a given reactivity, and in increasing the breadth of chemical transformations performed by proteins. For instance, replacement of heme binding His residues in de novo designed four-α-helical bundles with 4-β-(pyridyl)-L-alanine and 1-methyl-L-His has resulted in an increase in the reduction potential of the metal site for the pyridyl system and a preference for five-coordinate low-spin heme in the 1-methyl-L-His system72. In another system, introduction of bulky penicillamine ligands in place of cysteines led to the creation of an open metal-binding site in a ferredoxin model peptide73. This methodology resulted in the isolation of the first water-stable CdS3 complex through subtle manipulation of steric crowding around the metal center74. Furthermore substitution of L-amino acids for their D-enantiomers resulted in fine control between 3- and 4-coordinate Cd(II) complexes in a de novo designed metallopeptide75. Finally, the role of a conserved Tyr residue near the active site of rubredoxin was probed via a set of unnatural Tyr analogs in which the para -OH group was replaced with -H (native Phe), -F, -NO2, and -CN, spanning a range of electron withdrawing strengths (Fig. 3a)76. Interestingly, a linear correlation was found between the reduction potential of the nearby heme and the Hammett σp value, but no such trend is observed with dipole moment or residue size.
Figure 3. Site specific incorporation of unnatural amino acids into a protein scaffold for tuning of metal properties.

Crystal structures of a, rubredoxin (PDB: 1CAA) and b, azurin (PDB: 4AZU) showing variant residue location for unnatural amino acid incorporation.
A powerful advantage to the introduction of unnatural amino acids is the ability to probe the role of the amide backbone, since site-directed mutagenesis using natural amino acids cannot modify the protein backbone. Both the carbonyl and nitrogen functionalities of the protein backbone have been implicated in critical and specific hydrogen bonding interactions, either directly to the metal, or to the metal ligands. For example, the Fe4S4 cluster of high potential iron proteins (HiPIPs) has such an amide interaction, which was found to be responsible for stabilizing the reduced form through attenuation of a metal bound sulfur77. Replacement of this amide linkage with an ester linkage indeed resulted in a lowering of the reduction potential by ~100 mV.
Through the use of EPL, the metal bound Cys and Met residues in the electron transfer protein azurin were replaced with isostructurally similar unnatural amino acids (Fig. 3b)78,79. Replacing Cys with selenocysteine resulted in drastically different spectral features, but did little to affect the reduction potential. In contrast, replacing methionine with selenomethione and other isostructural variants resulted in spectral features similar to the wild type protein, but with drastically different reduction potentials. The use of unnatural amino acids helped to identify the hydrophobicity of the residues at this position as the major factor in tuning the reduction potential of the protein.
The repertoire of non-native functionalities has also been expanded through the incorporation of non-native metal containing cofactors into the protein scaffold. Biological systems incorporate metal cofactors such as heme through both non-covalent and covalent means. Among the covalent approach, Nature uses both single and dual point anchoring strategies to position the prosthetic group within the protein scaffold. These strategies have all been utilized for incorporation of non-native metal cofactors, with the non-covalent approach being the most extensively used strategy. For example, replacement of the native heme cofactors of myoglobin and horseradish peroxidase (HRP) with the less symmetrical structural isomer, porphycene (Fig. 4b) results in enhanced peroxidase activity, highlighted by a twelve-fold increase in oxidation of thioanisole when compared to native HRP80-82. In addition to native protein scaffolds, de novo designed α-helical bundles have also been engineered to bind non-biological heme cofactors preferentially over biological hemes83,84.
Figure 4. Strategies for non-native cofactor incorporation into a protein scaffold.

Native cofactor substitution of heme b in myoglobin for a, Fe-porphycene and b, 3,3-Cr salophen, exploiting the structural and dative-bonding similarities between the native cofactor and the surrogate. Affinity tagging of the catalyst with a protein-selective linker such as the biotin-streptavidin couple, c, allows for versatile attachment of non-native cofactors. Covalent strategies include a single attachment strategy as in d, showing the adipocyte lipid binding protein (ALBP) linked through Cys117 to a phenanthrolene complex; and dual covalent attachment as in e, showing dual anchoring of a Mn-salen complex into the myoglobin scaffold. Description of streptavidin complex obtained from ref91. Crystal structure of Fe porphycene, Cr(3,3′-Me2-salophen)-Mb complex, biotin-streptavidin complex, and phenanthroline-ALBP were obtained from the Protein Databank (PDB codes: 2D6C, 1J3F, 2QCB, and 1A1894, respectively). Computer model of Mb(L72C/Y103C) generated from an overlay with native heme b structure.
In addition to heme analogs, other planar moieties similar to heme have also been substituted into heme proteins. For example, strong binding of Mn, Fe, and Cr Schiff base complexes into a myoglobin scaffold was observed after careful design of the protein to relieve steric repulsion in the active site (Fig. 4b)57,85,86. Such rational design has resulted in novel asymmetric catalysts capable of oxidation rate enhancement and chiral induction.
To incorporate non-planar metal-containing cofactors into proteins where there is no native binding site is much more challenging. One way to meet such a challenge is through derivatization of the catalyst with a moiety exhibiting natural bio-affinity for a protein target, through, for example, the use of the high-affinity biotin-streptavidin couple (Ka = 1 ×1015 M−1). In this way, metal cofactors have been incorporated into the streptavidin scaffold through covalent attachment to a biotin moiety (Fig. 4c)87-89 resulting in artificial biocatalysts with new activities including hydrogenation of alkenes, transfer-hydrogenation of ketones, and C-C bond formation through allylic alkylation, and up to 99% ee88-91.
While the use of biotin and avidin/streptavidin to incorporate non-native metal cofactors is very powerful, not all cofactors are amenable to such a strategy, as binding affinity and location may not be readily controlled. Covalent attachment is an effective and versatile approach to attach a cofactor to the protein at a defined location through selective conjugation techniques involving engineered or native Cys or Lys residues. This strategy has been utilized to attach DNA cleaving agents such as 1,10-phenanthroline-copper and iron-EDTA to DNA binding proteins for the generation of artificial nucleases, 92, and 1,10-phenanthroline-copper (Fig. 4d) for enantioselective hydrolysis93,94.
While the single-point covalent approach mentioned above makes it possible to attach a non-native cofactor at a defined location, it does not necessarily fix the conformation of the cofactor inside the protein. For example, single covalent attachment of a 4-(2-methanesulfonylthio-ethoxy)salicylidene-1,2-ethanediamino-manganese(III) (Mn-Salen) cofactor to apo myoglobin through a single cysteine-selective linker resulted in only 12% ee toward sulfoxidation of thioanisole95, suggesting that the Mn-Salen conformation may be too flexible inside Mb to facilitate high enantioselectivity. Addition of a second linker (Fig. 4e) to the cofactor and selective cavity positioning led to a significant increase in catalytic efficiency (over 7-fold increase) and selectivity (from 12% to 51%)95. Furthermore, it has been shown that such incorporation of Mn-Salen into a myoglobin scaffold enhances significantly the chemoselectivity in oxidation of thioanisole with H2O2, virtually eliminating over oxidation to the sulfone product through electrostatic exclusion of the sulfoxide product from the site96.
Measures of success, lessons learned, and future challenges
Tremendous progress has been made in this exciting field of synthetic biology. Success is measured not only by how close the designed protein is to the native protein, but also by the insight gained from the process. For example, the crystal structure of a designed CuA site is almost identical to that of native CuA, including both primary and secondary coordination spheres (Fig. 5)49, indicating that metalloprotein design can lead to structures closely resembling those in Nature. More importantly, two metal-binding sites, native blue copper and the designed CuA center, were placed in the same azurin scaffold, allowing the electron transfer properties of two different metal centers to be compared in the same protein97, a task that is difficult, if not impossible to do by studying native proteins. Such a comparison provided deeper insight that the CuA center is a more efficient ET center than blue copper97.
Figure 5. Close match between a designed metalloprotein and its native target protein.
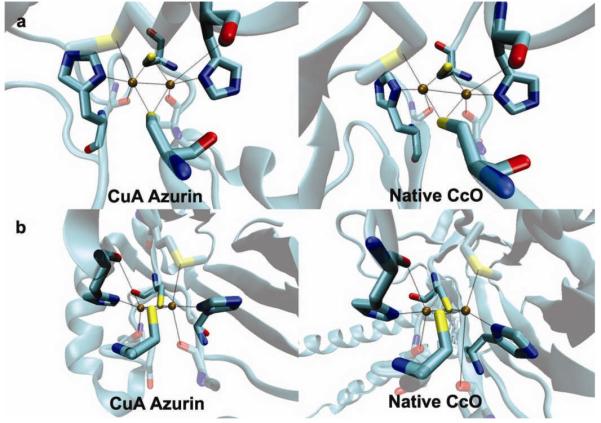
Crystal structures of a designed CuA site in azurin (PDB code 1CC3)49 and a CuA site in native CcO (PDB code: 1AR1) as viewed from above the Cu2(Scys)2 plane (a), and along the Cu2(Scys)2 plane.
Given the initial successes, it is a good time to summarize the lessons learned and to recognize future challenges. Essential to success is the recognition that structural biology must play a major role in the design process. Some early metalloprotein designs have generated much excitement in the field, only to be shown by x-ray crystallography to contain a different metal-binding site than originally designed. While spectroscopy is invaluable in providing support for the design, more concrete evidence from 3D structural characterization is essential. In addition to confirming the initial design, it provides detailed information on what has or has not been designed correctly, thus providing insights on the next level of design.
Furthermore, primary coordination sphere design is not enough in many cases, to confer structure and/or function. Consideration of the environment surrounding the primary coordination sphere is critical to successful design. One primary example is water inside the protein, where the associated hydrogen bonding interactions play essential roles in many metalloproteins, including Cyt P450, heme oxygenase (HO), and CcO33. For example, even though a CuB center was designed into myoglobin that structurally mimics the heme-copper center in CcO53, its function mimics HO rather than CcO when O2 is used as an oxidant54. When H2O2 is used as an oxidant, a ferryl species, a putative intermediate in CcO activity, was observed. These results suggest that the extra protons introduced through H2O2 are important for function54. The introduction of an additional hydrogen bonding donor through the replacement of the b-type heme in Mb with an o-type heme, resembling the heme found in CcO, allowed suppression of HO activity and promotion of CcO activity98.
Metalloprotein design has focused on the positive design of structural features responsible for correct metal-site geometry and function. However, negative design to prevent competing metal-site geometries and unwanted functions is also critical. For example, in designing dinuclear metalloproteins in a helical bundle, there was a possibility that undesirable heterotetrameric motifs and homooligomers would form since the protein was comprised of different peptides. To circumvent this problem, a computational algorithm was included to destabilize these interactions99.
The final lesson is that metalloprotein design often results in lower stability because of changes made to the protein scaffold. Sometimes, the stability is so low that even if the design is valid, not enough stable protein can be obtained to prove the design. The stability of the protein must be improved, even if function may not be improved directly. A beautiful example is demonstrated in cytochrome P450 where only after the protein was stabilized, could the highly destabilizing mutations introduced confer novel activity100.
The majority of the successes in the metalloprotein design field are derived from the construction of a geometrically correct, sterically compatible primary coordination sphere sufficient to reproduce the structure and function of a desired metal center, such as a bis-His heme or His2Cys2 tetrahedral Zn(II) site. However, many metal-binding sites in proteins, such as the type 1 copper and CuA centers, are not necessarily in their preferred metal ion geometric states. Therefore, one future challenge is to design metal sites and metal clusters with unique geometric requirements. Other future challenges include the design of metal sites at the interface of two or more proteins, which occur often in proteins but have not been designed until recently39,40, and the design of metal-binding sites from membrane proteins, which has also been rare. An even higher level of challenge is the design of protein metal-binding sites requiring helpers called chaperones. Consideration of the interactions between metal ions and protein hosts is not enough to confer function, making their interactions with chaperones necessary in the design process. In addition to metal-binding site design, functional metalloproteins also require the design of a substrate-binding site for catalysis. An even higher level of complexity includes coupling the redox reaction of a designed metal-binding site to proton transfer, conformational change, or charge separation.
To meet these challenges, advances in a number of fields are required. The most important requirement is the development of better computer programs for designing metal-binding sites in proteins, including consideration of structural features beyond the primary coordination sphere. In contrast to well-defined force fields for peptide bonds and amino acid side chains, most force fields for metal-binding sites are ill-defined because there are many more variables involved in defining a metal-binding site. These variables include variations on the identity of the metal ion, different oxidation states of the same metal ions, and different ligand and geometric preferences of different metal ions. Density functional theory has been excellent in defining small metal complexes. Extending its success to metal-binding sites in proteins, together with other leading algorithms, such as QM/MM (quantum mechanics/molecular mechanics), is required for the successful rational design of metalloproteins. Despite these challenges, metalloprotein design will remain a vibrant field where exciting discoveries of new insights and novel enzymes will occur every day.
Acknowledgment
we wish to thank Professor William F. DeGrado for providing Figure 1a, Ms. Nandini Nagraj for help in editing, and NSF and NIH for financial support.
REFERENCES
- 1.Lu Y, Berry SM, Pfister TD. Engineering novel metalloproteins: design of metal-binding sites into native protein scaffolds. Chem. Rev. 2001;101:3047–3080. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]
- 2.Barker PD. Designing redox metalloproteins from bottom-up and top-down perspectives. Curr. Opin. Struct. Biol. 2003;13:490–499. doi: 10.1016/s0959-440x(03)00108-8. [DOI] [PubMed] [Google Scholar]
- 3.Lu Y. Metalloprotein and metallo-DNA/RNAzyme design: current approaches, success measures and future challenges. Inorg. Chem. 2006;45:9930–9940. doi: 10.1021/ic052007t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. De novo design and structural characterization of proteins and metalloproteins. Annu. Rev. Biochem. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 5.Reedy CJ, Gibney BR. Heme protein assemblies. Chem. Rev. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 6.Choma CT, et al. Design of a heme-binding four-helix bundle. J. Am. Chem. Soc. 1994;116:856–65. [Google Scholar]
- 7.Robertson DE, et al. Design and synthesis of multi-heme proteins. Nature. 1994;368:425–32. doi: 10.1038/368425a0. [DOI] [PubMed] [Google Scholar]
- 8.Case MA, McLendon GL. Metal-assembled modular proteins: toward functional protein design. Acc. Chem. Res. 2004;37:754–762. doi: 10.1021/ar960245+. [DOI] [PubMed] [Google Scholar]
- 9.Huang SS, Koder RL, Lewis M, Wand AJ, Dutton PL. The HP-1 maquette: from an apoprotein structure to a structured hemoprotein designed to promote redox-coupled proton exchange. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5536–5541. doi: 10.1073/pnas.0306676101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sasaki T, Kaiser ET. Helichrome: Synthesis and enzymic activity of a designed hemeprotein. J. Am. Chem. Soc. 1989;111:380–381. [Google Scholar]
- 11.Das A, Hecht MH. Peroxidase activity of de novo heme proteins immobilized on electrodes. J. Inorg. Biochem. 2007;101:1820–1826. doi: 10.1016/j.jinorgbio.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monien BH, Drepper F, Sommerhalter M, Lubitz W, Haehnel W. Detection of heme oxygenase activity in a library of four-helix bundle proteins: towards the de novo synthesis of functional heme proteins. J. Mol. Biol. 2007;371:739–753. doi: 10.1016/j.jmb.2007.05.047. [DOI] [PubMed] [Google Scholar]
- 13.Koder RL, et al. Design and engineering of an O2 transport protein. 2009;458:305–309. doi: 10.1038/nature07841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klemba M, Regan L. Characterization of metal binding by a designed protein: single ligand substitutions at a tetrahedral Cys2His2 site. Biochemistry. 1995;34:10094–100. doi: 10.1021/bi00031a034. [DOI] [PubMed] [Google Scholar]
- 15.Handel T, DeGrado WF. De novo design of a Zn2+-binding protein. J. Am. Chem. Soc. 1990;112:6710–11. [Google Scholar]
- 16.Touw DS, Nordman CE, Stuckey JA, Pecoraro VL. Identifying important structural characteristics of arsenic resistance proteins by using designed three-stranded coiled coils. Proc. Natl. Acad. Sci. U.S.A. 2007;104:11969–11974. doi: 10.1073/pnas.0701979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lombardi A, et al. Retrostructural analysis of metalloproteins: application to the design of a minimal model for diiron proteins. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6298–6305. doi: 10.1073/pnas.97.12.6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaplan J, DeGrado WF. De novo design of catalytic proteins. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calhoun JR, et al. Oxygen reactivity of the biferrous site in the de novo designed four helix bundle peptide DFsc: nature of the “intermediate” and reaction mechanism. J. Am. Chem. Soc. 2008;130:9188–9189. doi: 10.1021/ja801657y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matzapetakis M, et al. Comparison of the binding of cadmium(II), mercury(II), and arsenic(III) to the de novo designed peptides TRI L12C and TRI L16C. J. Am. Chem. Soc. 2002;124:8042–8054. doi: 10.1021/ja017520u. [DOI] [PubMed] [Google Scholar]
- 21.Kiyokawa T, et al. Binding of Cu(II) or Zn(II) in a de novo designed triple-stranded alpha-helical coiled-coil toward a prototype for a metalloenzyme. J. Pept. Res. 2004;63:347–353. doi: 10.1111/j.1399-3011.2004.00109.x. [DOI] [PubMed] [Google Scholar]
- 22.Petros AK, Reddi AR, Kennedy ML, Hyslop AG, Gibney BR. Femtomolar Zn(II) affinity in a peptide-based ligand designed to model thiolate-rich metalloprotein active sites. Inorg. Chem. 2006;45:9941–9958. doi: 10.1021/ic052190q. [DOI] [PubMed] [Google Scholar]
- 23.Dieckmann GR, et al. De Novo design of mercury-binding two- and three-helical bundles. J. Am. Chem. Soc. 1997;119:6195–6196. [Google Scholar]
- 24.Ghosh D, Pecoraro VL. Probing metal-protein interactions using a de novo design approach. Curr. Opin. Chem. Biol. 2005;9:97–103. doi: 10.1016/j.cbpa.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 25.Kharenko OA, Ogawa MY. Metal-induced folding of a designed metalloprotein. J. Inorg. Biochem. 2004;98:1971–1974. doi: 10.1016/j.jinorgbio.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 26.Farrer BT, Pecoraro VL. Hg(II) binding to a weakly associated coiled coil nucleates an encoded metalloprotein fold: a kinetic analysis. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3760–3765. doi: 10.1073/pnas.0336055100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iranzo O, Jakusch T, Lee K-H, Hemmingsen L, Pecoraro VL. The correlation of 113Cd NMR and 111mCd PAC spectroscopies provides a powerful approach for the characterization of the structure of CdII-substituted ZnII proteins. Chem. Eur. J. 2009;15:3761–3772. doi: 10.1002/chem.200802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peacock AFA, Iranzo O, Pecoraro VL. Harnessing natures ability to control metal ion coordination geometry using de novo designed peptides. Dalton Trans. 2009:2271–2280. doi: 10.1039/b818306f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nanda V, et al. De novo design of a redox-active minimal rubredoxin mimic. J. Am. Chem. Soc. 2005;127:5804–5805. doi: 10.1021/ja050553f. [DOI] [PubMed] [Google Scholar]
- 30.Kono H, Saven JG. Statistical theory for protein combinatorial libraries. Packing interactions, backbone flexibility, and the sequence variability of a main-chain structure. J. Mol. Biol. 2001;306:607–628. doi: 10.1006/jmbi.2000.4422. [DOI] [PubMed] [Google Scholar]
- 31.Williams RJP. Energised (entatic) states of groups and of secondary structures in proteins and metalloproteins. Eur. J. Biochem. 1995;234:363–81. doi: 10.1111/j.1432-1033.1995.363_b.x. [DOI] [PubMed] [Google Scholar]
- 32.Ueno T, Ohki T, Watanabe Y. Molecular engineering of cytochrome P 450 and myoglobin for selective oxygenations. J. Porphyrins Phthalocyanines. 2004;8:279–289. [Google Scholar]
- 33.Yeung N, Lu Y. One heme, diverse functions: using biosynthetic myoglobin models to gain insights into heme copper oxidases and nitric oxide reductases. Chem. Biodivers. 2008;5:1437–1454. doi: 10.1002/cbdv.200890134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ozaki S.-i., Matsui T, Watanabe Y. Conversion of myoglobin into a highly stereospecific peroxygenase by the L29H/H64L mutation. J. Am. Chem. Soc. 1996;118:9784–9785. [Google Scholar]
- 35.Jensen KK, Martini L, Schwartz TW. Enhanced fluorescence resonance energy transfer between spectral variants of green fluorescent protein through zinc-site engineering. Biochemistry. 2001;40:938–945. doi: 10.1021/bi001765m. [DOI] [PubMed] [Google Scholar]
- 36.Evers TH, Appelhof MA, de Graaf-Heuvelmans PT, Meijer EW, Merkx M. Ratiometric detection of Zn(II) using chelating fluorescent protein chimeras. J. Mol. Biol. 2007;374:411–425. doi: 10.1016/j.jmb.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 37.Mizuno T, Murao K, Tanabe Y, Oda M, Tanaka T. Metal-ion dependent GFP emission in vivo by combining a circularly permutated green fluorescent protein with an engineered metal ion-binding coiled-coil. J. Am. Chem. Soc. 2007;129:11378–11383. doi: 10.1021/ja0685102. [DOI] [PubMed] [Google Scholar]
- 38.Wegner SV, Boyaci H, Chen H, Jensen MP, He C. Engineering a uranyl-specific binding protein from NikR. Ang. Chem. Int. Ed. 2009;48:2339–2341. doi: 10.1002/anie.200805262. [DOI] [PubMed] [Google Scholar]
- 39.Salgado EN, Faraone-Mennella J, Tezcan FA. Controlling protein-protein interactions through metal coordination: assembly of a 16-helix bundle protein. J. Am. Chem. Soc. 2007;129:13374–13375. doi: 10.1021/ja075261o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matthews JM, Loughlin FE, Mackay JP. Designed metal-binding sites in biomolecular and bioinorganic interactions. Curr. Opin. Struct. Biol. 2008;18:484–490. doi: 10.1016/j.sbi.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 41.Park HS, et al. Design and evolution of new catalytic activity with an existing protein scaffold. Science. 2006;311:535–538. doi: 10.1126/science.1118953. [DOI] [PubMed] [Google Scholar]
- 42.Vita C, Roumestand C, Tom F, Menez A. Scorpion toxins as natural scaffolds for protein engineering. Proc. Natl. Acad. Sci. U.S.A. 1995;92:6404–8. doi: 10.1073/pnas.92.14.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Müller HN, Skerra A. Grafting of a high-affinity Zn(II)-binding site on the beta-barrel of retinol-binding protein results in enhanced folding stability and enables simplified purification. Biochemistry. 1994;33:14126–35. doi: 10.1021/bi00251a023. [DOI] [PubMed] [Google Scholar]
- 44.Desjarlais JR, Clarke ND. Computer search algorithms in protein modification and design. Curr. Opin. Struct. Biol. 1998;8:471–475. doi: 10.1016/s0959-440x(98)80125-5. [DOI] [PubMed] [Google Scholar]
- 45.Benson DE, Wisz MS, Liu W, Hellinga HW. Construction of a novel redox protein by rational design: conversion of a disulfide bridge into a mononuclear iron-sulfur center. Biochemistry. 1998;37:7070–6. doi: 10.1021/bi980583d. [DOI] [PubMed] [Google Scholar]
- 46.Yang W, et al. Rational design of a calcium-binding protein. J. Am. Chem. Soc. 2003;125:6165–6171. doi: 10.1021/ja034724x. [DOI] [PubMed] [Google Scholar]
- 47.Shete VS, Benson DE. Protein design provides lead(II) ion biosensors for imaging molecular fluxes around red blood cells. Biochemistry. 2009;48:462–470. doi: 10.1021/bi801777h. [DOI] [PubMed] [Google Scholar]
- 48.Hay M, Richards JH, Lu Y. Construction and characterization of an azurin analog for the purple copper site in cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 1996;93:461–464. doi: 10.1073/pnas.93.1.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robinson H, et al. Structural basis of electron transfer modulation in the purple CuA center. Biochemistry. 1999;38:5677–5683. doi: 10.1021/bi9901634. [DOI] [PubMed] [Google Scholar]
- 50.Dennison C, Vijgenboom E, de Vries S, van der Oost J, Canters GW. Introduction of a CuA site into the blue copper protein amicyanin from Thiobacillus versutus. FEBS Lett. 1995;365:92–94. doi: 10.1016/0014-5793(95)00429-d. [DOI] [PubMed] [Google Scholar]
- 51.Jones LH, Liu A, Davidson VL. An engineered CuA amicyanin capable of intermolecular electron transfer reactions. J. Biol. Chem. 2003;278:47269–47274. doi: 10.1074/jbc.M308863200. [DOI] [PubMed] [Google Scholar]
- 52.Franklin SJ, Welch JT. The helix-turn-helix as a scaffold for chimeric nuclease design. Comments Inorg. Chem. 2005;26:127–164. [Google Scholar]
- 53.Sigman JA, Kwok BC, Lu Y. From myoglobin to heme-copper oxidase: design and engineering of a CuB Center into sperm whale myoglobin. J. Am. Chem. Soc. 2000;122:8192–8196. [Google Scholar]
- 54.Sigman JA, Kim HK, Zhao X, Carey JR, Lu Y. The role of copper and protons in heme-copper oxidases: kinetic study of an engineered heme-copper center in myoglobin. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3629–3634. doi: 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao X, Yeung N, Russell BS, Garner DK, Lu Y. Catalytic reduction of NO to N2O by a designed heme copper center in myoglobin: implications for the role of metal ions. J. Am. Chem. Soc. 2006;128:6766–6767. doi: 10.1021/ja058822p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yeung BKS, Wang X, Sigman JA, Petillo PA, Lu Y. Construction and characterization of a manganese-binding site in cytochrome c peroxidase: towards a novel manganese peroxidase. Chem. Biol. 1997;4:215–221. doi: 10.1016/s1074-5521(97)90291-x. [DOI] [PubMed] [Google Scholar]
- 57.Abe S, Ueno T, Watanabe Y. Artificial metalloproteins exploiting vacant space: preparation, structures, and functions. Top. Organomet. Chem. 2009;25:25–43. [Google Scholar]
- 58.Ueno T, et al. Size-selective olefin hydrogenation by a Pd nanocluster provided in an apo-ferritin cage. Ang. Chem. Int. Ed. 2004;43:2527–2530. doi: 10.1002/anie.200353436. [DOI] [PubMed] [Google Scholar]
- 59.Varpness Z, Peters JW, Young M, Douglas T. Biomimetic synthesis of a H2 catalyst using a protein cage architecture. Nano Lett. 2005;5:2306–2309. doi: 10.1021/nl0517619. [DOI] [PubMed] [Google Scholar]
- 60.Yin J, Mills JH, Schultz PG. A catalysis-based selection for peroxidase antibodies with increased activity. J. Am. Chem. Soc. 2004;126:3006–3007. doi: 10.1021/ja039198o. [DOI] [PubMed] [Google Scholar]
- 61.Rasmussen BS, et al. Enantioselective proteins: selection, binding studies and molecular modeling of antibodies with affinity towards hydrophobic BINOL derivatives. ChemBioChem. 2007;8:1974–1980. doi: 10.1002/cbic.200700295. [DOI] [PubMed] [Google Scholar]
- 62.Fasan R, Chen MM, Crook NC, Arnold FH. Engineered alkane-hydroxylating cytochrome P450BM3 exhibiting nativelike catalytic properties. Ang. Chem. Int. Ed. 2007;46:8414–8418. doi: 10.1002/anie.200702616. [DOI] [PubMed] [Google Scholar]
- 63.Lu Y. Design and engineering of metalloproteins containing unnatural amino acids or non-native metal-containing cofactors. Curr. Opin. Chem. Biol. 2005;9:118–126. doi: 10.1016/j.cbpa.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 64.Merrifield B. Concept and early development of solid-phase peptide synthesis. Methods Enzymol. 1997;289:3–13. doi: 10.1016/s0076-6879(97)89040-4. [DOI] [PubMed] [Google Scholar]
- 65.Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Synthesis of proteins by native chemical ligation. Science. 1994;266:776–9. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 66.Muir TW. Semisynthesis of proteins by expressed protein ligation. Annu. Rev. Biochem. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 67.Ikeda Y, et al. Synthesis of a novel histidine analog and its efficient incorporation into a protein in vivo. Protein Eng. 2003;16:699–706. doi: 10.1093/protein/gzg084. [DOI] [PubMed] [Google Scholar]
- 68.Qi D, Tann C-M, Haring D, Distefano MD. Generation of new enzymes via covalent modification of existing proteins. Chem. Rev. 2001;101:3081–3111. doi: 10.1021/cr000059o. [DOI] [PubMed] [Google Scholar]
- 69.Barrick D. Depletion and replacement of protein metal ligands. Curr. Opin. Biotechnol. 1995;6:411–418. doi: 10.1016/0958-1669(95)80070-0. [DOI] [PubMed] [Google Scholar]
- 70.Noren CJ, Anthony-Cahill SJ, Griffith MC, Schultz PG. A general method for site-specific incorporation of unnatural amino acids into proteins. Science. 1989;244:182–8. doi: 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- 71.Lee HS, Schultz PG. Biosynthesis of a site-specific DNA cleaving protein. J. Am. Chem. Soc. 2008;130:13194–13195. doi: 10.1021/ja804653f. [DOI] [PubMed] [Google Scholar]
- 72.Privett HK, Reedy CJ, Kennedy ML, Gibney BR. Nonnatural amino acid ligands in heme protein design. J. Am. Chem. Soc. 2002;124:6828–6829. doi: 10.1021/ja025534+. [DOI] [PubMed] [Google Scholar]
- 73.Petros AK, Shaner SE, Costello AL, Tierney DL, Gibney BR. Comparison of cysteine and penicillamine ligands in a Co(II) maquette. Inorg. Chem. 2004;43:4793–4795. doi: 10.1021/ic0497679. [DOI] [PubMed] [Google Scholar]
- 74.Lee K-H, Cabello C, Hemmingsen L, Marsh ENG, Pecoraro VL. Using nonnatural amino acids to control metal-coordination number in three-stranded coiled coils. Ang. Chem. Int. Ed. 2006;45:2864–2868. doi: 10.1002/anie.200504548. [DOI] [PubMed] [Google Scholar]
- 75.Peacock AFA, Hemmingsen L, Pecoraro VL. Using diastereopeptides to control metal ion coordination in proteins. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16566–16571. doi: 10.1073/pnas.0806792105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Low DW, Hill MG. Rational fine-tuning of the redox potentials in chemically synthesized rubredoxins. J. Am. Chem. Soc. 1998;120:11536–11537. [Google Scholar]
- 77.Low DW, Hill MG. Backbone-engineered high-potential iron proteins: effects of active-site hydrogen binding on reduction potential. J. Am. Chem. Soc. 2000;122:11039–11040. [Google Scholar]
- 78.Berry SM, Gieselman MD, Nilges MJ, Van der Donk WA, Lu Y. An engineered azurin variant containing a selenocysteine copper ligand. J. Am. Chem. Soc. 2002;124:2084–2085. doi: 10.1021/ja0169163. [DOI] [PubMed] [Google Scholar]
- 79.Berry SM, Ralle M, Low DW, Blackburn NJ, Lu Y. Probing the role of axial methionine in the blue copper center of azurin with unnatural amino acids. J. Am. Chem. Soc. 2003;125:8760–8768. doi: 10.1021/ja029699u. [DOI] [PubMed] [Google Scholar]
- 80.Hayashi T, Hisaeda Y. New functionalization of myoglobin by chemical modification of heme-propionates. Acc. Chem. Res. 2002;35:35–43. doi: 10.1021/ar000087t. [DOI] [PubMed] [Google Scholar]
- 81.Matsuo T, Hayashi T, Hisaeda Y. Reductive activation of dioxygen by a myoglobin reconstituted with a flavohemin. J. Am. Chem. Soc. 2002;124:11234–11235. doi: 10.1021/ja027291r. [DOI] [PubMed] [Google Scholar]
- 82.Hayashi T, et al. Crystal structure and peroxidase activity of myoglobin reconstituted with iron porphycene. Inorg. Chem. 2006;45:10530–10536. doi: 10.1021/ic061130x. [DOI] [PubMed] [Google Scholar]
- 83.Cochran FV, et al. Computational de novo design and characterization of a four-helix bundle protein that selectively binds a nonbiological cofactor. J. Am. Chem. Soc. 2005;127:1346–1347. doi: 10.1021/ja044129a. [DOI] [PubMed] [Google Scholar]
- 84.Bender GM, et al. De novo design of a single-chain diphenylporphyrin metalloprotein. J. Am. Chem. Soc. 2007;129:10732–10740. doi: 10.1021/ja071199j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ohashi M, et al. Preparation of artificial metalloenzymes by insertion of chromium schiff base complexes into apomyoglobin mutants. Angew. Chem. Int. Ed. 2003;42:1005–1008. doi: 10.1002/anie.200390256. [DOI] [PubMed] [Google Scholar]
- 86.Ueno T, et al. Crystal structures of artificial metalloproteins: tight binding of FeIII(schiff-base) by mutation of Ala71 to Gly in apo-myoglobin. Inorg. Chem. 2004;43:2852–2858. doi: 10.1021/ic0498539. [DOI] [PubMed] [Google Scholar]
- 87.Wilson ME, Whitesides GM. Conversion of a protein to a homogeneous asymmetric hydrogenation catalyst by site-specific modification with a diphosphinerhodium(I) moiety. J. Am. Chem. Soc. 1978;100:306–307. [Google Scholar]
- 88.Collot J, et al. Artificial metalloenzymes for enantioselective catalysis based on biotin-avidin. J. Am. Chem. Soc. 2003;125:9030–9031. doi: 10.1021/ja035545i. [DOI] [PubMed] [Google Scholar]
- 89.Steinreiber J, Ward TR. Artificial metalloenzymes as selective catalysts in aqueous media. Coord. Chem. Rev. 2008;252:751–766. [Google Scholar]
- 90.Letondor C, et al. Artificial transfer hydrogenases based on the biotin- (strept)avidin technology: fine tuning the selectivity by saturation mutagenesis of the host protein. J. Am. Chem. Soc. 2006;128:8320–8328. doi: 10.1021/ja061580o. [DOI] [PubMed] [Google Scholar]
- 91.Creus M, et al. X-ray structure and designed evolution of an artificial transfer hydrogenase. Ang. Chem. Int. Ed. 2008;47:1400–1404. doi: 10.1002/anie.200704865. [DOI] [PubMed] [Google Scholar]
- 92.Chen C.-h. B., Milne L, Landgraf R, Perrin DM, Sigman DS. Artificial nucleases. ChemBioChem. 2001;2:735–740. doi: 10.1002/1439-7633(20011001)2:10<735::aid-cbic735>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 93.Davies RR, Distefano MD. A semisynthetic metalloenzyme based on a protein cavity that catalyzes the enantioselective hydrolysis of ester and amide substrates. J. Am. Chem. Soc. 1997;119:11643–11652. [Google Scholar]
- 94.Ory J, et al. Structural characterization of two synthetic catalysts based on adipocyte lipid-binding protein. Protein Eng. 1998;11:253–261. doi: 10.1093/protein/11.4.253. [DOI] [PubMed] [Google Scholar]
- 95.Carey JR, et al. A site-selective dual anchoring strategy for artificial metalloprotein design. J. Am. Chem. Soc. 2004;126:10812–10813. doi: 10.1021/ja046908x. [DOI] [PubMed] [Google Scholar]
- 96.Zhang J, Garner DK, Liang L, Chen Q, Lu Y. Protein scaffold of a designed metalloenzyme enhances the chemoselectivity in sulfoxidation of thioanisole. Chem. Comm. 2008:1665–1667. doi: 10.1039/b718915j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Farver O, Lu Y, Ang MC, Pecht I. Enhanced rate of intramolecular electron transfer in an engineered purple CuA azurin. Proc. Natl. Acad. Sci. U.S.A. 1999;96:899–902. doi: 10.1073/pnas.96.3.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang N, Zhao X, Lu Y. Role of heme types in heme-copper oxidases: effects of replacing a heme b with a heme o mimic in an engineered heme-copper center in myoglobin. J. Am. Chem. Soc. 2005;127:16541–16547. doi: 10.1021/ja052659g. [DOI] [PubMed] [Google Scholar]
- 99.Summa CM, Rosenblatt MM, Hong J-K, Lear JD, DeGrado WF. Computational de novo design, and characterization of an A2B2 diiron protein. J. Mol. Biol. 2002;321:923–938. doi: 10.1016/s0022-2836(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 100.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5869–5874. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]