

Abstract
The dramatic biogeographical variations in the secondary metabolites from Psammocinia aff. bulbosa have complicated our efforts to re-isolate the two most cytotoxic of its metabolites, (+)-psymberin and (+)-cyclocinamide A. Reported now are the results of a new study that demonstrates our ability to repeatedly isolate these two compounds through targeted collection efforts. Additional study of the new sample of (+)-cyclocinamide A has enabled finalizing its biological activity and absolute stereochemistry as 4S, 7S, 11S, 14S.
In the early 1990s we began a campaign to survey the chemical ecology of Psammocinia aff. bulbosa communities from Papua New Guinea. It is now clear that a side-by-side chemical and taxonomic examination is obligatory to differentiate among populations of Psammocinia (Dictyoceratida, Irciniidae), Cacospongia (Dictyoceratida, Thorectidae), or Ircinia (Dictyoceratida, Irciniidae)1 having very similar morphologies. Recognizing this critical step made it possible to engage in a meaningful synopsis of the chemical variations observed for a small library of P. aff. bulbosa specimens. Reported now are the final results of a project whose goals were to: (a) obtain fresh sponge specimens containing both psymberin and cyclocinamide A in order to facilitate on-going molecular genetics studies on their biosynthetic pathways,2 (b) complete the full absolute chirality assignment for cyclocinamide A,3,4 and (c) accumulate additional biological activity data for cyclocinamide A.3
Outlined in Table 1 are an astonishing variety of biosynthetic products that we have observed to date from six distinct collections whose voucher material has each been identified as Psammocinia aff. bulbosa. The major constituents of specimens I – V range from sesterterpenes (variabilin5), polyketides (preswinholide A6 and swinholide A6,7), a cyano-sponge cyclic hexapeptide (psymbamide A7), a halogenated hexapeptide (cyclocinamide A3), to a NRPS-PKS mixed biogenetic derived compound (psymberin8). By contrast, meroterpenes (cacospongin B9 and chromarol D9) usually observed from sponges of Cacospongia- genus were isolated from sample VI. Though samples V and VI were obtained from adjacent sites, the chemical profiles of their major constituents clearly showed the biosynthetic machinery of these two sponge collections was distinct. These plus the other dramatic biogeographical variations shown in Table 1 complicated our efforts to re-isolate the two most structurally unique of these metabolites, psymberin and cyclocinamide A.
Table 1.
Varying Morphotypes of Psammocinia aff. bulbosa from Papua New Guinea Correlated with Biogeographical Variations in their Constituents. Underwater pictures of all samples have been previously published.1,7
| Specimen Type | Coll. no. | Location | Coordinates | Major Constituents |
|---|---|---|---|---|
| I | 931457 | Normanby | 9°43′S: 150°47′E | (+) cyclocinamide A (-) variabilin |
| IIa IIb |
023157 | Amphlett Milne Bay |
9°43′S: 150°44′E 10°13′S: 150°52′E |
(+) psymberin (+) swinholide A |
| III | 035261 | Amphlett | 9°37′S: 150°57′E to 9°14′S: 150°46′E |
(+) psymberin |
| IV | 054111 | Milne Bay to Pilkinton Reef | 10°41′S: 152°50′E 11°19′S: 154°16′E 11°26′S: 154°24′E |
(-) preswinholide A (+) swinholide A |
| V | 061217 | Milne Bay | 9°58′S: 150°57′E 10°15′S: 150°40′E |
(-) preswinholide A (-) psymbamide A |
| VI | 061227 | Milne Bay | 9°58′S: 150°57′E 10°15′S: 150°40′E |
(+) chromarol D (-) cacospongin B |
Before proceeding, it is important to briefly review the historical record highlighted in Figure 1 pertaining to absolute stereochemical assignments for psymberin and cyclocinamide A. The 5S, 8S, 9S, 11R, 13R, 15S, 16R, 17R structure we originally proposed for (+)-psymberin8 (1a) was reaffirmed from two independent total syntheses of 1c.10,11 The synthetic products also provided the basis for the 4S designation at the single unassigned carbon and verified that (+)-psymberin (1a) we isolated from P. aff. bulbosa was identical to (+)-ircinistatin A12 (1b) reported by Pettit from Ircinia cf. ramosa. Four additional diastereomers were also created during these synthesis projects and the most important included 1d and 1e.10,11,13 The stereostructure of (+)-cyclocinamide A (2a), we described in 1997, included firm assignments at 7S and 14S but the 4S and 11S designations were speculative.3 In 1998, a total synthesis by Grieco afforded the 4R, 7S, 11R, 14S structure 2b (no [α] reported), whose 1H NMR spectra was similar to but not identical to that of 2a.4 Rather fascinating is the report of (+)-4S, 7R, 11S, 14R-cyclocinamide B14 (3) by Ireland from a Fijian sponge Corticium sp. (Homoslerophida, Plakinidae) which would be enantiomeric to 2b if the C-36 substituents were identical.
Figure 1.
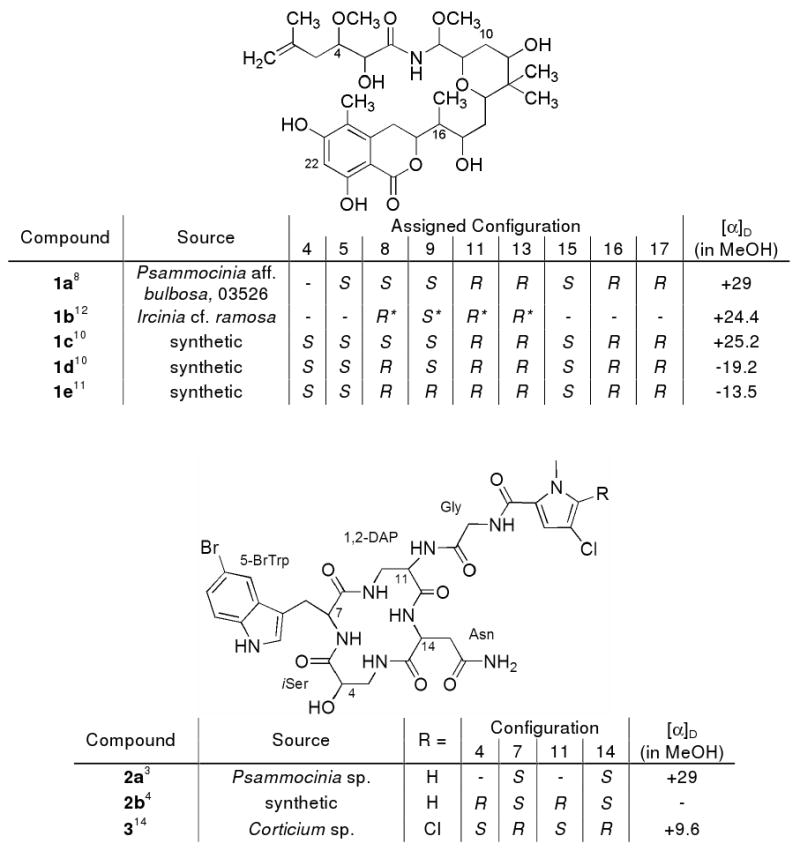
Psymberin (1a)/Ircinistatin A (1b) and Cyclocinamide (2-3) compounds
The depletion of both sponge material and authentic samples of psymberin and cyclocinamide A in our repository necessitated a recollection effort. Our plan to obtain taxa containing both psymberin and cyclocinamide A was formulated based on the clarifying chemotype patterns shown in Table 1. The best Papua New Guinea collection site for the new samples appeared to be in the Amphlett vicinity of 9°43′S: 150°50′E to 9°14′S: 150°46′E. Thus, in May 2007, recollections were conducted at two locations yielding: (a) sample VII: Milne Bay 10°16′S: 150°47′E (coll. no. 07202, 76 g dry weight); and (b) sample VIII: Amphlett 9°14′S: 150°46′E (coll. no. 07208, 21 g dry weight). Parallel extraction of both collections followed by LC-MS analyses of the resultant crude mixtures indicated the presence of major metabolites as follows: sample VII - swinholide A15 and preswinolide A6 but no psymberin; and sample VIII - (+)-psymberin (1a) (MW 609, [α]27D = +23.4, 3.2 mg, 1.5×10-2% of the dry weight), microgram quantities of two psymberin analogs of MW 595 and MW 625 previously observed,7 and (+)-cyclocinamide A (2a) (MW 772, [α]27D = +32, 0.5 mg, 2.4×10-3% of the dry weight). Although additional work on sample VII was discontinued, the preceding results reaffirmed the expectations shown in Table 1 pertaining to the best collection sites for 1a and 2a containing samples. Once these outcomes had been obtained, portions of sponge VIII were shuttled to the Piel group at the University of Bonn for molecular genetics experiments paralleling those completed for pederin16 and findings will be published in the near future.
The isolation work on sample VIII proceeded in a multi-step fashion. The dichloromethane (coded XFD) and methanol (coded XFM) fractions showed potent activity in the soft agar colony forming disk diffusion assay (DDA).3 However, due to the paucity of the biological material, a traditional round of prefractionation was forgone. Both these extract fractions were directly subjected to semipreparative HPLC. The new sample of (+)-psymberin (1a) was obtained from the XFD and XFM fractions, while (+)- cyclocinamide A (2a) came from the XFM fraction. The identity of both compounds were confirmed from 1H and 13C NMR data and included in the case of 2a the additional diagnostic ESI-TOF-MS BrCl pseudomolecular ion cluster at [M+Na]+ m/z 772.3 In the original structure elucidation of 2a, the iSer OH 1H NMR shift was reported at δH 3.27 from water suppression data, but further inspection of the published 1H NMR spectra of 2a reveals that in either DMSO-d6 or DMSO-d6/benzene-d6, there are exchangeable proton signals at δH 6.05 and δH 6.15, respectively; however, these are not detected under CD3OD conditions. The δH 6.01 (d, 3J = 5 Hz) iSer OH signal was clearly evident in the new sample of 2a and consistent with observation that in (+)-cyclocinamide B (3) the iSer OH occurs at δH 6.00 (d, 3J = 4 Hz).14
The Marfey's method was employed to establish the absolute configuration of the chiral carbons in the new sample leading to the structure assigned as 4S, 7S, 11S, 14S-cyclocinamide A (2a). This began by treating the acid hydrolysate of 2a with l-FDAA (N-α-(2,4-dinitro-5-flourophenyl)-l-alanine amide)17 followed by HPLC analysis. The reference complexes were prepared by a similar derivatization employing standard amino acids. The HPLC conditions gave profiles in which each l-amino acid-FDAA complex eluted before the d-amino acid analogue, except for the β-amino acid iSer whose l/d elution order was reversed.18 Acceptable retention time differences were observed for all HPLC runs. Thus, the comparative analysis by LC-MS of the derivatized hydrolysates obtained from 2a vs. that of the standard amino acids was the basis for the all l-amino acid configuration assigned.
The solid tumor selectivity of (+)-cyclocinamide A (2a), originally reported as selective against colon-38 vs. L1210 cells in the DDA,3 has stimulated sustained interest in this unusual cyclic tetrapeptide core structure possessing the unique dipeptide side chain. The Grieco total synthesis of 2b was relevant as a next step; unfortunately the small amount of material obtained was insufficient for further in vitro evaluation. Another development was that Ireland found (+)-cyclocinamide B (3) to be inactive against HCT-116 cells.14 Our further evaluation of 2a revealed non-potent, modest solid tumor selectivity with inhibition zone sizes (90 μg/disk) against HCT-125 (26 mm) and murine CFU-GM (16 mm) in the DDA, and the IC50 against HCT-116 was > 10 μg/mL. Thus, the previously encouraging solid tumor selectivity for 2a against colon-38 was not seen against HCT cell lines for both 2a and 3.
The results presented above have added a new dimension to the chemical ecology of P. aff. bulbosa sponges. First, neither 1a nor 2a can be considered as marker compounds for Psammocinia because of their occurrence in other unrelated sponges. Second, a specific population of P. aff. bulbosa has now been shown to repeatedly be a source of 1a and 2a, meaning that further examination of the microbial communities associated with these distinct populations could be rewarding.22 Third, adjacent populations of material identified as P. aff. bulbosa (see Samples V and VI, Table 1) have been shown to vary considerably in their terpene vs. their PKS and NRPS type major metabolites. Finally, our re-examination of the bioactivity of 2a offers additional clarifying data. Future efforts to prepare libraries of compounds based on the cyclocinamide framework both with and without the BrTrp moiety could be warrented.19
Experimental Section
General Experimental Procedures
1H, 13C NMR experiments were run in Shigemi Tubes on a Varian UNITY-500 spectrometer (500 and 125 MHz for 1H and 13C, respectively) using standard pulse sequences with residual solvent protons and carbons as references (DMSO-d6: 2.50 and 39.51 ppm for 1H and 13C, respectively). Mass measurements were obtained on a Mariner bench top ESI-TOF-MS. Crude extractions were obtained using an Accelerated Solvent Extractor (1500 psi, 100 °C). HPLC was performed with Phenomenex Luna reversed-phase semipreperative (10 mm × 250 mm) and Altima reversed-phase analytical (5 mm × 250 mm) C18 5 μm columns.
Collection and Identification
The sponge samples were collected in May 2007 using SCUBA at depths of 30 - 60 ft. Coll. no. 07202 (sample VII) was collected from Milne Bay, Papua New Guinea (GPS = 10° 15.830 S: 150° 46.191 E) and coll. no. 07208 (sample VIII) was collected in the Amphlett region of Papua New Guinea (GPS = 9° 14.280 S: 150° 46.946 E). The collected specimen were globular in morphology with a small apical oscule. The grey-colored ectosome is composed of a neat reticulation of foreign material. The cream-colored endosome is a loose network of cored primary fibers with sparce cored secondary fibers. The fine collagen filaments form a dense network in the endosome. Both collections were identified as a Psammocinia aff. bulbosa (Bergquist, 1995) by Dr. N. J. de Voogd. Voucher samples have been deposited at UCSC and Naturalis (RMNH); UCSC coll. no. 07202 = RMNH Por. 2980; UCSC coll. no. 07208 = RMNH Por. 2982. Topside and underwater photographs are also available from the Crews laboratory.
Extraction and Isolation
Biological material was handled using our standard laboratory protocol.1 Crude extracts of the dried sponge (21 g dry weight) were obtained employing an Accelerated Solvent Extractor (ASE) to yield hexanes (XFH, 344.8 mg), CH2Cl2 (XFD, 214.6 mg), and CH3OH (XFM, 839.1 mg) fractions. By LCMS, both the XFD and XFM showed trace amounts of (+)-psymberin (1a). The XFD was fractionated using semi-preparative RP-HPLC (25%aq to 100% CH3CN with 0.1% formic acid over 30 min) to afford 1.7 mg of pure 1a in fraction H17. The XFM was fractionated in a similar manner to yield 1.5 mg of 1a fraction H7. Fraction XFM-H5 contained (+)-cyclocinamide A (2a) which was purified using analytical RP-HPLC (isocratic 30%aq CH3CN with 0.1% formic acid) to yield 0.5 mg pure compound.
Hydrolysis and l-FDAA Derivatization of (+)-Cyclocinamide A (2a)
(+)-Cyclocinamide A (2a) (0.25mg) was hydrolyzed with 1 mL of 6 N HCl (0.1% phenol w/v)20 for 4 h in a sealed thick walled vial at 110 °C. The hydrolysate was evaporated under nitrogen and then derivatized by treatment first with 100 μL of 1 M NaHCO3 and second with 50 μL of N-α-(2,4-dinitro-5-flourophenyl)-l-alaninamide (l-FDAA) solution (10 mg/mL in acetone).17 The reaction mixture was heated to 80 °C for 30 min, cooled to room temperature, and finally quenched with 50 μL of 2 N HCl. CH3CN (300 μL) was then added for subsequent LC-MS analysis. Each standard amino acid (0.5mg) was derivatized under the same procedure with l-FDAA. For each reaction product, a linear gradient of 10% to 50% MeCN (0.1 M NH4OAc, pH = 6) over 60 min was used to separate the derivatized products by LC-MS (Phenomenex Luna C18 column, 5mm × 250mm, UV 340 nm, flow rate 1 mL/min).14 Retention times of the derivatized hydrolysate were monitored by mass and UV (340 nm) and compared to that of the derivatized commercially available standard amino acids. The HPLC of co-injected 2:1 derivatized standard amino acid-FDAA diastereomer complexes gave profiles for DAP and Asp in which the l-amino acid-FDAA complex eluted before the d-amino acid analogue, while the β-amino acid, iSer,18 demonstrated a d to l elution order.23 As a racemic mixture of BrTrp was used for derivatization, we assumed the standard elution order to be l vs. d elution. This was identical to that observed by Ireland14 using similar HPLC analysis conditions. Finally, the optical properties of the new sample of 2a ([α]27D = +32, in MeOH) matches that of our original compound ([α]27D = +29, in MeOH)3 in which l-BrTrp was established. Retention times for standard amino acid-FDAA derivatives: l-Asp 17.3 min, d-Asp, 20.0 min, d-iSer 30.6 min, l-iSer 31.3 min, l-DAP 49.1 min, l-5-BrTrp 49.8 min, d-DAP 50.1 min, d-5-BrTrp 53.2 min. Retention times for the FDAA hydrolysate derivatives of 2a: l-Asp 17.3 min, l-iSer 31.4 min, l-DAP 49.2 min, l-5-BrTrp 50.0 min.
Biological assays
The disk diffusion soft agar colony-formating assay (DDA)21 and IC50 determinations7 were performed as previously described.
(+)-Psymberin (1a)
Spectroscopic data in accordance with published data.8
(+)-Cyclocinamide A (2a)
Spectroscopic data in accordance with published data.3
Swinholide A
Spectroscopic data in accordance with published data.15
Preswinholide A
Spectroscopic data in accordance with published data.6
Supplementary Material
Acknowledgments
This work was supported by research grants from NSF (CHE-0617056), NIH (R01 CA 047135), by NMR equipment grants from NSF(CHE-0342912) and NIH (S10 RR19918), by a MS equipment grant from NIH (S10 RR20939), and by training grants from NIH (NIGMS R25GM058903) and NSF (REU 0552641). We also thank the Captain and Crew of the M/V Golden Dawn for their assistance in sponge collection and Dr. L. Matainaho of the University of Papua New Guinea for his continuing assistance.
Footnotes
Supporting Information Available: Sponge photographs and isolation scheme. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Rubio BK, van Soest RWM, Crews P. J Nat Prod. 2007;70:628–631. doi: 10.1021/np060633c. [DOI] [PubMed] [Google Scholar]
- 2.Piel J. Current Medicinal Chemistry. 2007;13:39–50. [PubMed] [Google Scholar]
- 3.Clark WD, Corbett T, Valeriote F, Crews P. J Am Chem Soc. 1997;119:9285–9286. [Google Scholar]
- 4.Grieco PA, Reilly M. Tetrahedron Lett. 1998;39:8925–8928. [Google Scholar]
- 5.Faulkner DJ. Tetrahedron Lett. 1973:3821–3822. [Google Scholar]
- 6.Todd JS, Alvi KA, Crews P. Tetrahedron Lett. 1992;33:441–442. [Google Scholar]
- 7.Robinson SJ, Tenney K, Yee DF, Martinez L, Media JE, Valeriote F, van Soest RWM, Crews P. J Nat Prod. 2007;70:1002–1009. doi: 10.1021/np070171i. [DOI] [PubMed] [Google Scholar]
- 8.Cichewicz RH, Valeriote FA, Crews P. Org Lett. 2004;6:1951–1954. doi: 10.1021/ol049503q. [DOI] [PubMed] [Google Scholar]
- 9.Cichewicz RH, Kenyon VA, Whitman S, Morales NM, Arguello JF, Holman TR, Crews P. J Am Chem Soc. 2004;126:14910–14920. doi: 10.1021/ja046082z. [DOI] [PubMed] [Google Scholar]
- 10.Jiang X, Garcia-Fortanet J, De Brabander JK. J Am Chem Soc. 2005;127:11254–11255. doi: 10.1021/ja0537068. [DOI] [PubMed] [Google Scholar]
- 11.Huang XH, Shao N, Palani A, Aslanian R, Buevich A. Org Lett. 2007;9:2597–2600. doi: 10.1021/ol071068n. [DOI] [PubMed] [Google Scholar]
- 12.Pettit GR, Xu JP, Chapuis JC, Pettit RK, Tackett LP, Doubek DL, Hooper JNA, Schmidt JM. J Med Chem. 2004;47:1149–1152. doi: 10.1021/jm030207d. [DOI] [PubMed] [Google Scholar]
- 13.Jiang X, Williams N, Brabander JK. Org Lett. 2007;9:227–30. doi: 10.1021/ol062656o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laird DW, Labarbera DV, Feng X, Bugni TS, Harper MK, Ireland CM. J Nat Prod. 2007;70:741–746. doi: 10.1021/np060489v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitagawa I, Kobayashi M, Katori T, Yamashita M, Tanaka J, Doi M, Ishida T. J Am Chem Soc. 1990;112:3710–3712. [Google Scholar]
- 16.Piel J. Proc Natl Acad Sci U S A. 2002;99:14002–14007. doi: 10.1073/pnas.222481399. [DOI] [PMC free article] [PubMed] [Google Scholar]; Piel J, Wen GP, Platzer M, Hui DQ. Chembiochem. 2004;5:93–98. doi: 10.1002/cbic.200300782. [DOI] [PubMed] [Google Scholar]
- 17.Harada K, Fujii K, Mayumi T, Hibino Y, Suzuki M, Ikai Y, Oka H. Tetrahedron Lett. 1995;36:1515–1518. [Google Scholar]
- 18.Fujii K, Ikai Y, Mayumi T, Oka H, Suzuki M, Harada K. Anal Chem. 1997;69:3346–3352. [Google Scholar]
- 19.Bittner S, Scherzer R, Harlev E. Amino Acids. 2007;33:19–42. doi: 10.1007/s00726-006-0441-8. [DOI] [PubMed] [Google Scholar]
- 20.Muramoto K, Kamiya H. Anal Biochem. 1990;189:223–230. doi: 10.1016/0003-2697(90)90112-m. [DOI] [PubMed] [Google Scholar]
- 21.Boot CM, Tenney K, Valeriote FA, Crews P. J Nat Prod. 2006;69:83–92. doi: 10.1021/np0503653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simmons TL, Coates RC, Clark BR, Engene N, Gonzalez D, Esquenazi E, Dorrestein PC, Gerwick WH. Proc Natl Acad Sci U S A. 2008;105:4587–4594. doi: 10.1073/pnas.0709851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.This l to d elution order for the iSer-FDAA complex is opposite of that reported under the HPLC conditions for the derivitized hydrolysates for cyclocinamide B.14 Due to the hydrophilic nature of the iSer side chain, this elution order can change with pH variance.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.