

Abstract
Caenorhabditis elegans was recently developed as a model system to study both pathogen virulence mechanisms and host defense responses. We previously demonstrated that C. elegans produces reactive oxygen species (ROS) in response to exposure to the important gram-positive nosocomial pathogen Enterococcus faecalis. We also presented evidence of oxidative stress and upregulation of stress responses after exposure to the pathogen. As in mammalian systems, this new work shows that production of ROS for innate immune functions occurs via an NADPH oxidase. Specifically, reducing expression of a dual oxidase, Ce-Duox1/BLI-3, causes a decrease in ROS production in response to E. faecalis. We also present evidence that reduction of expression of Ce-Duox1/BLI-3 increases susceptibility to this pathogen, specifically when expression is reduced in the intestine and the hypodermis. Ce-Duox1/BLI-3 was previously characterized as having a role in cuticle cross-linking. Two C. elegans mutants with point mutations in the peroxidase domain that exhibit severe cuticle defects were discovered to be unaffected in ROS production or pathogen susceptibility. These results demonstrate an important biological role for the peroxidase domain in cuticle cross-linking that is unrelated to ROS production. To further demonstrate the protective effects of the pathogen-induced ROS production, we show that antioxidants that scavenge ROS increase the sensitivity of the nematode to the infection, in stark contrast to their longevity-promoting effects under nonpathogenic conditions. In conclusion, we postulate that the generation of ROS by NADPH oxidases in the barrier epithelium is an ancient, highly conserved innate immune defense mechanism.
Caenorhabditis elegans has been developed as a proficient model to study host-pathogen interactions. A diverse collection of bacterial pathogens and some fungal pathogens infect and kill C. elegans, including the gram-positive bacterium Enterococcus faecalis (9, 13, 22). Multiple pathways are involved in innate immunity in the nematode, and C. elegans has become a valuable host with which to identify evolutionarily conserved innate immune responses (13, 22). In our previous work, C. elegans was found to produce reactive oxygen species (ROS) during infection (6).
Specifically, we found that C. elegans makes ROS in response to the pathogen E. faecalis but not in response to nonpathogenic Bacillus subtilis or Escherichia coli (6). ROS production was decreased when an NADPH oxidase inhibitor, diphenylene iodonium, was added, suggesting that proteins belonging to the NOX/DUOX family of proteins may be involved (37). NOX proteins have just an NADPH oxidase domain, while DUOX proteins have a peroxidase domain in addition to an NADPH oxidase domain. The C. elegans genome encodes two highly conserved DUOX proteins, Ce-Duox1/BLI-3 and Ce-Duox2 (7). A mutation in the Ce-Duox1/BLI-3 gene results in a blistered phenotype, providing the prefix for the name of the Ce-Duox1/BLI-3 gene, bli-3 (blistered) (4, 40). The topology of these proteins is predicted to exist in such a way that the carboxyl domain containing the NADPH oxidase activity is located in the membrane, oriented to release superoxide extracellularly. This domain is connected by EF hand motifs located in the cytoplasm to the peroxidase domain at the amino terminus of the protein, which is located extracellularly (26). The current model envisions activated Ce-Duox1/2 causing the oxidation of NADPH and the subsequent reduction of flavin adenine dinucleotide. Flavin adenine dinucleotide passes electrons through two heme groups in the NADPH oxidase domain to the cell surface, where oxygen is reduced to yield extracellular superoxide. Unstable superoxide is then thought to rapidly form H2O2. The H2O2 then acts as a substrate for the peroxidase domain, which forms tyrosyl radicals that cross-link to one another to create the cuticle of the worm (7). In this work, we explore whether Ce-Duox1 and/or Ce-Duox2 has an additional role in generating ROS during infection as a protective immune response.
The production of ROS by phagocytes in response to pathogens via the NADPH oxidase gp91phox is a well-studied process. However, in recent years, the NOX/DUOX family of proteins has garnered interest due to their possible roles in production of ROS in nonphagocytic cells (2, 27). Due to the presence of these proteins in the oral cavity, airway epithelial tissue, and gastrointestinal (GI) tract of humans, DUOX proteins are thought to play a significant role in mucosal innate immunity (11, 39). For example, the antimicrobial substance lactoperoxidase (LPO) is present in human saliva (24, 43). However, in order for LPO to exhibit antimicrobial qualities, hydrogen peroxide (H2O2) must be present to catalyze the oxidization of SCN− (thiocyanate), converting SCN− into antimicrobial hypothiocyanite anions (34). Human Duox2 was detected in the salivary submucosal glands and is thought to provide the H2O2 necessary for this antimicrobial system (11). A similar defense system is also active in the airway epithelium, as DUOX1 expression is found in the epithelia of human bronchial and tracheal sections, where LPO is also expressed (11, 39, 42). Rada and Leto recently showed, using airway epithelial cell types, that Pseudomonas aeruginosa is killed by this system (38). It appears that dual oxidases might also play a role in defense of the mucosal surfaces of the GI tract, as DUOX2 expression was identified in the GI tract (11). Recent studies showed that a reduction of dDuox in the gut of Drosophila melanogaster increased its susceptibility to pathogens (16). Despite the known and postulated links of NOX/DUOX enzymes and ROS production to innate immune processes in the mucosa, there are gaps in the mechanistic knowledge of how ROS production is caused and controlled.
In this work, we demonstrate that C. elegans Ce-Duox1/BLI-3 generates ROS induced by infection, using a combination of studies employing RNA interference (RNAi) and mutants to examine the effect of the loss of Ce-Duox1/BLI-3 and/or Ce-Duox2 on ROS production. Our data support Ce-Duox1/BLI-3 as the source of ROS production during infection. Additionally, two ROS-scavenging antioxidants were used to reduce ROS activity in order to determine if infection-induced ROS production is specifically protective. The presence of the antioxidants increased susceptibility in the worms. We also examined C. elegans with point mutations in the peroxidase domain and discovered that despite cuticle blistering, there were no effects on ROS production or immunity, suggesting that this domain is not involved in ROS production, a controversial point in the literature (28, 31). These experiments support our model that the ROS produced by Ce-Duox1/BLI-3 during infection are part of a protective immune response in the nematode. The results suggest that ROS production at mucosal surfaces is a conserved, ancient defense mechanism predating the evolution of the oxidative burst in specialized immune cells and that C. elegans may provide a convenient system with which to gain further insight into this phenomenon.
MATERIALS AND METHODS
C. elegans strains and growth conditions.
C. elegans strains were grown and maintained as previously described (19). All C. elegans strains used were obtained from the Caenorhabditis Genetics Center, except for the rde-1 strains. Strain VP303 with rde-1 under the expression of an intestinal promoter [rde-1(ne219); kbIs7(nhx-2::rde-1, rol-6)] was provided by K. Strange (8). Strains with rde-1 under the expression of a hypodermis-specific promoter (strain NR222) {rde-1(ne219)V; kzIs9 [pKK1260(lin-26p::nls::gfp), pKK1253(lin-26p::rde-1), pRF4 (rol-6)]} and a muscle-specific promoter (strain NR350) {rde-1(ne219)V; kzIs20 [pDM#715(hlh-1p::rde-1), pTG96(sur-5p::nls::gfp)]} were provided by H. Qadota (36). Strain RB1505 containing F53G12.3(ok1775) was backcrossed four times to the wild type to create GF41.
Amplex Red assay for H2O2 measurements.
We previously modified an assay to measure hydrogen peroxide, previously used to measure the oxidative burst in activated human leukocytes and adapted to C. elegans, using an Amplex Red hydrogen peroxide/peroxidase assay kit (6) (Molecular Probes). In the presence of hydrogen peroxide, Amplex Red is oxidized by horseradish peroxidase to form a red fluorescent oxidation product whose absorbance can be measured (29). Briefly, 200 to 300 L4 worms were exposed to each bacterial condition, as previously described (9), for 18 hours and then washed with reaction buffer. Concentrations were adjusted to approximately 100 nematodes/50 μl and pipetted into a 96-well plate. Fifty microliters of the Amplex Red reaction buffer was then added to the wells, and within 2 to 3 h, there was a measurable amount of hydrogen peroxide present, as observed by eye and by measuring the absorbance at 540 nm with a plate reader (Thermo Multiscan MCC plate reader), an absorbance acceptably close to the peak (560 nm) determined by examining the emission spectra (29). Each experiment was done in duplicate and repeated at least three times. The significance of differences between conditions was determined by an unpaired t test, with differences of <0.05 considered statistically significant. GraphPad Prism 5.0 was used for these calculations.
C. elegans killing and longevity assays.
RNAi exposure, killing assays, and longevity assays were performed as previously described (9, 10, 23). The following bacterial strains were used: OP50 (E. coli) and OG1RF (E. faecalis). RNAi constructs were obtained from an RNAi library (20). The antioxidants alpha-lipoic acid (LA) (Sigma) and epigallocatechin gallate (EGCG) (Sigma) were added to the agar of the NG plates to a final concentration of 25 μM.
A total of 60 to 90 worms were used in each experiment. The data were analyzed using GraphPad Prism 5.0. Survival was plotted by the Kaplan-Meier method and the curves compared using the log-rank test, which generates a P value testing the null hypothesis that the survival curves are identical. P values of 0.05 or less were considered significantly different from the null hypothesis. In Fig. 4, the data were also fitted to a Boltzmann sigmoidal curve, from which the time it took 50% of the worms to die (LT50) was determined. The average LT50s from the longevity and killing assays were used to calculate the relative mortality, as previously described (6, 41) Briefly, the formula used was as follows: value for rde-1 mutant fed vector control RNAi and exposed to E. faecalis/value for rde-1 mutant fed bli-3 RNAi and exposed to E. faecalis. Again, the unpaired t test, using GraphPad Prism 5.0, was used to test the statistical significance of any differences between the tissue-specific rde-1 mutants and the control, with a P value of 0.05 considered the cutoff.
FIG. 4.
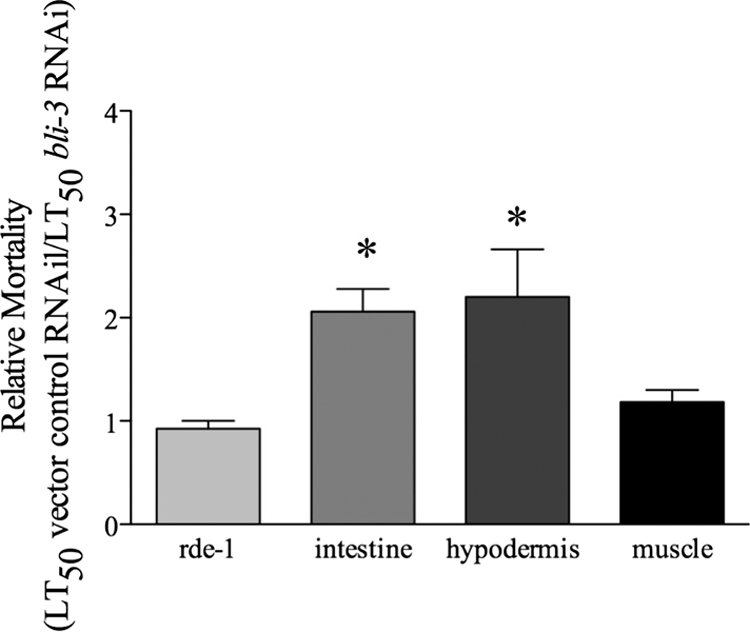
Reduction of bli-3 expression in the intestine and hypodermis increases susceptibility to infection. Reducing expression of bli-3 by RNAi in the intestine and the hypodermis, but not in the muscle, caused a significant increase in relative mortality. To determine relative mortality, the average LT50 on E. faecalis for each rde-1 strain previously exposed to vector control RNAi was divided by the LT50 for the same strain exposed to bli-3 RNAi. The averages from three independent experiments are shown. The error bars correspond to the standard errors, and asterisks indicate significant differences between the tissue-specific strain and the control strain (lacking rde-1 in all tissue) (P = 0.0019 [intestine], P = 0.0020 [hypodermis], and P = 0.3646 [muscle]).
RESULTS
Decreasing the expression of Ce-Duox1/2 reduces pathogen-induced ROS production.
NADPH oxidases are necessary for generating ROS in mammalian phagocytes and are of recent interest due to their antibacterial role in nonimmune cells (2, 26). The C. elegans genome encodes two NADPH oxidases, both of the dual oxidase variety, namely, Ce-Duox1/BLI-3 and Ce-Duox2. Ce-Duox1/BLI-3 was shown to be involved in cross-linking the cuticle of the worm, and defects in this protein cause a “blistered” phenotype (4, 40). No expression of Ce-Duox2 was detectable in previous studies (7, 18). Our laboratory previously reported ROS production by the worm after infection with the pathogen E. faecalis (6). We postulated that Ce-Duox1/BLI-3 and/or Ce-Duox2 is necessary to generate ROS in response to infection.
To test the role of Ce-Duox1/2 in pathogen susceptibility and ROS production, we reduced the expression of the genes encoding these proteins by RNAi. Due to a high degree of similarity between these genes, the RNAi construct based on bli-3 (gene encoding Ce-Duox1/BLI-3) used for feeding RNAi reduced the expression of both simultaneously. We discovered that the standard feeding RNAi procedure with this construct resulted in a severe blistered phenotype that did not allow for proper development. We mixed the RNAi bacteria with a vector control at a ratio of 1 to 10 and found that this mixture modified the severity of the phenotype enough to enable development. Some blistering was still visible, indicating at least partial reduction in the production of Ce-Duox1/BLI-3. When fed E. faecalis, the 1/10 bli-3 RNAi-exposed animals exhibited increased susceptibility compared to those exposed to the vector control RNAi (Fig. 1A). However, the longevity of the 1/10 bli-3 RNAi worms was also reduced, even on UV-killed E. coli, suggesting a loss of viability overall (data not shown). To determine if Ce-Duox1 and/or Ce-Duox2 was the source of the pathogen-induced ROS production, the worms were analyzed in an Amplex Red assay. The worms exposed to bli-3 RNAi produced significantly smaller amounts of ROS than those in worms fed only the vector control RNAi strain after infection with E. faecalis (Fig. 1B), strongly suggesting that Ce-Duox1/BLI-3 and/or Ce-Duox2 is the source of infection-induced ROS production. As shown in our previous work, noninfected worms (C. elegans fed only E. coli) did not produce significant amounts of ROS (6) (Fig. 1C). Of the little produced, there was no detectable difference between the vector control and bli-3 RNAi worms, suggesting that the bli-3 RNAi effect on ROS production is specific to infected worms.
FIG. 1.
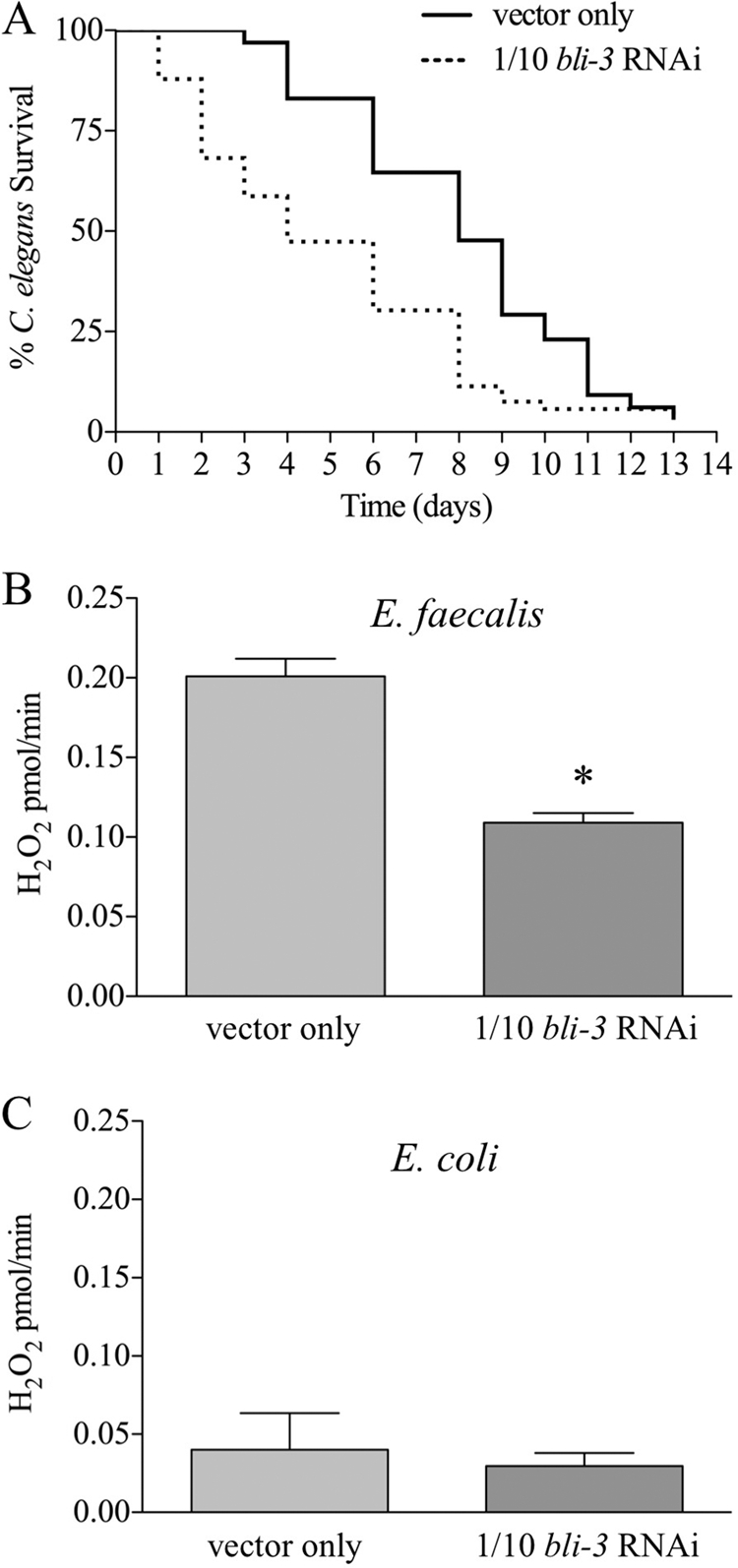
Decreasing expression of bli-3 causes increased susceptibility and decreased ROS production. RNAi experiments were carried out by mixing a bli-3 RNAi construct with E. coli containing an empty vector strain in a 1/10 dilution. (A) Worms exposed to E. faecalis were more susceptible to the infection when bli-3 expression was reduced (P < 0.0001). (B) Reducing bli-3 expression decreased ROS production when the worms were exposed to E. faecalis, in a statistically significant manner (P = 0.0180), suggesting that the expression of the bli-3 gene is necessary for optimal ROS generation. (C) Uninfected worms (exposed only to E. coli) produced very few ROS, on which the reduction of bli-3 expression had no effect (P = 0.7118). For panels B and C, the error bars indicate the standard errors, and the asterisk indicates a statistically significant difference (see Materials and Methods).
Ce-Duox1/BLI-3 generates ROS during infection.
A strain with a large deletion in the gene encoding Ce-Duox2, with the sequence name F53G12.3(ok1775), is publicly available from the Caenorhabditis Genetics Center. We obtained this strain, RB1505, and backcrossed it with the wild type to eliminate any other incidental mutations, creating strain GF41. In working with the strain, no phenotypic differences were seen compared to the wild type. Development and fecundity were completely normal, and no cuticle blisters were ever observed. We tested GF41 for sensitivity to pathogens and its ability to generate ROS in response to pathogens. As shown in Fig. 2A and B, the phenotypes of the Ce-Duox2 mutant were indistinguishable from those of the wild type (N2). (Note that the rate of killing of the wild-type N2 strain was higher in Fig. 2A than in Fig. 1A. This difference was consistent and attributable to differences in E. coli strains and conditions used to propagate the worms for RNAi knockdown [Fig. 1] compared to standard propagation conditions [Fig. 2].) Though an analogous deletion mutant in Ce-Duox1/BLI-3 does not exist to test directly, we concluded from these results and past studies demonstrating no detectable expression of Ce-Duox2 (7, 18) that a reduction in Ce-Duox1/BLI-3 was primarily responsible for the susceptibility and loss of ROS production phenotypes observed in the RNAi experiments (Fig. 1). Based on our observations with the bli-3 RNAi detailed above, we surmise that it will not be possible to obtain a deletion in the bli-3 gene that encodes Ce-Duox1/BLI-3 due to a severe developmental phenotype.
FIG. 2.
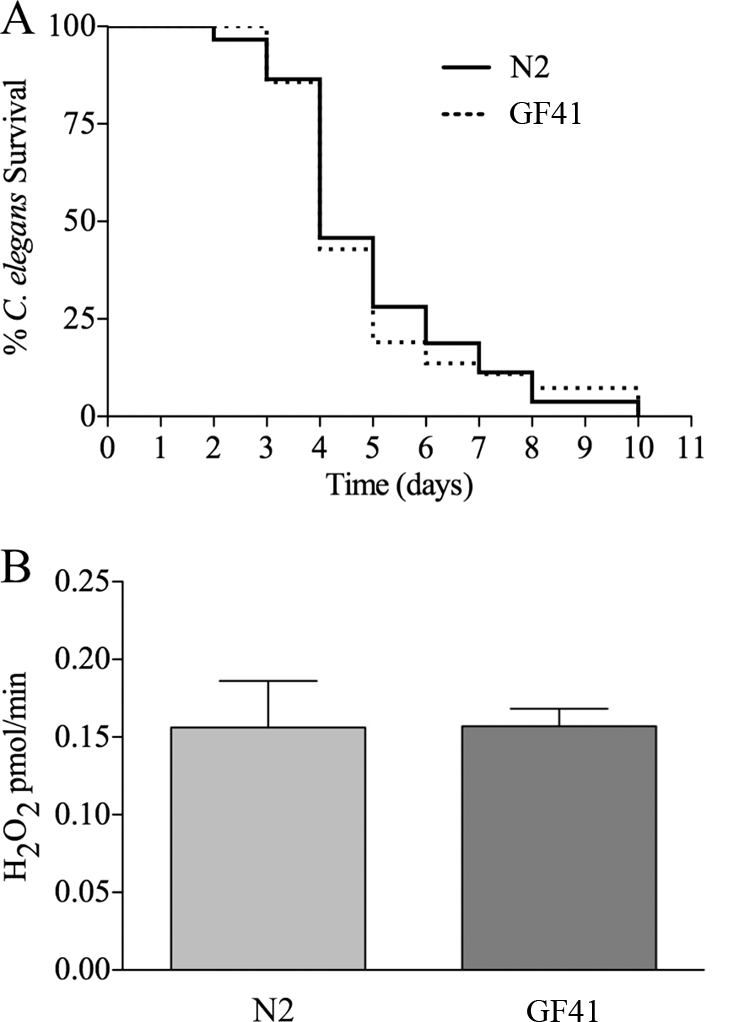
Ce-Duox2 is not involved in ROS production after infection. Experiments were carried out using C. elegans strain GF41, which has a deletion in the gene for Ce-Duox2. (A) Killing assays demonstrated that GF41 was as susceptible to infection as N2 (wild-type) nematodes (P = 0.7761). (B) Amplex Red assays did not demonstrate a difference in ROS production between GF41 and N2 (wild type) (P = 0.9734). The error bars indicate the standard errors.
Though a deletion in bli-3 does not exist, two point mutants are available from the Caenorhabditis Genetics Center, namely, the bli-3(e767) and bli-3(n529) mutants. Both of these mutations are located in the peroxidase domain (Fig. 3A) and are unlikely to affect the function of the NADPH oxidase domain, which catalytically produces ROS by generating superoxide (4, 40). We measured the amounts of ROS produced by these mutants in response to E. faecalis (Fig. 3C) and observed levels similar to that of the wild type. We next tested the mutants to see if they were more susceptible to E. faecalis than wild-type worms. The mutants displayed the same level of resistance to the pathogen as the wild-type strain did (Fig. 3B). These data suggest that the defects caused by the point mutations in the peroxidase domain do not affect the role of Ce-Duox1/BLI-3 in ROS production and pathogen defense. Additionally, because these mutants possessed a blistered phenotype substantially more severe than what we observed with wild-type worms exposed to 1/10 bli-3 RNAi but were fully resistant, we concluded that a blistered cuticle alone does not increase sensitivity to E. faecalis.
FIG. 3.
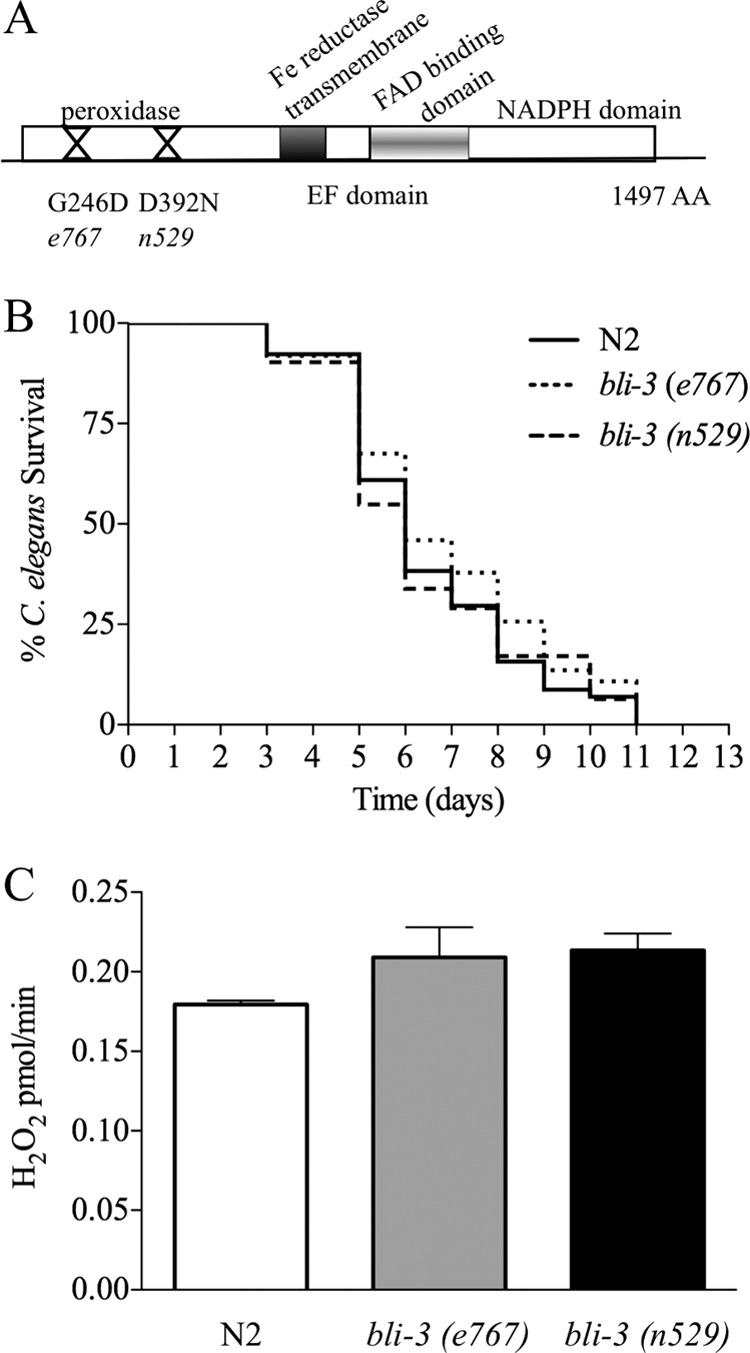
The peroxidase domain of Ce-Duox1 is not necessary for resistance to infection or ROS production. (A) Structure of Ce-Duox1 (BLI-3) and locations of tested point mutations in the peroxidase domain. (B) The bli-3 point mutants were no more susceptible to E. faecalis infection than the N2 (wild-type) strain [for bli-3(e767) mutant, P = 0.246; for bli-3(n529) mutant, P = 0.9469]. (C) The bli-3 point mutants produced equal amounts of ROS to that expressed by the N2 (wild-type) strain [for bli-3(e767) mutant, P = 0.2636; for bli-3(n529) mutant, P = 0.0877]. The error bars indicate the standard errors.
Reduction of Ce-Duox1 in the intestine and the hypodermis, but not in the muscle, increases sensitivity to pathogens.
Based on the above data, we speculated that Ce-Duox1/BLI-3 would be present at the site of infection, i.e., the intestine. We were interested in examining the effects of the presence of Ce-Duox1/BLI-3 in different tissues during infection. Therefore, we employed a recently developed C. elegans system that can reduce the expression of certain genes under the control of tissue-specific promoters (8, 36). For this experiment, we obtained C. elegans strains that have a mutation in the rde-1 gene. This mutation renders the RNAi machinery inactive and the worm is not susceptible to RNAi (33). However, when this gene is expressed under the control of a tissue-specific promoter, RNAi can then occur in these specific tissues. The strains used in this experiment have the rde-1 gene expressed by tissue-specific promoters in the intestine, muscle, and hypodermis. We predicted that reducing bli-3 expression in the intestine would render the worm more susceptible to the pathogen, whereas reducing bli-3 expression in other tissues, such as the muscle, would not. Since we found that there were differences in susceptibility to E. faecalis in the different rde-1 backgrounds, we compared the susceptibility of each strain exposed to bli-3 RNAi to that of the strain exposed to vector control RNAi and calculated the relative mortality (Fig. 4). The rde-1 mutant, which is completely resistant to RNAi, had the same sensitivity to E. faecalis after exposure to bli-3 RNAi as it did after exposure to the control RNAi, for a relative mortality of about 1. As predicted, we found that reducing bli-3 expression in the intestinal tissue increased susceptibility such that the relative mortality was much higher, at 2.06, while RNAi of the muscle tissue resulted in a relative mortality not statistically different from that of the control. However, we found that reducing expression in the hypodermis also caused the worms to be more sensitive to the pathogen. It has previously been demonstrated that Ce-Duox1/BLI-3 is present in the hypodermal cells, where it is necessary for cross-linking the cuticle (7). It is possible that the enzyme also functions in immune defense at this location. Interestingly, Pujol et al. found evidence of upregulated defense mechanisms in the worm hypodermis in response to infection (35). The increase in relative mortality found by decreasing expression of bli-3 in the intestine is suggestive of Ce-Duox1/BLI-3 additionally being present in this tissue during infection. We also examined the longevity of the RNAi-exposed worms and found that a loss of bli-3 in the intestine and the hypodermis reduced the life span of the worms on E. coli (data not shown).
Antioxidants that enhance longevity reduce survival during infection.
Because reducing the levels of bli-3 by RNAi in our experiments also affected longevity, we were not able to conclude definitively that the enzyme and its concomitant ROS production during infection are protective. The caveat remains that the increase we observed in susceptibility could be due to a loss in general viability due to pleiotropic effects from the loss of bli-3 expression. To address whether or not ROS production during pathogen challenge is specifically protective, we employed a different approach. Well-characterized antioxidants that are known to scavenge ROS, i.e., LA and EGCG (3, 14), were added to our assays.
Previous work demonstrated that the addition of LA to the food source of C. elegans significantly increased longevity (5, 44). The addition of EGCG did not increase the life span but did modulate other markers of age-related decline (5). Our previous work showed that these compounds protected against the accumulation of protein damage, as assayed by polyglutamine aggregation (30). These antioxidants are believed to exert their effects in part by scavenging ROS. If the ROS released during infection are part of a protective immune response, we postulated that neutralizing this response by adding antioxidants would be deleterious. Because E. faecalis is not a preferred food source, we impregnated the agar medium with LA and EGCG rather than adding it to the bacterial culture spread on the plate, as done in previous work, to ensure exposure to the compounds (5, 44). To verify the effects of the antioxidants on longevity despite the difference in methodology, we carried out a longevity assay (Fig. 5A). The addition of LA caused the worms to have a significantly longer life span (P < 0.0001), as previously reported (5, 44). However, we found that EGCG also significantly increased the life span (P < 0.0001). This could be due to the worms being exposed to a higher overall concentration of the antioxidant due to its addition to the agar medium rather than to the bacterial food source. The effects of these antioxidants during interaction with a pathogen were then assayed by measuring survival during exposure to E. faecalis, with and without the presence of the antioxidants (Fig. 5B). In stark contrast to the longevity assay, the presence of the antioxidants significantly reduced survival (for EGCG, P < 0.0001; for LA, P = 0.0005).
FIG. 5.
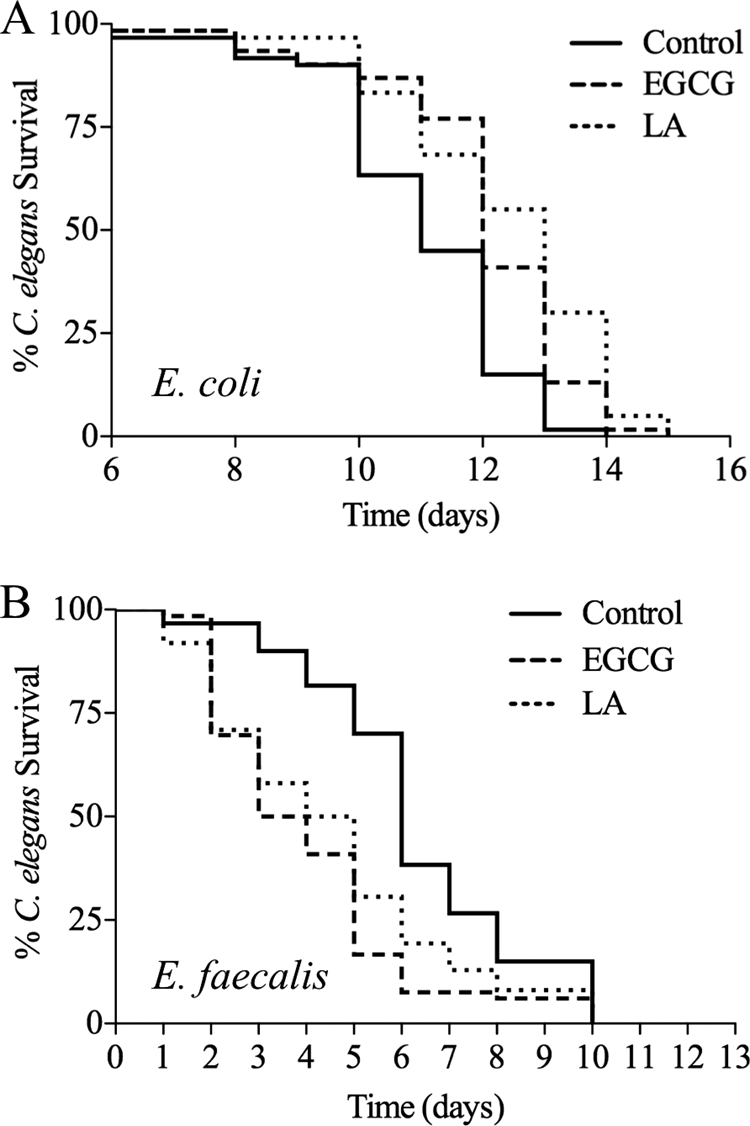
Antioxidants increase longevity but decrease survival during infection. (A) The addition of 25 μM antioxidants to the plates increased life span in the standard longevity assay (for EGCG, P < 0.0001; for LA, P < 0.0001). (B) In contrast, the addition of 25 μM antioxidants to the killing assay plates caused increased susceptibility to the pathogen (for EGCG, P < 0.0001; for LA, P = 0.0005).
These experiments provide support for our model that the ROS produced by Ce-Duox1/BLI-3 during infection are part of a protective immune response. To reduce this response by adding ROS scavengers increases susceptibility. The interesting contrast in the effects of these antioxidants depending on the context (longevity assay versus pathogen assay) emphasizes the dual nature of ROS in biological systems—they are sometimes helpful and sometimes harmful.
DISCUSSION
In this report, we provide evidence that Duox1/BLI-3 is necessary for the infection-induced ROS generation previously elucidated by our lab (6). We tested two strains that have single point mutations in the peroxidase domain of the dual oxidase, which causes a defect in the tyrosine cross-linking needed for the development of the hypodermis, resulting in a blistered cuticle (Fig. 3A) (4, 7, 40). Interestingly, these mutants with defective cuticles were just as resistant to infection and produced as many ROS as the wild type, suggesting that the loss of cuticle integrity is not the source of the susceptible phenotype. Both mutated residues are conserved in human Duox1 and Duox2. The G246 residue is not conserved among the other peroxidases, while D392 mostly is (7). The G246D mutation lies near two important active-site residues, the catalytic arginine residue (R236) and one of the heme-binding residues (E239) (28). Though the exact biochemical nature of the peroxidase mutants has not been investigated, it can be postulated by the mutant phenotype, the location of the mutations (in the peroxidase domain), and the fact that they generate wild-type levels of ROS (Fig. 3) that they are likely deficient in the step that uses H2O2 to create the tyrosyl radical necessary for cross-linking the cuticle.
The predicted role of the peroxidase domain in dual oxidases is controversial (28, 31). Morand et al. proposed that the peroxidase domain of the human dual oxidases generates H2O2 from superoxide, essentially acting as a superoxide dismutase. The basis for this postulate was their inability to detect peroxidase activity when measuring oxidation of several substrates (31). Additionally, readily detectable H2O2 seems to be the primary oxidant produced, as very little superoxide could be detected in studies of these dual oxidases (11, 31). The human and C. elegans peroxidase domains are not conventional in that they are missing some of the conserved residues necessary to bind heme. However, the Ce-Duox1/BLI-3 peroxidase domain was recently shown to form two covalent bonds with heme, causing some catalytic function in vitro, and no superoxide dismutase activity was detected (28) Additionally, there is in vitro evidence that the peroxidase domain of Ce-Duox1/BLI-3 behaves in the predicted manner and uses H2O2 to produce the dityrosines necessary for cuticle formation (7, 28). Our work provides the first in vivo evidence that the peroxidase domain of Ce-Duox1/BLI-3 acts in the traditional manner, because mutations in the peroxidase domain have no effect on ROS production but still result in severe cuticle blistering.
Interestingly, the peroxidase domain of the dDuox protein in Drosophila is predicted to act similarly to myeloperoxidase (MPO) in humans, as it was shown to generate potent antimicrobial activity in the presence of chloride and H2O2, presumably through the production of hydrochlorous acid (HOCl) (16). For the C. elegans immune defense, it is yet unclear whether superoxide or hydrogen peroxide is the primary oxidant produced by Ce-Duox1/BLI-3. Our current study used an assay to measure H2O2, which could be generated directly or result from the breakdown of superoxide. Determining the presence or absence of superoxide will require an assay that can directly detect this species. It is also unknown how ROS generated by Ce-Duox1/BLI-3 are translated into antimicrobial activity. Answering these questions will require further investigation.
Other components that contribute to ROS production by dual oxidases, such as cofactors and regulators, are beginning to be discovered. In a Drosophila infection model, yeast-induced DUOX activity was shown to require the Gαq-phospholipase C-β-Ca2+ pathway (15). The activation of phospholipase C-β, possibly caused by Gαq, causes the hydrolysis of the phospholipid phosphatidylinositol 4,5-bisphosphate into inositol 1,4,5-triphosphate, which can bind to an inositol 1,4,5-triphosphate receptor in the endoplasmic reticulum membrane. This interaction will then induce the release of Ca2+, positively activating DUOX through the EF hand domain (15). This regulation may provide insight into the regulation of Ce-Duox1/BLI-3 in C. elegans. Further studies will need to be conducted to determine the role of Ca2+ in Ce-Duox1/BLI-3 activation.
In mammalian systems, a factor shown to enable the activity of dual oxidases is a maturation factor called DUOXA, which is necessary for proper DUOX localization to the cytoplasmic membrane as well as for ROS-generating activity (12). Preliminary experiments in our lab show that reducing expression of duoxa by RNAi increases the susceptibility to E. faecalis and decreases the amount of H2O2 generated after infection (data not shown). In human respiratory and epithelial cells, Duox1 expression and H2O2 production were increased when the cells were treated with interleukin-4 and interleukin-13, while gamma interferon caused increased Duox2 expression and H2O2 production (17). C. elegans does not have these specialized immune signals, implicating other factors. It is possible that host recognition of an infection occurs by way of some of the well-known innate immune pathways in the worm, including the transforming growth factor beta-like pathway (32), the p38 mitogen-activated protein kinase pathway (23), the programmed cell death pathway (1), and/or the insulin-like signaling pathway (10). However, our previous study suggested that insulin signaling was not required for ROS production, though this pathway did contribute the expression of oxidative stress response genes (6). It is possible that the release of ROS is an upstream event that contributes to turning on these pathways (25). The many remaining open questions about the regulation of DUOX activity provide fertile ground for future investigations. The use of the C. elegans RNAi library (21) in combination with our assay for pathogen-induced ROS generation provides one potential tool set with which to discover these unknowns.
Acknowledgments
We thank the Caenorhabditis Genetics Center, K. Strange, and H. Qadota for strains used in this study. We gratefully acknowledge the technical support of the J. Schumacher laboratory.
This work was funded by National Institutes of Health grant R01AI076406 to D.A.G. and by a Ralph H. and Ruth J. McCullough Foundation award to V.C.
Editor: A. Camilli
Footnotes
Published ahead of print on 17 August 2009.
REFERENCES
- 1.Aballay, A., and F. M. Ausubel. 2001. Programmed cell death mediated by ced-3 and ced-4 protects Caenorhabditis elegans from Salmonella typhimurium-mediated killing. Proc. Natl. Acad. Sci. USA 98:2735-2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bedard, K., and K. H. Krause. 2007. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87:245-313. [DOI] [PubMed] [Google Scholar]
- 3.Biewenga, G. P., G. R. Haenen, and A. Bast. 1997. The pharmacology of the antioxidant lipoic acid. Gen. Pharmacol. 29:315-331. [DOI] [PubMed] [Google Scholar]
- 4.Brenner, S. 1974. The genetics of Caenorhabditis elegans. Genetics 77:71-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown, M. K., J. L. Evans, and Y. Luo. 2006. Beneficial effects of natural antioxidants EGCG and alpha-lipoic acid on life span and age-dependent behavioral declines in Caenorhabditis elegans. Pharmacol. Biochem. Behav. 85:620-628. [DOI] [PubMed] [Google Scholar]
- 6.Chavez, V., A. Mohri-Shiomi, A. Maadani, L. A. Vega, and D. A. Garsin. 2007. Oxidative stress enzymes are required for DAF-16-mediated immunity due to generation of reactive oxygen species by Caenorhabditis elegans. Genetics 176:1567-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edens, W. A., L. Sharling, G. Cheng, R. Shapira, J. M. Kinkade, T. Lee, H. A. Edens, X. Tang, C. Sullards, D. B. Flaherty, G. M. Benian, and J. D. Lambeth. 2001. Tyrosine cross-linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J. Cell Biol. 154:879-891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Espelt, M. V., A. Y. Estevez, X. Yin, and K. Strange. 2005. Oscillatory Ca2+ signaling in the isolated Caenorhabditis elegans intestine: role of the inositol-1,4,5-trisphosphate receptor and phospholipases C beta and gamma. J. Gen. Physiol. 126:379-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garsin, D. A., C. D. Sifri, E. Mylonakis, X. Qin, K. V. Singh, B. E. Murray, S. B. Calderwood, and F. M. Ausubel. 2001. A simple model host for identifying gram-positive virulence factors. Proc. Natl. Acad. Sci. USA 98:10892-10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garsin, D. A., J. M. Villanueva, J. Begun, D. H. Kim, C. D. Sifri, S. B. Calderwood, G. Ruvkun, and F. M. Ausubel. 2003. Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science 300:1921. [DOI] [PubMed] [Google Scholar]
- 11.Geiszt, M., J. Witta, J. Baffi, K. Lekstrom, and T. L. Leto. 2003. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 17:1502-1504. [DOI] [PubMed] [Google Scholar]
- 12.Grasberger, H., and S. Refetoff. 2006. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J. Biol. Chem. 281:18269-18272. [DOI] [PubMed] [Google Scholar]
- 13.Gravato-Nobre, M. J., and J. Hodgkin. 2005. Caenorhabditis elegans as a model for innate immunity to pathogens. Cell. Microbiol. 7:741-751. [DOI] [PubMed] [Google Scholar]
- 14.Guo, Q., B. Zhao, M. Li, S. Shen, and W. Xin. 1996. Studies on protective mechanisms of four components of green tea polyphenols against lipid peroxidation in synaptosomes. Biochim. Biophys. Acta 1304:210-222. [DOI] [PubMed] [Google Scholar]
- 15.Ha, E. M., K. A. Lee, S. H. Park, S. H. Kim, H. J. Nam, H. Y. Lee, D. Kang, and W. J. Lee. 2009. Regulation of DUOX by the Galphaq-phospholipase Cbeta-Ca2+ pathway in Drosophila gut immunity. Dev. Cell 16:386-397. [DOI] [PubMed] [Google Scholar]
- 16.Ha, E. M., C. T. Oh, Y. S. Bae, and W. J. Lee. 2005. A direct role for dual oxidase in Drosophila gut immunity. Science 310:847-850. [DOI] [PubMed] [Google Scholar]
- 17.Harper, R. W., C. Xu, J. P. Eiserich, Y. Chen, C. Y. Kao, P. Thai, H. Setiadi, and R. Wu. 2005. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett. 579:4911-4917. [DOI] [PubMed] [Google Scholar]
- 18.Hill, A. A., C. P. Hunter, B. T. Tsung, G. Tucker-Kellogg, and E. L. Brown. 2000. Genomic analysis of gene expression in C. elegans. Science 290:809-812. [DOI] [PubMed] [Google Scholar]
- 19.Hope, I. A. 1999. C. elegans: a practical approach. Oxford University Press, Oxford, United Kingdom.
- 20.Kamath, R. S., A. G. Fraser, Y. Dong, G. Poulin, R. Durbin, M. Gotta, A. Kanapin, N. Le Bot, S. Moreno, M. Sohrmann, D. P. Welchman, P. Zipperlen, and J. Ahringer. 2003. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421:231-237. [DOI] [PubMed] [Google Scholar]
- 21.Kamath, R. S., M. Martinez-Campos, P. Zipperlen, A. G. Fraser, and J. Ahringer. 2001. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2:RESEARCH0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim, D. 2008. Studying host-pathogen interactions and innate immunity in Caenorhabditis elegans. Dis. Model Mech. 1:205-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim, D. H., R. Feinbaum, G. Alloing, F. E. Emerson, D. A. Garsin, H. Inoue, M. Tanaka-Hino, N. Hisamoto, K. Matsumoto, M. W. Tan, and F. M. Ausubel. 2002. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297:623-626. [DOI] [PubMed] [Google Scholar]
- 24.Klebanoff, S. J., W. H. Clem, and R. G. Luebke. 1966. The peroxidase-thiocyanate-hydrogen peroxide antimicrobial system. Biochim. Biophys. Acta 117:63-72. [DOI] [PubMed] [Google Scholar]
- 25.Kondo, M., S. Yanase, T. Ishii, P. S. Hartman, K. Matsumoto, and N. Ishii. 2005. The p38 signal transduction pathway participates in the oxidative stress-mediated translocation of DAF-16 to Caenorhabditis elegans nuclei. Mech. Ageing Dev. 126:642-647. [DOI] [PubMed] [Google Scholar]
- 26.Lambeth, J. D. 2004. NOX enzymes and the biology of reactive oxygen. Nat. Rev. 4:181-189. [DOI] [PubMed] [Google Scholar]
- 27.Lambeth, J. D., T. Kawahara, and B. Diebold. 2007. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 43:319-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meitzler, J. L., and P. Ortiz de Montellano. 2009. Caenorhabditis elegans and human dual oxidase 1 (DUOX1) “peroxidase” domains: insights into heme binding and catalytic activity. J. Biol. Chem. 284:18634-18643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohanty, J. G., J. S. Jaffe, E. S. Schulman, and D. G. Raible. 1997. A highly sensitive fluorescent micro-assay of H2O2 release from activated human leukocytes using a dihydroxyphenoxazine derivative. J. Immunol. Methods 202:133-141. [DOI] [PubMed] [Google Scholar]
- 30.Mohri-Shiomi, A., and D. A. Garsin. 2008. Insulin signaling and the heat shock response modulate protein homeostasis in the Caenorhabditis elegans intestine during infection. J. Biol. Chem. 283:194-201. [DOI] [PubMed] [Google Scholar]
- 31.Morand, S., T. Ueyama, S. Tsujibe, N. Saito, A. Korzeniowska, and T. L. Leto. 2009. Duox maturation factors form cell surface complexes with Duox affecting the specificity of reactive oxygen species generation. FASEB J. 23:1205-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newfeld, S. J., R. G. Wisotzkey, and S. Kumar. 1999. Molecular evolution of a developmental pathway: phylogenetic analyses of transforming growth factor-beta family ligands, receptors and Smad signal transducers. Genetics 152:783-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parrish, S., and A. Fire. 2001. Distinct roles for RDE-1 and RDE-4 during RNA interference in Caenorhabditis elegans. RNA 7:1397-1402. [PMC free article] [PubMed] [Google Scholar]
- 34.Pruitt, K. M., J. Tenovuo, R. W. Andrews, and T. McKane. 1982. Lactoperoxidase-catalyzed oxidation of thiocyanate: polarographic study of the oxidation products. Biochemistry 21:562-567. [DOI] [PubMed] [Google Scholar]
- 35.Pujol, N., S. Cypowyj, K. Ziegler, A. Millet, A. Astrain, A. Goncharov, Y. Jin, A. D. Chisholm, and J. J. Ewbank. 2008. Distinct innate immune responses to infection and wounding in the C. elegans epidermis. Curr. Biol. 18:481-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qadota, H., M. Inoue, T. Hikita, M. Koppen, J. D. Hardin, M. Amano, D. G. Moerman, and K. Kaibuchi. 2007. Establishment of a tissue-specific RNAi system in C. elegans. Gene 400:166-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quinn, M. T., and K. A. Gauss. 2004. Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J. Leukoc. Biol. 76:760-781. [DOI] [PubMed] [Google Scholar]
- 38.Rada, B., and T. L. Leto. 2009. Redox warfare between airway epithelial cells and Pseudomonas: dual oxidase versus pyocyanin. Immunol. Res. 43:198-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwarzer, C., T. E. Machen, B. Illek, and H. Fischer. 2004. NADPH oxidase-dependent acid production in airway epithelial cells. J. Biol. Chem. 279:36454-36461. [DOI] [PubMed] [Google Scholar]
- 40.Simmer, F., C. Moorman, A. M. van der Linden, E. Kuijk, P. V. van den Berghe, R. S. Kamath, A. G. Fraser, J. Ahringer, and R. H. Plasterk. 2003. Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. PLoS Biol. 1:E12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tenor, J. L., B. A. McCormick, F. M. Ausubel, and A. Aballay. 2004. Caenorhabditis elegans-based screen identifies Salmonella virulence factors required for conserved host-pathogen interactions. Curr. Biol. 14:1018-1024. [DOI] [PubMed] [Google Scholar]
- 42.Wijkstrom-Frei, C., S. El-Chemaly, R. Ali-Rachedi, C. Gerson, M. A. Cobas, R. Forteza, M. Salathe, and G. E. Conner. 2003. Lactoperoxidase and human airway host defense. Am. J. Respir. Cell Mol. Biol. 29:206-212. [DOI] [PubMed] [Google Scholar]
- 43.Zeldow, B. J. 1963. Studies on the antibacterial action of human saliva. III. Cofactor requirements of Lactobacillus bactericidin. J. Immunol. 90:12-16. [PubMed] [Google Scholar]
- 44.Zhang, L., G. Jie, J. Zhang, and B. Zhao. 2009. Significant longevity-extending effects of EGCG on Caenorhabditis elegans under stress. Free Radic. Biol. Med. 46:414-421. [DOI] [PubMed] [Google Scholar]