

Abstract
The intracellular bacterium Francisella tularensis survives and replicates within macrophages, ultimately killing the host cell. Resolution of infection requires the development of adaptive immunity through presentation of F. tularensis antigens to CD4+ and CD8+ T cells. We have previously established that F. tularensis induces macrophage prostaglandin E2 (PGE2) production, leading to skewed T-cell responses. PGE2 can also downregulate macrophage major histocompatibility complex (MHC) class II expression, suggesting that F. tularensis-elicited PGE2 may further alter T-cell responses via inhibition of class II expression. To test this hypothesis, gamma interferon (IFN-γ)-activated reporter macrophages were exposed to supernatants from F. tularensis-infected macrophages, and the class II levels were measured. Exposure of macrophages to infection supernatants results in essentially complete clearance of surface class II and CD86, compromising the macrophage's ability to present antigens to CD4 T cells. Biochemical analysis revealed that infection supernatants elicit ubiquitin-dependent class II downregulation and degradation within intracellular acidic compartments. By comparison, exposure to PGE2 alone only leads to a minor decrease in macrophage class II expression, demonstrating that a factor distinct from PGE2 is eliciting the majority of class II degradation. However, production of this non-PGE2 factor is dependent on macrophage cyclooxygenase activity and is induced by PGE2. These results establish that F. tularensis induces the production of a PGE2-dependent factor that elicits MHC class II downregulation in IFN-γ-activated macrophages through ubiquitin-mediated delivery of class II to lysosomes, establishing another mechanism for the modulation of macrophage antigen presentation during F. tularensis infection.
The facultative intracellular pathogen Francisella tularensis is a gram-negative coccobacillus and the causative agent of the disease tularemia (18). The highly infectious F. tularensis subsp. tularensis (type A) strain has been labeled a category A select agent by the Centers for Disease Control since it can be weaponized and due to its mass production by the United States and the former Soviet Union (14). Although attenuated in humans, the type B subspecies holarctica derived live vaccine strain (LVS) induces fulminate infection in mice and has been used extensively in models of F. tularensis infection (21). Published studies have revealed multiple mechanisms by which F. tularensis can modulate the cell biology of host macrophages in order to survive and replicate intracellularly. Among these are the ability to prevent endocytic maturation of the phagolysosome, escape into the host cell cytosol (9), and inhibit inflammatory signaling (50). F. tularensis has also been shown to disrupt the assembly of neutrophil NADPH oxidase and inhibit the oxidative burst (41).
Survival of mice infected with F. tularensis requires NK cell-derived gamma interferon (IFN-γ) (36), and non-IFN-γ-activated macrophages fail to prevent the cytosolic entry and replication of F. tularensis (20). Intracellular killing of F. tularensis by activated macrophages is further enhanced if bacteria are antibody opsonized and targeted to Fcγ receptors (31). Moreover, low doses of F. tularensis LVS can induce adaptive immunity in mice, and the development of F. tularensis reactive CD4+ or CD8+ is critical to the control of infection (17, 59). Interaction of infected macrophages with F. tularensis-specific CD4+ T cells requires macrophage presentation of F. tularensis antigens on major histocompatibility (MHC) class II molecules, and the suppression of macrophage class II expression could promote bacterial survival. The mechanisms underlying F. tularensis processing and MHC class II-restricted presentation are not well understood.
Generation of peptide-MHC class II complexes requires antigen endocytosis and delivery to class II-rich intracellular multivesicular bodies (MVB). After endocytosis, membrane proteins can also be delivered to late endosomes and sorted into the intraluminal vesicles (ILV) of MVB via the ESCRT protein complex, which functions during the inward budding of the MVB-limiting membrane (1, 46). Delivery of proteins into ILV is dependent on the mono- or multiubiquitination of lysine residues within the cytoplasmic domain of the target protein (26, 39). Proteins incorporated into ILV are eventually degraded upon fusion of MVB with terminal lysosomes (30). This process regulates delivery of the antigen binding B-cell receptor and FcγRIIA to late endocytic compartments for efficient antigen processing by B cells and macrophages (7, 15).
A recent report by Shin et al. established that ubiquitination of the cytoplasmic domain of MHC class II regulates class II surface expression during murine dendritic cell (DC) maturation. Although the majority of class II in immature DCs is ubiquitinated and thus localized to the MVB, lipopolysaccharide-induced activation leads to a loss of class II ubiquitination and an increase in class II surface expression (49). The anti-inflammatory cytokine interleukin-10 (IL-10) induces class II ubiquitination and subsequent intracellular sequestration in human monocytes via induction of the mammalian E3 ubiquitin ligase MARCH-I (51). Furthermore, overexpression of the E3 ubiquitin ligase c-MIR (MARCH-VIII) in multiple B-cell lines leads to reduced expression of MHC class II and the costimulatory molecule CD86 (23, 45).
Multiple pathogens utilize ubiquitination to suppress activation of host cells by targeting critical signaling molecules or immunologically relevant surface receptors. Using a type III secretion system, the Shigella spp. and Salmonella enterica inject the effectors IpaH9.8 and SspH1, respectively, into host cells that act as ubiquitin ligases to direct the proteasomal degradation of host mitogen-activated protein kinases (47). Kaposi's sarcoma-associated herpesvirus (KSHV) encodes the E3 ubiquitin ligase K5, which mediates ubiquitination of MHC class I (10, 29), the costimulatory molecule CD86 (11), the adhesion molecule ICAM-1 (11), and ligands of the NK cell receptors NKG2D and NKp80 (52).
This report establishes a mechanism by which F. tularensis modulates the function of IFN-γ-activated macrophages by indirectly inducing the degradation of macrophage MHC class II protein. The induced class II degradation involves the ubiquitin-dependent delivery of internalized class II to macrophage lysosomal compartments. Furthermore, macrophage class II degradation is induced by a high-molecular-weight non-prostaglandin protease-resistant factor, the production of which is dependent on F. tularensis-induced prostaglandin E2 (PGE2). This is the first study to describe a pathogen-induced PGE2-dependent factor that induces degradation of host macrophage MHC class II via ubiquitin-dependent mechanism.
MATERIALS AND METHODS
Bacteria.
F. tularensis LVS (ATCC 29684; American Type Culture Collection) strain was provided by Karen Elkins (U.S. Food and Drug Administration, Bethesda, MD). F. tularensis SchuS4 was provided by the U.S. Army Medical Research Institute for Infectious Diseases (Frederick, MD). All experiments utilizing SchuS4 were performed within a Center for Disease Control-certified ABSL-3/BSL-3 laboratory at Albany Medical College. Both strains of F. tularensis were cultured on chocolate agar plates and resuspended in cell culture medium at 2 × 108 total bacteria/ml as determined by measuring the optical density. Bacterial concentrations were confirmed by serial dilution on chocolate agar. For experiments utilizing nonviable bacteria, F. tularensis LVS was resuspended in phosphate-buffered saline (PBS) at 2 × 108 bacteria/ml and inactivated by using a Spectroline UV Cross-Linker or by resuspension in a 4% formaldehyde solution for 20 min, followed by multiple washes before infection. Both treatments lead to >99% killing, which was confirmed by plating on chocolate agar.
Mice.
B10.BR/SgSnJ (B10.BR) mice were purchased from Jackson Laboratory and housed in the Animal Resource Facility at Albany Medical College under specific-pathogen-free conditions. The appropriate institutional review committee approved all reported protocols.
BMDM generation.
Bone marrow cells were flushed from B10.BR mouse femurs, and 106 viable cells were incubated for 7 days on non-tissue-culture-treated 15-cm2 dishes with Dulbecco modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 0.5 U of penicillin/ml, 0.5 μg of streptomycin/ml, 1.8 mg of NaHCO3/ml, and 33% L-cell conditioned medium containing granulocyte-macrophage colony-stimulating factor. After differentiation, nonadherent cells were removed by multiple washes with PBS, and adherent bone marrow-derived macrophages (BMDM) were removed from plates by incubation with 1 mM EDTA in PBS. Experiments using BMDM were performed in DMEM supplemented with 10% heat-inactivated FBS, 1 mM sodium pyruvate, 10 mM HEPES buffer, and 50 μM 2-mercaptoethanol.
Cell line.
I-Ak-HEL46-61-specific murine T-cell hybridoma line, h4Ly50.5 (provided by W. Wade, Dartmouth Medical School, Lebanon, NH), was cultured in DMEM supplemented with 10% heat-inactivated FBS, 1 mM sodium pyruvate, 2 mM l-glutamine, and 50 μM 2-mercaptoethanol.
Infection of cells and preparation of conditioned medium.
Adherent macrophages were plated at 2 × 105 viable cells per well of a 96-well plate and infected with F. tularensis at a multiplicity of infection (MOI) of 100:1. At 2 h after infection, the extracellular F. tularensis organisms were killed by the addition of 50 μg of gentamicin/ml for 45 min. After multiple washes with PBS, fresh antibiotic-free complete medium was added, and the cells were incubated overnight. Supernatants were collected and filtered with a 0.2-μm-pore-size filter to remove bacteria and eukaryotic cells. Samples of supernatants were plated onto chocolate agar to verify the removal of F. tularensis. For macrophage cyclooxygenase inhibition experiments, BMDM were pretreated with 0.001% ethanol (solvent control) or 10 μM indomethacin (catalog no. I7378; Sigma, St. Louis, MO) prior to infection. Indomethacin was added again after a final wash with PBS and remained in the cultures throughout supernatant generation. The supernatants were stored at −80°C until needed.
Flow cytometry.
BMDM were treated with 100 U of IFN-γ (Sigma)/ml for 24 h to induce MHC class II expression. After multiple washes to remove the IFN-γ, BMDM were then incubated with infection supernatant (diluted 1:2) or PGE2 (catalog no. 14010; Cayman Chemical, Ann Arbor, MI) for 15 h, suspended at 2.5 × 105 viable cells (vc)/ml, and stained with anti-I-Ak-FITC (11-5.2, catalog no. 553536; BD Pharmingen, San Diego, CA), anti-mouse CD11b (catalog no. 557397; BD Pharmingen), anti-mouse CD86-PE (catalog no. 553692; BD Pharmingen), or anti-mouse CD16/32-FITC (2.4G2, catalog no. 553144; BD Pharmingen) for 20 min on ice. After multiple washes with Hanks balanced salt solution-0.1% bovine serum albumin, the BMDM were stained with 1 μg of propidium iodide/ml to label the dead cells. The data were collected by flow cytometry using FACSCanto (BD Biosciences) and analyzed with CellQuest Pro software (version 4.0.4; BD Biosciences).
Antigen presentation assay.
BMDM were pretreated for 24 h with 100 U of IFN-γ/ml. After multiple washes, the BMDM were placed into a 96-well plate at 2 × 105/well. After a 2-h incubation at 37°C to allow adherence to the plate, the BMDM were cocultured with 10 μM concentrations of filter-sterilized hen egg lysozyme (HEL) and F. tularensis LVS infection supernatants for 15 h at 37°C. The BMDM were fixed with 1% paraformaldehyde in PBS, washed, and cultured with 105 h4Ly50.5 T cells for 15 h at 37°C. The supernatants were removed, and the IL-2 levels were determined by using a mouse IL-2 Flex cytokine kit and cytometric bead array according to the manufacturer's instructions (catalog no. 551287; BD Biosciences).
Detection of MHC class II levels by Western blotting.
The BMDM were activated with IFN-γ and treated with infection supernatants as previously described. Cells were lysed at 2 × 107 vc/ml by a 10-min incubation on ice with radioimmunoprecipitation assay buffer containing protease inhibitors (1 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, and Complete Mini EDTA-free protease inhibitor cocktail [Roche Applied Science, Indianapolis, IN]). Lysates were cleared of cellular debris by centrifugation for 15 min at 16,000 × g at 4°C, diluted 1:3 with 4× reducing sample buffer, and boiled. Lysates were analyzed by reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotted as previously described (42) with rabbit anti-MHC class II (murine I-A beta chain cytoplasmic sequence RHRSQKGPRGPPPAGLLQ; Invitrogen, Carlsbad, CA) or anti-GAPDH (catalog no. MA4300; Ambion, Austin, TX). After incubation of goat anti-rabbit immunoglobulin G (IgG; heavy and light chain specific) peroxidase conjugate (catalog no. 401353; Calbiochem, San Diego, CA) or goat anti-mouse-IgG (heavy and light chain specific) peroxidase conjugate (catalog no. 401253; Calbiochem), blots were washed and developed using SuperSignal West Femto chemiluminescent peroxidase substrate (catalog no. 34095; Pierce Biotechnology, Rockford, IL). Blue sensitive X-ray film (catalog no. AF5700; Greentree Scientific, Penfield, NY) was exposed to blots and analyzed for densitometry using the imaging software ImageJ (version 1.32j; National Institutes of Health).
P62-ubiquitin immunoprecipitation.
To determine the level of MHC class II ubiquitination, BMDM were treated with infection supernatants in the presence or absence of 50 mM NH4Cl, lysed at 2 × 107 vc/ml, and cleared as previously described. A portion of the lysates was isolated prior to the precipitation step for the detection of total MHC class II levels. Cell lysates were precipitated by an overnight incubation at 4°C with ubiquitin-binding UBA-P62 agarose beads (catalog no. UW9010; Biomol, Plymouth Meeting, PA). The two sets of lysates were boiled in reducing sample buffer and analyzed for MHC class II expression by SDS-PAGE and Western blotting as previously described.
Proteasome inhibition assays.
To inhibit proteasome function during exposure of infection supernatants, cells were pulsed for 6 h with supernatants from producer BMDM (infected with F. tularensis) and chased for an additional 6 h with 5 μM proteasome inhibitor MG-132 (catalog no. W5035o; BioMol). After treatment, the BMDM lysates were analyzed for MHC class II expression by SDS-PAGE and Western blotting as previously described.
Statistical analysis.
A Student t test was used to analyze differences in MHC class II expression using Prism version 4.0c (GraphPad Software, Inc.). Statistical significance is indicated as P ≤ 0.05, P ≤ 0.01, or P ≤ 0.001.
RESULTS
F. tularensis elicits a soluble factor that induces downregulation of IFN-γ induced macrophage antigen presentation molecules.
We previously reported that upon infection with F. tularensis, macrophages produce PGE2, which results in a skewed T-cell cytokine response to antigen presentation (56). Since PGE2 can also suppress MHC class II expression by inhibiting the activity of class II transactivator CIITA and subsequent class II transcription (33), we hypothesized that PGE2 in supernatants from cultures of F. tularensis-infected macrophages might downregulate MHC class II levels. To test this hypothesis, infection supernatants were harvested from “producer” BMDM infected with F. tularensis LVS at an MOI of 100:1. IFN-γ-preactivated “reporter” BMDM (as a model of the activated macrophages that would be found in vivo subsequent to Francisella infection and production of IFN-γ by NK cells [36]) were treated with these infection supernatants for 15 to 20 h, and the cell surface MHC class II expression was analyzed by staining and flow cytometry. F. tularensis infection supernatants elicited reduced surface expression of MHC class II in IFN-γ-activated reporter BMDM to the levels present on non-IFN-γ-treated control BMDM (Fig. 1A). Interestingly, expression of the costimulatory molecule CD86 is also reduced, whereas control (mock) supernatants have little to no effect on MHC class II or CD86 expression. This effect was specific for MHC class II and CD86 and not due to cell death since CD32 and CD11b expression remains unaltered and no increase in propidium iodide staining is observed (Fig. 1A). Furthermore, downregulation of MHC class II and CD86 is not due to blocking of IFN-γ stimulation since the IFN-γ-activated reported BMDM had been pretreated with IFN-γ and washed prior to supernatant exposure.
FIG. 1.

Downregulation of MHC class II in IFN-γ-activated macrophages by F. tularensis infection supernatants. (A) BMDM were infected with F. tularensis LVS at an MOI of 100:1 or treated with medium alone (mock) for 2 h. Cells were treated with gentamicin for 1 h, washed, and incubated overnight to generate supernatants. Fresh reporter BMDM (pretreated or not pretreated 100 U of IFN-γ/ml for 24 h) were exposed to cleared infection supernatants for 15 h and analyzed for cell surface protein expression by staining for flow cytometry. Exposure of reporter BMDM to LVS infection supernatants leads to reduced surface expression of MHC class II and CD86, while FcγR and CD11b expression remains high. A lack of propidium iodide (PI) uptake indicates no decrease in cell viability. A vertical line represents the mean fluorescence intensity of the “+IFN” control for each surface marker. (B) After exposure to LVS or SchuS4 infection supernatants, the BMDM were lysed, and the MHC class II levels were analyzed by Western blotting. Exposure of macrophages to LVS or SchuS4 supernatants leads to a loss of total MHC class II, suggesting degradation after internalization. Shown are representative results from one of three independent experiments. (C) IFN-γ-pretreated BMDM were cultured with 10 μM HEL and F. tularensis LVS infection supernatant for 15 h. The BMDM were fixed with 1% paraformaldehyde, washed, and cocultured with the HEL peptide-specific h4Ly50.5 T-cell hybridoma line for 15 h. IL-2 levels were determined by cytometric bead array. Treatment of macrophages with LVS infection supernatants leads to a >90% loss of antigen presentation and resultant T-cell activation. The results are representative of one of three independent experiments ± the standard error of the mean (SEM). ***, P ≤ 0.001 (statistical difference from mock supernatant treatment).
To determine whether the loss of class II surface expression corresponds to a loss in total cellular class II, whole-cell lysates from supernatant-treated macrophages were analyzed for total MHC class II by Western blotting. Exposure of IFN-γ-preactivated reporter BMDM to supernatants from F. tularensis LVS-infected BMDM dramatically reduces the expression of total MHC class II protein levels (Fig. 1B, lane 4), demonstrating that infection supernatants not only induce class II surface clearance but the degradation of total macrophage class II. Infection supernatants were generated using the human virulent F. tularensis SchuS4 strain to confirm and extend the experiments with F. tularensis LVS. The exposure of responder BMDM to supernatants from SchuS4-infected producer macrophages also leads to pronounced downregulation of total macrophage class II. These results demonstrate that, like F. tularensis LVS, F. tularensis SchuS4 can elicit soluble factors that downregulate MHC class II protein expression in IFN-γ-activated macrophages.
To establish the immunological impact of macrophage class II downregulation, the impact of LVS infection supernatant on the ability of IFN-γ-preactivated BMDM to process and present the soluble protein antigen HEL was determined. As shown by the results presented in Fig. 1C, supernatant-induced MHC class II downregulation results in a >90% inhibition of macrophage antigen presentation.
The dramatic (i.e., >70%) reduction in macrophage MHC class II expression in response to exposure to Francisella infection supernatant was somewhat unexpected since the half-life of MHC class II mRNA in BMDM is >8 h (58). If the PGE2 in the LVS infection supernatant was the only molecule responsible for class II downregulation by the established mechanism of eliciting cyclic AMP-induced protein kinase A (PKA) activation and subsequent CIITA phosphorylation and inhibition (33), this would not be expected to result in a >70% inhibition of MHC class II protein expression in 12 h. This finding is consistent with mathematic modeling studies that indicate it would require >10 h to observe a decrease in class II expression after a block in mRNA translation (8) and suggests the presence of an additional factor in the F. tularensis BMDM supernatants that is eliciting macrophage MHC class II downregulation via a distinct mechanism.
F. tularensis infection supernatants induce MHC class II degradation in lysosomal compartments.
In order to test the hypothesis that macrophage class II downregulation is not simply due to a block in class II transcription and to better characterize the mechanism of class II downregulation, the kinetics of class II clearance in IFN-γ-activated reporter BMDM exposed to infection supernatants was determined. As shown in Fig. 2A, total MHC class II levels are significantly (P < 0.003) reduced in reporter BMDM at 10 h posttreatment with infection supernatants and further decreased by 12 h. Exposure times of 4 to 8 h fail to induce statistically significant (P > 0.25) levels of clearance, demonstrating that MHC class II is strongly expressed in IFN-γ-activated BMDM until 10 to 12 h of exposure to infection supernatants.
FIG. 2.

Infection supernatant-mediated MHC class II degradation occurs in acidic vesicles. (A) BMDM pretreated or not pretreated with 100 U of IFN-γ/ml for 24 h were exposed to F. tularensis LVS infection supernatants for the indicated time points and analyzed by Western blotting for MHC class II expression. Statistically significant class II degradation was observed at 10 h postexposure to infection supernatants. Densitometry values are normalized to the “+IFN-γ” medium control. The results are representative of one of three independent experiments ± the SEM. **, P ≤ 0.01; ***, ≤ 0.001 (statistical difference from the control treatment). (B) BMDM pretreated or not pretreated with 100 U of IFN-γ/ml for 24 h were pulsed with infection supernatant for 6 h and cocultured with 10 or 50 mM ammonium chloride (NH4Cl) for an additional 10 h before being lysed and analyzed by Western blotting. Blocking of macrophage lysosomal activity prevents infection supernatant-induced degradation of MHC class II in a dose-dependent manner, demonstrating the degradation of internalized class II in lysosomes. Shown are representative results from one of three independent experiments.
Since activated macrophages have a high proteolytic capacity (13), it is possible that F. tularensis infection supernatants could cause degradation of MHC class II by inducing trafficking of class II to acidic compartments after internalization. To test this hypothesis, BMDM were treated with F. tularensis infection supernatants in the presence or absence of ammonium chloride (NH4Cl), which neutralizes intracellular acidic compartments and prevents late endosomal/lysosomal degradation of proteins without blocking other degradative pathways such as proteasome-mediated cytosolic proteolysis (5). Neutralization of acidic compartments prevents infection supernatant-induced class II degradation (Fig. 2B), firmly establishing a role for lysosomes in F. tularensis-induced MHC class II clearance. Consistent with reports utilizing lysosomal neutralizing agents to prevent class II turnover (49), exposure of untreated macrophages to increasing concentrations of NH4Cl leads to an increase in MHC class II levels above the “+IFN-γ medium” control, suggesting that neutralization of late endosomes/lysosomes also blocks the normal turnover of class II. These results suggest that F. tularensis exploits a host pathway normally used in protein turnover to preferentially downregulate macrophage MHC class II expression.
F. tularensis induces MHC class II ubiquitination for MVB/lysosome targeting in macrophages.
Although polyubiquitination is associated with proteasomal degradation of many cytosolic proteins, mono- and multiubiquitination of the cytoplasmic domain of many membrane proteins (e.g., B-cell receptor, FcγR, epidermal growth factor receptor, and MHC class II) allows for the ESCRT-mediated delivery of these membrane proteins to the ILV of MVB for subsequent trafficking to lysosomal compartments (7, 15, 35, 49). Induction of macrophage MHC class II ubiquitination, resulting in the ESCRT-mediated delivery class II molecules to MVB ILV and ultimately lysosomal trafficking, would be beneficial to F. tularensis survival, since uncontrolled intramacrophage bacterial replication occurs in the absence of MHC-restricted macrophage-T-cell interactions.
To determine whether the class II degradation elicited by the F. tularensis infection supernatants occurs via the ubiquitin-dependent delivery of class II to MVB, ubiquitinated proteins from infection-supernatant-treated and mock-supernatant-treated macrophages were precipitated and probed for class II. IFN-γ-activated BMDM were exposed to mock or infection supernatants for the indicated times, and lysates were incubated with ubiquitin-binding P62-UBA beads. After pulldown with P62-UBA beads, both total and ubiquitinated proteins were fractionated by SDS-PAGE and immunoblotted for MHC class II. As shown in Fig. 3 (lanes 1 to 6), little to no MHC class II was detected in the ubiquitin pulldown from supernatant-treated cells. However, it is possible that ubiquitinated MHC class II is rapidly degraded in lysosomes, preventing detection of the ubiquitinated form of the protein. In order to determine whether this is the case, lysosomal degradation of class II was blocked by the addition of ammonium chloride, and MHC class II ubiquitination was monitored. The amount of MHC class II in the ubiquitin pulldown is increased substantially in BMDM treated with infection supernatants in which lysosomal degradation was blocked by ammonium chloride (Fig. 3, lanes 7 to 9). Although the addition of ammonium chloride alone leads to an increase in class II in the ubiquitin pulldown (Fig. 3, lane 10), the effect was modest compared to the combined effect of ammonium chloride and supernatant treatment. Surprisingly, while ubiquitin monomers are 8 kDa in size and the covalent attachment of ubiquitin to MHC class II α or β chains should result in a subsequent increase in the molecular weight of class II, no molecular weight shift was detected for class II in the ubiquitin pulldown. This finding suggests that MHC class II may be ubiquitinated as a protein complex with MHC-associated molecules being the direct target of ubiquitination. These results suggests that ubiquitin regulates MHC class II degradation in activated macrophages via ESCRT-mediated lysosomal delivery during steady-state conditions and that factors elicited during F. tularensis infection can increase this process to preferentially reduce class II expression in IFN-γ-activated macrophages.
FIG. 3.
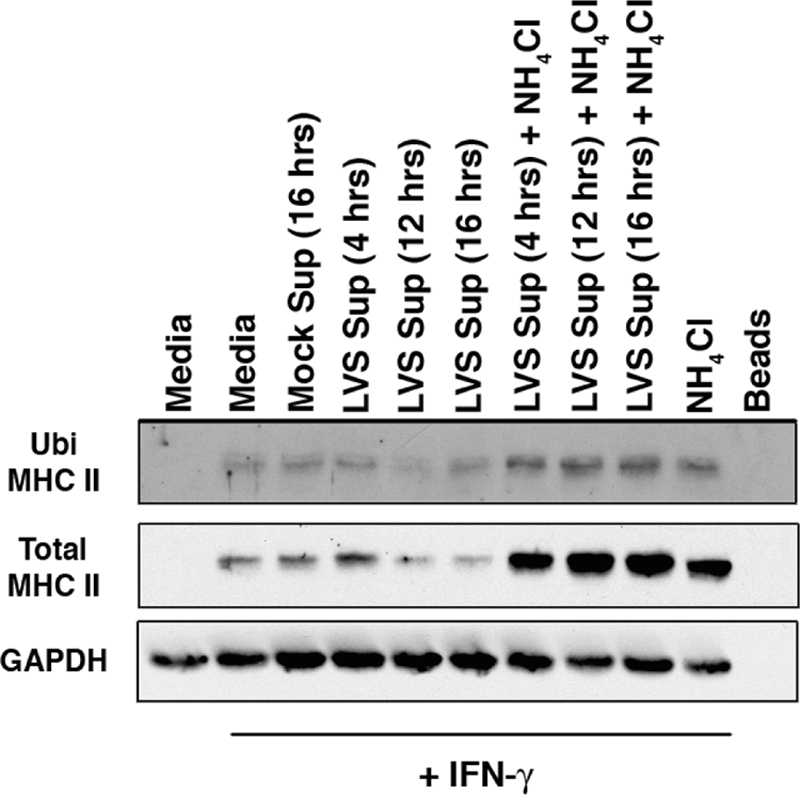
F. tularensis infection supernatants induce ubiquitination of MHC class II. BMDM were pretreated or not pretreated with 100 U of IFN-γ/ml for 24 h and then treated with infection supernatant with or without 50 mM NH4Cl as described in the text. Ubiquitinated MHC class II from cell lysates was precipitated with P62-UBA-ubiquitin-binding agarose beads. Ubiquitinated and total MHC class II was analyzed by Western blotting. Exposure of BMDM to infection supernatant leads to increased ubiquitination of MHC class II, observable only with the addition of NH4Cl to prevent rapid class II degradation. Shown are representative results from one of two independent experiments.
Although there are no pharmacological inhibitors that directly inhibit membrane protein ubiquitination, this process can been disrupted through acute inhibition of the proteasome due to the resultant depletion of free cytosolic ubiquitin (43). This approach has been reported to prevent of ESCRT-mediated sorting of epidermal growth factor receptor into MVB and its subsequent degradation (35). Moreover, this approach has been used by our laboratory to disrupt the ubiquitin-dependent intracellular sorting and subsequent processing of BCR-antigen complexes (15). Therefore, the proteasome inhibitor MG-132 was used to deplete the free ubiquitin pool in BMDM in order to determine whether inhibition of MHC class II ubiquitination prevents class II sorting and lysosomal degradation and to determine whether F. tularensis infection supernatant-induced MHC class II ubiquitination is responsible for the observed class II degradation. After pretreatment of BMDM with F. tularensis infection supernatants for 6 h, the cells were incubated for an additional 6 h with the reversible proteasome inhibitor MG-132 (5 μM) and analyzed for expression of total MHC class II by Western blotting. These time points were used due to the cell toxicity associated with MG-132 treatment beyond 6 h (not shown) and the lack of class II downregulation by infection supernatants before 6 h of incubation time (Fig. 2A). Consistent with a role for ubiquitination in class II downregulation, BMDM subjected to proteasome inhibition during treatment with infection supernatants display robust levels of MHC class II, similar to the “+IFN-γ control” (Fig. 4). Although MG-132 has been demonstrated to inhibit lysosomal proteases (22), MG-132 treatment failed to increase MHC class II expression beyond what was observed in the “+IFN-γ control” (as observed with NH4Cl treatment in Fig. 3 and 4), suggesting the effect of MG-132 was due to a block in class II ubiquitination, which would result in an inability of the class II molecules to enter into the ubiquitin-dependent MVB sorting pathway. The results presented in Fig. 4 demonstrate an ubiquitin-dependent mechanism for MHC class II downregulation during F. tularensis infection.
FIG. 4.

Ubiquitination is required for MHC class II degradation by F. tularensis infection supernatants. IFN-γ-pretreated BMDM were pulsed with infection supernatants for 6 h and chased with or without proteasome inhibitor MG-132 (5 μM) for an additional 6 h. MHC class II levels were analyzed by Western blotting. Densitometry results are normalized to the “+IFN-γ” medium control and expressed as the mean of three independent experiments ± the SEM. **, P ≤ 0.01 (statistical difference from the LVS supernatant plus MG-132 treatment).
MHC class II downregulation is mediated by a >10-kDa protease-resistant molecule, induced by F. tularensis-elicited PGE2.
We originally hypothesized that F. tularensis-elicited PGE2 would lead to the downregulation of macrophage MHC class II expression via inhibition of class II transcription. Interestingly, supernatants from F. tularensis-infected macrophages were found to induce ubiquitin-dependent degradation of MHC class II. These results suggest either a novel mechanism for PGE2-induced class II downregulation or the presence of one or more non-PGE2 factor(s) in the F. tularensis infection supernatants that are directly responsible for induction of the ubiquitin-dependent degradation of macrophage class II. To address this issue, the role of PGE2 in the observed macrophage class II downregulation was more precisely defined.
PGE2 is a short-lived molecule, derived from arachidonic acid by the action of the enzyme cyclooxygenase 1/2 (COX1/2) (24). We have previously reported that BMDM produce 10 to 20 ng of PGE2/ml in response to F. tularensis infection (56). Since infection supernatants are diluted 1:2 during the treatment of reporter BMDM, the final concentration of PGE2 in these experiments is in the range of 5 to 10 ng/ml, which is ∼10-fold below the dose of 0.25 μM (88 ng/ml) PGE2 that is required to elicit PKA-mediated CIITA inactivation (33) and block class II expression. To establish the direct contribution of PGE2 to infection supernatant-mediated MHC class II surface clearance, IFN-γ-activated reporter BMDM were treated with 1 to 50 ng of PGE2/ml and analyzed for class II surface expression by flow cytometry. Although the addition of 1 to 50 ng of PGE2/ml leads to a modest reduction of MHC class II surface expression, this reduction was not as extensive as that observed during treatment of BMDM with infection supernatants (Fig. 5A). Treatment of activated BMDM with LVS infection supernatants resulted in a 76.8% ± 5.2% inhibition of class II surface expression, whereas treatment of the BMDM with 10 ng of pure PGE2 (the maximum level of PGE2 present in the diluted supernatants)/ml only elicits 31.0% ± 5.4% downregulation (which is consistent with the level of inhibition observed upon inhibition of MHC class II transcription in BMDM [8]). These results strongly suggest that a significant portion of MHC class II downregulation elicited by exposure to LVS infection supernatant is not driven by PGE2 alone and suggest that the presence of one or more additional factors is responsible for inducing the lysosomal degradation of preexisting MHC class II.
FIG. 5.

The factor responsible for MHC class II downregulation is distinct from PGE2 but requires macrophage COX activity. (A) BMDM were exposed to infection supernatants or the indicated concentrations of PGE2 for 15 h without IFN-γ treatment or after treatment with 100 U of IFN-γ/ml and then analyzed by flow cytometry. Concentrations of PGE2 10-fold greater than that observed during infection failed to elicit the same level of MHC class II inhibition as that induced by infection supernatants, suggesting that a non-PGE2 factor is responsible for the majority of class II downregulation. The results represent the means of three independent experiments ± the SEM. *, Statistical difference (P ≤ 0.05) from the mock treatment. (B) F. tularensis infection supernatant was fractionated using a 10-kDa filter prior to addition to IFN-γ-activated BMDM. MHC class II levels were determined by Western blotting. Molecular mass filtration revealed that the factor responsible for MHC class II degradation is >10 kDa, suggesting the involvement of a nonprostaglandin factor. Shown are representative results from one of three independent experiments. (C) IFN-γ-activated reporter BMDM exposed to infection supernatants generated in the presence of 10 μM indomethacin or ethanol control. To account for the potential carryover effects of cyclooxygenase inhibition of reporter cells, supernatants generated in the absence of drug were “spiked” with indomethacin during incubation with reporter macrophages. Supernatants generated by infected BMDM with blocked COX1/2 activity failed to downregulate MHC class II levels, demonstrating the dependence of this enzyme in soluble-factor generation. A Western blot representative of one of three independent experiments with the relative densitometry plotted ± the SEM is shown. *, P ≤ 0.05; **, P ≤ 0.01 (statistical difference from the mock treatment).
To determine the size of the factor(s) responsible for the ubiquitin-dependent degradation of macrophage MHC class II, F. tularensis-elicited macrophage infection supernatants were fractionated using a 10-kDa cutoff molecular-weight filter. IFN-γ-activated responder BMDM were then treated with nonfractionated infection supernatants, the <10-kDa supernatant fraction, or the >10-kDa enriched supernatant fraction for 15 h. Reporter BMDM class II levels were then measured by Western blotting. Although the <10-kDa infection supernatant fraction failed to induce MHC class II protein degradation, this ability was fully retained in the >10-kDa enriched infection supernatant fraction. These results demonstrate the factor in the F. tularensis-elicited macrophage supernatants that is directly responsible for inducing class II protein downregulation is >10 kDa in size (Fig. 5B) and thus not PGE2 (which is <400 Da in size).
To determine whether there is any role for prostaglandins in the ubiquitin-dependent downregulation of macrophage class II protein, infection supernatants were generated from F. tularensis-infected producer BMDM in the presence of the COX inhibitor indomethacin and tested for their ability to elicit macrophage class II downregulation. (This approach was favored over the addition of anti-PGE2 neutralizing antibodies to the BMDM-Francisella cultures since anti-PGE2 antibody might not have complete access to autocrine/paracrine PGE2 before it binds to BMDM PGE2 receptors.). IFN-γ-activated reporter macrophages were treated with these supernatants for 15 h, and the MHC class II levels were determined by Western blotting. Supernatants generated in the presence of indomethacin lost the ability to downregulate MHC class II expression (Fig. 5C, lane 6). This lack of class II downregulation by infection supernatant generated in the presence of indomethacin was not due to inhibition of COX activity in reporter BMDM (i.e., carry over effects of indomethacin) since active infection supernatant spiked with indomethacin after generation induced the full extent of class II downregulation (Fig. 5C, lane 7).
Based on these findings, it appears that macrophage COX activity is required for the production of F. tularensis-elicited prostaglandin (PGE2) that, while incapable of directly eliciting the ubiquitin-dependent downregulation of macrophage class II, acts in an autocrine/paracrine fashion to induce or facilitate the production of a >10-kDa factor that is directly responsible for inducing class II clearance. Although our previous report that indomethacin treatment, while augmenting Francisella uptake, dampens the intramacrophage growth of F. tularensis (56) might suggest that the PGE2-supported intramacrophage growth of F. tularensis is necessary for the production of the class II downregulatory factor, the finding that dead Francisella can also elicit class II downregulation (see below) strongly argues against this possibility.
To determine whether Francisella-elicited PGE2 is the sole factor necessary for the production of the >10-kDa factor(s) directly responsible for inducing class II degradation (or whether other Francisella-derived signals are involved), supernatants from PGE2-treated producer BMDM were compared to supernatants from F. tularensis-infected BMDM for their ability to elicit downregulation of macrophage MHC class II. IFN-γ-activated reporter BMDM were treated with these supernatants for 15 h, and the MHC class II levels were determined by Western blotting. As shown in Fig. 6, treatment of producer macrophages with PGE2 leads to the dose-dependent generation of supernatants capable of eliciting macrophage class II downregulation. Using this approach, it takes ∼100 ng of PGE2/ml (which is similar to the dose of 0.25 μM [88 ng/ml] PGE2 that is required to elicit PKA-mediated CIITA inactivation [33]) to generate supernatants that downregulate class II to the level induced by F. tularensis infection supernatants, whereas the level of PGE2 produced by F. tularensis-infected BMDM is ∼5-fold lower (∼20 ng/ml, see Fig. 4A of reference 55). The need for this slightly greater level of exogenously added PGE2 is most likely due to the need to bring the entire culture to the PGE2 concentration that macrophages experience locally upon Francisella-induced PGE2 production. Nevertheless, these results suggest that the COX dependency of the generation of active F. tularensis infection supernatants capable of inducing class II degradation (Fig. 5B) is due to the necessary role of Francisella-elicited PGE2, which in turn acts in an autocrine/paracrine fashion to lead to production of the >10-kDa factor(s) responsible for MHC class II downregulation. These results are also consistent with the observation that TLR-2 knockout macrophages can both produce and respond to Francisella infection supernatants, suggesting no significant role for Francisella TLR-2 ligands (38) in this phenomenon (data not shown).
FIG. 6.

PGE2 indirectly responsible for MHC class II degradation. (A) BMDM were mock infected, infected with F. tularensis LVS, or treated with the indicated concentrations of PGE2. After a 15-h incubation, supernatants were collected and added to IFN-γ-activated BMDM. BMDM were lysed, and the MHC class II levels were analyzed by Western blotting. Exposure of macrophages to LVS or PGE2 supernatants leads to a loss of total MHC class II, suggesting that PGE2 can induce the production of the factor responsible for class II degradation. Shown are representative results from one of three independent experiments. (B) Proposed mechanism of Francisella-induced macrophage class II downregulation. Macrophage invasion by F. tularensis results in the production of PGE2, which acts in an autocrine/paracrine fashion to elicit the production of a high-molecular-weight (MW) protease-resistant factor that elicits the ubiquitin-dependent degradation of MHC class II molecules in acidic intracellular compartments (L, lysosomes) of non-Francisella-infected “reporter” macrophages.
Production of the F. tularensis-elicited factors responsible for eliciting class II downregulation does not depend on macrophage activation or F. tularensis viability.
IFN-γ-activated macrophages can restrict the growth of F. tularensis (20), and mice deficient for IFN-γ rapidly succumb to F. tularensis infection (17). Therefore, it might be expected that this IFN-γ activation could also block the production of the Francisella-induced soluble factor(s) responsible for the downregulation of macrophage class II expression. To test this possibility, infection supernatants were generated from either resting or IFN-γ-preactivated producer BMDM, and these supernatants were tested for the ability to downregulate MHC class II surface expression on reporter BMDM. Infection supernatants from IFN-γ-preactivated BMDM reduced class II surface expression as efficiently as supernatants from non-IFN-γ-activated controls (Fig. 7A), demonstrating that IFN-γ fails to block the ability of F. tularensis to elicit the production of the factor(s) that induce class II clearance.
FIG. 7.
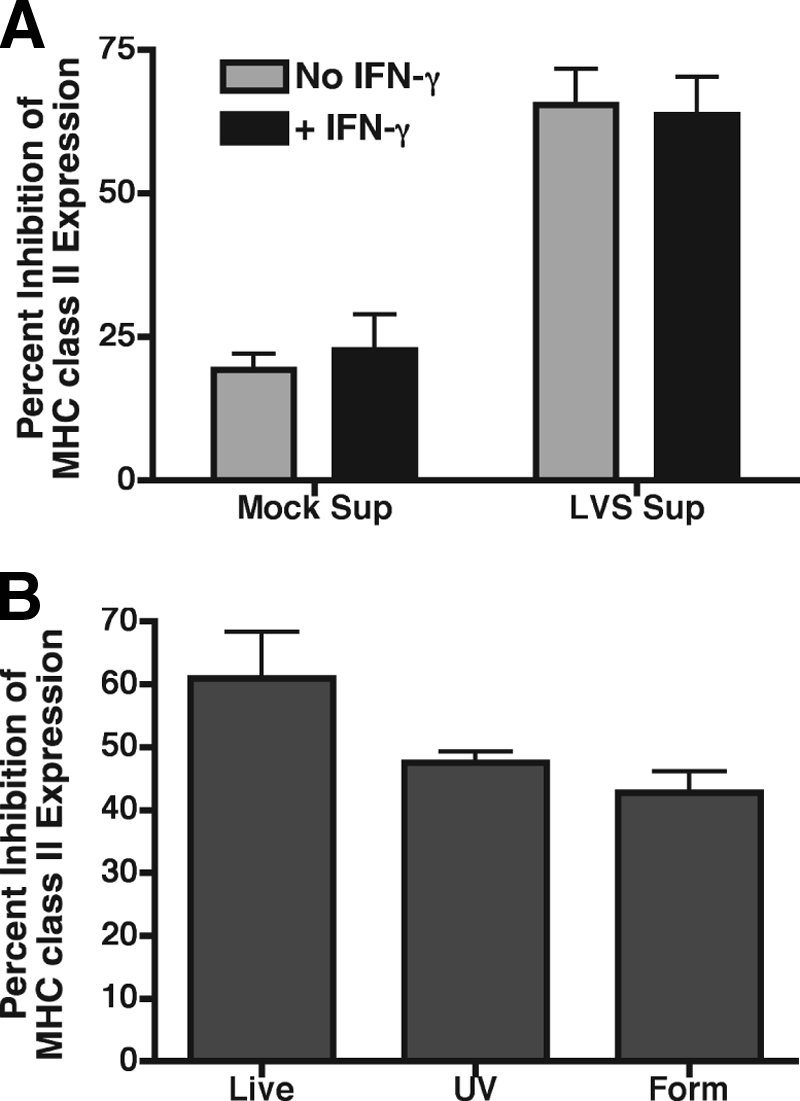
Production of F. tularensis-elicited factor independent of macrophage activation and F. tularensis viability. (A) Producer BMDM activated with 100 U of IFN-γ/ml or left untreated prior to infection with F. tularensis LVS were used to generate infection supernatants. Reporter BMDM were incubated with infection supernatants for 15 h and analyzed for MHC class II expression by flow cytometry. IFN-γ activation of producer macrophages failed to block production of the factor responsible for class II downregulation. (B) Infection supernatants were generated from producer BMDM infected with live, UV-killed, or formaldehyde-fixed F. tularensis LVS and exposed to IFN-γ-activated reporter BMDM for analysis of class II expression by flow cytometry. Inactivation of F. tularensis LVS had no effect on the elicitation of the factor responsible for class II downregulation. The data are expressed as the means of three independent experiments ± the SEM.
Nonviable forms of F. tularensis fail to escape macrophage phagolysosomes and do not enter the host cell cytosol (9). To further characterize the requirements for the production of the F. tularensis-elicited inhibitory factor(s), nonviable forms of F. tularensis LVS were used to generate “infection” supernatants. IFN-γ-activated reporter BMDM were exposed to supernatants from producer BMDM exposed to live, UV-killed, or formaldehyde-fixed LVS and then analyzed for MHC class II expression by flow cytometry. Although supernatants elicited by UV- and formaldehyde-killed F. tularensis LVS were slightly reduced in their ability to downregulate MHC class II surface expression, this level of inhibition was not significantly different (P > 0.08) than the level of inhibition elicited by supernatant produced by BMDM infected with live bacteria (Fig. 7B). These results demonstrate that the interaction of F. tularensis with macrophages alone elicits production of the factor responsible for inducing class II degradation and also demonstrates that bacterial replication and cytosolic entry are not required for the induction of these factor(s). Furthermore, these findings are consistent with the idea that PGE2 elicits the production of this >10-kDa factor since F. tularensis-induced PGE2 production also occurs independently of macrophage activation and bacterial viability (56).
F. tularensis-elicited factor responsible for MHC class II degradation is heat sensitive but protease resistant.
Although PGE2 is involved (both directly and indirectly) in the downregulation of macrophage MHC class II, it is still unclear what >10-kDa factor(s) are being induced by F. tularensis-elicited PGE2 that are directly responsible for driving the ubiquitin-dependent downregulation of macrophage class II. In order to further characterize the factor(s) directly responsible for inducing class II degradation, F. tularensis infection supernatants were boiled at 100°C for 10 or 30 min and then used to treat IFN-γ-activated reporter BMDM for 15 h. MHC class II levels were then determined by Western blotting. Heat treatment rendered F. tularensis-elicited macrophage infection supernatants incapable of inducing MHC class II downregulation (Fig. 8A, lanes 5 and 6), demonstrating the >10-kDa factor responsible for driving class II degradation is a heat-sensitive molecule or a molecule that gets trapped in heat-induced protein aggregates.
FIG. 8.

Factor responsible for class II degradation is heat sensitive but resistant to PK treatment. (A) Infection supernatants were boiled for the indicated times or incubated with 10, 50, or 100 mg of PK-beads/ml for 30 min at 37°C prior to exposure to IFN-γ-activated BMDM. MHC class II levels were determined by Western blotting. Infection supernatants boiled at 100°C failed to downregulate macrophage class II, whereas PK-treated infection supernatants retained the ability to induce class II degradation. A representative Western blot from one of three independent experiments is shown. (B) To confirm the PK activity, infection supernatants were treated with PK-beads as described above, and the presence of protein was detected by SDS-PAGE and silver staining. A silver-stained gel representative of one of two experiments is shown.
Infection supernatants were also treated with acrylic-bead-conjugated proteinase K (PK) at a dose of 10 to 100 mg/ml for 30 min at 37°C and then cleared of PK beads by centrifugation and filtration. PK-treated and untreated infection supernatants were then incubated with IFN-γ-activated reporter BMDM for 15 h, and the MHC class II levels were determined by Western blotting. Unlike heat treatment, PK treatment failed to reverse the ability of the infection supernatants to downregulate MHC class II in activated BMDM (Fig. 8A, lanes 7 to 9). To confirm the PK treatment was mediating protein degradation, the PK-treated supernatants were subjected to SDS-PAGE and silver staining. With the exception of a high-molecular-mass band (>170 kDa), several protein bands visible in the LVS supernatant lane (Fig. 8B, lane 2) were no longer detectable upon PK treatment (Fig. 8B, lanes 3 to 5), demonstrating that the PK treatment was effective in protein digestion. These results demonstrate that the factor(s) responsible for inducing MHC class II degradation is PK resistant and suggests that identification of this factor via mass spectrometry will be difficult due to the low levels of intact protein in active PK-treated infection supernatants. Furthermore, in combination with the findings that this factor is >10 kDa in size and heat sensitive, these results suggest this factor is a highly protease-resistant protein such as IL-1β (25), which we ruled out as a possibility (see Discussion), or a nonprotein molecule that gets trapped in heat-induced protein aggregates.
Overall, these combined results demonstrate that F. tularensis infection of macrophages leads to the production of PGE2 (which can directly downregulate class II expression via a block in class II transcription), which then induces the production of a >10-kDa heat-sensitive and PK-resistant macrophage-derived factor that induces ubiquitin-dependent MHC class II MVB targeting and subsequent degradation in activated macrophages and demonstrates an additional mechanism of F. tularensis to exploit host processes to subvert recognition by CD4+ T cells.
DISCUSSION
The results presented here demonstrate that the intracellular pathogen F. tularensis elicits a host factor that selectively targets MHC class II for degradation in lysosomes of IFN-γ-activated macrophages. This process occurs via a ubiquitin-dependent mechanism and is mediated by the production of one or more yet-to-be-identified factor(s), the production of which are dependent on the activity of host macrophage-derived PGE2. This represents an additional mechanism by which F. tularensis can modulate the cell biology of host macrophages, leading to the subversion of immune responses.
The survival of mice infected with F. tularensis LVS is dependent on the activity of CD4+ or CD8+ T cells (17, 59). Any suppression of T-cell function would potentially lead to the enhanced survival of the F. tularensis bacteria. A previous report from this laboratory demonstrated the skewing of T-cell responses by F. tularensis-induced macrophage PGE2 and illustrates one mechanism by which this suppression may be accomplished. PGE2 also inhibits the transcription of MHC class II genes by inactivating CIITA (33), suggesting that T-cell responses can be further blunted during F. tularensis infection via inhibition of macrophage MHC class II expression. Although culture supernatants from F. tularensis-infected macrophages were found to be highly efficient in downregulating MHC class II expression (Fig. 1A), PGE2 alone did not fully recapitulate this phenomenon. Although PGE2 reduces the expression of surface class II (Fig. 5A), the results presented here establish a mechanism of posttranslational clearance of MHC class II mediated by soluble factor(s) distinct from but induced by PGE2. Previous studies have established that 10 h of inhibition of class II transcription results in ∼25% reduction of surface MHC class II (8); thus, it is unlikely that exposure of IFN-γ-activated macrophages to PGE2 would lead to the rapid and extensive clearance of class II that we observed in response to the infection supernatants. Treatment of macrophages with PGE2 beyond 10 to 15 h would undoubtedly lead to a greater loss of class II expression since nascent class II molecules fail to arrive at the cell surface and since normal protein turnover continues. A report by Chang et al. supports this possibility with the finding that surface MHC class II is reduced by ∼66% at 100 h after transcriptional inhibition (8). Multiple mechanisms of class II inhibition may be required to efficiently prevent T-cell recognition of infected cells at different stages of infection, with our results suggesting that F. tularensis accomplishes this by inducing the trafficking of class II protein to acidic compartments for degradation by a currently unknown PGE2-elicited factor in conjunction with direct inhibition of class II transcription by PGE2 directly. The utilization of multiple mechanisms to reduce class II expression and function to avoid immune detection has been well characterized during chronic infection with the intracellular bacterium Mycobacterium tuberculosis (8).
F. tularensis-specific T lymphocytes are generated following sublethal challenge, and these cells can prevent bacterial growth in infected macrophages in vitro (16). However, these T cells fail to completely clear the infection in vivo since stable low levels of bacteria can be detected in surviving mice at least 2 months after secondary challenge (57). It is possible that by eliciting PGE2, F. tularensis prevents the proper T-cell recognition required for complete bacterial clearance by targeting multiple mechanisms involved in MHC class II presentation as described above. Moreover, F. tularensis may utilize PGE2 and PGE2-dependent factors to inhibit MHC class II expression in uninfected neighboring macrophages in order to create a more hospitable environment for bacteria released from initially infected macrophages. This possibility is supported by a report by Woolard et al. that describes the role of PGE2 in F. tularensis-infected mice (55). In that report, the authors show that the administration of the COX1/2 inhibitor indomethacin after intranasal infection with F. tularensis results in reduced production of PGE2, increased numbers of IFN-γ-producing T cells, and decreased bacterial burdens in the lungs of infected animals (55). Although the relative contribution of PGE2 on direct T-cell skewing and downregulation of macrophage antigen presentation was not determined, it is clear that the presence of PGE2 dampens the ability of the adaptive immune system to effectively recognize F. tularensis-infected macrophages in vivo. Future studies will be necessary to determine the contribution of the regulation of MHC class II expression during F. tularensis infection in vivo and to further characterize the nature of the factor(s) responsible for the downregulation of MHC class II.
The results presented here demonstrate that F. tularensis elicits a PGE2-dependent macrophage factor that induces ubiquitination of the MHC class II. Very little is known about the regulation of ubiquitin and ubiquitin ligases by soluble factors. Stimulation of immature DCs with lipopolysaccharide negatively regulates the expression of the ubiquitin ligase MARCH-I, leading to decreased class II ubiquitination and enhanced surface expression (12, 49). To date, IL-10 is the only cytokine that has been shown to induce ubiquitination of MHC class II (51). However, addition of anti-IL-10 neutralizing antibody to F. tularensis infection supernatants had no effect on class II downregulation, and F. tularensis infection supernatants from IL-10 knockout BMDM downregulate class II as efficiently as infections supernatants from wild-type macrophages (data not shown). The results presented in Fig. 5 and 6 demonstrate that PGE2 is indirectly responsible for class II downregulation since infection supernatants from BMDM treated with the COX inhibitor indomethacin fail to inhibit class II expression and PGE2 can elicit BMDM supernatants that are competent for inducing class II downregulation in the absence of F. tularensis infection. However, PGE2 alone has only a modest direct effect on macrophage class II expression, and fractionation of macrophage infection supernatants revealed that the factor responsible for inducing class II downregulation is >10 kDa, which is substantially larger than any known prostaglandin. These results demonstrate that PGE2 exerts its effects on the responding macrophage in a paracrine/autocrine fashion by inducing the production of a second factor that is >10 kDa in size, which is directly responsible for inducing class II ubiquitination.
PGE2 has been well documented to act in an autocrine manner, acting back on the producer cell to induce the secretion of nonprostaglandin factors such as matrix metalloproteinase-9 (60) and IL-6 (27), which are involved in multiple types of infection (3, 37, 44). Using gene chip analysis, Zhu et al. have reported a number of macrophage genes that are induced after stimulation by PGE2 (61). Only one of the induced genes (i.e., IL-1β) is associated with downregulation of MHC class II expression, although via CIITA-mediated inhibition of class II transcription (48, 61). Interestingly, like the factor responsible for class II downregulation by F. tularensis infection supernatants, IL-1β is >10 kDa and resistant to PK treatment (25). However, caspase-1 knockout macrophages (which are deficient for IL-1β activation and secretion) produce supernatants in response to F. tularensis infection that are fully capable of inducing degradation of MHC class II (not shown), demonstrating that IL-1β is not the factor responsible for class II downregulation in F. tularensis infection supernatants. These results also suggest that approaches other than gene chip analysis of PGE2-treated or F. tularensis-infected macrophages will be required to identify the factor responsible for inducing class II degradation. Furthermore, analysis of PK-treated infection supernatants (which still retain activity) by SDS-PAGE and silver staining revealed little to no detectable protein (Fig. 8), demonstrating that the factor responsible for inducing class II degradation may be active in extremely low concentrations that would be difficult to detect by techniques such as mass spectrometry.
Ubiquitination of the BCR and FcγR plays an important role in trafficking bound antigen to lysosomal compartments for efficient processing and presentation (7, 15). However, pathogens may exploit these cellular processes to evade immune surveillance by targeting critical immune molecules to these same degradative pathways. The gammaherpesvirus KSHV encodes its own ubiquitin ligase (K5) that utilizes host ubiquitin machinery to induce lysosomal degradation of CD86, ICAM-1, MHC class I, and ligands of NKG2D and NKp80 (10, 11, 29, 52). The mammalian MARCH family of ubiquitin ligases was discovered based on homology to K5 and have since been characterized to target many of the same immunologically relevant molecules (4, 23, 28), including MHC class II for ubiquitination and degradation (45). Although macrophages remain understudied, multiple reports have demonstrated the MHC class II regulatory role of MARCH-I and MARCH-VIII (c-MIR) in B cells (40, 45) and MARCH-I in DCs (12). The results presented in Fig. 3 demonstrate that macrophage MHC class II can undergo ubiquitination and subsequent lysosomal degradation. Interestingly, neutralization of macrophage lysosomes was required in order to observe class II ubiquitination (Fig. 3). This lysosomal inactivation step is not required for the detection of ubiquitinated MHC class II in B cells and DCs (15, 49, 54), suggesting that ubiquitination of class II in macrophages may lead to more rapid and efficient degradation compared to the other professional antigen-presenting cells. IFN-γ-activated macrophages have been established to have the greatest proteolytic capacity among antigen presenting cells (13), with IFN-γ-induced enzymes such as cathepsins mediating efficient protein degradation in lysosomal compartments (6). Thus, activated macrophages will likely have a greater capacity to also degrade ubiquitinated class II compared to B lymphocytes and DCs, which are generally thought to be more effective at antigen presentation (2). Treatment of macrophages with ammonium chloride alone increased the pool of both total and ubiquitinated MHC class II beyond that observed in untreated cells (Fig. 3, lane 10), demonstrating a role for acidic compartments in the normal turnover of MHC class II and suggesting that ubiquitination may direct class II for degradation under steady-sate conditions.
Also distinct from DCs, no shift in the molecular weight of the “ubiquitinated” form of class II was observed in macrophages (Fig. 3). This apparent lack of direct class II ubiquitination has also been observed in B cells (J. R. Drake et al., unpublished results) and suggests that MHC class II-associated proteins, rather than the MHC class II α/β-chain, may be the target of ubiquitination. It is currently unclear what MHC class II-associated proteins may be the target of direct ubiquitination. A number of molecules associate with MHC class II, including the invariant chain, DM, and the tetraspanins CD9, CD53, CD63, and CD81 (19, 53). CD9, CD53, and CD81 localize exclusively with MHC class II at the plasma membrane, while CD63 can bind to internalized class II (19). Moreover, CD9 enhances the clustering of MHC molecules (thought to allow for more efficient presentation to CD4+ T cells [53]), which may generate complexes for ubiquitination. A recent report by Lineberry et al. describes the ubiquitination of the tetraspanin CD81 (34), providing evidence that these molecules may be the direct target of ubiquitination in the MHC class II protein complex.
Although it is yet to be determined what ubiquitin ligase(s) are being activated or induced by F. tularensis infection supernatants, MARCH-I or MARCH-VIII are the prime candidates. The observation that significant class II clearance does not occur until after 10 to 12 h of exposure to infection supernatants (Fig. 2A) would be consistent with infection supernatant-induced synthesis of a nascent protein such as MARCH-I/VIII that could lead to class II ubiquitination and rapid degradation within a few hours. A similar rapid clearance of MHC class I occurs following expression of KSHV ubiquitin ligase K5 (32). Since MARCH-I is the predominant DC class II-specific ubiquitin ligase, it seems most likely that this protein may have a similar function in macrophages, especially since both DC and macrophage MHC class II expression is highly dependent on the activation status of the antigen-presenting cells.
This report establishes a previously undescribed mechanism by which F. tularensis (or any bacterial pathogen) can induce the production of one or more PGE2-dependent factor(s) that can downregulate MHC class II in neighboring macrophages via ubiquitin-dependent delivery and degradation in lysosomes. Furthermore, these results suggest that F. tularensis has developed the ability to exploit the cell biology involved in the regulation of macrophage class II expression in order to avoid recognition by CD4+ T cells for enhanced survival. Future studies will focus on the characterization and identification the factor(s) in question, as well as the nature and localization of the ubiquitin ligase(s) involved.
Acknowledgments
This study was supported by NIH grant PO1 AI-056321 to the Center for Immunology and Microbial Disease. J.E.W. was supported by a T32 training grant (AI-49822) to the Center for Immunology and Microbial Disease.
We have no conflicting financial interests.
We thank Lisa Drake and the Albany Medical College Immunology Core Facility and BSL-3 Core Facility for technical assistance, as well as Michelle Lennartz and Jonathan Harton for critical reading of reading of the manuscript and helpful discussions.
Editor: S. R. Blanke
Footnotes
Published ahead of print on 24 August 2009.
REFERENCES
- 1.Babst, M., D. J. Katzmann, W. B. Snyder, B. Wendland, and S. D. Emr. 2002. Endosome-associated complex, ESCRT-II, recruits transport machinery for protein sorting at the multivesicular body. Dev. Cell 3:283-289. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau, J., and R. M. Steinman. 1998. Dendritic cells and the control of immunity. Nature 392:245-252. [DOI] [PubMed] [Google Scholar]
- 3.Barrionuevo, P., J. Cassataro, M. V. Delpino, A. Zwerdling, K. A. Pasquevich, C. Garcia Samartino, J. C. Wallach, C. A. Fossati, and G. H. Giambartolomei. 2008. Brucella abortus inhibits major histocompatibility complex class II expression and antigen processing through interleukin-6 secretion via Toll-like receptor 2. Infect. Immun. 76:250-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartee, E., M. Mansouri, B. T. Hovey-Nerenberg, K. Gouveia, and K. Fruh. 2004. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J. Virol. 78:1109-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartholomeusz, G., M. Talpaz, W. Bornmann, L. Y. Kong, and N. J. Donato. 2007. Degrasyn activates proteasomal-dependent degradation of c-Myc. Cancer Res. 67:3912-3918. [DOI] [PubMed] [Google Scholar]
- 6.Boehm, U., T. Klamp, M. Groot, and J. C. Howard. 1997. Cellular responses to interferon-gamma. Annu. Rev. Immunol. 15:749-795. [DOI] [PubMed] [Google Scholar]
- 7.Booth, J. W., M. K. Kim, A. Jankowski, A. D. Schreiber, and S. Grinstein. 2002. Contrasting requirements for ubiquitylation during Fc receptor-mediated endocytosis and phagocytosis. EMBO J. 21:251-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang, S. T., J. J. Linderman, and D. E. Kirschner. 2005. Multiple mechanisms allow Mycobacterium tuberculosis to continuously inhibit MHC class II-mediated antigen presentation by macrophages. Proc. Natl. Acad. Sci. USA 102:4530-4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clemens, D. L., B. Y. Lee, and M. A. Horwitz. 2004. Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect. Immun. 72:3204-3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coscoy, L., and D. Ganem. 2000. Kaposi's sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. USA 97:8051-8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coscoy, L., and D. Ganem. 2001. A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T-cell costimulation. J. Clin. Investig. 107:1599-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Gassart, A., V. Camosseto, J. Thibodeau, M. Ceppi, N. Catalan, P. Pierre, and E. Gatti. 2008. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc. Natl. Acad. Sci. USA 105:3491-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delamarre, L., M. Pack, H. Chang, I. Mellman, and E. S. Trombetta. 2005. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science 307:1630-1634. [DOI] [PubMed] [Google Scholar]
- 14.Dennis, D. T., T. V. Inglesby, D. A. Henderson, J. G. Bartlett, M. S. Ascher, E. Eitzen, A. D. Fine, A. M. Friedlander, J. Hauer, M. Layton, S. R. Lillibridge, J. E. McDade, M. T. Osterholm, T. O'Toole, G. Parker, T. M. Perl, P. K. Russell, and K. Tonat. 2001. Tularemia as a biological weapon: medical and public health management. JAMA 285:2763-2773. [DOI] [PubMed] [Google Scholar]
- 15.Drake, L., E. M. McGovern-Brindisi, and J. R. Drake. 2006. BCR ubiquitination controls BCR-mediated antigen processing and presentation. Blood 108:4086-4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elkins, K. L., S. C. Cowley, and C. M. Bosio. 2003. Innate and adaptive immune responses to an intracellular bacterium, Francisella tularensis live vaccine strain. Microbes Infect. 5:135-142. [DOI] [PubMed] [Google Scholar]
- 17.Elkins, K. L., T. R. Rhinehart-Jones, S. J. Culkin, D. Yee, and R. K. Winegar. 1996. Minimal requirements for murine resistance to infection with Francisella tularensis LVS. Infect. Immun. 64:3288-3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellis, J., P. C. Oyston, M. Green, and R. W. Titball. 2002. Tularemia. Clin. Microbiol. Rev. 15:631-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engering, A., and J. Pieters. 2001. Association of distinct tetraspanins with MHC class II molecules at different subcellular locations in human immature dendritic cells. Int. Immunol. 13:127-134. [DOI] [PubMed] [Google Scholar]
- 20.Fortier, A. H., T. Polsinelli, S. J. Green, and C. A. Nacy. 1992. Activation of macrophages for destruction of Francisella tularensis: identification of cytokines, effector cells, and effector molecules. Infect. Immun. 60:817-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fortier, A. H., M. V. Slayter, R. Ziemba, M. S. Meltzer, and C. A. Nacy. 1991. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect. Immun. 59:2922-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuertes, G., J. J. Martin De Llano, A. Villarroya, A. J. Rivett, and E. Knecht. 2003. Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem. J. 375:75-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goto, E., S. Ishido, Y. Sato, S. Ohgimoto, K. Ohgimoto, M. Nagano-Fujii, and H. Hotta. 2003. c-MIR, a human E3 ubiquitin ligase, is a functional homolog of herpesvirus proteins MIR1 and MIR2 and has similar activity. J. Biol. Chem. 278:14657-14668. [DOI] [PubMed] [Google Scholar]
- 24.Harris, S. G., J. Padilla, L. Koumas, D. Ray, and R. P. Phipps. 2002. Prostaglandins as modulators of immunity. Trends Immunol. 23:144-150. [DOI] [PubMed] [Google Scholar]
- 25.Hazuda, D. J., J. Strickler, P. Simon, and P. R. Young. 1991. Structure-function mapping of interleukin 1 precursors: cleavage leads to a conformational change in the mature protein. J. Biol. Chem. 266:7081-7086. [PubMed] [Google Scholar]
- 26.Hicke, L., and R. Dunn. 2003. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu. Rev. Cell Dev. Biol. 19:141-172. [DOI] [PubMed] [Google Scholar]
- 27.Hinson, R. M., J. A. Williams, and E. Shacter. 1996. Elevated interleukin 6 is induced by prostaglandin E2 in a murine model of inflammation: possible role of cyclooxygenase-2. Proc. Natl. Acad. Sci. USA 93:4885-4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoer, S., L. Smith, and P. J. Lehner. 2007. MARCH-IX mediates ubiquitination and downregulation of ICAM-1. FEBS Lett. 581:45-51. [DOI] [PubMed] [Google Scholar]
- 29.Ishido, S., C. Wang, B. S. Lee, G. B. Cohen, and J. U. Jung. 2000. Downregulation of major histocompatibility complex class I molecules by Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 74:5300-5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katzmann, D. J., G. Odorizzi, and S. D. Emr. 2002. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell. Biol. 3:893-905. [DOI] [PubMed] [Google Scholar]
- 31.Kirimanjeswara, G. S., J. M. Golden, C. S. Bakshi, and D. W. Metzger. 2007. Prophylactic and therapeutic use of antibodies for protection against respiratory infection with Francisella tularensis. J. Immunol. 179:532-539. [DOI] [PubMed] [Google Scholar]
- 32.Lehner, P. J., S. Hoer, R. Dodd, and L. M. Duncan. 2005. Downregulation of cell surface receptors by the K3 family of viral and cellular ubiquitin E3 ligases. Immunol. Rev. 207:112-125. [DOI] [PubMed] [Google Scholar]
- 33.Li, G., J. A. Harton, X. Zhu, and J. P. Ting. 2001. Downregulation of CIITA function by protein kinase A (PKA)-mediated phosphorylation: mechanism of prostaglandin E, cyclic AMP, and PKA inhibition of class II major histocompatibility complex expression in monocytic lines. Mol. Cell. Biol. 21:4626-4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lineberry, N., L. Su, L. Soares, and C. G. Fathman. 2008. The single subunit transmembrane E3 ligase gene related to anergy in lymphocytes (GRAIL) captures and then ubiquitinates transmembrane proteins across the cell membrane. J. Biol. Chem. 283:28497-28505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Longva, K. E., F. D. Blystad, E. Stang, A. M. Larsen, L. E. Johannessen, and I. H. Madshus. 2002. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J. Cell Biol. 156:843-854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lopez, M. C., N. S. Duckett, S. D. Baron, and D. W. Metzger. 2004. Early activation of NK cells after lung infection with the intracellular bacterium, Francisella tularensis LVS. Cell. Immunol. 232:75-85. [DOI] [PubMed] [Google Scholar]
- 37.Malik, M., C. S. Bakshi, K. McCabe, S. V. Catlett, A. Shah, R. Singh, P. L. Jackson, A. Gaggar, D. W. Metzger, J. A. Melendez, J. E. Blalock, and T. J. Sellati. 2007. Matrix metalloproteinase 9 activity enhances host susceptibility to pulmonary infection with type A and B strains of Francisella tularensis. J. Immunol. 178:1013-1020. [DOI] [PubMed] [Google Scholar]
- 38.Malik, M., C. S. Bakshi, B. Sahay, A. Shah, S. A. Lotz, and T. J. Sellati. 2006. Toll-like receptor 2 is required for control of pulmonary infection with Francisella tularensis. Infect. Immun. 74:3657-3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marmor, M. D., and Y. Yarden. 2004. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene 23:2057-2070. [DOI] [PubMed] [Google Scholar]
- 40.Matsuki, Y., M. Ohmura-Hoshino, E. Goto, M. Aoki, M. Mito-Yoshida, M. Uematsu, T. Hasegawa, H. Koseki, O. Ohara, M. Nakayama, K. Toyooka, K. Matsuoka, H. Hotta, A. Yamamoto, and S. Ishido. 2007. Novel regulation of MHC class II function in B cells. EMBO J. 26:846-854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCaffrey, R. L., and L. A. Allen. 2006. Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J. Leukoc. Biol. 80:1224-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGovern, E. M., A. E. Moquin, A. Caballero, and J. R. Drake. 2004. The effect of B-cell receptor signaling on antigen endocytosis and processing. Immunol. Investig. 33:143-156. [DOI] [PubMed] [Google Scholar]
- 43.Melikova, M. S., K. A. Kondratov, and E. S. Kornilova. 2006. Two different stages of epidermal growth factor (EGF) receptor endocytosis are sensitive to free ubiquitin depletion produced by proteasome inhibitor MG132. Cell Biol. Int. 30:31-43. [DOI] [PubMed] [Google Scholar]
- 44.Nagabhushanam, V., A. Solache, L. M. Ting, C. J. Escaron, J. Y. Zhang, and J. D. Ernst. 2003. Innate inhibition of adaptive immunity: Mycobacterium tuberculosis-induced IL-6 inhibits macrophage responses to IFN-γ. J. Immunol. 171:4750-4757. [DOI] [PubMed] [Google Scholar]
- 45.Ohmura-Hoshino, M., Y. Matsuki, M. Aoki, E. Goto, M. Mito, M. Uematsu, T. Kakiuchi, H. Hotta, and S. Ishido. 2006. Inhibition of MHC class II expression and immune responses by c-MIR. J. Immunol. 177:341-354. [DOI] [PubMed] [Google Scholar]
- 46.Raiborg, C., T. E. Rusten, and H. Stenmark. 2003. Protein sorting into multivesicular endosomes. Curr. Opin. Cell Biol. 15:446-455. [DOI] [PubMed] [Google Scholar]
- 47.Rohde, J. R., A. Breitkreutz, A. Chenal, P. J. Sansonetti, and C. Parsot. 2007. Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe 1:77-83. [DOI] [PubMed] [Google Scholar]
- 48.Rohn, W., L. P. Tang, Y. Dong, and E. N. Benveniste. 1999. IL-1 beta inhibits IFN-gamma-induced class II MHC expression by suppressing transcription of the class II transactivator gene. J. Immunol. 162:886-896. [PubMed] [Google Scholar]
- 49.Shin, J. S., M. Ebersold, M. Pypaert, L. Delamarre, A. Hartley, and I. Mellman. 2006. Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature 444:115-118. [DOI] [PubMed] [Google Scholar]
- 50.Telepnev, M., I. Golovliov, and A. Sjostedt. 2005. Francisella tularensis LVS initially activates but subsequently down-regulates intracellular signaling and cytokine secretion in mouse monocytic and human peripheral blood mononuclear cells. Microb. Pathog. 38:239-247. [DOI] [PubMed] [Google Scholar]
- 51.Thibodeau, J., M. C. Bourgeois-Daigneault, G. Huppe, J. Tremblay, A. Aumont, M. Houde, E. Bartee, A. Brunet, M. E. Gauvreau, A. de Gassart, E. Gatti, M. Baril, M. Cloutier, S. Bontron, K. Fruh, D. Lamarre, and V. Steimle. 2008. Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. Eur. J. Immunol. 38:1225-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomas, M., J. M. Boname, S. Field, S. Nejentsev, M. Salio, V. Cerundolo, M. Wills, and P. J. Lehner. 2008. Down-regulation of NKG2D and NKp80 ligands by Kaposi's sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 105:1656-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trombetta, E. S., and I. Mellman. 2005. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 23:975-1028. [DOI] [PubMed] [Google Scholar]
- 54.van Niel, G., R. Wubbolts, T. Ten Broeke, S. I. Buschow, F. A. Ossendorp, C. J. Melief, G. Raposo, B. W. van Balkom, and W. Stoorvogel. 2006. Dendritic cells regulate exposure of MHC class II at their plasma membrane by oligoubiquitination. Immunity 25:885-894. [DOI] [PubMed] [Google Scholar]
- 55.Woolard, M. D., L. L. Hensley, T. H. Kawula, and J. A. Frelinger. 2008. Respiratory Francisella tularensis live vaccine strain infection induces Th17 cells and prostaglandin E2, which inhibits generation of gamma interferon-positive T cells. Infect. Immun. 76:2651-2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Woolard, M. D., J. E. Wilson, L. L. Hensley, L. A. Jania, T. H. Kawula, J. R. Drake, and J. A. Frelinger. 2007. Francisella tularensis-infected macrophages release prostaglandin E2 that blocks T-cell proliferation and promotes a Th2-like response. J. Immunol. 178:2065-2074. [DOI] [PubMed] [Google Scholar]
- 57.Wu, T. H., J. A. Hutt, K. A. Garrison, L. S. Berliba, Y. Zhou, and C. R. Lyons. 2005. Intranasal vaccination induces protective immunity against intranasal infection with virulent Francisella tularensis biovar A. Infect. Immun. 73:2644-2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xaus, J., M. Comalada, M. Barrachina, C. Herrero, E. Gonalons, C. Soler, J. Lloberas, and A. Celada. 2000. The expression of MHC class II genes in macrophages is cell cycle dependent. J. Immunol. 165:6364-6371. [DOI] [PubMed] [Google Scholar]
- 59.Yee, D., T. R. Rhinehart-Jones, and K. L. Elkins. 1996. Loss of either CD4+ or CD8+ T cells does not affect the magnitude of protective immunity to an intracellular pathogen, Francisella tularensis strain LVS. J. Immunol. 157:5042-5048. [PubMed] [Google Scholar]
- 60.Yen, J. H., T. Khayrullina, and D. Ganea. 2008. PGE2-induced metalloproteinase-9 is essential for dendritic cell migration. Blood 111:260-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu, X., M. S. Chang, R. C. Hsueh, R. Taussig, K. D. Smith, M. I. Simon, and S. Choi. 2006. Dual ligand stimulation of RAW 264.7 cells uncovers feedback mechanisms that regulate TLR-mediated gene expression. J. Immunol. 177:4299-4310. [DOI] [PubMed] [Google Scholar]