

Abstract
Vascular fibrosis is a major complication of hypertension and atherosclerosis, yet it is largely untreatable. Natriuretic peptides (NPs) repress fibrogenic activation of vascular smooth muscle cells (VSMCs), but the intracellular mechanism mediating this effect remains undetermined. Here we show that inhibition of RhoA through phosphorylation at Ser188, the site targeted by the NP effector cyclic GMP (cGMP)-dependent protein kinase I (cGK I), is critical to fully exert antifibrotic potential. cGK I+/− mouse blood vessels exhibited an attenuated P-RhoA level and concurrently increased RhoA/ROCK signaling. Importantly, cGK I insufficiency caused dynamic recruitment of ROCK into the fibrogenic programs, thereby eliciting exaggerated vascular hypertrophy and fibrosis. Transgenic expression of cGK I-unphosphorylatable RhoAA188 in VSMCs augmented ROCK activity, vascular hypertrophy, and fibrosis more prominently than did that of wild-type RhoA, consistent with the notion that RhoAA188 escapes the intrinsic inhibition by cGK I. Additionally, VSMCs expressing RhoAA188 became refractory to the antifibrotic effects of NPs. Our results identify cGK I-mediated Ser188 phosphorylation of RhoA as a converging node for pro- and antifibrotic signals and may explain how diminished cGMP signaling, commonly associated with vascular malfunction, predisposes individuals to vascular fibrosis.
The synthesis and deposition of extracellular matrix (ECM) are fundamental processes in tissue repair, but when excessive, they can cause the development of a fibrosis that progressively perturbs normal tissue architecture and function (65). In cardiovascular diseases such as hypertension, fibrosis occurs when large and small arteries undergo adaptive structural remodeling in response to hemodynamic stress, such as increased arterial pressure. Vascular remodeling is characterized by increased wall thickness and stiffness, mediated by hypertrophic growth of vascular smooth muscle cells (VSMCs) and overproduction of ECM. The fibrosis associated with vascular remodeling leads to elevated peripheral resistance, to impaired tissue perfusion, and, consequently, to the sequelae of hypertension, such as myocardial infarction and stroke (2, 13).
Vascular fibrosis is regulated by a plethora of profibrotic stimuli. These include mechanical stretch, vasoactive substances like angiotensin II (Ang II), and cytokines such as transforming growth factor beta (TGF-β) and connective tissue growth factor (CTGF) (45). Although these represent potential targets for antifibrotic therapy, their potential success for translation in humans has been called into question (54). Pharmacological blockade of Ang II showed some benefit in clinics, with the magnitude of this effect likely being small. Additionally, inhibition of TGF-β1 reduced fibrosis in animals but revealed a risk of T-cell-dependent hyperinflammation, as presaged from a mouse genetic model. These problems are attributed to the intimate cross talk between profibrogenic cues and their pleiotropic effects on various cell types, thus providing a rationale to investigate the intracellular pathways exploitable for specific and efficacious antifibrotic therapy.
Growing biochemical and developmental evidence has identified a repertoire of pathways that cross talk with each other and participate in profibrogenic programs. These comprise Smads, extracellular signal-regulated kinases, p38 mitogen-activated protein kinase, the small GTP-binding protein RhoA, and its effectors, Rho-associated protein kinases (ROCK) (45). Notably, RhoA and ROCK have emerged as critical regulators of multiple facets of cardiovascular cell functions (30), including VSMC contraction, migration, proliferation, hypertrophy, gene expression, and fibrosis (18, 43, 48, 53). Importantly, recent evidence presented the therapeutically relevant notions that the RhoA/ROCK pathway mediates TGF-β signaling with variable cooperativity with other pathways, such as Smads (4, 6, 24), and that the context-specific contribution of the RhoA/ROCK pathway underlies the magnitude of the fibrotic response (17, 68). However, the molecular mechanism that enables or disables RhoA/ROCK commitment to the fibrogenic machinery remains incompletely understood.
Considerably less is known about the intrinsic mechanisms in tissues that slow or halt the fibrogenic response. There is ample evidence that natriuretic peptides (NPs) and endothelium-derived nitric oxide (NO) play key roles in vascular homeostasis, through vasorelaxation and inhibition of vascular remodeling (20, 42). In animal models, NPs and NO also showed a defined antifibrotic potential. Indeed, pharmacological and genetic blockade of NO synthase, brain NP (BNP), or atrial NP culminated in an increased propensity for ECM deposition in the perivascular or cardiac interstitial area (26, 29, 56). Additionally, transcriptional profiling of cardiac fibroblasts revealed a prominent potential for BNP to oppose TGF-β-induced gene expression (25). Many effects of NO/NP signaling are mediated via stimulation of guanylyl cyclases and intracellular production of the second messenger cyclic GMP (cGMP), although the signaling cascades downstream of cGMP remain to be established (21). Recent evidence obtained with mutant mouse models identified cGMP-dependent protein kinase type I (cGK I), a serine-threonine kinase, as a principal effector of the NP/NO/cGMP pathway for vasorelaxation (41). Numerous cGK I substrates have been identified to date, encompassing regulators of calcium mobilization (inositol 1,4,5-trisphosphate receptor-associated cGK I substrate [IRAG] and regulator of G-protein signaling 2 [RGS2]) (51, 57) and the acto-myosin cytoskeleton (myosin phosphatase targeting subunit [MYPT1], vasodilator-stimulated phosphoprotein [VASP], and RhoA) (3, 47, 49, 64). However, the role of cGK I and its targets in antifibrotic signaling has not been determined in vivo.
We and others previously demonstrated that cGK I phosphorylates RhoA at Ser188 in vitro and in cultured cells (9, 47, 49). The classic model of Rho protein regulation involves a cycle between active GTP-bound and inactive GDP-bound conformations (11). Various extracellular signals, such as G-protein-coupled receptor agonists, activate guanine nucleotide exchange factors and increase the level of GTP-RhoA. Activated GTP-RhoA is readily prenylated at its carboxy terminus and then translocated to the cell membrane, where it interacts with effector proteins like ROCK to generate downstream signaling. Ser188 phosphorylation of RhoA is thought to uncouple this cycle and to terminate RhoA signaling by allowing the guanine dissociation inhibitors to bind and sequester GTP-RhoA in the cytosol (27, 44). The in vivo occurrence of this RhoA phosphorylation, however, has not been demonstrated directly in tissues, nor has its definitive role in vascular pathophysiology been explored conclusively.
An emerging theme on the pathogenesis of fibrosis is the interplay between pro- and antifibrotic signals. Although it is reasonable to assume that this counterregulation may occur intracellularly, such possibilities and underlying mechanisms have not been explored fully in vivo. In this study, we developed a mouse model of cGK I haploinsufficiency and mice with VSMC-specific expression of cGK I-unphosphorylatable RhoA. These distinct mouse models allowed us to address our hypothesis that cGK I phosphorylation of RhoA mediates inhibitory inputs from the NP/cGMP cascade into the RhoA/ROCK pathway in tissues and thereby serves as a critical determinant of vascular remodeling and fibrosis.
MATERIALS AND METHODS
Development of phospho-specific antibody that recognizes RhoA phosphorylated at Ser188.
Phospho-specific antibodies against RhoA phosphorylated at Ser188 (P-RhoA) were raised by immunizing rabbits with a synthetic peptide containing phosphoserine [CRRGKKKS(PO3H2)G-NH2; the underlined sequence corresponds to amino acids 182 to 189 of human RhoA] emulsified in complete Freund's adjuvant. The antibodies were purified by positive-affinity chromatography using the phosphoserine peptide, followed by negative-affinity chromatography with a nonphosphorylated peptide (RRGKKKSG). To verify the specificity of the antibody, 1 μg of human recombinant His6-tagged RhoA (Cytoskeleton, Denver, CO) was phosphorylated by 36.5 units of bovine cGMP-dependent protein kinase alpha isozyme (Promega, Madison, WI) in 75 μl of reaction buffer (40 mM Tris [pH 7.4], 20 mM magnesium acetate, 0.2 mM ATP) in the presence or absence of 13.3 μM cGMP. One-tenth of the reaction mix was subjected to immunoblotting with anti-RhoA antibody 119 (Santa Cruz Biotechnology, Santa Cruz, CA) or P-RhoA antibody.
SMC culture.
Human aortic SMCs (Biowhittaker, Walkersville, MD) were cultured in smooth muscle basal medium supplemented with 5% fetal calf serum and used at passage 5 or 6. Cells were preincubated for 30 min with vehicle, 0.5 mM 3-isobutyl-1-methylxanthine, or 2 μM RQIKIWFQNRRMKWKK-LRK5H-amide (DT-3), a peptide-based inhibitor of cGK Iα (Biolog Life Science Institute, Bremen, Germany). Subsequently, cells were treated with vehicle, 0.1 mM 8-(4-chlorophenylthio)-cGMP (8-pCPT cGMP), or 0.5 mM 8-bromo-cGMP (8-Br cGMP) for 2 h and then harvested for analysis. Mouse aortic SMCs (MASMCs) were prepared as described previously (46). Briefly, thoracic aortas were pooled from four to eight mice and digested with 1 mg/ml collagenase type II (Worthington Biochemical Corp., Lakewood, NJ) for 15 min. After removal of adventitia and endothelia, the aortas were minced and digested with 1 mg/ml collagenase type I (Worthington) and 0.125 mg/ml elastase type III for 1 h. The cells were mechanically dispersed, plated, cultured in Dulbecco's modified Eagle's medium containing 20% fetal calf serum, and used for experiments at passages 4 to 6.
cGK I null mice and BNP-Tg mice.
Mice were used for experiments at 2 to 5 months of age. All animal experiments were conducted in accordance with the institutional guidelines of the Kyoto University Graduate School of Medicine. cGK I null mice (129sv background) were generated as described previously (62). The null mutation of the cGK I gene caused deletion of both the Iα and Iβ isoforms. Because ∼80% of cGK I−/− mice were hypomorphic and died by 2 months of age, cGK I+/− and wild-type (WT) littermates were used for the study. We generated BNP transgenic (BNP-Tg) mice as described previously, with the use of the human serum amyloid P component promoter, whose activity is postnatally limited to the liver (38). We used line 55, with 20 copies of the transgene per genome, as these transgenic mice exhibit a plasma BNP concentration at least 2 orders of magnitude higher than that of nontransgenic (NTg) littermates.
Generation of transgenic mice that express either WT or phosphorylation-resistant RhoA in smooth muscle.
WT and A188 human RhoA cDNA constructs with a 5′ Kozak motif and a myc epitope tag were constructed as described previously (49). Briefly, a point mutation was introduced at the Ser188 position of WT RhoA cDNA (TCT→GCT), to create Ala188 RhoA cDNA. The XbaI-NotI fragment containing RhoA cDNA was subcloned into the XhoI-EcoRV site of pAd SM22α-loxP (a generous gift from L. M. Akyurek and E. G. Nabel) (1). pAd SM22α-loxP contains 441 bp of the murine SM22α promoter, the 231-bp bovine growth hormone (bGH) polyadenylation signal, and the 471-bp human 4F2 enhancer. A BglII site was created at the 5′ end of the SM22α promoter, using PCR-based site-directed mutagenesis, to allow for the release of the SM22α-RhoA-bGH polyA-4F2 enhancer transgenic cassette from the vector as a BglII-BglII fragment. Transgenic constructs were injected into the pronuclei of fertilized oocytes derived from C57BL/6NCrj mice. Transgenic founders were identified by Southern blot analysis of SpeI-digested genomic DNA, using an XbaI-HindIII probe comprising the SM22α promoter region. PCR-based genotyping was performed using the following forward and reverse primers, corresponding to the myc epitope tag and the 3′ end of the open reading frame of RhoA cDNA, respectively (the underlined nucleotide denotes the mutated nucleotide at Ser/Ala188): WT RhoA-F, 5′-GCAGAAGCTGATCTCCGAGGA; WT RhoA-R, 5′-CCGCTCACAAGACAAGGCAACCAGA; A188-F, 5′-GCAGAAGCTGATCTCCGAGGA; and A188-R, 5′-TCACAAGACAAGGCAACCAGC.
Assessment of transgene expression.
Transgene mRNA expression in the thoracic aortas of SMC-specific RhoA-Tg mice was assessed by semiquantitative reverse transcription-PCR (RT-PCR). The total RNA was isolated using an RNeasy fibrous tissue kit (Qiagen, Valencia, CA). Aortic RNA (0.5 μg) was reverse transcribed with SuperScript III and an oligo(dT)23 primer (Invitrogen, Carlsbad, CA), and 1/20 of the RT product was subjected to PCR with AccuPrime Taq polymerase (Invitrogen) and the same primer sets used for genotyping. The cycle conditions were optimized to detect amplification in the logarithmic phase. Transgene protein expression was determined by immunoblotting analysis of aortic extracts with anti-RhoA antibody 119 (Santa Cruz) and anti-myc-tag antibody clone 9B11 (Cell Signaling Technology, Danvers, MA). Transgene expression was further confirmed by immunohistochemistry. Aorta and heart cryosections (8 μm) were fixed with 4% paraformaldehyde for 20 min at 4°C. Anti-myc-tag antibody clone 4A6 (Upstate Biotechnology, Lake Placid, NY) was incubated with Envision+ anti-mouse immunoglobulin G-horseradish peroxidase conjugate (IgG-HRP) (Dako, Kyoto, Japan) to allow for polymer immune complex formation and subsequently absorbed with normal mouse serum to remove free anti-mouse IgG-HRP. The myc tag expression was detected by incubation of the cryosections with the immune complex followed by visualization with 3-amino-9-ethylcarbazol. Counterstaining was conducted with hematoxylin.
Morphometric analysis of blood vessels.
Mice were euthanized with an excess dose of pentobarbital and perfused with phosphate-buffered saline. The heart was dissected and cut horizontally into three parts, for histological analysis (upper third) and extraction of protein (middle third) and RNA (lower third). The thoracic and abdominal sections of the aorta were dissected from the aortic arch to the bifurcation of the iliac arteries. Segments from the descending thoracic aorta were used for histological analysis, and the remaining parts of the aorta were used for protein and RNA extraction. For morphometric assessment, the tissues were fixed in 10% neutral buffered formaldehyde, embedded in paraffin, and cut at a thickness of 8 μm. Sections were stained with Sirius red to visualize collagen fibers. For each heart, 20 to 40 coronary arteries from two separate sections were analyzed using a KS400 imaging system (Carl Zeiss, Oberkochen, Germany). Sirius red staining reproducibly visualized the three vascular components, i.e., the perivascular fibrotic area (dark red), the media (pink to pale red), and the lumen (no staining), owing to their noncontiguous staining patterns. In preliminary experiments, the identity and integrity of these structures determined by Sirius red staining were validated in serial sections stained with hematoxylin and eosin and by the elastica Van Gieson method. While the perivascular fibrotic area, with discrete and intense stainability, was recognized digitally by the KS400 software, the media and lumen were manually demarcated. The morphometric parameters used in this study were calculated according to the following formulas: (i) medial thickness (MT) = medial area/luminal area and (ii) perivascular fibrosis (PVF) = perivascular fibrotic area/(luminal area + medial area + perivascular fibrotic area). In addition, the interstitial fibrosis was assessed using KS400 software on six fields per heart section stained with Sirius red and is represented as the percentage of the total area.
Measurement of BP.
Blood pressure (BP) and heart rate were measured in conscious mice by use of a computerized tail-cuff method (Softron Co. Ltd., Tokyo, Japan) as described elsewhere (50).
Administration of Ang II and Y-27632.
Systemic administration of Ang II (400 to 1,500 ng/kg of body weight/min) and Y-27632 (50 mg/kg/day) was conducted by continuous infusion, using Alzet osmotic minipumps (model 1002; Durect Corporation, Cupertino, CA). Mice were anesthetized by intraperitoneal injection of pentobarbital (50 mg/kg). Osmotic pumps were implanted subcutaneously through an interscapular incision. The BP and heart rate were measured at days 7 to 10. The mice were euthanized at day 18 for organ harvest. Some mice were sacrificed at day 14, and the plasma and organ homogenates were used to determine the concentration of Y-27632 by high-performance liquid chromatography.
Immunoblotting, immunoprecipitation, and subcellular fractionation.
For protein extraction, cultured cells and mouse organs were homogenized in lysis buffer (Tris 20 mM [pH 7.5], 150 mM NaCl, 1 mM EGTA, 10 mM MgCl2, 25 mM NaF, 1 mM 4-amidinophenylmethanesulfonyl fluoride, 1% Triton X-100) containing protease and phosphatase inhibitor cocktails (Calbiochem, San Diego, CA). After centrifugation at 10,000 × g for 20 min, lysates containing 40 to 80 μg protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Immunoblotting was performed with the following primary antibodies: P-RhoA, RhoA 119, phospho-VASP (Ser239) clone 16C2 (Upstate Biotechnology), VASP (Biolegend, San Diego, CA), protein kinase G C terminus (amino acids 657 to 671) (Calbiochem), β-actin clone AC-15 (Sigma, St. Louis, MO), phospho-moesin (Thr558) (Santa Cruz Biotechnology), moesin clone 38 (Transduction Laboratories, Lexington, KY), and ROCK (a generous gift from S. Narumiya) (22). The immunoblot was incubated with HRP-conjugated secondary antibody, and the chemiluminescent signals were developed by ECL-Plus (Amersham, Buckinghamshire, United Kingdom). The signals were quantified with an LAS-1000 system (Fujifilm, Tokyo, Japan) or by scanning densitometry. For assessment of the phosphorylation level of RhoA at Ser188, total lysate containing 350 to 500 μg protein was used for immunoprecipitation with 1 μg of anti-RhoA antibody clone 26C4 (Santa Cruz) followed by 20 μl protein G-Sepharose. The immune complex was eluted by boiling in Laemmli buffer with 50 mM dithiothreitol and used for immunoblotting with RhoA 119 or P-RhoA antibody. To assess the membrane translocation of RhoA, subcellular fractionation was performed as described previously (49). Mouse aortas were homogenized in lysis buffer without Triton X-100 and then subjected to ultracentrifugation at 100,000 × g for 60 min. The supernatant was saved as the cytosolic fraction. The pellet was sonicated in lysis buffer with 1% Triton X-100 and centrifuged at 10,000 × g for 30 min. The supernatant was saved as the membrane fraction. Cytosolic and membrane fractions were subjected to immunoblotting analysis with RhoA 119 antibody.
Quantitative RT-PCR.
Total RNAs were isolated from the heart and aorta by use of an RNeasy fibrous tissue kit. The mRNA levels of TGF-β1, CTGF, fibronectin, and collagen type I and type III were quantified by real-time RT-PCR, using TaqMan EZ RT-PCR core reagents and an ABI Prism 7700 sequence detector (ABI Prism, Applied Biosystems, Foster City, CA). The specific primer pairs and fluorescence resonance energy transfer probes used for detection of each gene are listed in Table S1 in the supplemental material. The glyceraldehyde-3-phosphate dehydrogenase mRNA level was quantified with a specific TaqMan primer-probe set from ABI Prism. Either glyceraldehyde-3-phosphate dehydrogenase mRNA or 18S rRNA was used for normalization.
Immunofluorescence.
MASMCs were cultured on Lab-Tek II chamber slides (Nunc, Roskilde, Denmark) to 50 to 80% confluence. To assess the integrity of the MASMC preparation, cells cultured under standard conditions were fixed in 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. The cells were subsequently stained with anti-α smooth muscle actin antibody clone 1A4 (Sigma) or anti-myc-tag antibody clone 4A6, treated with Hoechst 33258 for nuclear staining, and probed with Alexa546-conjugated anti-mouse IgG (Molecular Probe). To study the effect of RhoA transgene expression on actin polymerization, cells were serum starved overnight, left untreated or treated with 0.5 mM 8-Br cGMP for 2 h, stimulated with 10 μM lysophosphatidic acid (LPA) for 15 min, and fixed. F-actin was detected by Alexa488-conjugated phalloidin. The images were captured with a fluorescence microscope.
Assessment of protein and DNA synthesis.
MASMCs were plated in 24-well plates at 20,000 cells/well, serum starved overnight, and treated with vehicle, 8-Br cGMP (0.5 mM), or Y-27632 (10 μM) for 2 h. Subsequently, the cells were stimulated with Ang II (100 nM) for 16 h and pulsed for 8 h with [3H]leucine (1 μCi) to assess protein synthesis. Alternatively, the cells were stimulated with 10% serum for 16 h and pulsed for 8 h with [3H]thymidine (1 μCi) to assess DNA synthesis. The cells were serially treated with 10% trichloroacetic acid and ethanol. The incorporated radioactivity was extracted with 0.25 N NaOH and measured by a liquid scintillation counter.
Luciferase assay.
MASMCs were plated in 12-well plates at 60,000 cells/well and transfected with 2 μg of pSRE-Luc or pAP-1-Luc cis-reporter plasmid (Stratagene) and 4 ng of internal control plasmid (pCMV-SPL), using Lipofectamine 2000 (Invitrogen). The enhancer element sequences for the reported plasmids are as follows: for pAP-1-Luc, (TGACTAA)7; and for pSRE-Luc, (AGGATGTCCATATTAGGACATCT)5. After 24 h, the cells were serum starved for 20 h, treated for 2 h with vehicle, 8-Br cGMP (0.5 mM), or Y-27632 (10 μM), stimulated for 8 h with TGF-β (10 ng/ml), and harvested to assess the luciferase activity by use of a dual-luciferase reporter assay system (Promega).
Statistics.
All data are expressed as means ± standard errors of the means (SEM). Analysis of variance followed by post hoc Fisher's test was used for comparison between groups. Values of P of <0.05 were considered significant. For the correlation analysis between P-RhoA/RhoA and P-moesin/moesin ratios, Spearman's rank correlation test was used.
RESULTS
Serine 188 phosphorylation of RhoA is inversely correlated with ROCK activity in mouse blood vessels.
We developed an antiserum that specifically recognizes RhoA phosphorylated at serine 188 (P-RhoA) (Fig. 1A). Immunoblotting with P-RhoA antibody revealed the occurrence of RhoA Ser188 phosphorylation in cultured human aortic SMCs (Fig. 1B). Membrane-permeating cGMP analogs increased the P-RhoA level, and this increase was prevented by the specific cGK I inhibitor DT-3 (8). The level of P-RhoA paralleled that of VASP phosphorylated at Ser237, the site selectively phosphorylated by cGK I (37). These results indicate that cGK I phosphorylates RhoA at Ser188 in VSMCs.
FIG. 1.

Inverse correlation between RhoA serine 188 phosphorylation and ROCK activity in mouse blood vessels. (A) Specificity of P-RhoA antibody (Ser188). Recombinant RhoA protein was phosphorylated or left unphosphorylated by cGK Iα and immunoblotted with P-RhoA antibody. (B) Immunoblot showing concurrent phosphorylation of RhoA Ser188 and VASP Ser237 in VSMCs treated with cGMP analogs. cGK I inh, a membrane-permeating cGK I inhibitory peptide (DT-3). (C to F) Immunoblotting analysis of cGK I+/− and WT mouse aortas. (C) (Left) cGK I protein level. (Right) Levels of P-RhoA and RhoA in immunoprecipitates with RhoA antibody. (D) Level of P-moesin Thr558, without or with Ang II administration for 2 weeks. (E) ROCK protein level. (F) Level of membrane-associated and cytosolic RhoA separated by cellular fractionation. (G) Spearman's rank correlation test between the P-RhoA/RhoA and P-moesin/moesin ratios in cGK I+/− and WT mouse aortas. (H and I) Immunoblotting analysis of aortas of BNP-Tg mice and NTg littermates. (H) (Top and middle) Levels of P-RhoA and RhoA in immunoprecipitates with RhoA antibody. IgGL, IgG light chain. (Bottom) cGK I protein level. (I) Level of P-moesin Thr558. Values are means ± SEM for three or four replicates. *, P < 0.05; **, P < 0.01.
To address the physiological relevance of RhoA phosphorylation, we used mouse models for loss of function (cGK I hemizygous null mice [cGK I+/−]) and gain of function (BNP-Tg mice) of the cGMP/cGK I pathway (38, 62). The cGK I protein level in cGK I+/− mouse aortas was reduced to 40% of that in WT mice (Fig. 1C). Concomitantly, the phosphorylated fraction of RhoA in cGK I+/− aortas was 50% less than that in WT aortas. This is consistent with the idea that cGK I is the major kinase in mouse blood vessels that phosphorylates RhoA at Ser188. We then examined RhoA signaling through assessment of the immediate downstream effector ROCK as an indicator (22). ROCK activity was determined by the phosphorylation status of moesin Thr558, which is the ROCK-specific site (23, 39). The ROCK activity in cGK I+/− aortas was 1.7-fold higher than that in WT aortas (Fig. 1D). This increase in ROCK activity became significant following 18 days of infusion of Ang II (2.4-fold). Despite the increased ROCK activity in cGK I+/− aortas, ROCK protein expression was not altered between the genotypes (Fig. 1E). A cell fractionation assay demonstrated augmented accumulation of RhoA in the cGK I+/− versus WT aortic cell membrane (Fig. 1F), consistent with the enhanced ROCK signaling in cGK I+/− vessels. Spearman's rank correlation test revealed a statistically significant reciprocal correlation between P-RhoA level and ROCK activity (Fig. 1G).
The BNP-Tg mice that we used exhibited three- to fivefold increases in plasma cGMP level and the upregulation of cGK I protein (twofold) and P-VASP (Ser237) in the aortas (Fig. 1H and data not shown). Consistent with the activation of the cGK I pathway, BNP-Tg aortas showed a significant increase in P-RhoA level (1.5-fold) compared to aortas from NTg mice. On the other hand, the ROCK activity was 54% lower in BNP-Tg mice than in NTg mice (Fig. 1I). These results unveil an inverse relationship between P-RhoA level and ROCK activity and agree with the notion that RhoA phosphorylation at Ser188 serves as a critical determinant of ROCK activity in vivo.
cGK I insufficiency facilitates ROCK activation and enhances ROCK-dependent vascular hypertrophy and fibrosis.
To investigate the effect of cGK I haploinsufficiency on ROCK activity and vascular pathophysiology, we subjected the cGK I+/− and WT mice to systemic Ang II treatment, with or without the ROCK inhibitor Y-27632 (60). The ROCK activities of Ang II-treated cGK I+/− and WT mouse aortas were 230% and 38% higher, respectively, than that of the nontreated WT mouse aortas (Fig. 1D and 2B). Y-27632 penetrated mouse aortas and other organs to achieve optimal inhibitory concentrations (see Table S2 in the supplemental material) and significantly inhibited the ROCK activity of Ang II-treated mouse aortas, to or below that of nontreated WT aortas (Fig. 2A and B). Because cGK I and ROCK have been implicated in the regulation of BP (41, 60), we assessed the systolic BP of these mice (Fig. 2C; see Table S3 in the supplemental material). Consistent with the antihypertensive effect of cGK I, Ang II elevated the BP of cGK I+/− mice more potently than that of WT mice (50 versus 30 mm Hg). However, the Ang II-induced BP rise did not parallel the increase of ROCK activity in cGK I+/− and WT mice (Fig. 2B). Additionally, Y-27632 had little effect on the Ang II-induced BP rise, despite the suppression of ROCK activity to or below the baseline. Collectively, these results indicate that Ang II-induced BP elevation and its countersuppression by cGK I in our mouse model are mediated through ROCK-independent, and presumably Ca2+-dependent, mechanisms (21).
FIG. 2.

cGK I hemizygosity facilitates ROCK activation and ROCK-dependent vascular hypertrophy and fibrosis. cGK I+/− and WT mice were left untreated or treated with Ang II and Y-27632 before analysis. (A) Immunoblot showing levels of P-moesin and moesin in aortas. (B) Bar graphs showing ROCK activity, as determined by the P-moesin/moesin ratio, in the aortas (n = 3). (C) Systolic BPs (sBP) of mice (n = 4 to 10). (D to F) Morphological assessment of coronary arteries (n = 13 to 92). (D) Representative images of Sirius red-stained heart sections. Bar, 50 μm. (E and F) Morphometric analysis of coronary artery MT (E) and PVF (F). (Left) MT and PVF were determined by the following formulas: (i) MT = medial area/luminal area and (ii) PVF = perivascular fibrotic area/(luminal area + medial area + perivascular fibrotic area). (Middle) Absolute values of reduction by Y-27632 in Ang II-treated mice. (Right) Y-27632-inhibitable fractions of Ang II-induced increases of MT and PVF. Values are means ± SEM. *, P < 0.05; **, P < 0.01; #, P < 0.001; NS, not significant.
To address the roles of cGK I and ROCK in vascular remodeling and fibrotic response, we next performed morphometric analysis of the coronary arteries from the mice by measuring the MT and PVF (Fig. 2D to F). Although there was no significant difference at baseline between cGK I+/− and WT arteries, Ang II increased the MT and PVF more profoundly in cGK I+/− than in WT mice (for MT, 4.2-fold versus 1.9-fold [P < 0.01]; and for PVF, 2.0-fold versus 1.3-fold [P < 0.001]), to a comparable degree for the induction of ROCK activity (3.3-fold versus 1.38-fold; P < 0.05) (Fig. 2B). More importantly, in contrast to the effect on BP, Y-27632 selectively and substantially reduced the MT and PVF of cGK I+/− mice (Fig. 2E and F, middle panels). The Ang II-induced increases of MT and PVF were almost completely blunted by Y-27632 in cGK I+/− mice (for MT, 98%; and for PVF, 92%), but only partially in WT mice (for MT, 29%; and for PVF, 33%) (Fig. 2E and F, right panels). Taken together, the results show that cGK I insufficiency provokes a robust activation of ROCK and commits it to the major pathways mediating vascular hypertrophy and fibrosis. Hypertrophic and fibrotic responses of WT vessels are less prominent than those of cGK I+/− vessels and are mostly mediated via ROCK-independent pathways.
cGK I insufficiency recruits ROCK as a major mediator for profibrotic gene expression.
To further elucidate the role of cGK I in the fibrotic response, we examined the mRNA expression of profibrotic genes (TGF-β1 [Tgfb1], CTGF [Ctgf], fibronectin [Fn1], type I collagen [Col1a1], and type III collagen [Col3a1] genes) in cGK I+/− and WT mouse tissues treated with Ang II and Y-27632. The Ang II-mediated induction of these genes was significantly more profound in cGK I+/− aortas and hearts than in those of WT mice (Fig. 3A and B). This was further corroborated by the salient fibrosis formation seen in the cGK I+/− myocardial interstitium following Ang II infusion compared to that of WT mice (see Fig. S1 in the supplemental material). Importantly, Y-27632 had little effect on the Ang II-induced gene expression in WT mice but significantly inhibited that in cGK I+/− mice (Fig. 3A to C). Furthermore, the increment of gene expression in cGK I+/− mice over that in WT mice was almost completely abolished by Y-27632 (Fig. 3D), as assessed by Y-27632 inhibitability, calculated by the formula shown in the legend for Fig. 3. Together, these results indicate that ROCK participates in profibrotic gene expression preferentially in cGK I+/− mouse blood vessels and myocardial tissues and underlies their exaggerated fibrogenic response to Ang II.
FIG. 3.

cGK I hemizygosity recruits ROCK as a major mediator for profibrotic gene expression. (A and B) Quantitative RT-PCR analysis of profibrotic gene mRNA expression in cGK I+/− and WT mouse aortas (A) (n = 5) and hearts (B) (n = 3 to 7) left untreated or treated with Ang II and Y-27632. (C) Absolute mRNA level reductions by Y-27632 in Ang II-treated cGK I+/− and WT mouse hearts. (D) Y-27632-inhibitable fractions of incremental gene expression of cGK I+/− over WT hearts under Ang II treatment, as calculated by the following formula: Y-27632 inhibitability (%) = [(expression in Ang II-treated cGK I+/− hearts) − (expression in Ang II-plus-Y-27632-treated cGK I+/− hearts)]/[(expression in Ang II-treated cGK I+/− hearts) − (expression in Ang II-treated WT hearts)] × 100. *, P < 0.05; **, P < 0.01; #, P < 0.001; NS, not significant.
Generation of transgenic mice that specifically express either WT or phosphorylation-resistant RhoA in arterial smooth muscle.
To directly determine the in vivo significance of Ser188 phosphorylation of RhoA, we created transgenic mice that express either WT RhoA (RhoAWT) or a cGK I-unphosphorylatable RhoA mutant with a Ser→Ala188 substitution (RhoAA188) (49) (Fig. 4A). The construct was designed so that transgene expression was driven by the proximal region of the SM22α promoter (positions −441 to −1), which is reported to direct arterial smooth muscle-specific gene expression (1, 28, 35). Of the 10 RhoAWT-Tg and 11 RhoAA188-Tg founder mice obtained, 5 RhoAWT-Tg and 7 RhoAA188-Tg lines were able to reproduce transgenic progeny. The transgene copy number for each line was evaluated by Southern blotting (Fig. 4B). The expression of the transgene was assessed by semiquantitative RT-PCR (Fig. 4C), immunohistochemistry (Fig. 4D), and Western blotting (Fig. 4E; see Fig. S3A in the supplemental material).
FIG. 4.

Generation of transgenic mice that specifically express either WT or phosphorylation-resistant RhoA in arterial smooth muscle. (A) Transgene constructs for WT RhoA (RhoAwt) and phosphorylation-resistant RhoA (RhoAA188). Arrowheads, primer set used for genotyping and RT-PCR analysis in panel C; gray bar, SpeI-SpeI fragment recognized by the XbaI-HindIII probe (open bar) in the Southern blotting analysis in panel B. bGH, bovine growth hormone polyadenylation signal; 4F2, human 4F2 enhancer. (B) Genomic Southern blot analysis was performed with the F1 and F2 offspring of the RhoAWT (line 39) and RhoAA188 (line 30) transgenic mice. The transgenic vector (A) was used to quantify the copy number (first and second lanes). (C) Transgene mRNA expression was assessed by semiquantitative RT-PCR analysis of aortas between different founder lines (F2 offspring) of RhoAWT-Tg and RhoAA188-Tg mice. (D) Representative immunostaining with anti-myc-tag antibody showing transgene protein expression in NTg, RhoAWT-Tg (line 39), and RhoAA188-Tg (line 30) mouse heart and aorta sections (brownish signal). L, lumen. Bars, 50 μm. (E) Immunoblot with anti-RhoA and anti-myc-tag antibodies showing the ∼24-kDa transgene product expression in NTg, RhoAWT-Tg, and RhoAA188-Tg mouse aortas. Arrowhead (myc-RhoA), transgene product; arrow, endogenous RhoA. (F) Immunoblot showing the P-moesin level in NTg, RhoAWT-Tg, and RhoAA188-Tg mouse aorta lysates. Values are means ± SEM for triplicates. *, P < 0.05.
The RT-PCR analysis revealed noticeable transgene mRNA expression in the aortas from three RhoAWT-Tg and two RhoAA188-Tg lines (Fig. 4C). Because RhoAWT-Tg line 39 (5 to 10 copies) and RhoAA188-Tg line 30 (3 or 4 copies) showed comparable levels of transgene protein expression in the aorta and the aortic SMCs (MASMCs), they were selected for subsequent analysis (Fig. 4E; see Fig. S3A in the supplemental material). Immunohistochemistry of the heart and aorta showed arterial media-confined expression of the transgene, which is consistent with the arterial SMC-directed expression by the 441-bp SM22α promoter (Fig. 4D; see Fig. S2 in the supplemental material). Despite the modest expression of transgene proteins compared to endogenous RhoA (Fig. 4E; see Fig. S3A in the supplemental material), the ROCK activity in the aorta was significantly upregulated in both RhoAWT-Tg and RhoAA188-Tg mice compared to that in NTg controls (Fig. 4F). In agreement, the abundance of RhoA translocated to the cell membrane after stimulation with the RhoA agonist thrombin was two- to threefold higher in RhoAWT-Tg and RhoAA188-Tg MASMCs than in NTg cells (see Fig. S3B in the supplemental material). Although the mechanism is not clear, these findings indicate that the low level of exogenous RhoA facilitated membrane translocation and activation of RhoA and provoked downstream signaling. More importantly, although the myc-RhoAA188 protein level was 20% lower than that of myc-RhoAWT (Fig. 4E), the ROCK activity of RhoAA188-Tg was significantly higher than that of RhoAWT-Tg, by 1.4-fold (Fig. 4F).
Smooth muscle-targeted expression of RhoAA188 augments vascular ROCK activity, hypertrophy, and fibrosis and imparts unresponsiveness to the inhibitory effect of BNP.
NPs have been reported to potently prevent fibrosis. Given that insufficiency of cGK I, the downstream effector of NPs, caused exaggerated ROCK activation and vascular fibrotic lesions in cGK I+/− mice (Fig. 2), we hypothesized that Ser188 phosphorylation of RhoA by cGK I mediates the antifibrotic effects of the NP/cGMP/cGK I pathway. To address this question, we crossbred RhoAWT-Tg and RhoAA188-Tg mice with BNP-Tg mice. We first compared the ROCK activities of the aorta between NTg, RhoAWT-Tg, RhoAA188-Tg, BNP-Tg, RhoAWT/BNP-Tg, and RhoAA188/BNP-Tg mice (Fig. 5A; see Fig. S4B in the supplemental material). myc-RhoAWT and myc-RhoAA188 transgenes caused 55% and 111% increases in ROCK activity, respectively, over that in NTg mice. On the other hand, BNP-Tg mice showed a 59% reduction in ROCK activity compared to that in NTg mice. The BNP-mediated inhibitability of myc-RhoA-induced ROCK activation was assessed as the change in ROCK activity in RhoA/BNP-Tg versus RhoA-Tg mice, followed by normalization for the basal inhibition of ROCK activity by BNP (i.e., 59%), according to the formula shown in the legend to Fig. 5 (Fig. 5A, right panel). Strikingly, while crossbreeding with BNP-Tg mice substantially suppressed the induction of ROCK activity by myc-RhoAWT, by 75%, BNP had no effect on that induced by myc-RhoAA188. These results most likely suggest that NPs inhibit ROCK signaling in blood vessels almost exclusively through phosphorylation of RhoA Ser188. However, we cannot exclude the possibility that a potential alteration in tertiary structure caused by the Ser→Ala188 substitution might have rendered the RhoAA188 protein insensitive to BNP/cGMP-mediated inhibition through a change(s) in binding capacities to other proteins, intracellular localization, or accessibility by cGK I.
FIG. 5.

Smooth muscle-targeted expression of RhoAA188 augments vascular ROCK activity, hypertrophy, and fibrosis and imparts unresponsiveness to the inhibitory effect of BNP. NTg, RhoAWT-Tg, RhoAA188-Tg, BNP-Tg, RhoAWT/BNP-Tg, and RhoAA188/BNP-Tg mice were compared for analysis. (A) (Left) Bar graphs showing ROCK activity in the aortas (n = 3). (Right) Fraction of incremental ROCK activity in RhoAWT-Tg and RhoAA188-Tg aortas over NTg aortas that is inhibitable by crossbreeding with BNP-Tg mice. The original blot image is found in Fig. S2B in the supplemental material. (B) Systolic BPs (sBP) of mice (n = 5 to 11). (C and D) Morphological assessment of coronary arteries (n = 25 to 53). (C) Representative Sirius red staining of heart sections. Bar, 50 μm. (D) (Left) Morphometric analysis of coronary artery MT (top) and PVF (bottom). The MT and PVF were determined as described in the legend for Fig. 2. (Right) Fraction of incremental MT (top) and PVF (bottom) of RhoAWT-Tg or RhoAA188-Tg coronary arteries over NTg coronary arteries that is inhibitable by crossbreeding with BNP-Tg mice. In panels A and D, the BNP inhibitability of the myc-RhoA-induced increase in the indicated parameter was corrected for the baseline effect of BNP by the following formula: BNP inhibitability (%) = [(RhoAx-Tg value − RhoAx/BNP-Tg value) − (NTg value − BNP-Tg value)]/(RhoAx-Tg value − NTg value), where x is WT or A188. Values are means ± SEM. *, P < 0.05; **, P < 0.01; #, P < 0.001; NS, not significant.
Although the exogenous RhoA transgene expression caused significant increases in aortic ROCK activity, with the rank order RhoAA188-Tg > RhoAWT-Tg > NTg mice, these mice were equally normotensive, with similar systolic BPs (Fig. 5B; see Table S4 in the supplemental material). In addition, BNP-Tg, RhoWT/BNP-Tg, and RhoAA188/BNP-Tg mice showed significantly differential ROCK activity in aortas but had comparable BPs that were equally lower, by 20 to 30 mm Hg, than those of NTg, RhoAWT-Tg, and RhoAA188-Tg mice, respectively. Taken together, these results suggest that the Rho/ROCK signal does not suffice to elevate BP or to mediate the hypotensive effect of BNP (38).
Morphometric analysis of coronary arteries revealed that RhoAWT expression increased MT 1.7-fold and PVF 1.5-fold compared to those for NTg mice (Fig. 5C and D), suggesting that activation of the RhoA/ROCK pathway is sufficient to promote medial hypertrophy and PVF. The fact that RhoAA188 expression activated ROCK more potently than did that of RhoAWT envisaged a possibility that the threshold for activation of RhoAA188 was lower than that for RhoAWT by escaping the endogenous inhibitory mechanism, i.e., the Ser188 phosphorylation of RhoA by cGK I. In this regard, RhoAA188 expression could mimic certain aspects of cGK I-deficient conditions, as in cGK I+/− mice. Indeed, expression of RhoAA188 at a similar level to that of RhoAWT (Fig. 4E) caused significantly pronounced increases in MT (2.5-fold higher than that of NTg mice) and PVF (1.9-fold) compared to those for RhoAWT. These results indicate that RhoAA188-Tg mice recapitulate the cGK I+/− phenotype in promoting vascular hypertrophy and fibrosis (Fig. 2E and F).
The inhibitability by BNP of RhoAWT- and RhoAA188-induced increases in MT and PVF was evaluated using the double-transgenic mouse system, using the formula shown in the legend for Fig. 5. Crossbreeding with BNP-Tg mice totally eliminated RhoAWT induction of MT and significantly reduced that of PVF, by 60%. In contrast, RhoAA188-induced MT and PVF were only partially negated by BNP (inhibition of MT, 38%; that of PVF, 16%) (Fig. 5D, right panels), suggesting that BNP inhibits RhoA-induced vascular hypertrophy and fibrosis mostly through Ser188 phosphorylation of RhoA.
Ser188 phosphorylation of myc-RhoAWT and myc-RhoAA188 proteins was undetectable using P-RhoA antibody, presumably owing to their relatively low abundance. Assessment of the endogenous RhoA protein, however, revealed paradoxical increases of P-RhoA in RhoAA188-Tg (2-fold compared to NTg and RhoWT-Tg mice) and RhoA188/BNP-Tg (1.7-fold compared to BNP-Tg and RhoWT/BNP-Tg mice) mice (see Fig. S4C in the supplemental material). The increased phosphorylation of endogenous RhoA in RhoAA188-expressing mice was associated with augmentation of the P-VASP (Ser237) level, and hence, it most likely represents the upregulation of cGK I activity by a negative feedback loop (see Fig. S4D in the supplemental material). The compensatory upregulation of cGK I activity, however, seems unable to overcome the resistance of RhoAA188-induced MT and PVF against BNP, underscoring the importance of RhoA Ser188 phosphorylation by cGK I as the pivotal mechanism for BNP-mediated inhibition of vascular hypertrophy and fibrosis.
The inhibitory effect of BNP on profibrotic gene expression is abrogated by the smooth muscle-targeted expression of RhoAA188.
Ang II treatment of NTg mice (C57BL/6 background) led to a 1.3-fold increase in aortic ROCK activity (see Fig. S5A in the supplemental material) and a 1.4- to 4-fold induction of the profibrotic genes in the heart (Fig. 6). Overexpression of BNP completely prevented the ROCK activation and profibrotic gene induction by Ang II. Interestingly, inhibition of ROCK by Y-27632 also repressed Ang II-induced gene expression in NTg mice, albeit to a lesser extent than did BNP overexpression (see Fig. S5B in the supplemental material). This contrasts with the minimal effect of Y-27632 on the gene expression of WT mice in the 129sv background (Fig. 3A and B). Moreover, these results suggest that the repression by BNP of profibrotic gene expression involves both ROCK-dependent and -independent pathways (Fig. 7C and D).
FIG. 6.
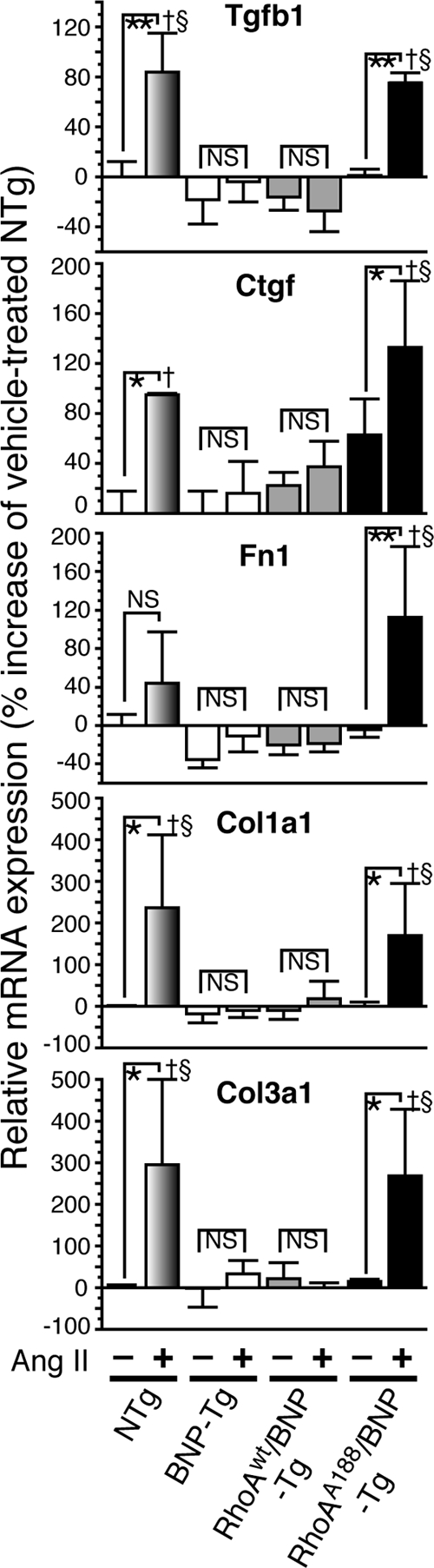
The inhibitory effect of BNP on profibrotic gene expression is abolished by the smooth muscle-targeted expression of RhoAA188. Quantitative RT-PCR analysis of profibrotic gene mRNA expression was performed with the hearts of the indicated mouse groups left untreated or treated with Ang II. Values are means ± SEM for three or four replicates. *, P < 0.05; **, P < 0.01; †, P < 0.01 versus BNP-Tg mice treated with Ang II; §, P < 0.01 versus RhoAWT/BNP-Tg mice treated with Ang II; NS, not significant.
FIG. 7.

The inhibitory effect of cGMP on RhoA-mediated protein synthesis and SRE-dependent gene transcription is reversed by the smooth muscle-targeted expression of RhoAA188. (A and B) MASMCs were derived from NTg, RhoAWT-Tg, and RhoAA188-Tg mice. (A) Effects of 8-Br cGMP and Y-27632 on Ang II-induced protein synthesis, as assessed by [3H]leucine uptake (left), and on serum-induced DNA synthesis, as assessed by [3H]thymidine uptake (right). Values are means ± SEM for six replicates. #, P < 0.0001; †, P < 0.05 versus serum-starved NTg cells; §, P < 0.0001 versus Ang II-treated NTg cells; ¶, P < 0.0001 versus Ang II- and Y-27632-treated RhoAA188-Tg cells; NS, not significant. (B) Effects of 8-Br cGMP and Y-27632 on TGF-β-induced SRE (left)- and AP-1 (right)-dependent transcriptional activity. Values are means ± SEM for triplicates. #, P < 0.0001; †, P < 0.05 versus serum-starved NTg cells; §, P < 0.0001 versus TGF-β-treated NTg cells; ¶, P < 0.0001 versus TGF-β- and Y-27632-treated RhoAA188-Tg cells. (C) Proposed model showing in vivo pathways whereby pro- and antifibrotic signals converge intracellularly. (D) cGK I is a major determinant of RhoA activity and controls the relative contributions of RhoA to the cellular profibrotic pathways in normal (left) and cGK I-deficient (right) blood vessels.
To investigate the role of RhoA phosphorylation in the repression by BNP of RhoA-mediated gene expression, BNP-Tg mice were crossbred with RhoAWT-Tg and RhoAA188-Tg mice and subjected to Ang II treatment. BNP was able to block the ROCK activation and gene induction elicited by Ang II and RhoAWT (Fig. 6; see Fig. S5A in the supplemental material). In contrast, forced expression of RhoAA188 promoted activation of ROCK by Ang II and almost completely abolished the ability of BNP to inhibit gene induction by Ang II and RhoA. These results suggest that RhoAA188 inhibits BNP signaling in a dominant-negative fashion and derepresses profibrotic gene transcription.
RhoAA188 blunts the ability of cGMP to inhibit Ang II-induced protein synthesis and serum response element (SRE)-dependent gene transcription in VSMCs.
To obtain mechanistic insights into the relevance of RhoA phosphorylation at the cellular level, we derived MASMCs from NTg, RhoAWT-Tg, and RhoAA188-Tg mice (see Fig. S6A and B in the supplemental material). Serum-starved MASMCs were stimulated with LPA, a classical agonist of RhoA, to induce actin polymerization as an indicator of RhoA activity (see Fig. S6C in the supplemental material). cGMP abolished the LPA-induced actin polymerization in NTg and RhoAWT-Tg MASMCs but failed to do so with RhoAA188-Tg MASMCs. This finding confirms an obligatory role for P-RhoA in cGMP-mediated inhibition of RhoA signaling in VSMCs.
Ang II-induced protein synthesis, the critical process involved in vascular hypertrophy, was determined by [3H]leucine uptake of MASMCs (12) (Fig. 7A). Upon Ang II stimulation, RhoAWT-Tg and RhoAA188-Tg MASMCs underwent four- to fivefold more protein synthesis than NTg cells did, and this augmentation was diminished by Y-27632. These results suggest that the RhoA/ROCK pathway promotes hypertrophic growth of VSMCs. The membrane-permeating analog 8-Br cGMP significantly blocked the Ang II-induced protein synthesis of RhoAWT-Tg MASMCs as efficiently as Y-27632 did, but it had little effect on that of RhoAA188-Tg MASMCs. Because the proliferation of VSMCs also contributes to vascular wall thickening, we assessed the DNA synthesis of MASMCs by [3H]thymidine uptake (Fig. 7A). Although cGMP and Y-27632 have been shown to inhibit the proliferation of rat and human VSMCs (5, 48, 52), they did not affect the serum-induced DNA synthesis of MASMCs, perhaps because of the interspecies difference in cell signaling. Collectively, these data indicate that cGMP inhibits the synthesis of proteins, but not DNA, through Ser188 phosphorylation and inhibition of RhoA.
TGF-β is induced by Ang II and pivotally mediates fibrogenic and hypertrophic responses of VSMCs through upregulation of profibrogenic genes and SMC contractile proteins (6, 13, 14, 40, 59). While our data suggest that RhoA/ROCK mediates Ang II-induced TGF-β expression, it has also been demonstrated that the RhoA/ROCK pathway is an important downstream mediator of TGF-β signaling (4, 24, 45). Indeed, the transcriptional program that is initiated by TGF-β and regulates the expression of smooth muscle α-actin, a contractile protein associated with VSMC hypertrophy, involves serum response factor/SRE, which is an established transcriptional mediator downstream of RhoA (6, 19, 32, 34, 59). On the other hand, the NP/cGMP pathway has been shown to inhibit TGF-β-induced signaling and gene expression (25). To address the role of P-RhoA in cGMP-mediated inhibition of TGF-β signaling, we assessed SRE-dependent gene transcription in RhoAWT-Tg and RhoAA188-Tg MASMCs stimulated by TGF-β. We found that similar to the profile of protein synthesis, TGF-β-induced SRE-dependent transcription in RhoAWT-Tg and RhoAA188-Tg MASMCs was pronounced compared to that in NTg cells, and it was inhibitable by Y-27632 (Fig. 7B). cGMP was able to block the TGF-β-induced SRE activity in RhoAWT-Tg MASMCs but not in RhoAA188-Tg cells. Additionally, we examined the activity of AP-1 and NF-κB, transcription factors implicated in profibrotic gene expression (33); however, their transcriptional activity was not significantly altered by Y-27632 or cGMP, thus precluding their participation in the cross talk between the cGMP and Rho pathways (Fig. 7B and data not shown).
DISCUSSION
Distinct mouse models identify cGK I-RhoA axis as a critical pathway for vascular remodeling and fibrosis.
NPs and RhoA/ROCK pathways are negative and positive regulators of vascular fibrosis (30, 42). The present study aimed to investigate whether these pathways counteract directly in the cell. Our findings demonstrated that haploinsufficiency of cGK I powerfully potentiated Ang II induction of ROCK activation, medial thickening, PVF, and profibrotic gene expression in comparison to the case for WT mice. Additionally, the pharmacological inhibition of ROCK revealed that Ang II-induced increases in MT, PVF, and gene expression were largely ROCK dependent in cGK I+/− mice, while those in WT mice were ROCK independent, at least in the context of the 129sv genetic background. These data provide evidence that endogenous cGK I, presumably through phosphorylation and inhibition of RhoA, prevents ROCK activation and subsequent mediation of the hypertrophic and fibrogenic program of VSMCs (Fig. 7C).
To directly assess the pathophysiological role of RhoA Ser188 phosphorylation, we developed transgenic mice expressing either RhoAWT or cGK I-unphosphorylatable RhoAA188 specifically in VSMCs. Expression of RhoAWT or RhoAA188 caused ROCK activation in blood vessels and concurrently increased MT and PVF, suggesting that RhoA/ROCK activation suffices to induce vascular hypertrophy and fibrosis. Importantly, despite the similar expression levels of RhoAWT and RhoAA188 proteins, the degree of ROCK activation and increases in MT and PVF were significantly higher in RhoAA188-Tg mice than in RhoAWT-Tg mice. This phenotypic difference of RhoAA188-Tg versus RhoAWT-Tg mice recapitulated that of cGK I+/− versus WT mice, consistent with the notion that RhoAA188 escaped the intrinsic inhibition by cGK I. The observation that RhoAA188-Tg mice phenocopied cGK I+/− mice was further supported by the finding that Ang II profoundly activated ROCK in the aortas of both genotypes in comparison to those of the respective controls (Fig. 1D; see Fig. S4A in the supplemental material).
The key finding of our study is that ectopic expression of RhoAA188, but not RhoAWT, in VSMCs in vivo blunted the ability of BNP to inhibit ROCK activation, vascular hypertrophy, fibrosis, and gene expression, which clearly demonstrates that Ser188 phosphorylation of RhoA is an obligatory step for the NP/cGMP/cGK I cascade to counter vascular remodeling. Importantly, the ability of cGMP to inhibit Ang II-induced protein synthesis and TGF-β-induced SRE-dependent gene transcription in MASMCs derived from RhoAWT-Tg mice, but not RhoAA188-Tg mice, correlated well with and could thus be the underlying mechanism for the inhibitory effect of BNP on vascular remodeling in RhoAWT-Tg versus RhoAA188-Tg mice. Recent studies using cGK I-deficient mice revealed that cGK I promotes VSMC proliferation in hyperlipidemia-induced atherosclerosis (63) but does not affect neointimal formation in vascular injury (31). While being consistent with the previous finding that RhoA/ROCK blockade blunts vascular hypertrophy (18, 66), our data showing that cGK I-mediated RhoA inhibition diminishes VSMC hypertrophic growth add more complexity to the context-dependent roles of cGK I in VSMC growth.
Although cGK I+/− mice showed exaggerated BP elevation in response to Ang II compared to WT mice, this was a ROCK-independent effect. Moreover, RhoAA188-Tg and RhoAWT-Tg mice were normotensive despite the elevated ROCK activity, and their BPs were equally lowered by crossbreeding with BNP-Tg mice. These results suggest that the cGK I-RhoA axis is not involved in the antihypertensive/BP-lowering effects of the NP/cGMP/cGK I pathway. Although it is well established that the RhoA/ROCK pathway promotes vascular contractility by enhancing calcium sensitivity (53), its role in BP regulation is equivocal. Indeed, in agreement with our findings, genetic or pharmacological blockade of ROCK failed to correct Ang II-induced hypertension in previous studies (18, 43). Because substantial evidence suggests that cGK I targets the calcium mobilizing system (IRAG and RGS2) and MYPT1 to exert vasorelaxing and BP-lowering effects (51, 57, 64), these mechanisms most likely mediate the cGK I effect on BP in our mouse models.
Significance of RhoA Ser188 phosphorylation as a converging node for pro- and antifibrotic signals.
Our data using RhoAA188-Tg mouse-derived MASMCs suggested that the actin polymerization, protein synthesis, and SRE activation we observed were mediated by the RhoA/ROCK pathway and counteracted by cGMP, largely through Ser188 phosphorylation of RhoA. A previous study showed that cGK I inhibits RhoA-mediated SRE-dependent gene transcription through interference upstream of RhoA, at the level of guanine nucleotide exchange factors, and downstream of RhoA, distal to the Rho effectors ROCK, protein kinase C-related kinase 1 (PRK1; also called PKN), and PRK2 (15). More recently, the same group reported that Ser239 phosphorylation of VASP by cGK I is partially responsible for cGK I inhibition of RhoA-induced SRE activity (69). These studies argued that Ser188 phosphorylation of RhoA is not involved in the inhibition of SRE by cGK I. The discrepancy between their data and ours may be attributable to differences in experimental conditions. Their studies were based on a reconstructive approach on glioma cells through transduction of exogenous cGK I and RhoA. Furthermore, they used a constitutively active form of unphosphorylatable RhoA (RhoAL63A188) for SRE induction under serum-starved conditions. In contrast, our experiments used primary VSMCs isolated from transgenic mice expressing a low level of the “wild-type” version of RhoAA188, and the cells were stimulated with TGF-β. Therefore, we consider that our in vitro data may represent physiologically more relevant conditions. On the other hand, our in vivo data do not preclude the possibility that mechanisms other than RhoA phosphorylation mediate the antifibrotic effect of BNP. While BNP did not repress ROCK activity in RhoAA188-Tg vessels, it was able to partially inhibit RhoA-induced increases in MT (38%) and PVF (16%). This suggests the possibility that NPs may inhibit the RhoA pathway, in part, by targeting downstream signals of RhoA independent of or distal to ROCK.
Previous in vitro evidence showed that Ser188 phosphorylation causes binding of GTP-RhoA to guanine dissociation inhibitors and subsequent sequestration in the cytosol (27). This uncouples the canonical Rho-GTP/GDP cycle and leads to RhoA inactivation. Cell culture studies demonstrated the functional significance of P-RhoA as the mediator for protein kinase A (PKA) and cGK I inhibition of actin stress fiber formation and cell spreading (7, 49); however, so far, the pathophysiological relevance of P-RhoA in vivo has not been addressed. In addition, previous studies were unable to directly detect the Ser188 phosphorylation of endogenous RhoA because of the apparently low abundance of P-RhoA in cells, which made it difficult to decipher its physiological roles (10, 47). In the present study, we developed a specific antiserum that allowed us to directly detect and quantify P-RhoA expression in mouse blood vessels. The inverse relationship between the P-RhoA level and ROCK activity, as demonstrated using this antibody, strongly suggests the negative regulatory effect of P-RhoA on Rho/ROCK activity. Although the transgene products in RhoAWT-Tg and RhoAA188-Tg vessels were significantly less abundantly expressed than endogenous RhoA (Fig. 4E; see Fig. S3A in the supplemental material), they were still capable of inducing potent signals. This may be explained partly by the propensity of exogenous myc-RhoA proteins to be translocated preferentially to the cell membrane due to unknown mechanisms (see Fig. S3B in the supplemental material). Our observation is supported by a study showing that exogenous myc-RhoA protein, stably expressed in cultured cells at a modest level comparable to those of the transgene products in MASMCs, prominently provoked RhoA-dependent events such as reduced cell spreading and increased stress fiber formation (9). Given that GTP-RhoA per se accounts for a small fraction of total RhoA in cells, these lines of evidence support the idea that P-RhoA existing at a low level in cells can exert a physiologically significant function for RhoA regulation.
Although our data show that cGK I is the major kinase that phosphorylates RhoA in mouse aortas, evidence suggests that RhoA Ser188 is also phosphorylated by PKA and Ste20-related kinase (16, 27). Notably, activation of the cAMP/PKA pathway with agonists such as forskolin, adrenomedullin, and beraprost or gene transfer of adenylyl cyclase led to potent inhibition of ECM synthesis (55, 67). Therefore, it is reasonable to assume that Ser188 phosphorylation of RhoA serves as a common target for cGK I and PKA to exert antifibrotic effects. Recent studies reported that Ser188 phosphorylation protects RhoA from ubiquitin/proteasome-mediated degradation and increases the RhoA protein level (44). Consistent with this finding, our data showed that RhoA/BNP-Tg mice tended to express higher levels of RhoA protein than did RhoA-Tg mice (see Fig. S4C in the supplemental material). However, the physiological relevance of this finding is uncertain in our study.
cGK I insufficiency converts RhoA/ROCK pathway into a profibrotic mediator.
The blood vessels of NTg mice displayed basal ROCK activity. Although BNP-Tg mice showed a 60% reduction in ROCK activity, the MT and PVF were not altered. In contrast, the increased ROCK activity in RhoAWT-Tg and RhoAA188-Tg vessels correlated with the degree of vascular lesions. These findings indicate that there is a threshold effect for ROCK participation in profibrotic signaling and that the incremental ROCK activity over the baseline is involved in the regulation of MT and PVF. This notion was strengthened by the findings that Ang II activated ROCK robustly in cGK I+/− mice but only modestly in WT mice in a 129sv genetic background and that the pronounced increase of profibrotic gene induction in cGK I+/− versus WT mice was fully ascribable to the magnitude of incremental ROCK activity, as evidenced by its complete abolition by Y-27632. Whereas the ROCK-independent pathway was also involved in Ang II-induced gene transcription in both WT and cGK I+/− blood vessels, reduced cGK I signaling in cGK I+/− vessels facilitated profibrotic gene expression preferentially through the ROCK pathway (Fig. 7C and D). The increased participation of ROCK in the cellular fibrotic machinery resulted in the exacerbation of vascular lesions in cGK I+/− mice. We did not determine the identity of the ROCK-independent mechanism, but the Smad proteins, the canonical mediators of fibrotic signaling, may conceivably represent this activity (29, 61).
There is accumulating evidence that the RhoA/ROCK pathway impacts various forms of fibrotic diseases. Its relative contribution, however, is context dependent and seems to vary in a spatiotemporal and disease state-specific fashion (17, 18, 26, 43, 68). These lines of evidence favor the view that an upstream mechanism(s) may exist that controls the commitment of Rho/ROCK to profibrotic signaling. We envision a scheme in which cGK I and PKA fulfill this role by networking with RhoA Ser188. For example, NO, constitutively synthesized from healthy endothelium, diffuses to the VSMC layer, activates the cGMP/cGK I cascade, and maintains the P-RhoA level, which in turn may contribute to vascular homeostasis by keeping the RhoA/ROCK activity below a threshold level. This protective mechanism may be turned off sequentially by reduced NO synthesis in hypertension, which results in the aberrant activation of ROCK in VSMCs that is known to mediate hypertensive vascular remodeling (36). More recently, evidence suggested that RhoA/ROCK activation is a key factor in myofibroblast formation, an important step for the initiation and progression of fibrosis (58). cAMP and cGMP cascades have received attention for their capacity to oppose this process (29, 55). Therefore, it may be worth investigating whether Ser188 phosphorylation and inhibition of RhoA may play a role in the regulation of fibroblast-to-myofibroblast conversion.
In conclusion, the present study highlighted the key roles for cGK I and its target, RhoA Ser188, in the mechanism that integrates pro- and antifibrotic signals associated with vascular remodeling. Our data provide clues to the long-standing question of how the NP/NO/cGMP cascade counters vascular fibrosis and insights into the development of antifibrotic means targeting intracellular pathways.
Supplementary Material
Acknowledgments
We are grateful to Robert Feil for his contribution to the development of cGK I+/− mice, to Elizabeth G. Nabel and Levent M. Akyürek for pAd SM22α-loxP plasmid, and to Shuh Narumiya for anti-ROCK antibody. We also thank Naomi Takeba and Nobuharu Goto (Mitsubishi Tanabe Pharma) for their contribution to the high-performance liquid chromatography analysis of the Y-27632 concentration in mouse organs.
This work was supported by a grant-in-aid for scientific research from the Ministry of Health, Labor, and Welfare and the Ministry of Education, Culture, Sports, Science, and Technology; by the Japan Foundation for Applied Enzymology; by the Kanae Foundation for the Promotion of Medical Science; by the Research Foundation for Community Medicine; and by the Japan Heart Foundation/Pfizer Grant for Research on Hypertension, Hyperlipidemia and Vascular Metabolism.
We have no conflict of interest to disclose.
Footnotes
Published ahead of print on 8 September 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Akyurek, L. M., Z. Y. Yang, K. Aoki, H. San, G. J. Nabel, M. S. Parmacek, and E. G. Nabel. 2000. SM22alpha promoter targets gene expression to vascular smooth muscle cells in vitro and in vivo. Mol. Med. 6:983-991. [PMC free article] [PubMed] [Google Scholar]
- 2.Berk, B. C., K. Fujiwara, and S. Lehoux. 2007. ECM remodeling in hypertensive heart disease. J. Clin. Investig. 117:568-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butt, E., K. Abel, M. Krieger, D. Palm, V. Hoppe, J. Hoppe, and U. Walter. 1994. cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J. Biol. Chem. 269:14509-14517. [PubMed] [Google Scholar]
- 4.Chen, S., M. Crawford, R. M. Day, V. R. Briones, J. E. Leader, P. A. Jose, and R. J. Lechleider. 2006. RhoA modulates Smad signaling during transforming growth factor-beta-induced smooth muscle differentiation. J. Biol. Chem. 281:1765-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiche, J. D., S. M. Schlutsmeyer, D. B. Bloch, S. M. de la Monte, J. D. Roberts, Jr., G. Filippov, S. P. Janssens, A. Rosenzweig, and K. D. Bloch. 1998. Adenovirus-mediated gene transfer of cGMP-dependent protein kinase increases the sensitivity of cultured vascular smooth muscle cells to the antiproliferative and pro-apoptotic effects of nitric oxide/cGMP. J. Biol. Chem. 273:34263-34271. [DOI] [PubMed] [Google Scholar]
- 6.Deaton, R. A., C. Su, T. G. Valencia, and S. R. Grant. 2005. Transforming growth factor-beta1-induced expression of smooth muscle marker genes involves activation of PKN and p38 MAPK. J. Biol. Chem. 280:31172-31181. [DOI] [PubMed] [Google Scholar]
- 7.Dong, J. M., T. Leung, E. Manser, and L. Lim. 1998. cAMP-induced morphological changes are counteracted by the activated RhoA small GTPase and the Rho kinase ROKalpha. J. Biol. Chem. 273:22554-22562. [DOI] [PubMed] [Google Scholar]
- 8.Dostmann, W. R., M. S. Taylor, C. K. Nickl, J. E. Brayden, R. Frank, and W. J. Tegge. 2000. Highly specific, membrane-permeant peptide blockers of cGMP-dependent protein kinase Ialpha inhibit NO-induced cerebral dilation. Proc. Natl. Acad. Sci. USA 97:14772-14777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellerbroek, S. M., K. Wennerberg, and K. Burridge. 2003. Serine phosphorylation negatively regulates RhoA in vivo. J. Biol. Chem. 278:19023-19031. [DOI] [PubMed] [Google Scholar]
- 10.Essler, M., J. M. Staddon, P. C. Weber, and M. Aepfelbacher. 2000. Cyclic AMP blocks bacterial lipopolysaccharide-induced myosin light chain phosphorylation in endothelial cells through inhibition of Rho/Rho kinase signaling. J. Immunol. 164:6543-6549. [DOI] [PubMed] [Google Scholar]
- 11.Etienne-Manneville, S., and A. Hall. 2002. Rho GTPases in cell biology. Nature 420:629-635. [DOI] [PubMed] [Google Scholar]
- 12.Geisterfer, A. A., M. J. Peach, and G. K. Owens. 1988. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ. Res. 62:749-756. [DOI] [PubMed] [Google Scholar]
- 13.Gibbons, G. H., and V. J. Dzau. 1994. The emerging concept of vascular remodeling. N. Engl. J. Med. 330:1431-1438. [DOI] [PubMed] [Google Scholar]
- 14.Gibbons, G. H., R. E. Pratt, and V. J. Dzau. 1992. Vascular smooth muscle cell hypertrophy vs. hyperplasia. Autocrine transforming growth factor-beta 1 expression determines growth response to angiotensin II. J. Clin. Investig. 90:456-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gudi, T., J. C. Chen, D. E. Casteel, T. M. Seasholtz, G. R. Boss, and R. B. Pilz. 2002. cGMP-dependent protein kinase inhibits serum-response element-dependent transcription by inhibiting rho activation and functions. J. Biol. Chem. 277:37382-37393. [DOI] [PubMed] [Google Scholar]
- 16.Guilluy, C., M. Rolli-Derkinderen, L. Loufrani, A. Bourge, D. Henrion, L. Sabourin, G. Loirand, and P. Pacaud. 2008. Ste20-related kinase SLK phosphorylates Ser188 of RhoA to induce vasodilation in response to angiotensin II type 2 receptor activation. Circ. Res. 102:1265-1274. [DOI] [PubMed] [Google Scholar]
- 17.Haydont, V., D. Mathe, C. Bourgier, J. Abdelali, J. Aigueperse, J. Bourhis, and M. C. Vozenin-Brotons. 2005. Induction of CTGF by TGF-beta1 in normal and radiation enteritis human smooth muscle cells: Smad/Rho balance and therapeutic perspectives. Radiother. Oncol. 76:219-225. [DOI] [PubMed] [Google Scholar]
- 18.Higashi, M., H. Shimokawa, T. Hattori, J. Hiroki, Y. Mukai, K. Morikawa, T. Ichiki, S. Takahashi, and A. Takeshita. 2003. Long-term inhibition of Rho-kinase suppresses angiotensin II-induced cardiovascular hypertrophy in rats in vivo: effect on endothelial NAD(P)H oxidase system. Circ. Res. 93:767-775. [DOI] [PubMed] [Google Scholar]
- 19.Hill, C. S., J. Wynne, and R. Treisman. 1995. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81:1159-1170. [DOI] [PubMed] [Google Scholar]
- 20.Hofmann, F., A. Ammendola, and J. Schlossmann. 2000. Rising behind NO: cGMP-dependent protein kinases. J. Cell Sci. 113:1671-1676. [DOI] [PubMed] [Google Scholar]
- 21.Hofmann, F., R. Feil, T. Kleppisch, and J. Schlossmann. 2006. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol. Rev. 86:1-23. [DOI] [PubMed] [Google Scholar]
- 22.Ishizaki, T., M. Maekawa, K. Fujisawa, K. Okawa, A. Iwamatsu, A. Fujita, N. Watanabe, Y. Saito, A. Kakizuka, N. Morii, and S. Narumiya. 1996. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 15:1885-1893. [PMC free article] [PubMed] [Google Scholar]
- 23.Jeon, S., S. Kim, J. B. Park, P. G. Suh, Y. S. Kim, C. D. Bae, and J. Park. 2002. RhoA and Rho kinase-dependent phosphorylation of moesin at Thr-558 in hippocampal neuronal cells by glutamate. J. Biol. Chem. 277:16576-16584. [DOI] [PubMed] [Google Scholar]
- 24.Kamaraju, A. K., and A. B. Roberts. 2005. Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-beta-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. J. Biol. Chem. 280:1024-1036. [DOI] [PubMed] [Google Scholar]
- 25.Kapoun, A. M., F. Liang, G. O'Young, D. L. Damm, D. Quon, R. T. White, K. Munson, A. Lam, G. F. Schreiner, and A. A. Protter. 2004. B-type natriuretic peptide exerts broad functional opposition to transforming growth factor-beta in primary human cardiac fibroblasts: fibrosis, myofibroblast conversion, proliferation, and inflammation. Circ. Res. 94:453-461. [DOI] [PubMed] [Google Scholar]
- 26.Kataoka, C., K. Egashira, S. Inoue, M. Takemoto, W. Ni, M. Koyanagi, S. Kitamoto, M. Usui, K. Kaibuchi, H. Shimokawa, and A. Takeshita. 2002. Important role of Rho-kinase in the pathogenesis of cardiovascular inflammation and remodeling induced by long-term blockade of nitric oxide synthesis in rats. Hypertension 39:245-250. [DOI] [PubMed] [Google Scholar]
- 27.Lang, P., F. Gesbert, M. Delespine-Carmagnat, R. Stancou, M. Pouchelet, and J. Bertoglio. 1996. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 15:510-519. [PMC free article] [PubMed] [Google Scholar]
- 28.Li, L., J. M. Miano, B. Mercer, and E. N. Olson. 1996. Expression of the SM22alpha promoter in transgenic mice provides evidence for distinct transcriptional regulatory programs in vascular and visceral smooth muscle cells. J. Cell Biol. 132:849-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li, P., D. Wang, J. Lucas, S. Oparil, D. Xing, X. Cao, L. Novak, M. B. Renfrow, and Y. F. Chen. 2008. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ. Res. 102:185-192. [DOI] [PubMed] [Google Scholar]
- 30.Loirand, G., P. Guerin, and P. Pacaud. 2006. Rho kinases in cardiovascular physiology and pathophysiology. Circ. Res. 98:322-334. [DOI] [PubMed] [Google Scholar]
- 31.Lukowski, R., P. Weinmeister, D. Bernhard, S. Feil, M. Gotthardt, J. Herz, S. Massberg, A. Zernecke, C. Weber, F. Hofmann, and R. Feil. 2008. Role of smooth muscle cGMP/cGKI signaling in murine vascular restenosis. Arterioscler. Thromb. Vasc. Biol. 28:1244-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mack, C. P., A. V. Somlyo, M. Hautmann, A. P. Somlyo, and G. K. Owens. 2001. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J. Biol. Chem. 276:341-347. [DOI] [PubMed] [Google Scholar]
- 33.Manabe, I., T. Shindo, and R. Nagai. 2002. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ. Res. 91:1103-1113. [DOI] [PubMed] [Google Scholar]
- 34.Masszi, A., C. Di Ciano, G. Sirokmany, W. T. Arthur, O. D. Rotstein, J. Wang, C. A. McCulloch, L. Rosivall, I. Mucsi, and A. Kapus. 2003. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am. J. Physiol. Renal Physiol. 284:F911-F924. [DOI] [PubMed] [Google Scholar]
- 35.Moessler, H., M. Mericskay, Z. Li, S. Nagl, D. Paulin, and J. V. Small. 1996. The SM 22 promoter directs tissue-specific expression in arterial but not in venous or visceral smooth muscle cells in transgenic mice. Development 122:2415-2425. [DOI] [PubMed] [Google Scholar]
- 36.Mukai, Y., H. Shimokawa, T. Matoba, T. Kandabashi, S. Satoh, J. Hiroki, K. Kaibuchi, and A. Takeshita. 2001. Involvement of Rho-kinase in hypertensive vascular disease: a novel therapeutic target in hypertension. FASEB J. 15:1062-1064. [DOI] [PubMed] [Google Scholar]
- 37.Oelze, M., H. Mollnau, N. Hoffmann, A. Warnholtz, M. Bodenschatz, A. Smolenski, U. Walter, M. Skatchkov, T. Meinertz, and T. Munzel. 2000. Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circ. Res. 87:999-1005. [DOI] [PubMed] [Google Scholar]
- 38.Ogawa, Y., H. Itoh, N. Tamura, S. Suga, T. Yoshimasa, M. Uehira, S. Matsuda, S. Shiono, H. Nishimoto, and K. Nakao. 1994. Molecular cloning of the complementary DNA and gene that encode mouse brain natriuretic peptide and generation of transgenic mice that overexpress the brain natriuretic peptide gene. J. Clin. Investig. 93:1911-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oshiro, N., Y. Fukata, and K. Kaibuchi. 1998. Phosphorylation of moesin by rho-associated kinase (Rho-kinase) plays a crucial role in the formation of microvilli-like structures. J. Biol. Chem. 273:34663-34666. [DOI] [PubMed] [Google Scholar]
- 40.Owens, G. K., A. A. Geisterfer, Y. W. Yang, and A. Komoriya. 1988. Transforming growth factor-beta-induced growth inhibition and cellular hypertrophy in cultured vascular smooth muscle cells. J. Cell Biol. 107:771-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pfeifer, A., P. Klatt, S. Massberg, L. Ny, M. Sausbier, C. Hirneiss, G. X. Wang, M. Korth, A. Aszodi, K. E. Andersson, F. Krombach, A. Mayerhofer, P. Ruth, R. Fassler, and F. Hofmann. 1998. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 17:3045-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Potter, L. R., S. Abbey-Hosch, and D. M. Dickey. 2006. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr. Rev. 27:47-72. [DOI] [PubMed] [Google Scholar]
- 43.Rikitake, Y., N. Oyama, C. Y. Wang, K. Noma, M. Satoh, H. H. Kim, and J. K. Liao. 2005. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/− haploinsufficient mice. Circulation 112:2959-2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rolli-Derkinderen, M., V. Sauzeau, L. Boyer, E. Lemichez, C. Baron, D. Henrion, G. Loirand, and P. Pacaud. 2005. Phosphorylation of serine 188 protects RhoA from ubiquitin/proteasome-mediated degradation in vascular smooth muscle cells. Circ. Res. 96:1152-1160. [DOI] [PubMed] [Google Scholar]
- 45.Ruiz-Ortega, M., J. Rodriguez-Vita, E. Sanchez-Lopez, G. Carvajal, and J. Egido. 2007. TGF-beta signaling in vascular fibrosis. Cardiovasc. Res. 74:196-206. [DOI] [PubMed] [Google Scholar]
- 46.Sakata, Y., F. Xiang, Z. Chen, Y. Kiriyama, C. N. Kamei, D. I. Simon, and M. T. Chin. 2004. Transcription factor CHF1/Hey2 regulates neointimal formation in vivo and vascular smooth muscle proliferation and migration in vitro. Arterioscler. Thromb. Vasc. Biol. 24:2069-2074. [DOI] [PubMed] [Google Scholar]
- 47.Sauzeau, V., H. Le Jeune, C. Cario-Toumaniantz, A. Smolenski, S. M. Lohmann, J. Bertoglio, P. Chardin, P. Pacaud, and G. Loirand. 2000. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J. Biol. Chem. 275:21722-21729. [DOI] [PubMed] [Google Scholar]
- 48.Sawada, N., H. Itoh, K. Ueyama, J. Yamashita, K. Doi, T. H. Chun, M. Inoue, K. Masatsugu, T. Saito, Y. Fukunaga, S. Sakaguchi, H. Arai, N. Ohno, M. Komeda, and K. Nakao. 2000. Inhibition of rho-associated kinase results in suppression of neointimal formation of balloon-injured arteries. Circulation 101:2030-2033. [DOI] [PubMed] [Google Scholar]
- 49.Sawada, N., H. Itoh, J. Yamashita, K. Doi, M. Inoue, K. Masatsugu, Y. Fukunaga, S. Sakaguchi, M. Sone, K. Yamahara, T. Yurugi, and K. Nakao. 2001. cGMP-dependent protein kinase phosphorylates and inactivates RhoA. Biochem. Biophys. Res. Commun. 280:798-805. [DOI] [PubMed] [Google Scholar]
- 50.Sawada, N., S. Salomone, H. H. Kim, D. J. Kwiatkowski, and J. K. Liao. 2008. Regulation of endothelial nitric oxide synthase and postnatal angiogenesis by Rac1. Circ. Res. 103:360-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schlossmann, J., A. Ammendola, K. Ashman, X. Zong, A. Huber, G. Neubauer, G. X. Wang, H. D. Allescher, M. Korth, M. Wilm, F. Hofmann, and P. Ruth. 2000. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature 404:197-201. [DOI] [PubMed] [Google Scholar]
- 52.Seasholtz, T. M., M. Majumdar, D. D. Kaplan, and J. H. Brown. 1999. Rho and Rho kinase mediate thrombin-stimulated vascular smooth muscle cell DNA synthesis and migration. Circ. Res. 84:1186-1193. [DOI] [PubMed] [Google Scholar]
- 53.Somlyo, A. P., and A. V. Somlyo. 2003. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83:1325-1358. [DOI] [PubMed] [Google Scholar]
- 54.Sorescu, D. 2006. Smad3 mediates angiotensin II- and TGF-beta1-induced vascular fibrosis: Smad3 thickens the plot. Circ. Res. 98:988-989. [DOI] [PubMed] [Google Scholar]
- 55.Swaney, J. S., D. M. Roth, E. R. Olson, J. E. Naugle, J. G. Meszaros, and P. A. Insel. 2005. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc. Natl. Acad. Sci. USA 102:437-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tamura, N., Y. Ogawa, H. Chusho, K. Nakamura, K. Nakao, M. Suda, M. Kasahara, R. Hashimoto, G. Katsuura, M. Mukoyama, H. Itoh, Y. Saito, I. Tanaka, H. Otani, and M. Katsuki. 2000. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc. Natl. Acad. Sci. USA 97:4239-4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang, K. M., G. R. Wang, P. Lu, R. H. Karas, M. Aronovitz, S. P. Heximer, K. M. Kaltenbronn, K. J. Blumer, D. P. Siderovski, Y. Zhu, and M. E. Mendelsohn. 2003. Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat. Med. 9:1506-1512. [DOI] [PubMed] [Google Scholar]
- 58.Tomasek, J. J., G. Gabbiani, B. Hinz, C. Chaponnier, and R. A. Brown. 2002. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3:349-363. [DOI] [PubMed] [Google Scholar]
- 59.Turla, M. B., M. M. Thompson, M. H. Corjay, and G. K. Owens. 1991. Mechanisms of angiotensin II- and arginine vasopressin-induced increases in protein synthesis and content in cultured rat aortic smooth muscle cells. Evidence for selective increases in smooth muscle isoactin expression. Circ. Res. 68:288-299. [DOI] [PubMed] [Google Scholar]
- 60.Uehata, M., T. Ishizaki, H. Satoh, T. Ono, T. Kawahara, T. Morishita, H. Tamakawa, K. Yamagami, J. Inui, M. Maekawa, and S. Narumiya. 1997. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389:990-994. [DOI] [PubMed] [Google Scholar]
- 61.Wang, W., X. R. Huang, E. Canlas, K. Oka, L. D. Truong, C. Deng, N. A. Bhowmick, W. Ju, E. P. Bottinger, and H. Y. Lan. 2006. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 98:1032-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wegener, J. W., H. Nawrath, W. Wolfsgruber, S. Kuhbandner, C. Werner, F. Hofmann, and R. Feil. 2002. cGMP-dependent protein kinase I mediates the negative inotropic effect of cGMP in the murine myocardium. Circ. Res. 90:18-20. [DOI] [PubMed] [Google Scholar]
- 63.Wolfsgruber, W., S. Feil, S. Brummer, O. Kuppinger, F. Hofmann, and R. Feil. 2003. A proatherogenic role for cGMP-dependent protein kinase in vascular smooth muscle cells. Proc. Natl. Acad. Sci. USA 100:13519-13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wooldridge, A. A., J. A. MacDonald, F. Erdodi, C. Ma, M. A. Borman, D. J. Hartshorne, and T. A. Haystead. 2004. Smooth muscle phosphatase is regulated in vivo by exclusion of phosphorylation of threonine 696 of MYPT1 by phosphorylation of serine 695 in response to cyclic nucleotides. J. Biol. Chem. 279:34496-34504. [DOI] [PubMed] [Google Scholar]
- 65.Wynn, T. A. 2007. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Investig. 117:524-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamakawa, T., S. Tanaka, K. Numaguchi, Y. Yamakawa, E. D. Motley, S. Ichihara, and T. Inagami. 2000. Involvement of Rho-kinase in angiotensin II-induced hypertrophy of rat vascular smooth muscle cells. Hypertension 35:313-318. [DOI] [PubMed] [Google Scholar]
- 67.Yokoyama, U., H. H. Patel, N. C. Lai, N. Aroonsakool, D. M. Roth, and P. A. Insel. 2008. The cyclic AMP effector Epac integrates pro- and anti-fibrotic signals. Proc. Natl. Acad. Sci. USA 105:6386-6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang, Y. M., J. Bo, G. E. Taffet, J. Chang, J. Shi, A. K. Reddy, L. H. Michael, M. D. Schneider, M. L. Entman, R. J. Schwartz, and L. Wei. 2006. Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. FASEB J. 20:916-925. [DOI] [PubMed] [Google Scholar]
- 69.Zhuang, S., G. T. Nguyen, Y. Chen, T. Gudi, M. Eigenthaler, T. Jarchau, U. Walter, G. R. Boss, and R. B. Pilz. 2004. Vasodilator-stimulated phosphoprotein activation of serum-response element-dependent transcription occurs downstream of RhoA and is inhibited by cGMP-dependent protein kinase phosphorylation. J. Biol. Chem. 279:10397-10407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.