

Abstract
The guanine N7 adduct of aflatoxin B1exo-8,9-epoxide hydrolyzes to form the formamidopyrimidine (AFB-FAPY) adduct, which interconverts between α and β anomers. The β anomer is highly mutagenic in Escherichia coli, producing G → T transversions; it thermally stabilizes the DNA duplex. The AFB-α-FAPY adduct blocks replication; it destabilizes the DNA duplex. Herein, the structure of the AFB-α-FAPY adduct has been elucidated in 5′-d(C1T2A3T4X5A6T7T8C9A10)-3′·5′-d(T11G12A13A14T15C16A17T18A19G20)-3′ (X = AFB-α-FAPY) using molecular dynamics calculations restrained by NMR-derived distances and torsion angles. The AFB moiety intercalates on the 5′ face of the pyrimidine moiety at the damaged nucleotide between base pairs T4·A17 and X5·C16, placing the FAPY C5−N5 bond in the Ra axial conformation. Large perturbations of the ε and ζ backbone torsion angles are observed, and the base stacking register of the duplex is perturbed. The deoxyribose orientation shifts to become parallel to the FAPY base and displaced toward the minor groove. Intrastrand stacking between the AFB moiety and the 5′ neighbor thymine remains, but strong interstrand stacking is not observed. A hydrogen bond between the formyl group and the exocyclic amine of the 3′-neighbor adenine stabilizes the E conformation of the formamide moiety. NMR studies reveal a similar 5′-intercalation of the AFB moiety for the AFB-α-FAPY adduct in the tetramer 5′-d(C1T2X3A4)-3′, involving the Ra axial conformation of the FAPY C5−N5 bond and the E conformation of the formamide moiety. Since in duplex DNA the AFB moiety of the AFB-β-FAPY adduct also intercalates on the 5′ side of the pyrimidine moiety at the damaged nucleotide, we conclude that favorable 5′-stacking leads to the Ra conformational preference about the C5−N5 bond; the same conformational preference about this bond is also observed at the nucleoside and base levels. The structural distortions and the less favorable stacking interactions induced by the AFB-α-FAPY adduct explain its lower stability as compared to the AFB-β-FAPY adduct in duplex DNA. In this DNA sequence, hydrogen bonding between the formyl oxygen and the exocyclic amine of the 3′-neighboring adenine stabilizing the E configuration of the formamide moiety is also observed for the AFB-β-FAPY adduct, and suggests that the identity of the 3′-neighbor nucleotide modulates the stability and biological processing of AFB adducts.
Introduction
Aflatoxins are metabolites of Aspergillus flavus and related fungi that contaminate food crops.1−3 Aflatoxin B1 (AFB) is a mutagen in bacteria,2,4−6 a carcinogen in fish,7,8 a carcinogen in rodents,9,10 and is implicated in the etiology of human liver cancer.11−15 Aflatoxin exposures are implicated in mutations to the p53 tumor suppressor gene.16−22 The metabolism of AFB involves oxidation to AFB-exo-8,9-epoxide by cytochromes P45023−26.
The short-lived AFB-exo-8,9-epoxide(27) reacts efficiently with duplex DNA to yield the N7-dGuo adduct trans-8,9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin B1 (Scheme 1).2,28 This is attributed to intercalation of the epoxide above the 5′ face of deoxyguanosine in duplex DNA,(29) facilitating the nucleophilic attack by the N7 nitrogen at the C8 carbon of the epoxide.(30) The cationic N7-dGuo adduct undergoes hydrolysis of the imidazole ring to form the AFB formamidopyrimidine adduct (AFB-FAPY).(31) The chemistry of the FAPY adduct is complex (Scheme 1). It can interconvert between α and β deoxyribose anomers.(32) In duplex DNA, the β-anomer is strongly favored, but in single-strand DNA, a 2:1 α/β equilibrium mixture of anomers is observed.(32) Additionally, the FAPY adduct can undergo conformational interconversions involving atropisomers about the C5−N5 bond and geometrical isomers of the formyl moiety.(32)
Scheme 1. Chemistry of the FAPY Adduct Following Base-Catalyzed Ring-Opening of the AFB-N7-dGuo Cationic Adduct.

The adduct interconverts between AFB-α-FAPY and AFB-β-FAPY anomers; the equilibrium is dependent on single strand vs duplex DNA environments.(32) Geometrical isomers of the formamide group and atropisomers about the C5−N5 bond are also observed.(32)
The genotoxicity of AFB has been ascribed primarily to the AFB-FAPY adduct, which is strongly mutagenic and induces the G → T transitions(33) associated with AFB mutagenesis in Escherichia coli,(5) whereas the cationic AFB-dGuo adduct is less mutagenic.(34) Moreover, the AFB-FAPY adduct is persistent in vivo.35−37 Smela et al.(33) have demonstrated that an oligodeoxynucleotide containing an AFB-FAPY adduct equilibrates between two separable species, one of which is mutagenic, whereas the other blocks DNA replication. These correspond to the α- and β-anomers of the AFB-FAPY adduct; the mutagenic species is the AFB-β-FAPY adduct.(32)
In duplex DNA, the AFB-β-FAPY adduct intercalates with the AFB moiety on the 5′ face of the pyrimidine moiety of the adducted nucleotide,38,39 similar to the initially formed cationic adduct.40−43 This results in the Ra axial conformation for the C5−N5 bond linking the pyrimidine ring to the formamido nitrogen. The stability of the AFB-β-FAPY adduct in duplex DNA is attributed to interstrand stacking interactions.38,39 In the 5′-d(TXA)-3′ sequence, a hydrogen bond between the exocyclic amino group of the 3′-neighbor adenine and the formamido carbonyl group is consistent with the downfield shift of the amino proton in the 1H spectrum.38,39 The similar structures of the AFB-dGuo40−43 and AFB-β-FAPY(38) adducts in duplex DNA are consistent with each producing G → T transversions.
Here, the structure of the AFB-α-FAPY adduct in 5′-d(C1T2A3T4X5A6T7T8C9A10)-3′·5′-d(T11G12A13A14T15C16A17T18A19G20)-3′ (X =AFB-α-FAPY) is elucidated using molecular dynamics calculations restrained by NMR-derived distances and torsion angles.44−47 The AFB moiety intercalates above the 5′ face of the damaged base. The FAPY C5−N5 bond is in the Ra axial conformation, as observed for the AFB-β-FAPY adduct,38,39 and the nucleoside and the FAPY base,(32) which is attributed to stacking preferences of the AFB moiety. Hydrogen bond stabilization of the E conformation of the formamide moiety, also seen for the AFB-β-FAPY adduct,38,39 suggests DNA sequence modulates the stability and biological processing of AFB adducts. The lower stability of the AFB-α-FAPY as compared to the AFB-β-FAPY adduct38,39 is attributed to structural perturbations in the DNA and reduced interstrand stacking interactions.
Results
Sample Preparation
The oligodeoxynucleotide 5′-d(CTATGATTCA)-3′ contained one guanine to avoid formation of regioisomeric AFB-dGuo adducts.(38) It was desired to utilize dsDNA for the preparation of the adducted sample since the alkylation mechanism involves intercalation of AFB-exo-8,9-epoxide.29,30 To simplify purification of the 5′-d(CTATXATTCA)-3′ alkylation product, the scaffold 5′-d(AATCATA)-3′ was annealed with 5′-d(CTATGATTCA)-3′, forming a 7-mer duplex with dangling ends. The reaction was maintained at 5 °C and pH 7.8 to minimize depurination of the initially formed cationic N7-dGuo adduct. Alkaline treatment of the cationic adduct yielded a mixture of oligodeoxynucleotides containing either the AFB-α-FAPY or AFB- β-FAPY adducts, identified by mass spectrometry. The FAPY-modified oligodeoxynucleotides were separated by HPLC (Figure 1). The AFB-β-FAPY modified oligodeoxynucleotide eluted as a single peak. The peak containing the AFB-α-FAPY-modified oligodeoxynucleotide exhibited a shoulder. CD spectra showed a negative induced molar ellipticity between 310 and 400 nm for both anomers (Figure S1 in the Supporting Information). The UV spectra of the oligodeoxynucleotide containing the AFB-α-FAPY anomer and of its shoulder peak were identical in the 320−380 nm spectral region, but differed in the far-UV region (Figure S2 in the Supporting Information). The main peak and the shoulder were collected individually, but re-equilibrated in 30 min at room temperature. The re-equilibration was independent of pH. The oligodeoxynucleotides were maintained at 5 °C and pH 8.0 to minimize the rate of re-equilibration of the two anomers.(32) The samples were maintained for a period of days, sufficient for NMR experiments (Figure 1).
Figure 1.

HPLC analyses of the AFB-α-FAPY modified duplex, monitored by UV absorbance at 360 nm. (A) Freshly prepared sample. (B) Three days after preparing the sample. The oligodeoxynucleotide containing the AFB-α-FAPY adduct eluted at 20.1 min. The oligodeoxynucleotide containing the AFB-β-FAPY adduct eluted at 19.5 min.
Thermal Analyses of the AFB-FAPY Modified Duplexes
The thermal denaturation of AFB-α-FAPY and AFB-β-FAPY modified duplexes was monitored by UV hyperchromicity at 260 nm. The unmodified duplex produced a single transition; its Tm was 36 °C (Figure 2, panel A). The AFB-β-FAPY modified duplex (Figure 2, panel B) produced a single transition at 50 °C during the heating stage. During the cooling stage, a biphasic transition was observed at 50 and 23 °C. The AFB-α-FAPY modified duplex (Figure 2, panel C) produced biphasic transitions at 50 and 23 °C during both heating and cooling cycles.
Figure 2.

Thermal denaturation studies monitored by UV absorbance at 260 nm showing heating (▲) and cooling (Δ) transitions. (A) The unmodified duplex 5′-d(CTATGATTCA)-3′·5′-d(TGAATCATAG)-3′. (B) The duplex containing the AFB-β-FAPY adduct. (C) The duplex containing the AFB-α-FAPY adduct. The solid lines are first derivative curves.
NMR Spectroscopy of the AFB-α-FAPY Modified Duplex
(a) Non-Exchangeable DNA Protons
The resonances were assigned using established strategies (Figure S3 in the Supporting Information).48−50 For the modified strand, the C1 H6 → C1 H1′, C1 H1′ → T2 H6, T2 H6 → T2 H1′, T2 H1′ → A3 H8, A3 H8 → A3 H1′, A3 H1′ → T4 H6, and T4 H6 → T4 H1′ NOEs were observed. There was a break in the sequential NOE connectivity between T4 H1′ and X5 base protons. At X5, the formyl proton of the FAPY base exhibited an NOE to X5 H1′, but a NOE between X5 H1′ and A6 H8 was not observed. The anticipated NOE connectivity A6 H8 → T7 H1′, T7 H1′ → T8 H6, T8 H6 → T8 H1′, T8 H1′ → C9 H6, C9 H6 → A10 H8, A10 H8 → A10 H1′ was observed. For the complementary strand, a break in the sequential connectivity occurred between C16 H1′ and A17 H8. Two sets of resonances were observed for A14 H8 and T15 H6 and for several of the C16 deoxyribose protons. With the exception of several of the H4′ protons, and the stereotopic assignments of the H5′ and H5′′ sugar protons, assignments were made unequivocally. Comparisons between these NOE intensities and the canonical distances between the H4′, H5′, and H5′′ protons in B-form DNA were used to tentatively assign the H5′′ and H5′′ deoxyribose protons.
(b) Anomeric Configuration
The assignments of the H2′ and H2′′ resonances were based on their relative cross peak intensities to H3′ at NOE mixing times of 80 and 150 ms (Figure 3). The configuration of H1′ was determined by cross peak intensity to H2′ and H2′′.(38) The intensity of the X5 H1′ → X5 H2′′ NOE was less than the X5 H1′ → X5 H2′ NOE, which established the α configuration (Figure 3, panel A).
Figure 3.

Analysis of NOE intensities for the deoxyribose protons. (A) 5′-d(CTATXATTCA)-3′·5′-d(TGAATCATAG)-3′. (B) 5′-d(CTXA)-3′, X = AFB-α-FAPY.
(c) Exchangeable DNA Protons
The assignments of Watson−Crick hydrogen-bonded imino and amino protons were made using established methods (Figure S4 in the Supporting Information).(51) There was a break in sequential NOE connectivity between the T4 N3H and X5 N3H imino resonances. Both T4 N3H and X5 N3H exhibited NOEs to the AFB methoxy protons. The strong cross peaks from X5 N3H to C16N4H1 and C16N4H2 amino protons indicated that Watson−Crick hydrogen bonding between X5 and C16 was intact. Similarly, the imino resonances for base pairs A3·T18, T4·A17, X5·C16, A6·T15, T7·A14, T8·A13, and C9·G12 were observed, suggesting Watson−Crick hydrogen bonding was preserved. The imino resonances for the base pairs C1·G20, T2·A19, and A10·T11 were not observed, which was attributed to exchange broadening with water.
(d) Aflatoxin Protons
The AFB H5, H6a, H8, H9, H9a, and -OCH3 resonances were assigned from a combination of NOE and chemical shift data.(38) AFB H6a and H9a were identified from both COSY and NOESY experiments. AFB H8 and H9 were assigned based on NOEs between them and to H6a or H9a. A strong NOE from AFB H5 to AFB -OCH3 revealed that the latter resonance was at δ 3.51 ppm, while AFB H5 was at δ 5.7 ppm. The X5 CHO resonance was observed at δ 8.3 ppm. The assignment of X5 CHO was supported by NOEs to X5 H1′ and AFB H8. The cyclopentenone ring protons AFB H2α, H2β, H3α, and H3β were identified from a combination of COSY and NOESY spectra.
(e) Aflatoxin to DNA NOEs
A total of 28 NOEs between AFB and DNA protons were observed (Table 1). The AFB furan protons exhibited NOEs to major groove and imino protons; most were to the 5′-neighboring base-pair T4·A17 (Figure 4). Thus, H6a and H9a, which are located on the same face of the AFB moiety, both produced NOEs to T4 H6 and CH3. Weaker NOEs were observed for AFB H8 and H9. The AFB H5 and -OCH3 resonances produced NOEs with minor groove and imino protons. These were primarily with base pair T4·A17 in the 5′-direction and with the nucleotide X5. These included NOEs between AFB -OCH3 and T4 H1′, T4 H2′, T4 H2′′, T4 H5′, T4 H3, X5 H3, and A17 H2. The cyclopentenone ring H2α exhibited NOEs with H1′, H3′, H2′′, H4′, H5, and H6 of C16 in the complementary strand.
Table 1. NOEs Observed between the AFB-α-FAPY Adduct and DNA Protonsa.
| AFB-α-FAPY | 10-mer ds duplex | tetramer |
|---|---|---|
| H2α | C16 H1′, C16 H3′, C16 H2′′, C16 H4′, C16 H5, C16 H6, A17 H8 | |
| H2β | A17 H8 | |
| H3α | A17 H3′ | |
| H3β | A17 H8 | |
| H5 | A17 H2, T4 H6, T4 H4′ | T4 H2′, T4 H2′′ |
| H6a | T4 CH3, T4 H6, X5 H8 | |
| H8 | X5 CHO | X3 CHO |
| H9a | T4 CH3, T4 H6, T4 H1′, X5 H8 | |
| H9 | ||
| -OCH3 | A17 H2, T4 H1′, T4 H2′, T4 H2′′, T4 H5′, T4 H3, X5 H3 | T4 H1′ |
NOEs involving adenine H2 protons were not included in structural refinement due to the long T1 relaxation times associated with these protons.
Figure 4.

A tile plot of an 800 MHz NOESY spectrum obtained at 250 ms mixing time, showing the assignments of AFB-α-FAPY protons and NOEs to neighboring nucleotide protons. The black lines indicate the assignmnents of the AFB-α-FAPY protons and the gray lines indicate NOEs between the AFB moiety and DNA.
(f) Chemical Shift Perturbations
The 1H chemical shifts of the AFB-α-FAPY adducted duplex were compared to those of the nonadducted duplex (Figure 5, Tables S1 and S2 in the Supporting Information). Chemical shift perturbations were observed at the X5 adduct site and at nucleotides T4, A6, T7, A14, T15, C16, A17, and T18. The X5 N3H imino resonance was shielded 0.4 ppm as compared to the corresponding G5 N1H imino resonance in the unadducted duplex. The T7, T8, and T15 N3H imino resonances were deshielded an average of 0.15 ppm. Deshielding of the A6 non-Watson−Crick hydrogen bonded exocyclic amine proton is also observed in the AFB-α-FAPY NMR data. In the 31P spectrum, relative to other 31P resonances in the duplex, the resonance associated with the phosphate group located between C16 and A17 was deshielded approximately 0.6 ppm (Figure S5 in the Supporting Information).
Figure 5.

Chemical shift perturbation of AFB-α-FAPY modified duplex relative to the unmodified oligodeoxynucleotide. (A) The aromatic H6/H8 (black bars) and pyrimidine H5/CH3 (white bars) resonances of the modified strand. (B) The aromatic H6/H8 (black bars) and pyrimidine H5/CH3 (white bars) resonances of the complementary strand. (C) The deoxyribose resonances H1′ (black bars), H2′ (white bars), H2′′ (dark gray bars), and H3′ (light gray bars) of the modified strand. (D) The deoxyribose resonances H1′ (black bars), H2′ (white bars), H2′′ (dark gray bars), and H3′ (light gray bars) of the complementary strand. (E) The exchangeable N1H/N3H (black bars) resonances. (F) Comparison of deoxyribose resonances H1′ (black bars), H2′ (white bars), H2′′ (dark gray bars), and H3′ (light gray bars) for corresponding nucleotides of the duplex and single strand tetramer.
Refinement of the AFB-α-FAPY Modified Duplex
Structural refinement utilizing a simulated annealing protocol for molecular dynamics calculations restrained by experimentally derived distance and torsion angle restraints, with additional empirical restraints, was accomplished using established methods.44−47 A total of 322 distance restraints as summarized in Table 2 were obtained from NOESY data. There were 167 intranucleotide and 155 internucleotide restraints. These included 29 internucleotide restraints involving the AFB-α-FAPY adduct. Additional intradeoxyribose distances were not utilized during structural refinement, but the intensities of the intradeoxyribose NOEs were included in subsequent complete relaxation matrix analysis of the refined structures. A total of 78 backbone torsion restraints were applied; however, at the AFB-α-FAPY adduct, the backbone torsion angles α, β, γ, ε, and ζ were not restrained. With regard to deoxyribose pseudorotation, nucleotides A3, X5, A6, C9, G12, A14, A17 were allowed to explore both N and S conformations; the remainder were restrained to the S conformation. A total of 36 empirical Watson−Crick hydrogen bonding restraints were applied, but not at the AFB-α-FAPY adduct. Twenty structures that emerged from the simulated annealing rMD calculations were evaluated by pairwise rmsd analyses. They exhibited a maximum pairwise rmsd value of 2.1 Å for the six core base pairs. Evaluation of sixth root residuals (R1x values) between theoretical and experimental NOE intensities52,53 revealed residuals of 8.8 × 10−2 (Table 3), in good agreement with NOESY data.
Table 2. Distribution of Restraints Applied to Structural Refinement.
| restraints | |
|---|---|
| Assigned Distances | 233 |
| Watson−Crick | 36 |
| Backbone Torsion | 78 |
| Deoxyribose Torsion | 100 |
| Distance: | 322 |
| Inter-residue | 155 |
| Intraresidue | 167 |
| Total Restraints | 536 |
| Avg. Restraint per Residue | 26 |
Table 3. Structural Characterization of the AFB-α-FAPY Adduct in Duplex DNAa.
| intra R1X | inter R1X | overall R1X | |
|---|---|---|---|
| ds-α-FAPY-i1c | 11.4 | 12.9 | 11.8 |
| ds-α-FAPY-i2c | 11.2 | 10.9 | 11.1 |
| ds-α-FAPY-i3c | 11.1 | 11.3 | 11.2 |
| ds-α-FAPY-i4c | 11.2 | 10.9 | 11.1 |
| ds-α-<rMD i>d | 8.5 | 9.7 | 8.8 |
| ds-α-<rMD ensemble>e | 8.4 | 9.9 | 8.7 |
| ds-β-FAPY-iAb | 9.5 | 12.1 | 11.5 |
| ds-β-FAPY-iBb | 8.8 | 9.9 | 8.9 |
| ds-β-FAPY-<rMDavg>b,f | 8.5 | 9.5 | 8.7 |
Comparison of sixth root residual indices, R1X (×10−2), for starting models and structural ensembles emergent from rMD calculations.
Reported by Mao et al.(38)
Starting structures for rMD calculations using the simulated annealing protocol; i1, i2, i3, i4 represent four separate initial structures.
Starting structure used for isothermal rMD calculations in explicit solvent.
Ensemble of ten structures extracted from the isothermal rMD calculations, calculated with an equal occupancy of 10% for each member of the ensemble.
<rMDavg> reported as average of 12 rMD structures starting from either A-form or B-form structures as reported by Mao et al.(38)
Isothermal rMD Calculations in Explicit Solvent
To examine the dynamics of the refined structure and to analyze hydrogen bond occupancies involving the formamido group of the FAPY moiety, 5 ns of equilibrium rMD calculations were performed in explicit solvent, using a procedure similar to that of Aramini et al.(47) The 5 ns rMD trajectory was analyzed for occupancies of hydrogen bonding motifs. Hydrogen bond occupancies were calculated using a distance cutoff of 3.5 Å and an angle cutoff of 120°. On this basis, the A6 H61 non-Watson−Crick hydrogen bonded exocyclic amine proton satisfied hydrogen bond criteria with the X5 formyl oxygen for 95% of the trajectory.
Figure 6.

Stereoview of nine superimposed structures of the AFB-α-FAPY modified duplex extracted from isothermal rMD calculations in explicit solvent (PDB ID: 2KH3). The AFB moiety and the formyl oxygen are depicted in red. The R1x residual of 8.7 × 10−2 for the ensemble indicates agreement with NOESY data (Table 3).
Structure of the AFB-α-FAPY Modified Duplex
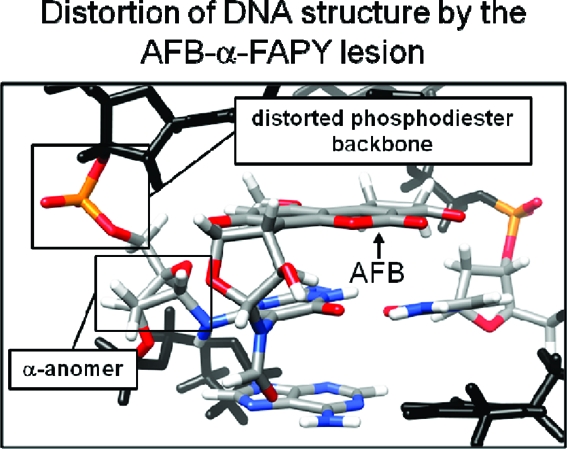
Expanded views of the structure are shown in Figure 7. The AFB moiety intercalated above the 5′-face of the modified nucleotide X5 and between base pairs T4·A17 and X5·C16, causing the rise between these base pairs to increase from 3.4 to 7.0 Å. In addition, the duplex unwound by 15° relative to the unmodified duplex. The FAPY moiety was in the Ra conformation about the C5−N5 bond. The A6 H61 non-Watson−Crick hydrogen bonded exocyclic amine proton was within hydrogen bonding distance of the X5 formyl oxygen; this positioned the formamide in the E conformation. The deoxyribose moiety of the AFB-α-FAPY adduct was also altered as compared to the AFB-β-FAPY adduct;(38) it was approximately parallel to the FAPY base (Figure 7). Perturbations of the AFB-α-FAPY duplex resulted from altered intercalation of the AFB moiety between base pairs T4·A17 and X5·C16 (Figure 8). The AFB moiety exhibited favorable stacking between AFB and the X5 pyrimidine moiety. However, there was reduced interstrand stacking between AFB and C16 of the complementary strand. At the AFB-FAPY modified X5 nucleotide, the relative differences in backbone torsion angles for the duplexes containing AFB-α-FAPY and AFB-β-FAPY38 adducts were as follows: α, 50°; β, 16°; γ, −21°; δ, 9°; ε, 243°; ζ, −215° (Table 4); the result was that the phosphate groups were oriented approximately orthogonal relative to each other (Figure 9).
Figure 7.
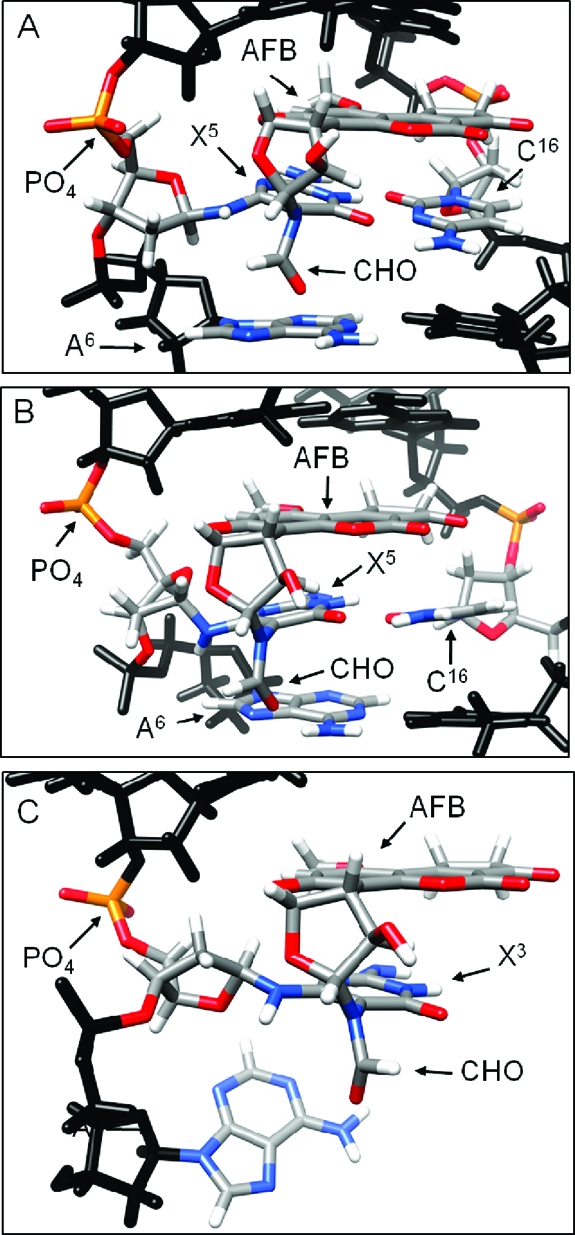
(A) Refined structure of the AFB-β-FAPY adduct in duplex DNA (PDB ID: 1HM1).(38) (B) Refined structure of the AFB-α-FAPY adduct in duplex DNA (PDB ID: 2KH3). (C) Molecular model of the AFB-α-FAPY adduct the single stranded tetramer (PDB ID: 2KH4).
Figure 8.

Stacking interactions of AFB-α-FAPY adduct in single-strand (molecular modeling) and duplex DNA (refined structure) compared with the stacking interactions of the AFB-β-FAPY adduct (refined structure) in duplex DNA. (A) The AFB-β-FAPY adduct in duplex DNA (PDB ID: 1HM1).(38) (B) The AFB-α-FAPY adduct in duplex (PDB ID: 2KH3). (C) The AFB-α-FAPY adduct as modeled in the tetramer 5′-d(CTXA)-3′ (PDB ID: 2KH4).
Table 4. Comparison of X5 FAPY Nucleotide Torsion Anglesa.
| angle | AFB-β-FAPY dsDNAb | AFB-α-FAPY dsDNA | AFB-α-FAPY ssDNA |
|---|---|---|---|
| α | −34 | −84 | −75 |
| β | −155 | −171 | 178 |
| γ | 24 | 46 | −46 |
| δ | 148 | 139 | 158 |
| ε | 138 | −105 | −101 |
| ζ | −98 | 118 | 82 |
| O4′-C1′-N6−H6 | 112 | −77 | −58 |
| H6−N6−C6-C5 | 33 | −129 | −44 |
Figure 9.
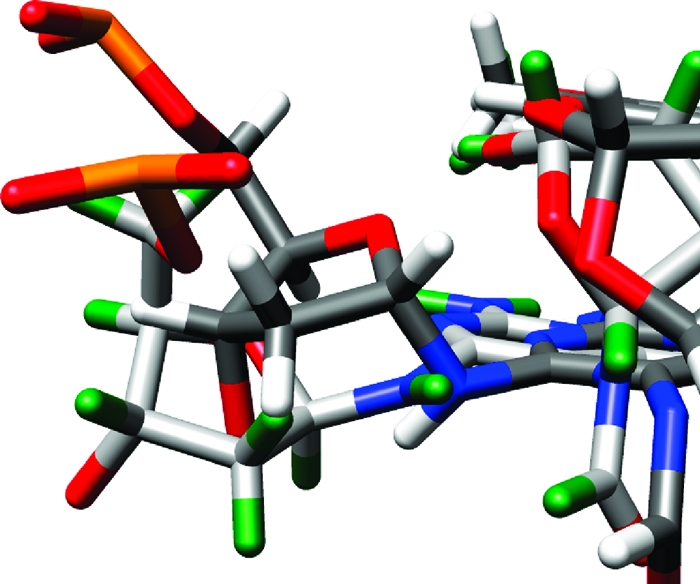
Overlay of AFB-α-FAPY and AFB-β-FAPY adducts, showing differences in phosphodiester backbone geometry. AFB-β-FAPY is depicted with light gray carbon atoms and green hydrogen atoms. AFB-α-FAPY is depicted with dark gray carbon atoms and white hydrogen atoms. In both structures, oxygen atoms are red and phosphorus atoms are orange.
The AFB-α-FAPY Modified Tetramer
The tetramer 5′-d(C1T2X3A4)-3′ modeled the AFB-α-FAPY adduct in the single strand. NMR experiments were conducted at 0.5 mM concentration, to minimize the potential for self-association by the tetramer, for example, as has been reported for 5′-d(CTGA)-3′(55) and 5′-d(CGA)-3′.(56) Under these conditions, no self-association of the 5′-d(C1T2X3A4)-3′ tetramer was observed. At 7 °C, no resonances from exchangeable protons were observed, suggesting that the tetramer did not form interstrand hydrogen bonds. Only intranucleotide NOEs were observed, which was attributed to the disorder of the single stranded oligomer. The C1 and T2 spin systems were identified from COSY spectra on the basis of 3JH5−H6 and 4JCH3-H6 couplings, respectively. The A4 assignment was based on the chemical shifts of the aromatic signals and was consistent with published values.(55) The X3 resonances were assigned via a process of elimination. When the chemical shifts of nucleotides T2, X3, and A4 of the tetramer were compared to the corresponding nucleotides T4, X5 and A6 of the duplex, the former were deshielded an average of 0.5 ppm relative to those of the duplex (Panel F of Figure 5). The deoxyribose spin systems were assigned by NOE correlations between deoxyribose H1′ and H2′ protons and pyrimidine H6 and purine H8 protons. The intensity of the X5 H1′ → X5 H2′′ NOE was less than the X5 H1′ → X5 H2′ NOE, which established the α configuration (Figure 3, panel B). The AFB H5, H6a, H8, H9, H9a, and -OCH3 resonances were assigned from COSY and NOESY data.(38) A strong NOE from AFB H5 to AFB -OCH3 revealed that the latter resonance was at δ 3.76 ppm. The AFB H5 and X5 CHO proton resonances were observed at δ 5.7 and 8.3, respectively. The X5 CHO proton had a NOE cross peak with AFB H8. A single resonance assigned as the formyl proton was observed at 8.3 ppm in spectra acquired at 5 °C; it exhibited a strong NOE to the AFB H8 proton (Figure S6 in the Supporting Information). At 30 °C, an additional sharp resonance was observed at δ 7.5 ppm, which produced a weak NOE to AFB H8. This was assigned as the formyl resonance arising from the Z configuration of the formamide. The two formyl resonances did not exhibit a chemical exchange cross peak.
Structure of the AFB-α-FAPY Adduct in Single-Strand DNA
The only AFB internucleotide NOEs involved the AFB H5 and -OCH3 to the T2 H1′, T2 H2′, and T2 H2′′ deoxyribose protons (Figure S7 in the Supporting Information). Internucleotide NOEs between the AFB moiety and the 3′-neighbor A4 were not observed. This established that the AFB moiety was located above the 5′-face of the pyrimidine moiety at the adducted nucleotide, and was located between nucleotides T2 and X3. This resulted in the C5−N5 bond of the FAPY adduct being in the Ra axial conformation. The E conformation for the AFB formamide was assigned on the basis of its 8.3 ppm chemical shift,(32) and was corroborated by a strong NOE between the formyl proton and AFB H8. The intensities of the NOEs for the deoxyribose resonances indicated that the nucleotide remained as the α-anomer (Figure 3).
It was not possible to refine the structure of the AFB-α-FAPY adduct in the tetramer due to the low number of NOEs that were observed. However, molecular modeling studies, in which the AFB moiety was placed above the 5′-face of the pyrimidine moiety at the damaged nucleotide, and in which the Ra axial conformation of the FAPY C5−N5 bond and the E conformation for the AFB formamide were maintained as suggested by the NMR data, indicated that in the tetramer the structure of the AFB-α-FAPY adduct was likely to be similar to that observed in the duplex (Figure 7). The A4 non-Watson−Crick exocyclic amine proton was within hydrogen bonding distance of the X5 formyl oxygen. This held the formyl group in the E geometrical configuration. The phosphodiester backbone torsion angles at the X5 nucleotide are listed in Table 4. The relative differences in backbone torsion angles between the AFB-α-FAPY adduct in duplex and in the tetramer were the following: α, −9°; β, −349°; γ, 91°; δ −19°; ε, −4.5°; ζ, 36°. At the adduct site X5, the deoxyribose was approximately orthogonal to the pyrimidine moiety, with O4′ oriented in the 3′ direction (Figure 7, panel C); this differed from the structure of the AFB-β-FAPY adduct in duplex DNA for which O4′ was oriented in the 5′ direction (Figure 7, panel A).
Discussion
The AFB-α-FAPY Adduct in Duplex DNA
The break in the sequential NOE connectivity for the adducted strand between T4 H6 and X5 H1′ and the corresponding break in connectivity for the complementary strand between C16 H1′ and A17 H8 provides evidence that the AFB moiety is intercalated between base pairs T4·A17 and X5·C16. Thus, the AFB moiety intercalates above the 5′-face of the adducted pyrimidine base in double-stranded DNA. In this duplex, the axial conformation of the pyrimidine C5−N5 bond is Ra. In the same oligodeoxynucleotide sequence, the AFB-β-FAPY adduct also exhibited the Ra conformation.(38) For the AFB-α-FAPY adduct, the formyl group is positioned to hydrogen bond with the A6 non-Watson−Crick exocyclic amine proton. No evidence is found for equilibration between Ra and Sa atropisomers. For the Sa isomer, Watson−Crick base-pairing would position the AFB moiety on the 3′ face, rather than on the 5′-face of the adducted pyrimidine. Thus, the preference for the Ra atropisomer and 5′-intercalation of the AFB moiety is observed irrespective of the anomeric configuration of the deoxyribose.
Comparison with the AFB-β-FAPY Adduct in Duplex DNA
While the AFB-α-FAPY and AFB-β-FAPY adducts maintain similar 5′-intercalated structures in duplex DNA, for the AFB-α-FAPY adduct, maintaining this structure comes at a high price. The α deoxyribose anomer disrupts the right-handed helical structure of DNA. Intercalation of the AFB moiety induces perturbations in the phosphodiester backbone of the AFB-α-FAPY modified duplex. Differences of >100° in the phosphodiester ε and ζ backbone torsion angles are observed when the oligodeoxynucleotide containing the AFB-α-FAPY is compared with that containing the AFB-β-FAPY adduct (Figure 9).(38) Germann and co-workers also concluded that the introduction of α anomeric nucleotides into DNA induces distortions of the phosphodiester backbone.47,57−60 Additionally, the backbone distortions are accompanied by differences in the orientations of the deoxyribose moieties of AFB-α-FAPY and AFB−β-FAPY adducts (Figure 8). For the AFB-α-FAPY adduct, the deoxyribose is parallel to the FAPY base and displaced toward the minor groove by 2 Å. The α deoxyribose of AFB-FAPY is not orthogonal to the modified base as was observed for the α-adenosine nucleotide in duplex DNA,(47) suggesting the distorted deoxyribose geometry results from the intercalated AFB and not the α-anomer itself. For the β-anomer, the deoxyribose is orthogonal to the plane of the FAPY base, similar to canonical DNA bases.(38) In the AFB-β-FAPY modified duplex, the AFB moiety is intercalated such that it stacks not only with T4 and the X5 pyrimidine ring in its own strand, but also with A17 and C16 of the complementary strand.38,39 In contrast, for the AFB-α-FAPY modified duplex, the AFB moiety maintains stacking between T4 and the X5 pyrimidine ring, but stacking is reduced with A17 and C16 in the complementary strand (Figure 8). This difference in stacking is consistent with differences in the CD (Figure S1 in the Supporting Information) and UV (Figure S2 in the Supporting Information) spectra relative to the duplex containing the AFB-β-FAPY adduct; the CD spectrum shows a greater negative induced molar ellipticity at 310−400 nm.61−63
The differences in the phosphodiester backbone conformations and in stacking between the AFB-α-FAPY and AFB-β-FAPY adducts explain the lower Tm value for the duplex containing the AFB-α-FAPY adduct. The biphasic melting transition of the duplex containing the AFB-α-FAPY adduct, with the initial Tm value of 23 °C followed by a second transition at 50 °C (Figure 2, panel C), is interpreted in terms of an initial melting of the duplex containing the less favored AFB-α-FAPY configuration with subsequent isomerization to the preferred AFB-β-FAPY configuration, which produces the 50 °C transition. The AFB-β-FAPY adduct differs from the AFB-α-FAPY adduct in that it stabilizes this duplex with respect to the unadducted duplex, leading to a Tm of 50 °C, that is, a 14 °C rise (Figure 2, panel B). Above the Tm, equilibration to a mixture of α- and β-anomers occurs, leading to a biphasic cooling transition in both the AFB-α-FAPY and AFB-β-FAPY samples. The role of base stacking in modulating nucleic acid stability is apparent.64−68 The stability of the AFB-β-FAPY modified duplex has been attributed to favorable stacking of the intercalated AFB moiety,38,39 and the reduction of AFB stacking is credited with reduced stability of the AFB-α-FAPY modified duplex.
The AFB-α-FAPY Adduct in Single-Strand DNA
The shielding of AFB H5, H6a, H8, and H9 resonances is consistent with the stacking of the AFB moiety with neighboring nucleotides. The chemical shift values are in agreement with those observed for the AFB-α-FAPY modified duplex, with the exception of AFB H9a (δ 3.66 tetramer; δ 3.47 duplex). The downfield shift of AFB H9a in the tetramer is indicative of reduced stacking of the furan portion of the AFB moiety in the tetramer. Several key NOEs are observed, locating the AFB moiety above the 5′ face of the pyrimidine moiety of the damaged nucleotide. Although the inability to observe many internucleotide NOEs diminishes the detail with which the structure of the AFB-α-FAPY can be defined in single-strand DNA, the data suggest that the structure of the AFB-α-FAPY adduct in single-stranded DNA is similar to that in double-stranded DNA. This suggests that stacking permits the AFB moiety to escape from the aqueous environment. The concept of ‘hydrophobic bonding’, introduced by Nemethy and Scheraga,(69) is important in protein folding70−74 but is equally important in stabilizing DNA.68,75,76 Interestingly, stacking of the AFB moiety on the 5′ face of the pyrimidine moiety at the adduct site is favored both for double- and single-stranded DNA, suggesting that this orientation produces better stacking with the neighboring nucleobases than would occur on the 3′ face of the pyrimidine moiety. This is a function of the asymmetry of AFB itself, not the configuration of the deoxyribose or structure of the oligodeoxynucleotide because the preference for 5′ stacking was observed in our previous study of the configuration of the AFB-β-FAPY adduct in the nucleoside and even in the base.(32) Thus, stacking interactions alone are sufficient to lead to the Ra preference.
Role of Hydrogen Bonding
In this DNA duplex, the formamide moiety of the AFB-α-FAPY adduct is held in the E configuration by a hydrogen bond between the formyl oxygen and the N6 non-Watson−Crick hydrogen bonded exocyclic amine proton of the 3′ neighboring A6. Analysis of rMD trajectories indicates 95% occupancy for this hydrogen bond (Table 4). Mao et al.(38) reported a similar hydrogen bond with the 3′-neighboring adenine for the AFB-β-FAPY adduct, and correlated this with the deshielding of the A6 exocyclic amine proton; deshielding of the A6 non-Watson−Crick hydrogen bonded exocyclic amine proton is also observed in the AFB-α-FAPY NMR data. Additional evidence for the presence of this hydrogen bond comes from the observation of a strong NOE between the formamide proton and AFB H8. In this duplex, the preference of both anomers for the E conformation differs from the situation at the nucleoside level, where the Z conformation is favored.(32)
HPLC analysis of the strand containing the AFB-α-FAPY adduct revealed a shoulder on the chromatographic peak (Figure 1). The shoulder species rapidly equilibrates with the major component in a pH-independent fashion and, therefore, is assigned as the non-hydrogen bonding Z geometrical isomer of the formamide. The oligomer containing the AFB-β-FAPY adduct did not show a shoulder meaning that either the adenine hydrogen bond is stronger for the β-anomer or the geometrical isomers coelute. Moreover, for the duplex containing the AFB-α-FAPY adduct, a second formamide resonance appears at 30 °C that has only a weak NOE to H8 and is assigned as the Z conformation (Figure S3 in the Supporting Information).
The discovery of this hydrogen bond and its effect on adduct structure suggests that other sequences should be studied. A neighboring cytosine might provide similar stabilization of the E conformation but a thymine would not. We have, as yet, little insight on the effects of 5′ neighbors, but the 5′-thymine may not be optimal due to steric interference with intercalation by the methyl group. Thus, a C:G pair on the 5′ side of the adduct might lead to improved duplex stability. Modifications of AFB that decrease planarity or increase bulk of the cyclopentenone ring interfere with equilibrium binding to DNA and reduce the efficiency of reactions of the epoxides with DNA.77,78
Biological Implications
A broad picture has emerged concerning the chemistry of AFB FAPY adducts in DNA and their biological ramifications. Opening of the imidazole ring of the initially formed cationic AFB-N7-dGuo adduct2,28 yields the AFB-β-FAPY adduct31,32 (Figure 8, panel A), which stabilizes duplex DNA by improved stacking interactions (Figure 9, panel A).38,39 The AFB-β-FAPY adduct is highly mutagenic in E. coli (36%) and is postulated to be predominantly responsible for the genotoxicity associated with AFB exposures.(33) In single strand DNA, the lack of the complementary strand facilitates the epimerization of the deoxyribose and the formation of an equilibrium mixture of α and β FAPY anomers, with the α-anomer predominating in this sequence context.(33) The position of the equilibrium is controlled by a number of factors, including optimization of intrastrand stacking interactions between the AFB moiety and its neighboring base pairs, and the shielding of the hydrophobic AFB moiety from solvent (Figure 9, panel C). The single strand DNA, the AFB moiety of the AFB-α-FAPY adduct lies on the 5′ face of the pyrimidine moiety at the adducted base with the deoxyribose ring orthogonal to the base and the 5′ phosphate group protruding into solvent (Figure 8, panel C); the deoxyribose conformation is similar to that of α-adenosine in duplex DNA.(47) Upon annealing with a complementary strand of DNA, the presence of the α-anomer distorts the phosphodiester backbone and base stacking register, thus, lowering the thermal stability of the resulting duplex. This is apparent in the inability of the deoxyribose moiety of the α-anomer to remain orthogonal to the FAPY base in duplex DNA (Figure 7). Consequently, the AFB-α-FAPY adduct reverts to AFB-β-FAPY (Figure 8, panel A).(32)
Anomerization in single strand DNA is slow at physiological pH;(32) it may be so slow that DNA polymerases will not encounter significant amounts of the AFB-α-FAPY adduct, which blocks DNA replication in E. coli.(33) However, in the presence of histones and multicomponent protein arrays involved in replication and repair, anomerization could be faster. Also, the study of replication across the two anomers of AFB-FAPY was carried out in E. coli;(33) eukaryotes may have alternative pathways available for bypass of the AFB-α-FAPY adduct. Finally, the mutagenesis studies utilized a sequence in which the 3′-neighbor nucleotide was an adenine. The present study of AFB-α-FAPY and the studies of the AFB-β-FAPY38,39 adduct reveal that the 3′-neighbor adenine holds the formamide in the normally less stable E configuration. This may not be true in other sequences, potentially enabling the bypass of the AFB-α-FAPY adduct. The effects of each of the two anomers of the AFB-FAPY adduct upon DNA repair also remains to be determined. In E. coli, the rate of incision by UvrABC endonuclease on AFB-FAPY adducts varies by sequence, with those containing a 3′-neighbor dA being more resistant to cleavage.(79) While not reported, the anomeric status of the AFB-FAPY adduct was probably β, because it involved double-stranded DNA.
Experimental Section
Chemicals
The oligodeoxynucleotides 5′-d(CTATGATTCA)-3′, 5′-d(TGAATCATAG)-3′, 5′-d(AATCATA)-3′, and 5′-d(CTGA)-3′ were purchased from Midland Certified Reagent Co. (Midland, TX) and purified by reverse-phase HPLC. Their concentrations were measured by UV absorbance at 260 nm.(80) Dimethyldioxirane was synthesized and assayed as described;81−83 solutions were stored over anhydrous MgSO4 at −20 °C and used within 1 week of preparation. AFB was purchased from Aldrich Chemical Co. (Milwaukee, WI). AFB exo-8,9-epoxide was prepared as described.(84)Caution: AFB is a potent liver toxin and is genotoxic, and it should be presumed that AFB exo-8,9-epoxide is highly toxic and genotoxic. Crystalline aflatoxins are particularly hazardous due to their electrostatic nature. AFB can be destroyed by oxidation with NaOCl. Manipulations should be carried out in a well-ventilated hood with suitable containment procedures.
Sample Preparation
The oligodeoxynucleotide 5′-d(CTATGATTCA)-3′, containing the targeted N7-dGuo alkylation site (underlined), was annealed with 5′-d(AATCATA)-3′ in 100 mL of 100 mM sodium phosphate buffer (pH 6.5) to form a partially double-stranded scaffold. AFB exo-8,9-epoxide (1 mg) was dissolved in 100 μL of anhydrous CH2Cl2 to produce a 3.2 mM solution. The epoxide was added in two 50 μL aliquots to the oligodeoxynucleotide solution at 5 °C, to create a 5:1 AFB exo-8,9-epoxide/oligodeoxynucleotide molar ratio. The biphasic mixture was stirred at 5 °C for 15 min. The product 5′-d(CTATXATTCA)-3′ (X = AFB-N7-dGuo) was separated from 5′-d(CTATGATTCA)-3′ and 5′-d(AATCATA)-3′ using HPLC (Gemini C-18 250 mm × 10 mm column, Phenomenex, Inc., Torrance, CA) at a flow rate of 2 mL/min, with a linear 25 min gradient of 6−30% CH3CN in 0.1 M ammonium formate (pH 8.0). The eluant was monitored by UV absorbance at 260 and 360 nm. The yield of 5′-d(CTATXATTCA)-3′ (X = AFB-N7-dGuo) was 90%. The 5′-d(CTATXATTCA)-3′ (X = AFB-N7-dGuo) was suspended in 500 mL of 100 mM Na2CO3 at 37 °C (pH 10). The AFB-α-FAPY and AFB-β-FAPY modified oligodeoxynucleotides were separated by HPLC as described above and collected in a saturated Na2HPO4 solution. The AFB-α-FAPY modified 5′-d(CTATXATTCA)-3′ oligodeoxynucleotide was desalted using a C-18 Sep-Pak cartridge (Waters Corp, Milford, MA) before annealing with 5′-d(TGAATCATAG)-3′ in a 1:1 molar ratio in 0.2 M sodium phosphate, 0.1 M NaCl, 10 mM NaN3, and 50 μM Na2EDTA (pH 8.7) to yield a duplex concentration of 1 mM. For the tetramer 5′-d(CTGA)-3′, the adduction reaction utilized a 5:1 epoxide/oligodeoxynucleotide ratio that provided a 40% yield of adducted tetramer at strand concentrations >1 mM. Its purification, conversion to the FAPY adduct, and isolation of the oligodeoxynucleotide containing the AFB-α-FAPY adduct followed the protocols described above. All samples were stored at 5 °C except during spectroscopic measurements.
Thermal Melting
UV thermal melting analyses were conducted in 1 mL of 10 mM sodium phosphate, 0.5 M NaCl, and 5 mM Na2EDTA (pH 7.0) and monitored at 260 nm. The concentration of all samples was maintained at 0.1 A260 unit under denaturing conditions. The Tm values were determined from first derivatives of the absorbance versus temperature curves. The experiments temperature range was 5−80 °C.
NMR Spectroscopy
Spectra were recorded at 1H frequencies of 500, 600, and 800 MHz. To examine nonexchangeable protons, samples were suspended in D2O. For observation of exchangeable protons, samples were suspended in 9:1 H2O/D2O. The programs XWINNMR and TOPSPIN (Bruker, Inc., Billerica, MA) were used for data processing. Resonance assignment and peak integration were performed using the program SPARKY.(85) COSY spectra were conducted in double quantum filtered (DQF)(86) and magnitude modes. A series of experiments were collected at NOE mixing times of 80, 150, 200, and 250 ms; the NOESY spectra for the single strand tetramer samples were collected at mixing times of 250 and 300 ms. NOESY87,88 experiments used for the assignment of exchangeable protons used the watergate pulse sequence.(89) Water suppression for heteronuclear 1H−31P correlation(90) experiments was achieved with the Dante pulse sequence.(91) Pulses were optimized for 12 Hz 3JPH coupling constants.
NMR Restraints
NOE intensities were determined from integration of cross-peak volumes using the program SPARKY.(85) Experimental intensities were combined with those generated from complete relaxation matrix analysis of a model structure, using established methodologies,(44) to produce a hybrid intensity matrix.52,53 The program MARDIGRAS,92,93 using the RANDMARDI94,95 function, was used to refine the hybrid intensity matrix. Calculations at NOE mixing times of 80, 150, 200, and 250 ms were conducted using isotropic correlation times of 2, 3, 4, and 5 ns; analysis of the resulting distances produced the distance restraints used in rMD calculations. Torsion angle restraints were determined from 31P−1H heterocorrelation, DQF-COSY, and NOESY spectra. Phosphodiester backbone restraints96−98 were applied to the α, β, γ, ν0−ν4 angles except that the backbone torsion angles at X5 and C16 were not restrained. The χ0 to χ4 deoxyribose torsion angles were determined by pseudorotation analysis.(99) For the N-type conformation, the phase angle ρ was restrained between 0° and 205°, and for the S-type conformation, the phase angle ρ was restrained between 110° and 205°.
Structural Refinement
The refinement used a simulated annealing rMD protocol involving established methods,44−47 and employing generalized Born solvation100,101 with the AMBER(102) force field. A 15 Å cutoff for nonbonded interactions was used, and bond lengths involving hydrogen were fixed using the SHAKE algorithm.(103) The emergent structures from 20 calculations with different starting velocities were analyzed as to pairwise rmsd deviations. This provided a measure of the precision of the calculations. Complete relaxation matrix analysis was performed using the program CORMA.52,53 This provided a measure of the accuracy of the calculations. The output from the simulated annealing calculations was also validated with respect to restraint violations. Graphical analysis was performed with InsightII (Accelrys, San Diego, CA) and CHIMERA.(54)
Isothermal rMD Calculations in Explicit Solvent
The starting structure was a representative potential energy minimized structure from the ensemble of refined structures for the duplex containing the AFB-α-FAPY adduct emergent from the simulated annealing calculations.(44) The DNA was neutralized with Na+ ions and placed in a truncated octahedral TIP3P water box with periodic boundaries at a distance of 8 Å from the solute. Sodium ions were constrained to be proximate to sequential backbone phosphates using restraints having a lower bound of 3.0 Å and an upper bound of 8.0 Å. The solvated system was energy minimized for 500 steps of steepest descents followed by 500 steps of conjugate gradients at constant volume with the solute held fixed. Next, the system was subjected to potential energy minimization at a constant volume for 2500 steps with no positional restraints. The system was subsequently heated to 300 K over 100 ps. The experimental distance and torsion angle restraints and empirical restraints (vide supra) were increased linearly during the heating. Isothermal rMD calculations were performed for 5 ns at 300 K using the AMBER(102) force field, restrained by experimental distance and torsion angle restraints and empirical restraints. The temperature was maintained using the Langevin thermostat104,105 with a collision frequency of 1 ps−1. Electrostatic interactions were treated with the particle mesh Ewald (PME) method.(106) A 15 Å cutoff for nonbonded interactions was used. Bond lengths involving hydrogen were fixed using the SHAKE algorithm.(103) The trajectories were analyzed with the programs PTRAJ and SUPPOSE.(102)
Acknowledgments
Dr. Nicholas Uylanov provided guidance with MARDIGRAS calculations and refinement. Dr. Jarrod A. Smith assisted with refinement. Professor Ivan Kozekov and Ms. Pamela Tamura assisted with mass spectrometry and HPLC. This work was funded by NIH grants RO1 CA-55678 (M.P.S.), the Vanderbilt University Center in Molecular Toxicology, P30 ES-00267, the Vanderbilt-Ingram Cancer Center, P30 CA-68485. Funding for NMR was supplied by Vanderbilt University and by NIH grant RR-05805.
Supporting Information Available
The Supporting Information includes Table S1, resonance assignments for the AFB-α-FAPY modified tetramer, and Figures S1, circular dichroism of AFB-FAPY anomers in oligodeoxynucleotides; S2, UV spectra of AFB-α-FAPY-modified 5′-d(CTATXATTCA)-3′; S3, sequential connectivity NOEs for the AFB-α-FAPY modified duplex; S4, imino and amino resonance assignments for AFB-α-FAPY modified duplex; S5, 31P NMR for the AFB-α-FAPY-modified duplex and tetramer; S6, 1H NMR spectra of tetramer formamide protons; and S7, NOE assignments of involving AFB for the AFB-α-FAPY modified tetramer. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Busby W. F. Jr.; Wogan G. N. In. Chemical Carcinogens; 2nd ed.; Searle C. E., Ed.; American Chemical Society: Washington, D.C., 1984; pp 945−1136. [Google Scholar]
- Smela M. E.; Currier S. S.; Bailey E. A.; Essigmann J. M. Carcinogenesis 2001, 22, 535–545. [DOI] [PubMed] [Google Scholar]
- Bennett J. W.; Klich M. Clin. Microbiol. Rev. 2003, 16, 497–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann J.; Spingarn N. E.; Koburi J.; Ames B. N. Proc. Natl. Acad. Sci. U.S.A. 1975, 72, 979–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster P. L.; Eisenstadt E.; Miller J. H. Proc. Natl. Acad. Sci. U.S.A. 1983, 80, 2695–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster P. L.; Groopman J. D.; Eisenstadt E. J. Bacteriol. 1988, 170, 3415–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey G. S.; Williams D. E.; Wilcox J. S.; Loveland P. M.; Coulombe R. A.; Hendricks J. D. Carcinogenesis 1988, 9, 1919–1926. [DOI] [PubMed] [Google Scholar]
- Bailey G. S.; Loveland P. M.; Pereira C.; Pierce D.; Hendricks J. D.; Groopman J. D. Mutat. Res. 1994, 313, 25–38. [DOI] [PubMed] [Google Scholar]
- McMahon G.; Davis E. F.; Huber L. J.; Kim Y.; Wogan G. N. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 1104–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soman N. R.; Wogan G. N. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 2045–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; Zhou H.; Kong R. Y.; Fong W. F.; Ren L. Q.; Liao X. H.; Wang Y.; Zhuang W.; Yang S. Mutat. Res. 1997, 381, 25–29. [DOI] [PubMed] [Google Scholar]
- Rashid A.; Wang J. S.; Qian G. S.; Lu B. X.; Hamilton S. R.; Groopman J. D. Br. J. Cancer 1999, 80, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y.; Zhu J. J.; Han F.; Ishikawa T.; Oda H. Int. J. Cancer 1999, 82, 187–190. [DOI] [PubMed] [Google Scholar]
- Katiyar S.; Dash B. C.; Thakur V.; Guptan R. C.; Sarin S. K.; Das B. C. Cancer 2000, 88, 1565–1573. [DOI] [PubMed] [Google Scholar]
- Kirk G. D.; Camus-Randon A. M.; Mendy M.; Goedert J. J.; Merle P.; Trepo C.; Brechot C.; Hainaut P.; Montesano R. J. Natl. Cancer. Inst. 2000, 92, 148–153. [DOI] [PubMed] [Google Scholar]
- Bressac B.; Kew M.; Wands J.; Ozturk M. Nature 1991, 350, 429–431. [DOI] [PubMed] [Google Scholar]
- Hsu I. C.; Metcalf R. A.; Sun T.; Welsh J. A.; Wang N. J.; Harris C. C. Nature 1991, 350, 427–428. [DOI] [PubMed] [Google Scholar]
- Greenblatt M. S.; Bennett W. P.; Hollstein M.; Harris C. C. Cancer Res. 1994, 54, 4855–4878. [PubMed] [Google Scholar]
- Shen H. M.; Ong C. N. Mutat. Res. 1996, 366, 23–44. [DOI] [PubMed] [Google Scholar]
- Soini Y.; Chia S. C.; Bennett W. P.; Groopman J. D.; Wang J. S.; DeBenedetti V. M.; Cawley H.; Welsh J. A.; Hansen C.; Bergasa N. V.; Jones E. A.; DiBisceglie A. M.; Trivers G. E.; Sandoval C. A.; Calderon I. E.; Munoz Espinosa L. E.; Harris C. C. Carcinogenesis 1996, 17, 1007–1012. [DOI] [PubMed] [Google Scholar]
- Lunn R. M.; Zhang Y. J.; Wang L. Y.; Chen C. J.; Lee P. H.; Lee C. S.; Tsai W. Y.; Santella R. M. Cancer Res. 1997, 57, 3471–3477. [PubMed] [Google Scholar]
- Mace K.; Aguilar F.; Wang J. S.; Vautravers P.; Gomez-Lechon M.; Gonzalez F. J.; Groopman J.; Harris C. C.; Pfeifer A. M. Carcinogenesis 1997, 18, 1291–1297. [DOI] [PubMed] [Google Scholar]
- Shimada T.; Guengerich F. P. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 462–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raney K. D.; Shimada T.; Kim D. H.; Groopman J. D.; Harris T. M.; Guengerich F. P. Chem. Res. Toxicol. 1992, 5, 202–210. [DOI] [PubMed] [Google Scholar]
- Ueng Y. F.; Shimada T.; Yamazaki H.; Guengerich F. P. Chem. Res. Toxicol. 1995, 8, 218–225. [DOI] [PubMed] [Google Scholar]
- Gallagher E. P.; Kunze K. L.; Stapleton P. L.; Eaton D. L. Toxicol. Appl. Pharmacol. 1996, 141, 595–606. [DOI] [PubMed] [Google Scholar]
- Johnson W. W.; Harris T. M.; Guengerich F. P. J. Am. Chem. Soc. 1996, 118, 8213–8220. [Google Scholar]
- Essigmann J. M.; Croy R. G.; Nadzan A. M.; Busby W. F. Jr.; Reinhold V. N.; Buchi G.; Wogan G. N. Proc. Natl. Acad. Sci. U.S.A. 1977, 74, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan S.; Byrd S.; Stone M. P.; Harris T. M. Biochemistry 1989, 28, 726–734. [DOI] [PubMed] [Google Scholar]
- Iyer R. S.; Coles B. F.; Raney K. D.; Thier R.; Guengerich F. P.; Harris T. M. J. Am. Chem. Soc. 1994, 116, 1603–1609. [Google Scholar]
- Hertzog P. J.; Smith J. R. L.; Garner R. C. Carcinogenesis 1982, 3, 723–725. [DOI] [PubMed] [Google Scholar]
- Brown K. L.; Deng J. Z.; Iyer R. S.; Iyer L. G.; Voehler M. W.; Stone M. P.; Harris C. M.; Harris T. M. J. Am. Chem. Soc. 2006, 128, 15188–15199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smela M. E.; Hamm M. L.; Henderson P. T.; Harris C. M.; Harris T. M.; Essigmann J. M. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 6655–6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey E. A.; Iyer R. S.; Stone M. P.; Harris T. M.; Essigmann J. M. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 1535–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzog P. J.; Lindsay Smith J. R.; Garner R. C. Carcinogenesis 1980, 1, 787–793. [DOI] [PubMed] [Google Scholar]
- Croy R. G.; Wogan G. N. Cancer Res. 1981, 41, 197–203. [PubMed] [Google Scholar]
- Groopman J. D.; Croy R. G.; Wogan G. N. Proc. Natl. Acad. Sci. U.S.A. 1981, 78, 5445–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H.; Deng Z.; Wang F.; Harris T. M.; Stone M. P. Biochemistry 1998, 37, 4374–4387. [DOI] [PubMed] [Google Scholar]
- Giri I.; Stone M. P. Biopolymers 2002, 65, 190–201. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan S.; Harris T. M.; Stone M. P. Biochemistry 1990, 29, 10438–10448. [DOI] [PubMed] [Google Scholar]
- Johnston D. S.; Stone M. P. Biochemistry 1995, 34, 14037–14050. [DOI] [PubMed] [Google Scholar]
- Jones W. R.; Johnston D. S.; Stone M. P. Chem. Res. Toxicol. 1998, 11, 873–881. [DOI] [PubMed] [Google Scholar]
- Giri I.; Jenkins M. D.; Schnetz-Boutaud N. C.; Stone M. P. Chem. Res. Toxicol. 2002, 15, 638–647. [DOI] [PubMed] [Google Scholar]
- Schmitz U.; James T. L. Methods Enzymol. 1995, 261, 3–44. [DOI] [PubMed] [Google Scholar]
- Konerding D.; James T. L.; Trump E.; Soto A. M.; Marky L. A.; Gmeiner W. H. Biochemistry 2002, 41, 839–846. [DOI] [PubMed] [Google Scholar]
- Tonelli M.; Ulyanov N. B.; Billeci T. M.; Karwowski B.; Guga P.; Stec W. J.; James T. L. Biophys. J. 2003, 85, 2525–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramini J. M.; Cleaver S. H.; Pon R. T.; Cunningham R. P.; Germann M. W. J. Mol. Biol. 2004, 338, 77–91. [DOI] [PubMed] [Google Scholar]
- Wuthrich K.NMR of Proteins and Nucleic Acids; John Wiley & Sons: New York, 1986. [Google Scholar]
- Patel D. J.; Shapiro L.; Hare D. Q. Rev. Biophys. 1987, 20, 35–112. [DOI] [PubMed] [Google Scholar]
- Reid B. R. Q. Rev. Biophys. 1987, 20, 2–28. [DOI] [PubMed] [Google Scholar]
- Boelens R.; Scheek R. M.; Dijkstra K.; Kaptein R. J. Magn. Reson. 1985, 62, 378–386. [Google Scholar]
- Keepers J. W.; James T. L. J. Magn. Reson. 1984, 57, 404–426. [Google Scholar]
- James T. L. Curr. Opin. Struct. Biol. 1991, 1, 1042–1053. [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Veselkov A. N.; Baranovskii S. F.; Dymant L. N.; Petrenko N. V.; Osetrov S. G.; Veselkov D. A.; Parkes X.; Davies D. Biofizika 1998, 43, 205–214. [PubMed] [Google Scholar]
- Robinson H.; van der Marel G. A.; van Boom J. H.; Wang A. H. Biochemistry 1992, 31, 10510–10517. [DOI] [PubMed] [Google Scholar]
- Aramini J. M.; Kalisch B. W.; Pon R. T.; van de Sande J. H.; Germann M. W. Biochemistry 1996, 35, 9355–9365. [DOI] [PubMed] [Google Scholar]
- Aramini J. M.; van de Sande J. H.; Germann M. W. Biochemistry 1997, 36, 9715–9725. [DOI] [PubMed] [Google Scholar]
- Aramini J. M.; Mujeeb A.; Germann M. W. Nucleic Acids Res. 1998, 26, 5644–5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramini J. M.; Germann M. W. Biochemistry 1999, 38, 15448–15458. [DOI] [PubMed] [Google Scholar]
- Schipper P. E.; Norden B.; Tjerneld F. Chem. Phys. Lett. 1980, 70, 17–21. [Google Scholar]
- Norden B.; Tjerneld F. Biopolymers 1982, 21, 1713–1734. [DOI] [PubMed] [Google Scholar]
- Lyng R.; Hard T.; Norden B. Biopolymers 1987, 26, 1327–1345. [DOI] [PubMed] [Google Scholar]
- Petersheim M.; Turner D. G. Biochemistry 1983, 22, 256–263. [DOI] [PubMed] [Google Scholar]
- Turner D. H.; Petersheim M.; Albergo D. D.; Dewey T. G.; Freier S. M. In Biomolecular Stereodynamics; Sarma R. H., Ed.; Adenine Press: New York, 1995; pp 429−438. [Google Scholar]
- Oostenbrink C.; van Gunsteren W. F. Chemistry 2005, 11, 4340–4348. [DOI] [PubMed] [Google Scholar]
- Yakovchuk P.; Protozanova E.; Frank-Kamenetskii M. D. Nucleic Acids Res. 2006, 34, 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen A.; Nielsen P. E. Biophys. Chem. 2009, 141, 29–33. [DOI] [PubMed] [Google Scholar]
- Nemethy G.; Scheraga H. A. J. Chem. Phys. 1962, 36, 3382–3400. [Google Scholar]
- Dill K. A. Biochemistry 1990, 29, 7133–7155. [DOI] [PubMed] [Google Scholar]
- Ruelle P.; Kesselring U. W. J. Pharm. Sci. 1998, 87, 987–997. [DOI] [PubMed] [Google Scholar]
- Ruelle P.; Kesselring U. W. J. Pharm. Sci. 1998, 87, 998–1014. [DOI] [PubMed] [Google Scholar]
- Ruelle P.; Kesselring U. W. J. Pharm. Sci. 1998, 87, 1015–1024. [DOI] [PubMed] [Google Scholar]
- Baldwin R. L. J. Mol. Biol. 2007, 371, 283–301. [DOI] [PubMed] [Google Scholar]
- Kool E. T.; Morales J. C.; Guckian K. M. Angew. Chem., Int. Ed. 2000, 39, 990–1009. [DOI] [PubMed] [Google Scholar]
- Kool E. T. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 1–22. [DOI] [PubMed] [Google Scholar]
- Asao T.; Buechi G.; Abdel-Kader M. M.; Chang S. B.; Wick E. L.; Wogan G. N. J. Am. Chem. Soc. 1965, 87, 882–886. [DOI] [PubMed] [Google Scholar]
- Baertschi S. W.; Raney K. D.; Shimada T.; Harris T. M.; Guengerich F. P. Chem. Res. Toxicol. 1989, 2, 114–122. [DOI] [PubMed] [Google Scholar]
- Oleykowski C. A.; Mayernik J. A.; Lim S. E.; Groopman J. D.; Grossman L.; Wogan G. N.; Yeung A. T. J. Biol. Chem. 1993, 268, 7990–8002. [PubMed] [Google Scholar]
- Cavaluzzi M. J.; Borer P. N. Nucleic Acids Res. 2004, 32, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray R. W.; Jeyaraman R. J. Org. Chem. 1985, 50, 2847–2853. [Google Scholar]
- Adam W.; Chan Y. Y.; Cremer D.; Gauss J.; Scheutzow D.; Schindler M. J. Org. Chem. 1987, 52, 2800–2803. [Google Scholar]
- Adam W.; Bialas J.; Hadjiarapoglou L. Chem. Ber. 1991, 124, 2377–2377. [Google Scholar]
- Baertschi S. W.; Raney K. D.; Stone M. P.; Harris T. M. J. Am. Chem. Soc. 1988, 110, 7929–7931. [Google Scholar]
- Goddard T. D.; Kneller D. G. SPARKY 3, University of California: San Francisco, 2006.
- Derome A. E.; Williamson M. P. J. Magn. Reson. 1990, 88, 177–185. [Google Scholar]
- Jeener J.; Meier B. H.; Bachmann P.; Ernst R. R. J. Chem. Phys. 1979, 71, 4546–4553. [Google Scholar]
- Wagner R.; Berger S. J. Magn. Res. A 1996, 123, 119–121. [DOI] [PubMed] [Google Scholar]
- Piotto M.; Saudek V.; Sklenar V. J Biomol. NMR 1992, 6, 661–665. [DOI] [PubMed] [Google Scholar]
- Wang H.; Zuiderweg E. R. P.; Glick G. D. J. Am. Chem. Soc. 1995, 117, 2981–2991. [Google Scholar]
- Blondet P.; Albrand J. P.; Vonkienlin M.; Decorps M.; Lavanchy N. J. Magn. Reson. 1987, 71, 342–346. [Google Scholar]
- Borgias B. A.; James T. L. Methods Enzymol. 1989, 176, 169–183. [DOI] [PubMed] [Google Scholar]
- Borgias B. A.; James T. L. J. Magn. Reson. 1990, 87, 475–487. [Google Scholar]
- Liu H.; Spielmann H. P.; Ulyanov N. B.; Wemmer D. E.; James T. L. J. Biomol. NMR 1995, 6, 390–402. [DOI] [PubMed] [Google Scholar]
- Spielmann H. P.; Dwyer T. J.; Hearst J. E.; Wemmer D. E. Biochemistry 1995, 34, 12937–12953. [PubMed] [Google Scholar]
- Mujeeb A.; Kerwin S. M.; Kenyon G. L.; James T. L. Biochemistry 1993, 32, 13419–13431. [DOI] [PubMed] [Google Scholar]
- Wijmenga S. S.; Mooren M. M. W.; Hilbers C. W. In NMR of Macromolecules; A Parctical Approach; Roberts G. C. K., Ed.; Oxford University Press: New York, 1993; pp 217−288. [Google Scholar]
- Pikkemaat J. A.; Altona C. Magn. Reson. Chem. 1996, 34, S33–S39. [Google Scholar]
- van Wijk J.; Huckriede B. D.; Ippel J. H.; Altona C. Methods Enzymol. 1992, 211, 286–306. [DOI] [PubMed] [Google Scholar]
- Bashford D.; Case D. A. Annu. Rev. Phys. Chem. 2000, 51, 129–152. [DOI] [PubMed] [Google Scholar]
- Tsui V.; Case D. A. Biopolymers 2000, 56, 275–291. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Cheatham T. E. 3rd; Darden T.; Gohlke H.; Luo R.; Merz K. M. Jr.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. J. Comput. Chem. 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychaert J. P.; Ciccotti G.; Berendsen H. J. C. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- Izaguirre J. A.; Catarello D. P.; Wozniak J. M.; Skeel R. D. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar]
- Loncharich R. J.; Brooks B. R.; Pastor R. W. Biopolymers 1992, 32, 523–535. [DOI] [PubMed] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.