

Abstract
Tumor cell–derived heat shock proteins are used as vaccines for immunotherapy of cancer patients. However, current approaches require the generation of custom-made products and are clinically ineffective. To improve the applicability of heat shock protein–based immunotherapy in cancers and to enhance clinical efficacy, we explored combinational treatments in a myeloma setting using pooled heterogeneous or allogeneic myeloma cell line–derived glycoprotein 96 (gp96) as universal vaccines, and clearly demonstrated that pooled but not single gp96 from heterogeneous or allogeneic myeloma cell lines was as effective as autologous gp96 in protecting mice from tumor challenge and rechallenge and in treating established myeloma. We showed that interferon γ and CD4+ and CD8+ T cells were required for gp96-induced antimyeloma responses and that pooled gp96 induced broader immune responses that protected mice from developing different myeloma. Furthermore, pooled gp96 plus CpG in combination with anti-B7H1 or anti–interleukin-10 monoclonal antibodies were effective in treating mice with large tumor burdens. Thus, this study strongly suggests that pooled gp96 vaccines from myeloma cell lines can replace gp96 vaccines from autologous tumors for immunotherapy and induce immune responses against broader tumor antigens that may protect against tumor recurrence and development of unrelated tumors in vaccinated myeloma patients.
Introduction
Tumor immunotherapy holds great promise in controlling or even eradicating residual diseases and may provide an alternative treatment modality to conventional chemotherapy for cancer patients.1,2 In multiple myeloma (MM) and other B-cell malignancies, tumor-specific immune and clinical responses can be induced by immunization using idiotype-based vaccines.3,4 However, the idiotype proteins represent a unique tumor-specific antigen and thus cannot provide shared immunity and protection to other patients with B-cell tumors. Therefore, in addition to optimizing immunotherapy methods, there is an urgent need to search for and use novel shared tumor antigens to efficiently stimulate antitumor cytotoxic T lymphocyte (CTL) responses in treated cancer patients.
Heat shock proteins (hsp) are a large family of both inducible and ubiquitously expressed protein chaperones involved in assisting protein folding and unfolding in cells. They include several different molecular- weight-class families, such as hsp27, hsp70, and hsp90. Hsps are among the most conserved proteins known in phylogeny with respect to both structure and function.5,6 Understanding the immunologic significance of hsps came from the observation that tumor cell–derived hsps could immunize against tumors, which is attributed to hsp-chaperoned peptides derived from tumor antigens.7–9 Studies in solid tumors have shown that tumor-derived hsps such as hsp70 and gp96 are immunogenic and potent in stimulating the generation of tumor-specific CTLs.10,11 Hsp70- and glycoprotein 96 (gp96)–based vaccines have been tested in early-phase clinical trials in solid tumors as well as in lymphoma and leukemias; all of these vaccines showed minimal toxicity and potential efficacy.12–14 Phase 3 clinical studies using tumor-derived gp96 as vaccines for melanoma and renal cell carcinoma have recently been completed, and clinical benefits were observed only in subsets of patients.15 However, these approaches require preparation of individualized hsp vaccines, which is not only laborious but also expensive. Moreover, in many types of tumors including MM, although myeloma-derived gp96 can efficiently stimulate an antimyeloma CTL response,16 the requirement of a relatively large amount of tumor-derived hsps17,18 as vaccines limits the applicability and feasibility of clinical trials in treating this and other malignancies. To improve the applicability and feasibility of hsp-based immunotherapy in cancers and to enhance clinical efficacy, we have explored the use of pooled, heterogeneous, or allogeneic myeloma cell line–derived gp96 as a universal vaccine for immunotherapy of MM. Furthermore, we combined the vaccines with immune adjuvants and other therapeutic strategies to break immune suppression to further improve the clinical efficacy of the immunotherapy.
Methods
Myeloma cell lines
The murine myeloma cell lines P3X63Ag8U.1, MOPC-315, MPC-11, HOPC-1F/12, and C1.18.4 (Table 1) were purchased from ATCC. The murine myeloma cell line 5TGM119,20 was derived from 5T33 myeloma cells developed in aged C57BL/KaLwRij mice.21 All cell lines were cultured in Iscove modified Dulbecco medium complete medium (Atlanta Biologicals) with 1% penicillin/streptomycin and 2 mM l-glutamine.
Table 1.
Murine myeloma tumor cell lines used in this study
| Designation | Idiotype | Strain of mouse | |
|---|---|---|---|
| A | P3X63Ag8U.1 | None | Balb/c |
| B | MOPC-315 | IgA | Balb/c |
| C | MPC-11 | IgG2b | Balb/c |
| D | HOPC-1F/12 | IgG2a | Balb/c |
| E | 5TGM1 | IgG2b | C57BL/KaLwRij |
| F | C1.18.4 | IgG2a | C3H |
Mice
Male mice at 6 to 8 weeks old were used. C57BL/KaLwRij mice were purchased from Harlan CPB, Balb/c mice were purchased from National Cancer Institute (NCI), and C3H/He and interferon γ (IFN-γ) knockout Balb/c mice were purchased from The Jackson Laboratory. The studies were approved by the Institutional Animal Care and Use Committee of the University of Texas M. D. Anderson Cancer Center.
Preparation of gp96
Gp96 was purified from each of the tumor cell lines and normal murine liver as described before.16,22,23 After sterile filtration, the preparations were aliquoted and stored at −20°C until use. No lipopolysaccharide contamination was detected in gp96 preparation by a limulus amebocyte lysate QCL-1000 kit (Cambrex Bioscience).
Generation of DCs
Dendritic cells (DCs) were generated from murine bone marrow stem cells as described previously.24 At day 8, immature DCs were collected, pooled, and pulsed with gp96 protein at a concentration of 100 μg per 106 DCs. TNF-α (10 ng/mL) and interleukin-1β (IL-1β; 10 ng/mL; R&D Systems) were added to the immature DCs, and after 48 hours of culture, gp96-pulsed mature DCs were collected and used in the study.
Mouse vaccination
In prophylaxis studies, vaccinations consisted of 2 subcutaneous injections (on days 0 and 7) of 10 μg/mouse gp96 derived from murine tumor cell lines. Control mice received injections of 10 μg/mouse gp96 derived from murine liver. On day 14, mice were subcutaneously or intravenously challenged with tumors (106 cells/mouse). In therapeutic studies, mice received subcutaneous injection of tumors (106 cells/mouse) on day 0, followed by 3 subcutaneous gp96 vaccinations (10 μg/mouse) at a 3-day interval when the subcutaneous tumors reached 25 mm2 or 100 mm2. As a control, mice were immunized with gp96 purified from murine liver. Mice were killed when subcutaneous tumors reached 225 mm2 or when mice became moribund. Tumor burdens and survival data are used for comparison. The schema of mouse vaccination experiments is shown in supplemental Figure 1 (available on the Blood website; see the Supplemental Materials link at the top of the online article).
For enhancing the efficacy of gp96 vaccines, various adjuvants including granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-12, or CpG were used. GM-CSF (200 ng/day per mouse) and IL-12 (40 ng/day per mouse), respectively, were injected subcutaneously adjacent to the vaccination sites for 3 consecutive days. CpG-ODN-1826 (CpG; TCCATGACGTTCCTGACGTT; DNA Technologies Inc) was administered at a dose of 50 μg/mouse, mixed with and injected together with gp96 vaccines.
In vivo depletion of lymphocytes
To deplete CD4+ or CD8+ T cells or natural killer (NK) cells in vivo, mice were first vaccinated twice with gp96 vaccines, followed by intraperitoneal injections of depleting monoclonal antibodies (mAbs; 100 μg per mouse) on days 1, 4, and 7 after the second vaccination. Myeloma cells were injected subcutaneously on the same day of the last mAb injection. The following mAbs were used: anti-CD4 (GK1.5; ATCC); anti-CD8 (2.43; ATCC); rabbit anti–asialo-GM1 (Wako Pure Chemical Inc), or rat immunoglobulin G (IgG). The efficacy of the treatment, evaluated by flow cytometry using T- and NK-cell markers, was shown to be greater than 90%.
In some experiments, anti-B7H1 (M1H5, kindly provided by Prof Miyuki Azuma, Tokyo Medical and Dental University, Tokyo, Japan), anti–IL-10 (JES052A5; R&D Systems), or anti-CD25 (PC61; ATCC) mAbs were used to block B7H1, neutralize IL-10, or deplete regulatory T cells (Tregs), respectively, in vivo. Normal rat IgG (Sigma-Aldrich) was used as a control. Mice were injected intraperitoneally (4 times at a 3-day interval) with anti-B7H1 (200 μg/mouse), IL-10 (100 μg/mouse), or anti-CD25 (100 μg/mouse) after the first vaccination.
Adoptive transfer of lymphocytes
Splenocytes from 8- to 10-week-old vaccinated or naive Balb/c mice were obtained and CD4+ or CD8+ T cells were isolated using anti-CD4 or anti-CD8 mAb-coated magnetic beads (Miltenyi Biotec). Purified CD4+ or CD8+ or splenocytes were resuspended in sterile phosphate-buffered saline at a concentration of 5 × 107 cells/mL, and intravenously injected to recipient mice (5 × 106 cells/mouse) one week after tumor inoculation.
To examine in vivo proliferation of adoptively transferred T cells, purified CD4+ or CD8+ T cells were incubated with 5μM carboxyfluorescein succinimidyl ester (CFSE; Molecular Probes) for 10 minutes at 37°C, followed by washing. CFSE-labeled cells were intravenously injected into recipient mice. Five days after the transfer, mice were killed and spleens and lymph nodes were collected to detect the T cells.
Antigen-specific T-cell proliferation
Splenocytes or cultured T cells were labeled with 5μM CFSE for 10 minutes at 37°C. After washing, labeled cells were seeded into 96-well, U-bottom plates and incubated with various stimulatory cells for 7 days. Flow cytometric analysis was used to detect dilution of CFSE, which represents T-cell proliferation.25
Cytotoxicity assay
The standard 4-hour 51Cr-release assay was performed to examine the cytotoxicity of T cells against myeloma cells, as described previously.26
Flow cytometric analysis
Phycoerythrin-, fluorescein isothiocyanate–, or allophycocyanin-conjugated mAbs were added to cell pellets, incubated for 30 minutes on ice, and washed 3 times before analysis. Intracellular cytokine staining was performed using the Cytofix/Cytoperm kit (BD PharMingen). Samples were analyzed using a flow cytometer (FACSCalibur; Becton Dickinson).
Immunohistochemistry
Formalin-fixed, paraffin-embedded sections of tissues or tumors from mice were used for the analysis as described in detail previously.27 All slides were observed with light microscopy (10×/0.30 objective lens, Axiovert 200; Carl Zeiss) and images were captured with digital image processing software (AxioVision AC; Carl Zeiss).
Statistical analysis
The Student t test was used to compare various experimental groups. A P value less than .05 was considered statistically significant. Unless otherwise indicated, means and SD are shown.
Results
Pooled heterogeneous or allogeneic gp96 vaccines protect mice from tumor challenge and rechallenge and are as effective as autologous gp96 vaccines
In this study, we used MM as a tumor model to test our hypothesis that pooled gp96 from heterogeneous or allogeneic tumor cell lines can be used as universal tumor vaccines for cancer patients. As shown in Table 1, 4 different murine myeloma cell lines that originated from Balb/c mice (tumors A-D) and 2 myeloma cell lines that originated from C57BL and C3H mice (tumors E and F) were obtained. gp96 proteins purified from these cell lines were used as vaccines and gp96 from mouse liver cells was used as normal protein control. Balb/c mice were vaccinated with 2 weekly subcutaneous injections of either normal gp96 (g96N); gp96 from tumor A (gp96A), tumor B (gp96B), or tumor C (gp96C); pooled gp96 from tumors B and C (gp96BC); or pooled gp96 from tumors B, C, and D (gp96BCD), followed by subcutaneous challenge with tumor A cells 1 week later. Tumor burdens were monitored by measuring tumor sizes, and mice were killed when subcutaneous tumors reached 15 mm in diameter. As shown in Figure 1A, whereas 100% of mice vaccinated with gp96N developed myeloma, 10%, 90%, 100%, 40%, and 20% of mice vaccinated with gp96A (autologous gp96), gp96B, gp96C, pooled gp96BC, or gp96BCD, respectively, developed myeloma. More than 80% of mice receiving gp96A or gp96BCD vaccines survived without tumor burden.
Figure 1.

Protective effects of gp96 vaccines against myeloma development. Results shown are measurements of tumor burdens and survival of mice that received different treatments. Tumor burdens were measured twice every week. Mice were killed when subcutaneous tumors reached 225 mm2 or when mice became moribund. (A) Balb/c mice (10 per group) were subcutaneously vaccinated twice with either normal gp96 (g96N); gp96 from tumor A (gp96A), tumor B (gp96B), or tumor C (gp96C); pooled allogeneic gp96 from tumors B and C (gp96BC); or pooled allogeneic gp96 from tumors B, C, and D (gp96BCD) followed by challenge with tumor A. (B) Mice were subcutaneously vaccinated twice with gp96A followed by challenge with tumor A, and surviving mice (5 per group) were rechallenged with tumors A, B, C, or D 8 weeks after the first challenge. (C) Mice were subcutaneously vaccinated twice with gp96BCD followed by challenge with tumor A, and surviving mice (5 per group) were rechallenged with tumors A, B, C, or D 8 weeks after the first challenge. (D) Staining for CD138+ myeloma cells (brown) in the tumor nodules of tumor-bearing or injection sites of tumor-free Balb/c mice immunized with autologous gp96A or pooled heterogeneous gp96BCD. Representative results of 1 of 3 independent experiments performed are shown. *P < .05; **P < .01 (compared with controls).
Figure 1B shows that vaccination also generated tumor-specific memory responses; gp96A-vaccinated and tumor A–inoculated surviving mice were protected from rechallenge with tumor A but not with tumors B, C, or D, indicating that gp96 vaccines and subsequent tumor challenge induced specific memory immune responses against the original tumor. gp96BCD-vaccinated and tumor A–inoculated surviving mice were protected or partially protected from rechallenge with tumors A, B, and D, but not tumor C (Figure 1C), which suggests that vaccination with pooled gp96 induced a broader immune responses against different tumors and subsequent tumor (A) inoculation expanded the response toward the challenging tumor cells. Immunohistochemical staining confirmed that there were fewer or no tumor cells in the subcutaneous nodules or tumor-injection sites in tumor-free mice immunized with gp96A or pooled gp96 (Figure 1D). Pooled heterogeneous gp96 vaccines were also as effective as autologous gp96 vaccines in protecting Balb/c mice from developing tumor B (supplemental Figure 2) or tumor D myeloma (supplemental Figure 3). However, gp96 vaccines, either autologous or heterogeneous, were less protective in mice against tumor C myeloma (supplemental Figure 4), indicating that tumor C cells are less immunogenic than the other tumors. These findings clearly show that pooled, but not single, heterologous gp96 vaccines, particularly the ones pooled from 3 cell lines, were as potent as autologous gp96 vaccines in protecting mice from developing myeloma.
We also examined whether gp96 from Balb/c mouse–derived myeloma cell lines could protect against myeloma in other strains of mice. As described previously, mice received 2 injections of gp96 vaccines followed by inoculation of tumor cells intravenously (tumor E) or subcutaneously (tumor F). Although gp96 vaccines were less efficient at protecting the mice from developing tumors E or F, pooled gp96BCD was as effective as gp96 from the autologous tumors in delaying the development of myeloma and prolonging the survival of tumor E–bearing C57BL (supplemental Figure 5a) and tumor F–bearing C3H (supplemental Figure 5b) mice. These results demonstrate the ability of pooled allogeneic gp96 vaccines in protecting mice with different major histocompatibility complex (MHC) haplotypes from developing myeloma. Taken together, these findings strongly suggest that pooled gp96 vaccines from heterogeneous or allogeneic myeloma cell lines can replace gp96 vaccines from autologous tumor cells as universal tumor vaccines for all patients. Moreover, pooled gp96 vaccines induce broader immune responses against different tumor antigens than autologous gp96 vaccines, which may be important for protecting against tumor recurrence and development of unrelated tumors in vaccinated patients.
gp96 vaccines induced CD4+ and CD8+ T-cell responses against tumor cells
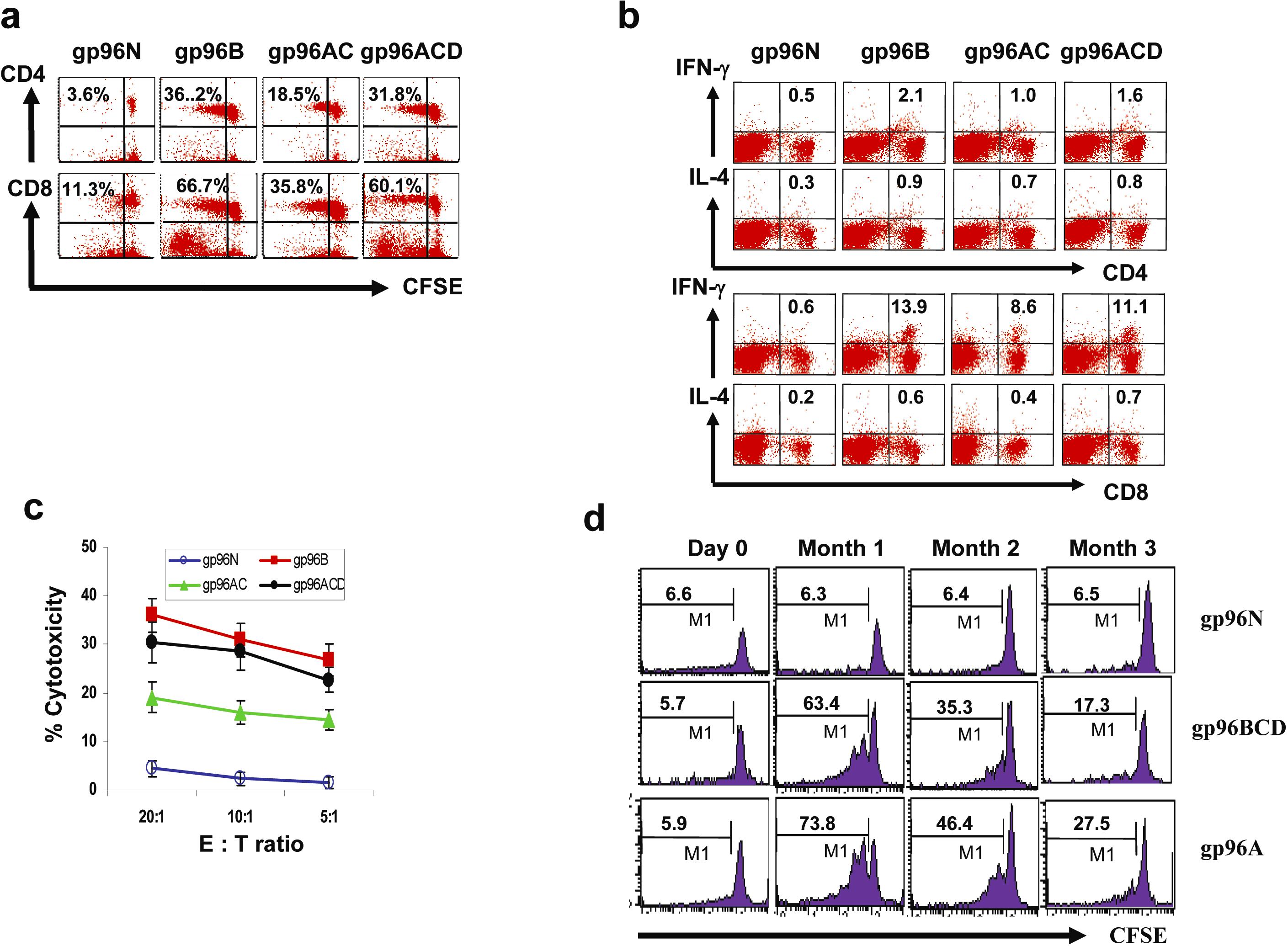
The mechanisms of tumor protection induced by tumor vaccines are numerous2,28 and are often associated with CTL and IFN-γ production. To assess whether CTL activity was induced by the vaccines and tumor challenge, Balb/c mice were first vaccinated with gp96N, gp96A, gp96BC, or gp96BCD, followed by inoculation with tumor A cells. One week after tumor injection, mice were killed and splenocytes were collected, restimulated with irradiated tumor A cells in vitro for 3 days, and subjected to analyses. In some cases, splenocytes were labeled with CFSE, followed by ex vivo restimulation with irradiated tumor-A cells. Flow cytometric analysis showed that restimulated CD8+ T cells exhibited the typical effector CTL phenotype (CD25+CD69+CD62LlowCD44highperforin+) in mice vaccinated with gp96A, gp96BC, or gp96BCD (Figure 2A). CFSE dilution analysis indicated that the tumor-specific T-cell proliferative response from mice vaccinated with pooled gp96BCD was the same as that of mice vaccinated with the autologous gp96A, and a stronger CD8+ T-cell response was observed compared with CD4+ T-cell response (Figure 2B). Intracellular cytokine staining showed that the restimulated CD4+ and CD8+ T cells secreted IFN-γ and a lower amount of IL-4 (Figure 2C). Cytotoxicity assays demonstrated that splenocytes from mice vaccinated with gp96A, gp96BC, and gp96BCD efficiently killed tumor A cells in vitro (Figure 2D). As expected, no tumor-specific T-cell response was detected in mice vaccinated with control gp96N and subsequently challenged with tumor-A cells, which underscores the requirement of gp96 vaccination in generating tumor-specific T-cell responses. Similar results were also obtained from mice vaccinated with gp96 vaccines and inoculated with tumor-B cells (supplemental Figure 6a-c). These results indicate that, similar to vaccination with autologous gp96, vaccination with pooled heterogeneous gp96BCD or gp96ACD induced predominantly tumor-specific, IFN-γ–producing CD4+ and CD8+ T-cell responses in mice inoculated with tumor cells. These results confirm the ability of pooled gp96 vaccines from heterogeneous tumor cell lines to induce tumor-specific T-cell responses in vivo.
Figure 2.

gp96 vaccines induce CD4+ and CD8+ tumor-specific T-cell responses. Balb/c mice (3 per group) were subcutaneously vaccinated twice with either normal gp96N or tumor-derived gp96A, gp96BC, or gp96BCD followed by challenge with tumor A. One week later, splenocytes were isolated, pooled, and restimulated with irradiated tumor-A cells for 5 days. Shown are the results of T cells from mice immunized with different gp96 vaccines. (A) Expression of CD25, CD69, CD62L, and CD44 by gated CD8+ T cells measured by flow cytometric analysis. Numbers inside represent mean fluorescence index (MFI). (B) Percentages of proliferative T cells measured by CFSE dilution assay on gated CD4+ and CD8+ T cells. (C) Percentages of IFN-γ or IL-4–positive T cells on gated CD4+ and CD8+ T cells. (D) Cytotoxicity of CTLs against tumor-A cells. Representative results of 3 independent experiments are shown. The error bars in panel D represent SD of 3 independent experiments.
We also examined the longevity of tumor-specific T-cell responses in mice vaccinated with autologous and pooled heterogeneous gp96 vaccines. Balb/c mice received 2 subcutaneous injections of gp96 vaccines and killed at 1, 2, or 3 months after the final gp96 vaccination. Splenocytes were labeled with CFSE and restimulated with irradiated tumor A cells for 5 days. As shown in supplemental Figure 6d, tumor-specific T-cell responses could be detected at 3 months after vaccination, although the percentages of proliferating T cells gradually decreased over time. Again, tumor-specific T-cell responses were comparable in mice vaccinated with the autologous gp96A and pooled gp96BCD.
IFN-γ and CD4+ and CD8+ CTLs are crucial for gp96 vaccine–induced antitumor immune responses
Next, we evaluated the role of IFN-γ, CD4+, and CD8+ T cells, and NK cells in gp96 vaccine-induced antitumor responses in vivo using Ab depletion and Balb/c mice deficient in IFN-γ (IFNγ−/−). First, Balb/c mice were vaccinated with 2 subcutaneous injections of gp96BCD, followed by depletion of CD4+ or CD8+ T cells or NK cells before challenge with tumor-A cells. Mice vaccinated with gp96N or injected with control rat IgG served as controls. As shown in Figure 3, whereas all mice vaccinated with gp96N died of myeloma and all mice vaccinated with gp96BCD and injected with rat IgG were protected from developing myeloma, depletion of CD4+ T cells by injecting anti-CD4 mAb led to a loss of protection in 3 of 5 mice. Depleting CD8+ T cells resulted in myeloma development in 4 of 5 mice, whereas depletion of NK cells resulted in myeloma development in 1 of 5 mice. Mice deficient in the production of IFN-γ (IFNγ−/− mice) lacked the ability to mount antitumor immunity after the vaccination and all died from myeloma. Similar results were also obtained in Balb/c–tumor-B model (supplemental Figure 7). These results demonstrate that, although NK cells may play a role, IFN-γ and both CD4+ and CD8+ T cells are the major effector molecules and cells in gp96 vaccine–induced protection against myeloma.
Figure 3.

Importance of T cells, NK cells, and IFN-γ in gp96 vaccine–induced tumor protection. Mice (5 per group) were subcutaneously vaccinated twice with pooled heterologous gp96BCD and depleted of CD4+ or CD8+ T cells or NK cells by specific mAbs before tumor-A challenge, or IFNγ−/− mice were subcutaneously vaccinated twice with gp96BCD and followed by tumor A challenge. Tumor burdens were measured twice each week. Mice were killed when subcutaneous tumors reached 225 mm2 or when mice became moribund. Results shown are measurements of tumor burdens and survival of mice that received different treatments. Representative results of 1 of 2 independent experiments performed are shown. *P < .05; **P < .01 (compared with controls).
Adoptive transfer of vaccinated splenic T cells protected naive mice from developing myeloma
To further confirm the importance of tumor-specific CD4+ and CD8+ T cells in antitumor responses, we investigated the in vivo efficacy of splenic T cells at mediating antimyeloma activity by adoptive transfer experiments. Balb/c mice were first challenged with tumor-A cells, and 7 days later, were infused intravenously with 107 splenocytes or purified CD4+ or CD8+ T cells from mice vaccinated with gp96BCD and survived from tumor A challenge. Splenic CD4+ and CD8+ T cells from naive Balb/c mice served ascontrols. As shown in Figure 4, panels A and B, all tumor-bearing mice receiving infusion of phosphate-buffered saline or T cells from naive mice died from myeloma within 1 month after tumor inoculation, whereas 3 of 5, 4 of 5, and 5 of 5 mice infused with immunized CD4+ T cells, CD8+ T cells, or splenocytes, respectively, survived with minimal or without tumor burdens. To examine the in vivo proliferation of these adoptively transferred T cells, we labeled splenocytes with CFSE and infused them into Balb/c mice 7 days after tumor-A inoculation. Five days later, lymph nodes and spleens were collected and T cells were analyzed for CFSE dilution. As shown in Figure 4C, in the spleens and lymph nodes of the recipient mice, CD4+ and CD8+ T cells from immunized mice proliferated, whereas T cells from naive mice did not. These results confirm the importance of gp96 vaccine–induced, tumor-specific CD4+ and CD8+ T cells in protecting mice from developing myeloma in vivo.
Figure 4.

Effects of adoptively transferred T cells on myeloma development. Mice (5 per group) were challenged with tumor A cells, followed by intravenous infusion of 107 splenocytes or purified CD4+ and CD8+ T cells from tumor-A surviving mice vaccinated with gp96BCD or naive Balb/c mice 7 days later. Shown are (A) tumor burdens and (B) survival of mice receiving different T cells; or (C) percentages of proliferating CD4+ and CD8+ T cells measured by CFSE dilution in the spleens or lymph nodes of mice receiving CFSE-labeled splenocytes from naive or gp96BCD-vaccinated mice. The error bars in panel A represent SD of 5 mice. Representative results of 1 of 3 independent experiments performed are shown. *P < .05; **P < .01 (compared with controls).
T cells from tumor-bearing mice secreted more IL-10
Next, we compared T-cell responses induced by gp96 vaccines in mice that either were cleared of or succumbed to myeloma to better understand the relationship of myeloma and the immune system. Balb/c mice were first vaccinated with different gp96 vaccines and subsequently challenged with tumor-A cells. Mice were followed for 3 weeks and tumor-bearing, tumor-free, or no-visible-tumor-burden mice were selected and killed. Splenocytes from the mice were restimulated with irradiated tumor A cells for 5 days, and supernatants of the cultures were collected and used for enzyme-linked immunosorbent assay to detect secreted cytokines. As shown in Figure 5A, splenocytes from mice that were tumor-free or those with no visible tumor burden secreted high amounts of IFN-γ and IL-4 but low IL-10, whereas splenocytes from tumor-bearing mice secreted high amounts of IL-4 and IL-10 but low IFN-γ. These results indicate that clearance of myeloma was accompanied by induction of tumor-specific type-1 and type-2 T-cell responses induced by gp96 vaccines, whereas tumor development led to a shift to tumor-specific type-2 and IL-10–secreting regulatory T-cell responses. Indeed, our results showed that more CD4+Foxp3+ Tregs were found in the spleens of tumor-bearing mice compared with tumor-free mice and in the tumors (Figure 5B). Furthermore, we showed that all the myeloma cells expressed a negative T-cell costimulatory molecule B7H1 (Figure 5C), and blocking surface B7H1 using a specific mAb (M5H1)29 enabled tumor-specific CTLs from vaccinated mice to be more efficient at lysing the tumor cells (Figure 5D). Thus, these results indicate that myeloma cells are able to induce or recruit regulatory T cells and actively inhibit the cytolytic function of tumor-specific CTLs by expressing B7H1.
Figure 5.

Myeloma cells recruit Tregs and express B7H1. (A) Cytokines secreted by tumor-specific T cells in tumor-free and tumor-bearing mice. Balb/c mice were subcutaneously vaccinated twice with either normal g96N, autologous gp96A, or pooled heterologous gp96BC or gp96BCD, followed by challenge with tumor-A cells. Three weeks later, splenocytes were isolated from tumor-bearing and tumor-free mice, and restimulated for 5 days with irradiated tumor-A cells. Culture supernatants were collected and assayed by enzyme-linked immunosorbent assay to quantify IFN-γ, IL-4, and IL-10. (B) Percentages of CD4+Foxp3+ Tregs in the spleens of tumor-free and tumor-bearing mice and in subcutaneous tumors. (C) B7H1 expression on murine tumors A, B, C, D, E, and F. (D) Blocking B7H1 enhanced the killing activity of tumor-specific CTLs. Shown is the cytolytic activity of splenocytes from mice vaccinated with gp96BCD and ex vivo restimulated with irradiated tumor-A cells for 5 days, in cultures with or without the addition of anti-B7H1 mAb (M5H1; 20 μg/mL) or an isotype control rat IgG. Target cells were tumors A, B, C, or D cells. Representative results of 3 independent experiments are shown. *P < .05; **P < .01 (compared with controls). The error bars in panels A and D represent SD of 3 independent experiments.
Pooled heterogeneous gp96 vaccines are as effective as autologous gp96 vaccines in the treatment of established myeloma
Finally, we investigated whether autologous or pooled heterogeneous gp96 vaccines could be used to treat established myeloma. Balb/c mice were first inoculated subcutaneously with tumor-A cells, and when subcutaneous tumors reached 25 mm2, vaccination with autologous gp96A, pooled gp96BC, or gp96BCD was initiated, which included 3 subcutaneous injections of gp96 proteins at a 3-day interval. Gp96N was used as a protein control. As shown in Figure 6A, vaccination with gp96N, gp96A, gp96BC, or gp96BCD led to tumor regression in 0 of 5, 3 of 5, 1 of 5, and 3 of 5 mice, respectively, indicating that pooled gp96 from 3 heterogeneous tumor cell lines (gp96BCD) was as effective as autologous gp96A in treating established myeloma. To enhance the therapeutic efficacy of gp96 vaccines, we explored combinational treatment of the vaccines (gp96BCD) with adjuvants GM-CSF,30,31 IL-12,32,33 or Toll-like receptor ligand CpG-ODN-1826 (CpG).34,35 As shown in Figure 6B, whereas gp96N, CpG alone, gp96BCD alone, gp96BCD plus GM-CSF, and gp96BCD plus IL-12 eradicated established myeloma in 0 of 5, 2 of 5, 3 of 5, 4 of 5, and 3 of 5 mice, respectively, gp96BCD plus CpG eradicated myeloma in all 5 mice. However, if the treatments were given when subcutaneous tumors reached 100 mm2 (large tumor burdens) in the mice, the immunotherapies were less effective (Figure 6C). Nevertheless, these findings clearly indicate that pooled heterogeneous gp96 vaccines can replace gp96 vaccines from autologous tumor cells to treat cancers, and among the adjuvants examined, CpG is most potent at enhancing the therapeutic effects of gp96 vaccines.
Figure 6.

Therapeutic effects of gp96 vaccines against established myeloma. Balb/c mice (5 per group) were inoculated subcutaneously with tumor-A cells, and when subcutaneous tumors reached 25 mm2, received 3 subcutaneous injections at 3-day intervals of (A) normal gp96N, autologous gp96A, or pooled heterologous gp96BC or gp96BCD; or (B) gp96N, CpG; gp96BCD, gp96BCD + GM-CSF, IL-12; or Balb/c mice (5 per group) were inoculated subcutaneously with tumor-A cells, and when subcutaneous tumors reached 100 mm2, received 3 subcutaneous injections at 3-day intervals of (C) gp96N, CpG; gp96BCD, gp96BCD + GM-CSF, IL-12; or (D) gp96N, rat control IgG (IgG-ctrl), gp96BCD or gp96BCD + CpG combined with anti-CD25, B7H1, or IL-10 mAbs. Tumor burdens were measured twice every week. Mice were killed when subcutaneous tumors reached 225 mm2 or when mice became moribund. Results shown are measurements of tumor burdens and survival of mice that received different treatments. Representative results of 1 of 2 independent experiments are shown. *P < .05; **P < .01 (compared with controls).
To cure mice with large tumor burdens with the pooled heterogeneous gp96 vaccines, we hypothesized that it is necessary not only to enhance the immunogenicity of the vaccines, but also to target the suppressive tumor microenvironment and break immune suppression on effector cells. Based on the results presented previously in this study, we examined whether depleting Tregs, blocking B7H1-negative T-cell signaling, and neutralizing IL-10 would further improve the efficacy of the vaccine (gp96BCD). In this study, we used anti-CD25 mAb to deplete CD25+ Tregs,36,37 anti-B7H1 (M5H1) mAb29 to block negative T-cell signaling, and anti–IL-10 mAb to neutralize IL-10 in mice bearing large tumor burdens. Mice were inoculated with tumor-A cells, and when tumors reached 100 mm2, vaccination and other treatments were given, which included 3 subcutaneous vaccinations followed by intraperitoneal injections of depleting, blocking, or neutralizing mAbs on days 1, 4, 7, and 10 after the first vaccination. As shown in Figure 6D, whereas all mice receiving gp96N and control IgG and 4 of 5 mice vaccinated with gp96BCD alone died from myeloma by 25 day after tumor injection, treatments with pooled gp96BCD plus CpG, or gp96BCD plus CpG in combination with anti-CD25, anti-B7H1, or anti–IL-10 mAbs, respectively, eradicated myeloma in 3 of 5, 3 of 5, 4 of 5, and 4 of 5 mice, respectively. These results indicate that pooled gp96BCD plus CpG in combination with anti-B7H1 or anti–IL-10 mAbs were more effective at eradicating established large myeloma in vivo. Taken together, these results lay a basis for future clinical trials in myeloma using pooled allogeneic gp96 from human tumor cell lines.
Discussion
The goals of this study were to develop hsp-based immunotherapy for cancer patients without the requirement of individualized vaccine preparation and to improve the therapeutic efficacy of the treatment. To overcome the first problem, we hypothesized that pooled hsps such as gp96 from established tumor cell lines can be used to replace gp96 purified from autologous tumor cells and can be used as universal vaccines for immunotherapy of all patients with the same type of cancers. By pooling hsps from several cell lines, a broader repertoire of tumor antigens may be obtained to cover the need of all potential patients. In this study, preclinical studies using murine myeloma cell lines and mouse models were performed. Our results clearly showed that pooled gp96 vaccines from heterogeneous or allogeneic myeloma cell lines protected mice against developing myeloma and treated myeloma-bearing mice as efficiently as gp96 from the autologous tumor cells. These results also showed that gp96 vaccines from individual heterologous tumor cell lines did not provide sufficient protection against other tumors, which is in line with results from previous studies showing that immunotherapy with autologous but not heterologous cancer (a single cell line)–derived gp96 is effective in therapy.9,38,39 Furthermore, pooled gp96 vaccines were able to protect mice with different MHC backgrounds from developing tumors. Thus, these findings strongly suggest that pooled gp96 vaccines from heterogeneous or allogeneic myeloma cell lines not only can replace gp96 vaccines from autologous tumor cells as universal tumor vaccines for all patients but also induce broader immune responses against different tumor antigens than autologous gp96 vaccines, which may be important for protecting against tumor recurrence and development of unrelated tumors in vaccinated patients.
The second problem associated with hsp-based immunotherapy in human cancers is the disappointing clinical response in treated patients.15,40 As we have a better understanding of tumor microenvironment and the presence of regulatory T cells, it has become clear that simple active vaccination with tumor antigens, whether combined with DCs or other adjuvants such as GM-CSF or IL-12,32,33 is insufficient to induce strong immune responses and overcome immune suppression to generate objective clinical responses. Thus, we explored combinational therapy of gp96 vaccines with more potent immune adjuvants such as CpG to improve the immunogenicity of the vaccines, and depletion of Tregs, blocking B7H1 signaling, and/or neutralizing IL-10 to overcome immune suppression on vaccine-induced, tumor-specific CTLs and Th1 cells. These treatments were chosen because similar to other tumor cells, myeloma cells recruit and expand Tregs (Figure 5B),41,42 express B7H1 (Figure 5C), and secrete IL-10.43,44 Our results showed that gp96 vaccines alone or in combination with GM-CSF or IL-12, which were the commonly used forms of hsp vaccines in clinical trials,45,46 were therapeutic in some mice bearing intermediate tumor burdens, whereas gp96 vaccines in combination with CpG eradiated all intermediate tumors in the mice, suggesting that CpG may be a better adjuvant for immunotherapy with gp96 vaccines. However, when large tumors developed in the mice, which may be similar to the situations for human cancers, the treatments (gp96 vaccines + CpG) alone were less therapeutic, indicating that strategies to break immune suppression are also required. Indeed, blocking B7H1 and/or neutralizing IL-10 further improved the therapeutic efficacy of gp96 vaccines in mice bearing large tumor burdens. Depletion of Tregs by anti-CD25 mAb did not improve the therapeutic efficacy, although our preliminary studies confirmed that the mAb indeed depleted most of Tregs in the blood and spleen after infusions (data not shown). Considering the facts that anti-CD25 mAbs also deplete effector T cells expressing CD2547 and Tregs can be converted from CD4+CD25− T cells after Treg depletion,48 it is not surprising that anti-CD25 mAb showed no effect to improve the therapeutic effect of gp96 vaccines. Nevertheless, these results support our hypothesis that the efficacy of hsp-based immunotherapies can be improved by combinational treatments and lay a basis for future clinical trials in myeloma and other tumors using pooled allogeneic gp96 from human tumor cell lines as universal tumor vaccines.
The ability of hsps to facilitate the cross-presentation of MHC class I–restricted epitopes and to prime CD8+ cell effector responses is well established.49 Interestingly, recent studies have also shown that hsp-peptide complexes can also lead to antigen presentation on MHC class II molecules, thus activating CD4+ T cells.50,51 In line with these findings, our studies clearly showed that gp96 vaccines induced tumor-specific CD4+ and CD8+ T-cell responses, although the CD8+ T-cell response was stronger than that of CD4+ T cells. In vivo depletion experiments further indicate that both CD4+ and CD8+ T cells play a role in protecting mice against myeloma and adoptive transfer of splenocytes containing both CD4+ and CD8+ T cells from vaccinated mice provided better protection than that of CD4+ or CD8+ T cells alone. The ability of gp96 to activate CD8+ and CD4+ T cells in vivo bears considerable potential for future clinical applications, especially with recent reports supporting the essential role of costimulated CD4+ T-cell responses in the induction of functionally competent memory CTLs.52,53 CD8+ T cells have previously been shown to have a greater proliferative potential than CD4+ T cells,54,55 and require only a short period of antigenic stimulation to become programmed to differentiate into effectors,56,57 whereas CD4+ T-cell effector differentiation generally requires longer periods of antigenic stimulation and/or extrinsic cytokines.57–59 Our study appears to be consistent with these previous observations by suggesting that CD4+ and CD8+ T cells might differ in their immunization requirements for developing the ability to express the effector cytokine IFN-γ. Thus, gp96 vaccines that induce cognate naive CD8+ T cells to proliferate and develop the potential to express IFN-γ can induce cognate naive CD4+ T cells to proliferate, but not to differentiate into effectors that have the potential to express IFN-γ.
In conclusion, our study suggests that pooled gp96 from established tumor cell lines can be used as universal tumor vaccines for immunotherapy of patients with the same type of cancers. This is supported by our results that pooled gp96 from 3 heterogeneous or allogeneic myeloma cell lines provided equally strong protection as gp96 from autologous tumor cells against mice from developing myeloma. Pooled gp96 was effective in treating established myeloma in the mouse models. Furthermore, using CpG, B7H1 blocking Abs, and IL-10 neutralizing mAbs, the therapeutic efficacy of gp96 vaccines can be greatly improved to eradicate large tumors developed in the mice. As established tumor cell lines are available to many if not most human cancers, and clinical-grade mAbs for B7H1, PD-1, or IL-10 are available, this novel approach is feasible and applicable to human cancer treatments.
Supplementary Material
Acknowledgments
We thank Ms Alison Woo for providing editorial assistance.
This work was supported by institutional start-up funds from the University of Texas M. D. Anderson Cancer Center, grants from the National Cancer Institute (R01 CA96569, R01 CA103978, and R01 CA138402), the Leukemia & Lymphoma Society, Multiple Myeloma Research Foundation, Commonwealth Foundation for Cancer Research, and the National Natural Science Foundation of China (no. 30828017).
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.Q., S.H., and S.W. performed the majority of experiments and contributed to the writing of the paper; L.Z., L.S., and J.Y. performed some experiments; M.W. and L.W.K. provided critical suggestions; J.H. contributed to the concept of the paper and provided critical discussion and suggestions; and Q.Y. contributed to the concept of the paper, experimental design, and writing and editing of the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Qing Yi, Department of Lymphoma and Myeloma, M. D. Anderson Cancer Center, 1515 Holcombe Blvd, Unit 0903, Houston, TX 77030; e-mail: qyi@mdanderson.org; or Jian Hou, Department of Hematology, Shanghai Chang Zheng Hospital, Shanghai 200003, PR China; e-mail: houjian@medmail.com.cn.
References
- 1.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lollini PL, Cavallo F, Nanni P, Forni G. Vaccines for tumour prevention. Nat Rev Cancer. 2006;6(3):204–216. doi: 10.1038/nrc1815. [DOI] [PubMed] [Google Scholar]
- 3.Wen YJ, Barlogie B, Yi Q. Idiotype-specific cytotoxic T lymphocytes in multiple myeloma: evidence for their capacity to lyse autologous primary tumor cells. Blood. 2001;97(6):1750–1755. doi: 10.1182/blood.v97.6.1750. [DOI] [PubMed] [Google Scholar]
- 4.Stevenson FK, Anderson KC. Preparing the ground for vaccination against multiple myeloma. Immunol Today. 2000;21(4):170–171. doi: 10.1016/s0167-5699(99)01579-0. [DOI] [PubMed] [Google Scholar]
- 5.Beliakoff J, Whitesell L. Hsp90: an emerging target for breast cancer therapy. Anticancer Drugs. 2004;15(7):651–662. doi: 10.1097/01.cad.0000136876.11928.be. [DOI] [PubMed] [Google Scholar]
- 6.Ciocca DR, Oesterreich S, Chamness GC, McGuire WL, Fuqua SA. Biological and clinical implications of heat shock protein 27,000 (Hsp27): a review. J Natl Cancer Inst. 1993;85(19):1558–1570. doi: 10.1093/jnci/85.19.1558. [DOI] [PubMed] [Google Scholar]
- 7.Srivastava PK, Udono H. Heat shock protein-peptide complexes in cancer immunotherapy. Curr Opin Immunol. 1994;6(5):728–732. doi: 10.1016/0952-7915(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 8.Udono H, Levey DL, Srivastava PK. Cellular requirements for tumor-specific immunity elicited by heat shock proteins: tumor rejection antigen gp96 primes CD8+ T cells in vivo. Proc Natl Acad Sci U S A. 1994;91(8):3077–3081. doi: 10.1073/pnas.91.8.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamura Y, Peng P, Liu K, Daou M, Srivastava PK. Immunotherapy of tumors with autologous tumor-derived heat shock protein preparations. Science. 1997;278(5335):117–120. doi: 10.1126/science.278.5335.117. [DOI] [PubMed] [Google Scholar]
- 10.Janetzki S, Blachere NE, Srivastava PK. Generation of tumor-specific cytotoxic T lymphocytes and memory T cells by immunization with tumor-derived heat shock protein gp96. J Immunother. 1998;21(4):269–276. doi: 10.1097/00002371-199807000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Rivoltini L, Castelli C, Carrabba M, et al. Human tumor-derived heat shock protein 96 mediates in vitro activation and in vivo expansion of melanoma- and colon carcinoma-specific T cells. J Immunol. 2003;171(7):3467–3474. doi: 10.4049/jimmunol.171.7.3467. [DOI] [PubMed] [Google Scholar]
- 12.Janetzki S, Palla D, Rosenhauer V, Lochs H, Lewis JJ, Srivastava PK. Immunization of cancer patients with autologous cancer-derived heat shock protein gp96 preparations: a pilot study. Int J Cancer. 2000;88(2):232–238. doi: 10.1002/1097-0215(20001015)88:2<232::aid-ijc14>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 13.Belli F, Testori A, Rivoltini L, et al. Vaccination of metastatic melanoma patients with autologous tumor-derived heat shock protein gp96-peptide complexes: clinical and immunologic findings. J Clin Oncol. 2002;20(20):4169–4180. doi: 10.1200/JCO.2002.09.134. [DOI] [PubMed] [Google Scholar]
- 14.Mazzaferro V, Coppa J, Carrabba MG, et al. Vaccination with autologous tumor-derived heat-shock protein gp96 after liver resection for metastatic colorectal cancer. Clin Cancer Res. 2003;9(9):3235–3245. [PubMed] [Google Scholar]
- 15.Testori A, Richards J, Whitman E, et al. Phase III comparison of vitespen, an autologous tumor-derived heat shock protein gp96 peptide complex vaccine, with physician's choice of treatment for stage IV melanoma: the C-100-21 Study Group. J Clin Oncol. 2008;26(6):955–962. doi: 10.1200/JCO.2007.11.9941. [DOI] [PubMed] [Google Scholar]
- 16.Qian J, Wang S, Yang J, et al. Targeting heat shock proteins for immunotherapy in multiple myeloma: generation of myeloma-specific CTLs using dendritic cells pulsed with tumor-derived gp96. Clin Cancer Res. 2005;11(24 pt 1):8808–8815. doi: 10.1158/1078-0432.CCR-05-1553. [DOI] [PubMed] [Google Scholar]
- 17.Li Z. Priming of T cells by heat shock protein-peptide complexes as the basis of tumor vaccines. Semin Immunol. 1997;9(5):315–322. doi: 10.1006/smim.1997.0087. [DOI] [PubMed] [Google Scholar]
- 18.Oki Y, Younes A. Heat shock protein-based cancer vaccines. Expert Rev Vaccines. 2004;3(4):403–411. doi: 10.1586/14760584.3.4.403. [DOI] [PubMed] [Google Scholar]
- 19.Garrett IR, Dallas S, Radl J, Mundy GR. A murine model of human myeloma bone disease. Bone. 1997;20(6):515–520. doi: 10.1016/s8756-3282(97)00056-2. [DOI] [PubMed] [Google Scholar]
- 20.Mundy G. Preclinical models of bone metastases. Semin Oncol. 2001;28(4) suppl 11:2–8. doi: 10.1016/s0093-7754(01)90225-8. [DOI] [PubMed] [Google Scholar]
- 21.Radl J, De Glopper ED, Schuit HR, Zurcher C. Idiopathic paraproteinemia, II: transplantation of the paraprotein-producing clone from old to young C57BL/KaLwRij mice. J Immunol. 1979;122(2):609–613. [PubMed] [Google Scholar]
- 22.Srivastava PK. Purification of heat shock protein-peptide complexes for use in vaccination against cancers and intracellular pathogens. Methods. 1997;12(2):165–171. doi: 10.1006/meth.1997.0464. [DOI] [PubMed] [Google Scholar]
- 23.Srivastava PK, Jaikaria NS. Methods of purification of heat shock protein-peptide complexes for use as vaccines against cancers and infectious diseases. Methods Mol Biol. 2001;156:175–186. doi: 10.1385/1-59259-062-4:175. [DOI] [PubMed] [Google Scholar]
- 24.Lutz MB, Kukutsch N, Ogilvie AL, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223(1):77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 25.Fulcher D, Wong S. Carboxyfluorescein succinimidyl ester-based proliferative assays for assessment of T cell function in the diagnostic laboratory. Immunol Cell Biol. 1999;77(6):559–564. doi: 10.1046/j.1440-1711.1999.00870.x. [DOI] [PubMed] [Google Scholar]
- 26.Qian J, Xie J, Hong S, et al. Dickkopf-1 (DKK1) is a widely expressed and potent tumor-associated antigen in multiple myeloma. Blood. 2007;110(5):1587–1594. doi: 10.1182/blood-2007-03-082529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Qian J, Wezeman M, et al. Targeting beta(2)-microglobulin for induction of tumor apoptosis in human hematological malignancies. Cancer Cell. 2006;10(4):295–307. doi: 10.1016/j.ccr.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 28.Yu M, Finn OJ. DNA vaccines for cancer too. Cancer Immunol Immunother. 2006;55(2):119–130. doi: 10.1007/s00262-005-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanai T, Totsuka T, Uraushihara K, et al. Blockade of B7-H1 suppresses the development of chronic intestinal inflammation. J Immunol. 2003;171(8):4156–4163. doi: 10.4049/jimmunol.171.8.4156. [DOI] [PubMed] [Google Scholar]
- 30.Kwak LW, Young HA, Pennington RW, Weeks SD. Vaccination with syngeneic, lymphoma-derived immunoglobulin idiotype combined with granulocyte/macrophage colony-stimulating factor primes mice for a protective T-cell response. Proc Natl Acad Sci U S A. 1996;93(20):10972–10977. doi: 10.1073/pnas.93.20.10972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang S, Hong S, Wezeman M, Qian J, Yang J, Yi Q. Dendritic cell vaccine but not idiotype-KLH protein vaccine primes therapeutic tumor-specific immunity against multiple myeloma. Front Biosci. 2007;12:3566–3575. doi: 10.2741/2335. [DOI] [PubMed] [Google Scholar]
- 32.Wüthrich M, Warner T, Klein BS. IL-12 is required for induction but not maintenance of protective, memory responses to Blastomyces dermatitidis: implications for vaccine development in immune-deficient hosts. J Immunol. 2005;175(8):5288–5297. doi: 10.4049/jimmunol.175.8.5288. [DOI] [PubMed] [Google Scholar]
- 33.Silla S, Fallarino F, Boon T, Uyttenhove C. Enhancement by IL-12 of the cytolytic T lymphocyte (CTL) response of mice immunized with tumor-specific peptides in an adjuvant containing QS21 and MPL. Eur Cytokine Netw. 1999;10(2):181–190. [PubMed] [Google Scholar]
- 34.Bernard MP, Phipps RP. CpG oligodeoxynucleotides induce cyclooxygenase-2 in human B lymphocytes: implications for adjuvant activity and antibody production. Clin Immunol. 2007;125(2):138–148. doi: 10.1016/j.clim.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klinman DM. Adjuvant activity of CpG oligodeoxynucleotides. Int Rev Immunol. 2006;25(3–4):135–154. doi: 10.1080/08830180600743057. [DOI] [PubMed] [Google Scholar]
- 36.Casares N, Arribillaga L, Sarobe P, et al. CD4+/CD25+ regulatory cells inhibit activation of tumor-primed CD4+ T cells with IFN-gamma-dependent antiangiogenic activity, as well as long-lasting tumor immunity elicited by peptide vaccination. J Immunol. 2003;171(11):5931–5939. doi: 10.4049/jimmunol.171.11.5931. [DOI] [PubMed] [Google Scholar]
- 37.Zhang M, Zhang Z, Garmestani K, et al. Pretarget radiotherapy with an anti-CD25 antibody-streptavidin fusion protein was effective in therapy of leukemia/lymphoma xenografts. Proc Natl Acad Sci U S A. 2003;100(4):1891–1895. doi: 10.1073/pnas.0437788100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Przepiorka D, Srivastava PK. Heat shock protein–peptide complexes as immunotherapy for human cancer. Mol Med Today. 1998;4(11):478–484. doi: 10.1016/s1357-4310(98)01345-8. [DOI] [PubMed] [Google Scholar]
- 39.Srivastava PK, Udono H. Heat shock protein-peptide complexes in cancer immunotherapy. Curr Opin Immunol. 1994;6(5):728–732. doi: 10.1016/0952-7915(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 40.Oki Y, McLaughlin P, Fayad LE, et al. Experience with heat shock protein-peptide complex 96 vaccine therapy in patients with indolent non-Hodgkin lymphoma. Cancer. 2007;109(1):77–83. doi: 10.1002/cncr.22389. [DOI] [PubMed] [Google Scholar]
- 41.Atanackovic D, Cao Y, Luetkens T, et al. CD4+CD25+FOXP3+ T regulatory cells reconstitute and accumulate in the bone marrow of patients with multiple myeloma following allogeneic stem cell transplantation. Haematologica. 2008;93(3):423–430. doi: 10.3324/haematol.11897. [DOI] [PubMed] [Google Scholar]
- 42.Prabhala RH, Neri P, Bae JE, et al. Dysfunctional T regulatory cells in multiple myeloma. Blood. 2006;107(1):301–304. doi: 10.1182/blood-2005-08-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kröning H, Tager M, Thiel U, et al. Overproduction of IL-7, IL-10 and TGF-beta 1 in multiple myeloma. Acta Haematol. 1997;98(2):116–118. doi: 10.1159/000203602. [DOI] [PubMed] [Google Scholar]
- 44.Urbańska-Ryś H, Wiersbowska A, Stepień H, Robak T. Relationship between circulating interleukin-10 (IL-10) with interleukin-6 (IL-6) type cytokines (IL-6, interleukin-11 (IL-11), oncostatin M (OSM)) and soluble interleukin-6 (IL-6) receptor (sIL-6R) in patients with multiple myeloma. Eur Cytokine Netw. 2000;11(3):443–451. [PubMed] [Google Scholar]
- 45.Pilla L, Patuzzo R, Rivoltini L, et al. A phase II trial of vaccination with autologous, tumor-derived heat-shock protein peptide complexes Gp96, in combination with GM-CSF and interferon-alpha in metastatic melanoma patients. Cancer Immunol Immunother. 2006;55(8):958–968. doi: 10.1007/s00262-005-0084-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ito A, Fujioka M, Tanaka K, Kobayashi T, Honda H. Screening of cytokines to enhance vaccine effects of heat shock protein 70-rich tumor cell lysate. J Biosci Bioeng. 2005;100(1):36–42. doi: 10.1263/jbb.100.36. [DOI] [PubMed] [Google Scholar]
- 47.Couper KN, Lanthier PA, Perona-Wright G, et al. Anti-CD25 antibody-mediated depletion of effector T cell populations enhances susceptibility of mice to acute but not chronic Toxoplasma gondii infection. J Immunol. 2009;182(7):3985–3994. doi: 10.4049/jimmunol.0803053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Long SA, Walker MR, Rieck M, et al. Functional islet-specific Treg can be generated from CD4+CD25- T cells of healthy and type 1 diabetic subjects. Eur J Immunol. 2009;39(2):612–620. doi: 10.1002/eji.200838819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Ann Rev Immunol. 2002;20:395–425. doi: 10.1146/annurev.immunol.20.100301.064801. [DOI] [PubMed] [Google Scholar]
- 50.Doody AD, Kovalchin JT, Mihalyo MA, Hagymasi AT, Drake CG, Adler AJ. Glycoprotein 96 can chaperone both MHC class I- and class II-restricted epitopes for in vivo presentation, but selectively primes CD8+ T cell effector function. J Immunol. 2004;172(10):6087–6092. doi: 10.4049/jimmunol.172.10.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.SenGupta D, Norris PJ, Suscovich TJ, et al. Heat shock protein-mediated cross-presentation of exogenous HIV antigen on HLA class I and class II. J Immunol. 2004;173(3):1987–1993. doi: 10.4049/jimmunol.173.3.1987. [DOI] [PubMed] [Google Scholar]
- 52.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300(5617):337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 53.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300(5617):339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Boer RJ, Homann D, Perelson AS. Different dynamics of CD4+ and CD8+ T cell responses during and after acute lymphocytic choriomeningitis virus infection. J Immunol. 2003;171(8):3928–3935. doi: 10.4049/jimmunol.171.8.3928. [DOI] [PubMed] [Google Scholar]
- 55.Foulds KE, Zenewicz LA, Shedlock DJ, Jiang J, Troy AE, Shen H. Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses. J Immunol. 2002;168(4):1528–1532. doi: 10.4049/jimmunol.168.4.1528. [DOI] [PubMed] [Google Scholar]
- 56.van Stipdonk MJ, Lemmens EE, Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol. 2001;2(5):423–429. doi: 10.1038/87730. [DOI] [PubMed] [Google Scholar]
- 57.Iezzi G, Karjalainen K, Lanzavecchia A. The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity. 1998;8(1):89–95. doi: 10.1016/s1074-7613(00)80461-6. [DOI] [PubMed] [Google Scholar]
- 58.Iezzi G, Scotet E, Scheidegger D, Lanzavecchia A. The interplay between the duration of TCR and cytokine signaling determines T cell polarization. Eur J Immunol. 1999;29(12):4092–4101. doi: 10.1002/(SICI)1521-4141(199912)29:12<4092::AID-IMMU4092>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 59.Jelley-Gibbs DM, Lepak NM, Yen M, Swain SL. Two distinct stages in the transition from naive CD4 T cells to effectors, early antigen-dependent and late cytokine-driven expansion and differentiation. J Immunol. 2000;165(9):5017–5026. doi: 10.4049/jimmunol.165.9.5017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}