

Abstract
The predominant outgrowth of malignant cells over their normal counterparts in a given tissue is a shared feature for all types of cancer. However, the impact of a cancer environment on normal tissue stem and progenitor cells has not been thoroughly investigated. We began to address this important issue by studying the kinetics and functions of hematopoietic stem and progenitor cells in mice with Notch1-induced leukemia. Although hematopoiesis was progressively suppressed during leukemia development, the leukemic environment imposed distinct effects on hematopoietic stem and progenitor cells, thereby resulting in different outcomes. The normal hematopoietic stem cells in leukemic mice were kept in a more quiescent state but remained highly functional on transplantation to nonleukemic recipients. In contrast, the normal hematopoietic progenitor cells in leukemic mice demonstrated accelerated proliferation and exhaustion. Subsequent analyses on multiple cell-cycle parameters and known regulators (such as p21, p27, and p18) further support this paradigm. Therefore, our current study provides definitive evidence and plausible underlying mechanisms for hematopoietic disruption but reversible inhibition of normal hematopoietic stem cells in a leukemic environment. It may also have important implications for cancer prevention and treatment in general.
Introduction
The predominant outgrowth of malignant cells over their normal counterparts in a given tissue is a shared feature for all types of cancer. Although many intrinsic and extrinsic factors have been implicated in cancer development, the impact of a specific cancer environment on normal tissue stem and progenitor cells has been poorly understood. Like other cancers, leukemia is caused by both intrinsic factors, such as the aberrant expression of oncogenes or tumor suppressors, and extrinsic factors, such as immune dysfunction, neovasculature and other tumor-promoting microenvironmental cues in the hematopoietic system. Leukemia stem cells (LSCs) are thought to play a key role in the initiation, and possibly the maintenance, of leukemia.1–3 During leukemogenesis, LSCs outcompete their normal counterparts, namely, hematopoietic stem cells (HSCs), and become dominant by acquiring an increased capacity for self-renewal coupled with decreased levels of cell death or a disrupted differentiation program.4 Thus, the clinical manifestations of leukemia are determined largely by the competition between LSCs and normal HSCs.
HSCs give rise to all types of mature blood and immune cells, and these mature cells provide environmental factors that can also influence leukemogenesis. Deregulated hematopoiesis can lead to significantly decreased numbers of blood cells and to anemia, infection, and hemorrhage, all of which are directly responsible for the poor life quality and increased mortality of cancer patients. Moreover, because autologous HSC transplantation (auto-HSCT) has been used with chemotherapy and radiotherapy to treat hematologic malignancies and other solid tumors, the quality of normal HSCs from a cancer patient may serve as a critical parameter for the ultimate success of auto-HSCT for the patient. Therefore, the impact of a leukemic environment on normal HSCs and hematopoietic progenitor cells (HPCs) during leukemogenesis is an issue of high significance. Studies along this line may have implications for other cancers as well.
In this study, we have examined the kinetics of normal HSCs and HPCs in the Notch1-induced murine T-cell leukemia model. Our results demonstrate that hematopoietic cells in the leukemic environment are progressively decreased as measured by both phenotype and function. However, the effects of the leukemic environment on HSCs and HPCs are distinct in that the repopulation potential of HSCs from the leukemic environment is preserved, whereas mature blood cells cannot be produced because of the exhaustion of HPCs. Therefore, these data yield new insights into the pathogenesis of Notch1-involved T-cell leukemia and may guide better clinical management for this type of malignancy.
Methods
Mice
Wild-type C57BL/6J mice were obtained from the Jackson Laboratory, and B6.SJL-PtprcaPepcb/BoyJ mice (B6.SJL) were purchased from either Taconic Farms or the Jackson Laboratory. CD45.1+/CD45.2+ mice were the F1 generation of C57BL/6J and B6.SJL-PtprcaPepcb/BoyJ mice. All mice were maintained in the certified animal facility of the Hillman Cancer Center, University of Pittsburgh. The procedures involved in the animal work were approved by the Institutional Animal Care and Use Committee at the institutions involved in this study.
Transduction of murine primary bone marrow cells
The retrovirus vector containing the cDNA-encoding intracellular domain of Notch1 (ICN1) was kindly provided by Dr David Scadden (Harvard University, Boston, MA). The plasmid (MSCV-ICN1-IRES-GFP) was cotransfected into package cell line 293T (bestowed by Dr Wan Yong, University of Pittsburgh, Pittsburgh, PA) with pCMV-VSV-G and pKAT, using lipofectamine 2000 (Invitrogen). Supernatant was harvested 48 hours and 72 hours after transfection.
Lineage-negative (Lin−) cells from the bone marrow (BM) of female C57BL/6J or B6.SJL mice were enriched with biotin-conjugated lineage antibodies (CD3 [RM3415-3], CD4 [RM2515-3], CD8 [RM2215-3], CD11b [Mac-1, RM2815-3], Gr-1 [Ly-6G, RM3015-3], CD45R [B220,RM2615-3], Ter-119 [MTER15-3]; Caltag Laboratories), and selected with Microbead-conjugated streptavidin (Miltenyi Biotec) using the manufacturer's protocol. Then the Lin− cells were reenriched for expression of Sca-1 using the EasySep murine Sca-1 positive selection kit (Miltenyi Biotec). Transduction of lineage-negative Sca-1 positive (Lin−Sca-1+) cells with ICN1 plasmid was performed as previously described with minor modifications.5 The transduction efficiency was measured by flow cytometry.
Transplantation of the transduced cells and analysis of leukemia-bearing mice
Bone marrow nucleated cells (BMNCs; 107/host) from B6.SJL mice at the age of 6 to 8 weeks were transplanted into lethally irradiated (10 Gy) female C57BL/6J recipients (6-8 weeks old) with or without 106 ICN1 plasmid-transduced Lin−Sca-1+ cells from C57BL/6J mice as indicated in Figure 1A. Because of the in vitro transduction and culture procedures, the majority of the cells transplanted into the recipients (detailed below) should be the progenies of Lin−Sca-1+ cells, not the Lin−Sca-1+ cells themselves. The recipients were followed up physically on a daily basis. Peripheral blood (PB) sampling from the lateral tail vein was collected starting at 1 week after transplantation for white blood cell (WBC) count, and the percentage of CD45.2+GFP+ cells was analyzed by flow cytometry using phycoerythrin (PE)–Cy5.5-CD45.1 and PE-CD45.2 antibodies (eBioscience).
Figure 1.

Experimental design and establishment of a murine leukemia model by Notch1 overexpression. Lin−Sca-1+ cells (CD45.2+) were isolated from BM of C57BL/6J mice. A total of 107 BMNCs (CD45.1+) from B6.SJL mice were transplanted into lethally irradiated recipients (10 Gy, C57BL/6J) either with (leukemia group) or without (the control group) 106 cells that were transduced with the MSCV-ICN1-IRES-GFP vector. The transplanted normal hematopoietic cells (CD45.1+GFP−) from the recipients were analyzed and sorted by flow cytometry at different time points (A). Resultant data are shown in all the figures except Figure 5. An additional control with a higher dose of normal congenic BMNCs was included in some experiments (supplemental Figure 2). A T-ALL feature of the affected mice is shown by an immature T-cell phenotype (supplemental Figure 1). The hosts receiving Notch1-transduced cells displayed leukemia symptoms, including increases of total leukemic cells (CD45.2+GFP+) in PB (B), WBCs in PB (C), and BM cellularity (D). Values are mean ± SD. *P < .05 (t test). The 100% penetrance of the leukemic development is shown in the survival curve (E). P < .01 (n = 15 each, Kaplan-Meier analysis).
Flow cytometric analysis and cell sorting
Mouse BM cells were obtained by flushing ilias, femurs, tibias, and humeri as described.6 The immunophenotypes for murine long-term repopulating HSCs (CD34−Lin−c-Kit+Sca-1+, CD34−LKS), short-term repopulating HSCs (CD34+Lin−c-Kit+Sca-1+, CD34+ LKS), and Lin−c-Kit+Sca-1− (LKS−)7,8 were used to quantify these different cell types within the normal cell populations that were separated with different congenic markers. All the antibodies were purchased from BD Biosciences unless otherwise noted. For detection of HSCs/HPCs, we used streptavidin conjugated with PE-TxRed (to stain for biotinylated CD34), PE-Cy7 conjugated with a mixture of lineage antibodies (anti-CD3, CD4, CD8, B220, Gr-1, Mac-1, Ter-119; eBioscience), PE-conjugated Sca-1, allophycocyanin (APC)–conjugated c-Kit, PE-Cy5.5–conjugated CD45.1, and fluorescein isothiocyanate–conjugated CD45.2 (eBioscience). The method used to quantitatively measure the frequency of primitive cells was as previously described.9 For cell-cycle analysis, cells were stained with Hoechst 33342 (Invitrogen) and Pyronin-Y (PY; Sigma-Aldrich), or APC-BrdU Flow kit with HSC/HPC cell surface markers. Analyses were performed on an LSR (BD Biosciences).
For HSC/HPC isolation, the cells were then enriched for c-Kit expression by immunoselection with CD117-conjugated micromagnetic beads (Miltenyi Biotec) according to the manufacturer's instructions. Then the enriched cells were stained with PE-Cy7 conjugated with a mixture of lineage antibodies, PE-Sca-1 and APC-c-Kit. CD45.1+ LKS cells were directly sorted into different tubes and lysed for gene expression analysis. For normal hematopoietic cell sorting, CD45.1+GFP− cells were sorted with BD FACSAria+ sorter (BD Biosciences). During the sorting procedure, 4,6-diamidino-2-phenylindole was used to exclude the dead cells. For normal hematopoietic cell (CD45.1+) sorting, the whole BM cells from mice in leukemia-bearing or control groups were each pooled together, respectively. To discriminate transplanted normal hematopoietic cells (CD45.1+GFP−) from endogenous (CD45.2+GFP−) and leukemia cells (CD45.2+GFP+), the cells were stained with PE-Cy5.5-conjugated CD45.1 and PE-conjugated CD45.2 antibodies. To exclude the possibility of leukemia cell contamination in CD45.1+GFP− cell sorting, double sorting was used in the sorting strategy. The purity of CD45.1+GFP− cells was more than 99.99% (Figure 3A).
Figure 3.

In vitro clonal growth of normal hematopoietic cells from a leukemic environment. Two weeks after transplantation, the mice were killed and BM was harvested. The normal hematopoietic cells (CD45.1+GFP−)were doubly sorted with near 100% purity for in vitro clonal assays (A). For the CFC assay to measure committed HPCs (B), the sorted CD45.1+GFP− cells were cultured in the defined methylcellulose medium supplemented with a cytokine cocktail. Mix, GM, G, M, and E represent CFC-mix (> 2 lineages), CFC-granulocyte/monocyte, CFC-granulocyte, CFC-monocyte, and BFU-erythrocyte, respectively. Values are mean ± SD. *P < .05 (t test). **P < .01 (t test). In addition, the CAFC assay with limiting dilution at day 35 during the long-term culture was used to measure the more primitive hematopoietic cells, and the result is shown as a representative dataset from 3 experiments with similar results (C).
In vitro clonal assay
CD45.1+GFP− cells from the leukemia and control groups were sorted at 2 weeks after transplantation for in vitro clonal assay and in vivo competitive BM transplantation (cBMT), respectively. For the colony-forming cell (CFC) assay, the cells were placed in methylcellulose medium M3234 (StemCell Technologies) supplemented with a mixture of recombinant cytokines, including 50 ng/mL murine stem cell factor, 20 ng/mL murine interleukin-3, 20 ng/mL murine interleukin-6 (PeproTech), and 3 U/mL human erythropoietin (Amgen). Cells were plated in 24-well plates with a 0.5-mL volume at a density of 2 × 104 cells/mL with 4 replicated wells. At day 10, the CFC colonies were counted under an inverted microscope and recorded as colony number in a specific lineage. For 5-fluorouracil (5-FU) exposure assay in vivo 72 hours after transplantation, 150 mg/kg 5-FU was given (intravenously) at 12 hours before BM was harvested. CD45.1+GFP− cells were sorted for CFC assay as described above.
The frequency of cobble-stone area forming cells (CAFCs) at day 35 was determined using limiting dilution assay as described.10 Briefly, sorted CD45.1+GFP− cells were plated on an irradiated (15 Gy) primary murine stroma in 96-well plates containing 100 μL M5300 (StemCell Technologies) supplemented with 10−6 M hydrocortisone (Sigma-Aldrich) at 6 different concentrations with 20 replicates per concentration. The cells were fed with half-medium change weekly. The cobblestones were counted at day 35 and recorded as negative or positive for each well. Based on the Poisson distribution of the negative wells, the frequency of long-term initiating cells was calculated with L-Calc software (StemCell Technologies).
Apoptosis assay
Two weeks after transplantation, the whole BM cells were isolated from each group and stained for PE-Cy7 conjugated with a mixture of lineage antibodies and PE–annexin V with congenic marker. The proportion of apoptosis cells was determined using flow cytometry.
Competitive and serial BM transplant
The cBMT procedure was detailed in our previous publication.11 Briefly, normal hematopoietic cells (CD45.1+GFP−) were sorted from the BM of leukemia or control mice 2 weeks after transplantation. A total of 2 × 106 sorted CD45.1+GFP− cells (test cells) along with an equal number of CD45.1+/CD45.2+ BMNCs (competitive cells) were cotransplanted into lethally irradiated (10 Gy) female C57BL/6J recipients (n = 5/group, 6-8 weeks old). After transplantation, blood was collected monthly. The endpoint was 6 months. The relative contribution of test cells (CD45.1+) and competitive cells (CD45.1+/CD45.2+) in reconstituted recipients was measured by flow cytometry using PE-conjugated anti–mouse CD45.1 and fluorescein isothiocyanate–conjugated CD45.2 antibodies (eBioscience).
BrdU detection and cell-cycle analysis
For in vivo analysis, the mice were given a single pulse administration of 5-bromo-2-deoxyuridine (BrdU; BD Biosciences) at 72 hours after transplantation; an intraperitoneal injection of 100 μg/g of BrdU was given at 2 hours before harvesting the BM cells. BrdU staining was quantitated using flow cytometry by combining surface staining to define the BM subset with intracellular staining using the BrdU-APC staining kit following the manufacturer's instructions (BD Biosciences). For in vitro cell-cycle analysis, the whole BM cells from the leukemic or control group were isolated at 14 days after transplantation. Then the cells were permeabilized and stained with 1.67 μM Hoechst 33342 followed by 1 μg/mL PY. The proportion of CD45.1+ LKS cells in G0 phase was determined by flow cytometry with quantitation of DNA and RNA.
Cell proliferation tracing in vivo
Whole BM cells from B6.SJL mice were labeled with 1 μm of carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen) as described.12 A total of 108 CFSE-labeled CD45.1+ cells were injected into lethally irradiated C57BL/6J recipients with or without 5 × 106 developed ICN1-overexpressing leukemic cells (CD45.2+). Seventy-two hours after transplantation, the recipients were killed and the BM cells were harvested and stained with PE-Cy5.5–conjugated CD45.1, PE-conjugated Sca-1, APC-conjugated c-Kit, and a mixture of PE-Cy7-conjugated lineage markers. Dead cells were identified with 4,6-diamidino-2-phenylindole. The BD LSRII was used for data acquisition, and ModFit LT software (version 3.0, Verity Software House) was used for cell proliferation analysis.
Real-time RT-PCR
A total of 3000 CD45.1+ LKS cells were sorted directly into the lysis buffer (Stratagene). Total RNA was extracted with the RNA nanoprep kit according to the manufacturer's instructions (Stratagene). Reverse transcription was achieved using oligo-dT(12-18) and M-MLV reverse transcriptase (Ambion). Real-time polymerase chain reaction (PCR) was done with SYBR green Master Mix (Finnzymes), 0.3 μM of specific forward and reverse primers, and normalized cDNA. The parameters for the thermal cycling of PCR were as follows: 15 seconds at 95°C and 60 seconds at 60°C, 45 cycles. All the primer sequences are listed in supplemental Table 2.
Results
Notch1 overexpression induces T-ALL with 100% penetrance
We chose the Notch1-induced T acute lymphoblastic leukemia (T-ALL) model because the Notch1 signaling is thought to be a conserved pathway for self-renewal in HSCs and LSCs,2 and the retrovirus-mediated overexpression of ICN1 was shown to be potent in T-cell leukemogenesis.13,14 We established the mouse T-ALL model in which lethally irradiated recipients (C57BL/6J) were transplanted with 106 cells that were transduced with the MSCV-ICN1-IRES-GFP vector (starting with Lin−Sca-1+ cells) from C57BL/6J and 107 BMNCs from B6.SJL mice (Figure 1A). Those recipients accumulated a double-positive population (CD4+CD8+) of immature T cells in the PB and BM from the Notch1-transduced cells as early as 7 days after transplantation (supplemental Figure 1, available on the Blood website; see the Supplemental Materials link at the top of the online article). Two to 4 weeks after transplantation, WBCs in PB and BM cellularity dominated by GFP+ cells were increased in a temporal fashion (Figure 1B-D). Gross examination of tissues from the sick mice revealed splenomegaly, hepatomegaly, and lymphadenopathy, resulting from extensive organ infiltration by CD4+CD8+ lymphoblasts. This robust leukemia model, in which 100% of the mice that received Notch1-overexpressing cells developed T-ALL within 6 weeks (Figure 1E), permitted us to study the kinetics of cotransplanted normal hematopoietic cells (CD45.1) in a leukemic host from the onset of the disease.
Hematopoietic suppression at the stem cell and progenitor cell levels in leukemic mice
To document the disrupted growth of normal hematopoietic cells during leukemia development, we monitored the kinetics of cotransplanted normal hematopoietic cells that expressed CD45.1 (Figure 1A). Using the Notch1-induced leukemia model, we examined the percentage of CD45.1+ cells in both PB and BM 1 week after transplantation. The results showed that, in contrast to the rapid proliferation of the GFP+ leukemic cells, the percentage of CD45.1+ cells declined dramatically (Figure 2A-B). The average percentage of CD45.1+ cells in PB was 43.67% plus or minus 4.14% at 2 weeks after transplantation and 10.83% plus or minus 5.67% at 4 weeks after transplantation. As expected, the level of CD45.1+ cells in the PB of the control group was stable at approximately 90%. A similar level was observed in BM.
Figure 2.

Growth kinetics of normal hematopoietic cells in leukemia-bearing mice. The percentages of normal hematopoietic cells (CD45.1+) in the leukemia group were monitored in both PB (A) and BM (B) 2 and 4 weeks after transplantation. In addition, the frequencies of GFP−CD45.2−CD45.1+Lin− and GFP−CD45.2−CD45.1+ LKS cells in BM were measured 1, 2, and 3 weeks after transplantation (C-D). P < .05 (n = 5/each, t test). Frequencies and absolute numbers of different hematopoietic cell populations within GFP−CD45.2−CD45.1+ BMNCs 4 weeks after transplantation are shown in the graphs (E-F). *P < .05 (t test). **P < .01 (t test). The BM cellularity of tibia, femur, iliac, and humerus was calculated as 40% of the total BM cellularity of a young adult C57BL/6J mouse (based on our own data). The data shown here represent 1 of 4 independent experiments (n = 6 or 7/group). Values are mean ± SD.
To investigate whether the underrepresentation of normal hematopoietic CD45.1+ cells was the result of the direct negative impact of the leukemic environment on HSCs and HPCs, we quantified the frequencies of hematopoietic primitive cells at different time points after transplantation (Figure 2C-F). At 1 week after transplantation, the frequency of the HSC-enriched Lin−c-Kit+Sca-1+ (LKS) cell population in the BM of leukemic hosts was similar to that of the control, whereas the frequency of the HPC-enriched Lin− population was statistically higher than that in the control group. After the second week, the frequencies of both the LKS and Lin− cell populations became lower in the leukemia group than that in the control group (Figure 2C-D), whereas LKS cells in both groups continued to proliferate. At the fourth week, CD34−LKS (long-term repopulating HSCs), CD34+LKS (short-term repopulating HSCs), and LKS− (differentiating HPC) were significantly lower in the leukemic BM than in the control group as measured by either the frequency or the absolute yield of each population (Figure 2E-F). The input (CD45.1+CD34−LKS) and output (CD45.1+LKS) cells harvested 2 weeks after transplantation were measured. The yield of LKS cells per initial CD34−LKS in the leukemic host was 5 times lower than that in the nonleukemic but irradiated recipients (supplemental Table 1). An independent assessment for HSC quantitation with the signaling lymphocytic activation molecule markers15 also confirmed the significant inhibition of HSC growth in the leukemic host (supplemental Figure 2). More specifically, to exclude the possibility that the decreased frequency of the primitive cells in the leukemic marrow could be solely attributed to the absence of competition, we transplanted 107 BMNCs from B6.SJL (CD45.1+) along with a 100-times higher dose of BMNCs from C57BL/6J (108, CD45.2+) than that of the leukemic cells into the lethally irradiated control recipients (C57BL/6J). The frequency of HSCs (defined as CD150+CD48−LKS) in the leukemic group was still statistically lower than that in the control group (supplemental Figure 2).
We also performed in vitro clonal functional assays. CD45.1+ cells from leukemic and control groups 4 weeks after transplantation were isolated for the assessment of CFCs and CAFCs (Figure 3A). Notably, the frequency of CFCs was dramatically lower in the leukemic group (Figure 3B). Moreover, both groups showed detectable CAFC activity in the long-term culture system, but the CAFC frequency at day 35 during the culture from leukemia-bearing mice was approximately 4 times lower than that of the cells from the control mice (P < .05; Figure 3C). Together with the phenotypic analysis (Figure 2C-D), these data provide direct evidence for the progressive suppression of hematopoietic cell growth during leukemia development, although the kinetics of HSC and HPC seemed to differ at the early stage.
Functional preservation of HSCs in leukemic marrow
HSC is best defined by its ability to reconstitute the hematopoiesis of a lethally irradiated recipient.16 Given the significant suppression of normal HSCs and HPCs during leukemia development as assessed by phenotypic analysis and in vitro culture, we next tested whether the repopulating ability of normal hematopoietic cells from a leukemic environment was decreased in a new nonleukemic host. We cotransplanted equal numbers of CD45.1+ cells isolated from either a leukemic or a control group (2 weeks after transplantation) along with competitor cells (BMNCs freshly isolated from mice with a CD45.1+/CD45.2+ phenotype) into lethally irradiated C57BL/6J recipients. Because the repopulation unit in an unmanipulated C57BL/6J mouse has been well documented,16 a fixed number of competitor cells (2 × 106) can serve as a standard measure against which the input cells from both groups can be compared. PB was collected 1 to 6 months after cBMT, and the relative contribution to hematopoiesis of CD45.1+ cells isolated from both the leukemic and control groups was quantified using flow cytometry based on distinct congenic surface markers (CD45.1+ vs CD45.2+). Unexpectedly, CD45.1+ cells isolated from the leukemic hosts engrafted better than the cells isolated from the nonleukemic control recipients. The level of CD45.1+ cells gradually increased after transplantation and stabilized at 3 months. Six months after cBMT, the level of CD45.1+ hematopoietic cells from the leukemic environment was more than 3 times higher than that from the control group (38.45% vs 11.40%; Figure 4A). This indicates no apparent defect in the hematopoietic potential of the residual normal HSCs isolated from the leukemic environment. A multilineage analysis of PB from cBMT recipients was performed to determine whether the HSCs in the leukemic environment maintained their ability to give rise to both myeloid and lymphoid lineages. The myeloid cells (granulocytes and monocytes) were immunophenotypically defined by Mac-1+, the T cells by CD3+, and the B cells by B220+ (Figure 4B). At the endpoint of 6 months after cBMT, the average level of CD45.1+ cells isolated from leukemic BM was approximately 10-fold higher than that of cells from the irradiated but nonleukemic environment (44.87% vs 4.68%; Figure 4C-D). Consistent with this finding, the number of HSCs/HPCs from the leukemic environment was significantly higher than that from the control environment in the new recipients (Figure 4E).
Figure 4.

Long-term reconstitution of normal hematopoietic cells from leukemic marrow in new nonleukemic hosts. The hematopoietic regeneration of the normal hematopoietic cells from leukemic or control mice were examined in secondary nonleukemic recipients using the cBMT assay, in which equal numbers of test (CD45.1+GFP−) and competitor cells (CD45.1+/.2+) were cotransplanted into lethally irradiated congenic recipients (CD45.2+). The overall reconstitution levels of normal HSCs from the primary recipients were monitored within 6 months after transplantation (A). Multilineage differentiation of the engrafted cells was analyzed 6 months after transplantation (B). GM, T, and B indicate lineages for myeloid (Mac-1+), T (CD3+), and B (B220+) cells, respectively. Six months after cBMT, the overall representation of CD45.1+GFP− cells, multilineage analysis, and different hematopoietic cell subsets in BM were also quantified (C-E). *P < .05; **P < .01 (n = 5-7/group, t test). Data are from 1 of 3 experiments with similar results.
Increased proliferation of normal hematopoietic progenitors on exposure to leukemic marrow
The marked decrease of normal hematopoietic cells in leukemic marrow may be the result of alterations in cell proliferation or apoptosis. We examined the proliferation of the affected HPC populations with multiple assays. We first measured cell divisions of an immunophenotypically defined HPC population in lethally irradiated recipients either in the presence or absence of leukemic cells. We used the dye 5-(and 6-)CFSE to label the normal BMNCs from B6.SJL animals before transplantation,17 and surface markers of hematopoietic primitive cells were used to costain BMNCs that were harvested 72 hours after transplantation. Because of the undetectable level of c-Kit expression on cells shortly after transplantation,12 we used the Sca-1 marker to define a relatively primitive state within the Lin− population and found that 72 hours after transplantation, the CD45.1+Lin−Sca-1+ cells cotransplanted with the leukemic cells underwent more cell divisions than the control cells (Figure 5A). This suggests that HPCs in the leukemic environment have a more active cycling status.
Figure 5.

Proliferative response of normal HPCs to leukemic cells in vivo. (A) CFSE assay. A total of 108 BMNCs (C57BL/6J, CD45.2+) labeled with CFSE were coinjected with or without 5 × 106 Notch1-induced leukemia cells (CD45.2+GFP+) into lethally irradiated recipient mice (SJL.B6, CD45.1+). BMNCs were harvested 72 hours after the transplantation to assess the number of cell divisions. BMNCs were stained with lineage markers and Sca-1, and CFSE-labeled cells were analyzed in the gate for CD45.1+Lin−Sca-1+. A representative figure of the flow cytometric analysis is shown. Blue peaks on the right represent undivided cells (parent cells); each peak toward the left side, one cell division or generation. The figure shown is from 1 of 4 experiments with similar results. The proliferation index12 in the CD45.1+Lin−Sca-1+ population is shown in the graph. *P < .05. (B) BrdU assay. BrdU was injected intraperitoneally 72 hours after transplantation. BM cells were analyzed 2 hours later, and the proportion of BrdU+CD45.1+Lin−Sca-1+ cells was analyzed with flow cytometry. A representative plot is shown, and the figure shown is from 1 of 6 experiments with similar results. Values are mean ± SD. **P < .01 (n = 3 mice/group, t test). (C) 5-FU assay. A total of 150 mg/g 5-FU was injected 12 hours before BM was harvested, and then CD45.1+GFP− cells were isolated for the CFC assay 72 hours after transplantation. The total CFC colonies were counted at day 14 with microscopy. The data shown are from 1 of 2 independent experiments (n = 3-5 wells). **P < .01.
To determine the actual cycling status of the hematopoietic primitive cells under leukemic stress, mice in the leukemia and control groups were then pulsed with BrdU 72 hours after transplantation of the leukemic cells. CD45.1+Lin−Sca-1+ cells were analyzed for BrdU incorporation 2 hours after injection. As shown in Figure 5B, BrdU+Lin−Sca-1+ cells were significantly more abundant in the leukemic group than in the control group. Furthermore, we used the antimetabolic reagent 5-FU to functionally measure the cells in S-phase. Seventy-two hours after transplantation, CD45.1+GFP− cells were isolated for CFC assay. Whereas the CFC frequency increased significantly in leukemic marrow, there was a more dramatic reduction of the CFC yield after 5-FU treatment (Figure 5C), thus confirming the result of BrdU incorporation (Figure 5B). These cell-cycle measurements, together with the CFC yields, are also consistent with the finding that CD45.1+Lin− cells were more abundant in leukemic mice at the early stage (Figure 2C).
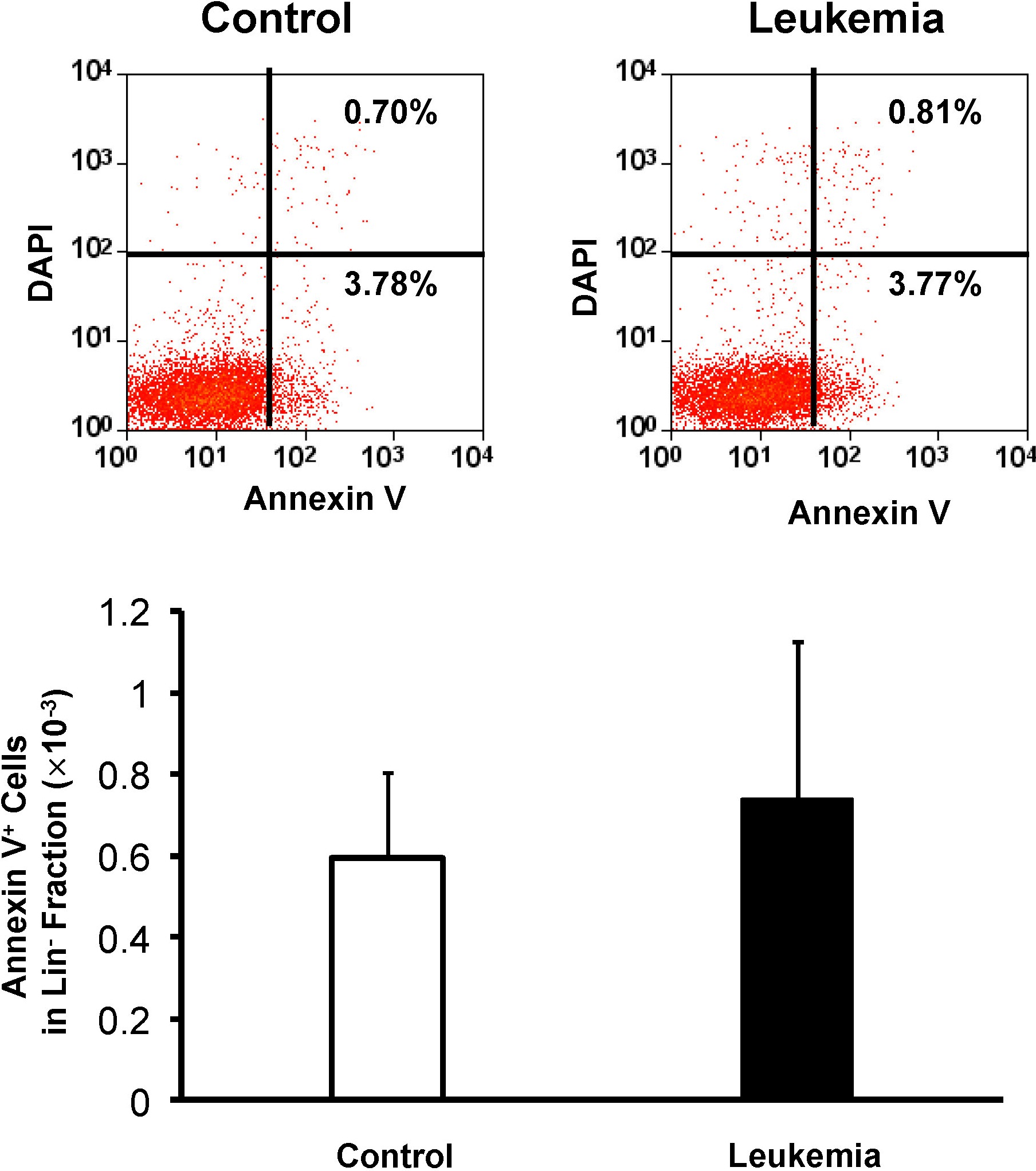
To exclude the contribution of apoptosis, we then stained for the apoptotic marker, annexin V, in the cell population after transplantation and found no statistical difference in the apoptotic fraction between the hematopoietic cells from the leukemic environment and the cells from the control environment (supplemental Figure 3). This suggests that apoptosis may not be the main reason for the decrease of hematopoietic cells in the leukemic environment.
Increased quiescence of normal HSCs in leukemic hosts
It is known that relative quiescence is associated with the quality of stem cells in adults.10,11 To understand why HSCs from leukemic BM displayed higher levels of reconstitution than those isolated from the control mice when transplanted into nonleukemic new recipients, the quiescent fraction of HSCs in cell cycle was examined using the RNA dye PY, which has been used as a measure for stem cell quiescence (G0 phase in cell cycle).19,20 Although there was no significant difference in the cycling fraction (S + G2/M) as assessed by the DNA dye propidium iodide (PI; Figure 6A), CD45.1+ LKS cells from leukemia-bearing hosts demonstrated a larger fraction in G0 phase (PYlow in the continuum) as opposed to that in G1 phase (Figure 6B), thereby suggesting that the leukemic environment is able to prevent quiescent HSCs from moving into cell cycle.
Figure 6.

Mitotic quiescence of the primitive hematopoietic cells in the leukemic hosts and expression of several cell-cycle regulators. As illustrated in Figure 1A, 107 BMNCs from B6.SJL mice were transplanted with or without 106 Notch1-induced leukemia cells (CD45.2+GFP+) into lethally irradiated C57BL/6J recipients. At the 2-week time point, LKS cells from CD45.1+ BMNCS were sorted for staining with PI to assess the general cell-cycle profile (G0/G1 vs S/G2 + M) or staining with PY in conjunction with Hoechst 33342 (HO) to specifically determine the portion of cells in G0 vs G1 with flow cytometry. An aliquot of the cells was also used to examine the expression of several cell-cycle regulators with real time RT-PCR. (A) PI staining. A representative figure is shown from 1 of 2 experiments with similar results. (B) PY staining. Cells residing in G0 appear at the bottom of the G0/G1 peak as shown in the representative plot. Values are mean ± SD. **P = .017 (t test). (C) Expression of cell-cycle regulators. The CD45.1+ LKS cells were sorted directly into lysis buffer for real-time RT-PCR analysis. Data shown are the ratios between leukemic and control groups from 1 of 2 independent experiments with similar results. Values are mean ± SD. *P < .05 (t test). **P < .01 (t test).
Potential molecular players underlying the increased quiescence of the HSC pool were explored by examining several known cell-cycle inhibitors, including p18INK4C, p21Cip/Waf1, and p27Kip1 (p18, p21, and p27 hereafter), in tissue stem and progenitor cells.10,12,21,22 As demonstrated in our previous studies,10,21 p21 maintains HSC quiescence in at least some mouse strains, whereas p27 primarily controls the proliferation of HPCs. However, p18 and p21 play opposite roles in regulating HSC self-renewal.11 CD45.1+ LKS cells were harvested 2 weeks after initiation of leukemogenesis (Figure 1A) for the analysis with real-time reverse-transcribed (RT)–PCR. There was a decrease in p18 expression inthe cells from leukemic hosts compared with the control cells. However, p21 expression was significantly increased in the LKS cells isolated from a leukemic environment. Consistent with this, its upstream regulator, Gfi-1,23 was more dramatically increased. In contrast, there was no statistical difference in p27 expression between the cells from leukemic and control marrow (Figure 6C). Therefore, the altered expression of these cell-cycle regulators may contribute to an underlying molecular basis for the more quiescent state and preserved self-renewal potential of HSCs in leukemic marrow. This mechanism could prevent normal HSCs from overly reacting to the leukemic environment and better maintain the potentiality of HSCs (supplemental Figure 4).
Discussion
In summary, normal HSCs and HPCs are progressively suppressed during leukemogenesis. Although HPCs rapidly overreact to leukemic BM with a more actively cycling status, it causes the exhaustion of HPC pools. In contrast, the function of HSC is better preserved at least in part because of increased quiescence in cell cycle. Therefore, HSCs and HPCs have distinct responses to a leukemic environment. Our findings may not only be able to model Notch1-induced leukemia, which is associated with more than 50% of T-ALL in children,24 they may also have implications for studying other types of leukemia or cancer via such a unique approach.
Our study demonstrates that the reduction in the number of blood cells in a leukemic environment is largely the result of the impairment of HSC/HPC repopulation. Evolving leukemic cells cause disruption to the BM structure, which may interrupt the physiologic interaction between HSCs and their niches. As a consequence, the responsiveness of normal HSCs and HPCs to cytokines may be altered.25,26 It has been reported that leukemic cell growth disrupts normal HPC niches in BM and creates an abnormal response to microenvironments that sequester transplanted human HPCs in a xenograft model.13 Based on our current study, this response could be heightened during the early stages of leukemogenesis (Figures 2C, 5) and decreased at a later stage (Figure 2C-F). Importantly, our study documents, for the first time, that normal HSCs can be reversibly suppressed by a leukemic environment. The compartment of normal HSCs in a leukemia host can be kept in a more quiescent state with preserved self-renewal potential, whereas normal HPCs may be exhausted after undergoing an accelerated proliferation. Therefore, when more quiescent HSCs from a leukemic environment are seeded into a nonleukemic or perhaps minimally leukemia-loaded hematopoietic microenvironment, they reenter the cell cycle and the self-renewal potential of HSCs can be fully revealed (supplemental Figure 4). This model underscores the importance of host environment or condition in dictating a specific functional state of tissue stem cells; and more specifically, it is also consistent with the rapid recovery of normal hematopoiesis in leukemia patients after an effective chemotherapy.
It should be noted that, in our current study, young mice were used and the period of latency of leukemia was short. Thus, we cannot generalize our conclusions to leukemia development in elderly mice. Our previous work, in which HSCs and T leukemic cells were transplanted for multiple rounds over a 2- to 3-year period, demonstrated that normal HSCs in leukemic mice were no longer functional at later stages as assessed by both in vitro and in vivo assays.9 In our present study, normal HSCs recovered from the leukemic environment were still highly functional after transplantation into new nonleukemic hosts. The difference in the performance between the HSCs in these 2 studies is probably the result of the different proliferation history of the stem cells. HSCs that remain in a leukemic environment may experience less proliferation than control cells, which may contribute to their ability to better repopulate after the second transplantation. In addition, in our current study, we used lethally irradiated recipients, which may impact the engrafted HSCs via the bystander effect of irradiation (H.S., H.Y., and T.C., manuscript in preparation). But this potential confounding factor should be minimized because of our proper control, in which normal hematopoietic cells alone were transplanted into irradiated recipients (Figure 1A).
Although hematopoietic suppression is a general feature of leukemogenesis, the mechanisms that lead to the suppression and especially to the better performance of donor HSCs in secondary recipients also may be context-dependent. A previous study by others showed that the size of the normal HSC compartment was unaffected in the B-ALL induced by ETV6-RUNX1 or p190 BCR-ABL, whereas it was significantly reduced in the B-ALL induced by p210 BCR-ABL.27 Therefore, the actual effects of a cancer environment on normal tissue stem cells are probably determined by multiple factors, including the type and stage of leukemia, the patient age, and the specific therapeutic regimen. Future studies on the underlying molecular pathways, especially in the context of a specific cancer environment, are needed. Such studies may eventually offer complementary approaches for the prevention and treatment of cancer.
Supplementary Material
Acknowledgments
The authors thank Jon Kilner for editorial assistance.
This work was supported by the Ministry of Science and Technology of China (2008CB517301 and 2009CB918900), the Tianjin Natural Science Foundation (07JCZDJC10600), the National Institutes of Health (RO1-HL70561; T.C.), and the Shanghai Science and Technology Committee (08JC1406500; J.W.). T.C. was a recipient of the Scholar Award from the Leukemia & Lymphoma Society (1027-09), the Yangzi River Scholarship from the Ministry of Education of China (2007-JGT-08), and the Outstanding Young Scholar Award from the National Natural Science Foundation of China (NSFC; 30825017). X.H. was a recipient of the Young Scholar Grant from NSFC (30800488).
Footnotes
Presented at the 2007 annual meeting of American Society of Hematology, Atlanta, GA, December 10, 2007.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: X.H. performed most of the experiments, analyzed the data, and wrote the paper; H.S. performed the flow cytometry work and analyzed the data; C.T. participated in leukemia modeling and data analyses; H.Y. performed the transplantation experiment; G.Z. contributed to the leukemia model and data analyses; R.X.F. helped perform the transplantation experiment; Z.J. performed some of the flow cytometry work; J.X. contributed to retroviral transduction and plasmid preparation; J.W. analyzed the data and cosupervised the research work of X.H.; and T.C. came up with the original idea, designed the experiments, analyzed the data, wrote the paper, and oversaw the research project.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for X.H. is Department of Hematology, Changhai Hospital, Shanghai, China.
Correspondence: Tao Cheng, Chinese Academy of Medical Sciences and Peking Union Medical College, 288 Nanjing Rd, Tianjin 300020, People's Republic of China; e-mail: chengt@pumc.edu.cn; or Jianmin Wang, Department of Hematology, Changhai Hospital, 168 Changhai Rd, Shanghai 200433, China; e-mail: jmwangch@hotmail.com.
References
- 1.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 2.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 3.Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- 4.Cheng T. Cell cycle inhibitors in normal and tumor stem cells. Oncogene. 2004;23:7256–7266. doi: 10.1038/sj.onc.1207945. [DOI] [PubMed] [Google Scholar]
- 5.Stier S, Cheng T, Dombkowski D, Carlesso N, Scadden DT. Notch1 activation increases hematopoietic stem cell self-renewal in vivo and favors lymphoid over myeloid lineage outcome. Blood. 2002;99:2369–2378. doi: 10.1182/blood.v99.7.2369. [DOI] [PubMed] [Google Scholar]
- 6.Spangrude GJ. Enrichment of murine haemopoietic stem cells: diverging roads. Immunol Today. 1989;10:344–350. doi: 10.1016/0167-5699(89)90192-8. [DOI] [PubMed] [Google Scholar]
- 7.Takano H, Ema H, Sudo K, Nakauchi H. Asymmetric division and lineage commitment at the level of hematopoietic stem cells: inference from differentiation in daughter cell and granddaughter cell pairs. J Exp Med. 2004;199:295–302. doi: 10.1084/jem.20030929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yilmaz OH, Kiel MJ, Morrison SJ. SLAM family markers are conserved among hematopoietic stem cells from old and reconstituted mice and markedly increase their purity. Blood. 2006;107:924–930. doi: 10.1182/blood-2005-05-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan Y, Yu H, Boyer MJ, et al. Hematopoietic stem cells are not the direct target of spontaneous leukemic transformation in p18(INK4C)-null reconstituted mice. Cancer Res. 2006;66:343–351. doi: 10.1158/0008-5472.CAN-05-2945. [DOI] [PubMed] [Google Scholar]
- 10.Cheng T, Rodrigues N, Shen H, et al. Hematopoietic stem cell quiescence maintained by p21(cip1/waf1). Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 11.Yu H, Yuan Y, Shen H, Cheng T. Hematopoietic stem cell exhaustion impacted by p18INK4C and p21Cip1/Waf1 in opposite manners. Blood. 2006;107:1200–1206. doi: 10.1182/blood-2005-02-0685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat Cell Biol. 2004;6:436–442. doi: 10.1038/ncb1126. [DOI] [PubMed] [Google Scholar]
- 13.Pear WS, Aster JC, Scott ML, et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med. 1996;183:2283–2291. doi: 10.1084/jem.183.5.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aster JC, Xu L, Karnell FG, Patriub V, Pui JC, Pear WS. Essential roles for ankyrin repeat and transactivation domains in induction of T-cell leukemia by notch1. Mol Cell Biol. 2000;20:7505–7515. doi: 10.1128/mcb.20.20.7505-7515.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 16.Harrison DE. Competitive repopulation: a new assay for long-term stem cell functional capacity. Blood. 1980;55:77–81. [PubMed] [Google Scholar]
- 17.Oostendorp RA, Audet J, Eaves CJ. High-resolution tracking of cell division suggests similar cell cycle kinetics of hematopoietic stem cells stimulated in vitro and in vivo. Blood. 2000;95:855–862. [PubMed] [Google Scholar]
- 18.Krause DS, Theise ND, Collector MI, et al. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369–377. doi: 10.1016/s0092-8674(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 19.Gothot A, Pyatt R, McMahel J, Rice S, Srour EF. Functional heterogeneity of human CD34(+) cells isolated in subcompartments of the G0/G1 phase of the cell cycle. Blood. 1997;90:4384–4393. [PubMed] [Google Scholar]
- 20.Gothot A, Pyatt R, McMahel J, Rice S, Srour EF. Assessment of proliferative and colony-forming capacity after successive in vitro divisions of single human CD34+ cells initially isolated in G0. Exp Hematol. 1998;26:562–570. [PubMed] [Google Scholar]
- 21.Cheng T, Rodrigues N, Dombkowski D, Stier S, Scadden D. Stem cell repopulation efficiency but not pool size is governed by p27. Nat Med. 2000;6:1235–1240. doi: 10.1038/81335. [DOI] [PubMed] [Google Scholar]
- 22.Walkley CR, Fero ML, Chien WM, Purton LE, McArthur GA. Negative cell-cycle regulators cooperatively control self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol. 2005;7:172–178. doi: 10.1038/ncb1214. [DOI] [PubMed] [Google Scholar]
- 23.Hock H, Hamblen MJ, Rooke HM, et al. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature. 2004;431:1002–1007. doi: 10.1038/nature02994. [DOI] [PubMed] [Google Scholar]
- 24.Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 25.Williams RT, den Besten W, Sherr CJ. Cytokine-dependent imatinib resistance in mouse BCR-ABL+, Arf-null lymphoblastic leukemia. Genes Dev. 2007;21:2283–2287. doi: 10.1101/gad.1588607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322:1861–1865. doi: 10.1126/science.1164390. [DOI] [PubMed] [Google Scholar]
- 27.Castor A, Nilsson L, Astrand-Grundstrom I, et al. Distinct patterns of hematopoietic stem cell involvement in acute lymphoblastic leukemia. Nat Med. 2005;11:630–637. doi: 10.1038/nm1253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}