

One of the principal objectives of supramolecular chemistry is to create nanoscale structures while exerting control over their sizes and shapes and to be able to utilize their unique structures to emulate biological systems in synthetic ones.1 To date, polymers and dendrimers have been successfully used as functional biomimetic systems, i.e., as macroscopic analogies of biopolymers and globular proteins with potential uses in catalysis and drug delivery.2 However, multifunctional polymers and dendrimers often require considerable synthetic effort to prepare and can be plagued by low yields and largely amorphous final structures. When issues such as control of functional groups and structural precision, the ability to perform selective encapsulation, and synthetic ease and building-block versatility come into question, coordination driven self-assembly3 emerges as a powerful alternative to the purely covalent synthesis of multifunctional molecules.
Though there have recently been reports on functionalized M12L24 Pd-based supramolecules by Fujita and co-workers,1c–f the self-assembly of multifunctionalized Pt-based metallosupramolecules has focused largely on two-dimensional ensembles.4 These investigations have demonstrated that the addition of functional groups to individual building blocks in no way disrupts the self-assembly of 2D metallocycles and leads to novel, multifunctional structures with precise control over the number, location, and relative distribution of functional moieties.4 The extension of these studies to 3D Pt-based supramolecular metallacages allows for even greater structural control and positioning of functional groups both on the exterior and interior of the nanoscopic structures. It is well-known that the proper folding of polypeptides into 3D protein structures is a prerequisite for their functionality and therefore for life. Furthermore, a multitude of biological 3D assemblies, such as vesicles and viral capsids, perform various biochemical operations throughout nature.5 In attempts to learn from and emulate such complex biological examples, here we extend our studies of multifunctional systems to higher dimensions. Supramolecular cuboctahedra6 have unique structures that allow them to play important roles in the fields of nanotechnology, pharmaceuticals, and medicinal chemistry. They can be formed by the combination of twelve 120° molecular subunits with eight complementary, planar tritopic subunits, as previously reported.6 This combination of rigid molecular subunits presents a number of different locations where functional moieties can be attached in such a way that they are positioned around the periphery or inside the cavity of cuboctahedral supramolecules. In this communication, we present the functionalization of 120° diPt(II) acceptors 1 and 2 that can be utilized to self-assemble peripherally functionalzed cuboctahedra.
We have recently shown that the addition of functional groups, such as crown ethers, Fréchet-type dendrons, and ferrocene moieties at the vertex of 120° building units enables the preparation of novel, functionalized, cavity-cored assemblies.4a–f Stirring a mixture of 120° acceptor 1 or 2 with the linear tritopic donor 3 in a 3:2 ratio resulted in the formation of cuboctahedral complexes 4 or 5 with 12 pendant ferrocene or crown ether groups, respectively, at the vertexes (Scheme 1).
Scheme 1.
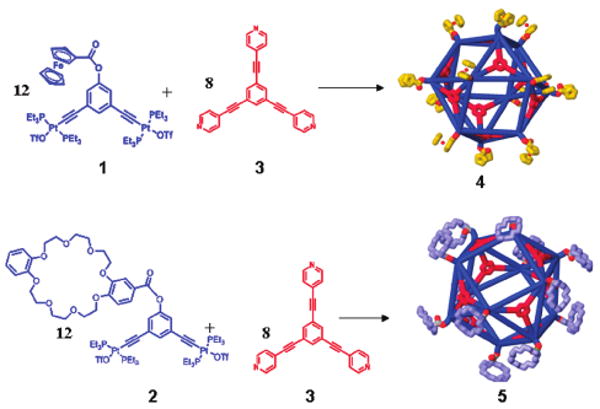
Graphical Representation of the Self-Assembly of Cuboctahedra 4 and 5
Multinuclear (1H and 31P) NMR analysis (Figure 1) of the reaction mixtures revealed the formation of discrete, highly symmetric species. The 31P{1H} NMR spectra of 4 and 5 each displayed a single sharp singlet (∼16.9 ppm for 4, ∼18.1 ppm for 5) shifted upfield from those of the corresponding starting platinum acceptors 1 and 2 by ∼6.0 and 4.4 ppm, respectively. This change, as well as the decrease in coupling of the flanking 195Pt satellites (ΔJ = −63.2 Hz for 4, ΔJ = −70.1 Hz for 5), is consistent with back-donation from the platinum atoms. Additionally, the protons of the pyridine rings exhibited downfield shifts (α-HPy, 0.22–0.54 ppm; β-HPy, 0.2–0.3 ppm) resulting from the loss of electron density upon coordination of the pyridine N atom with the Pt(II) metal center.
Figure 1.
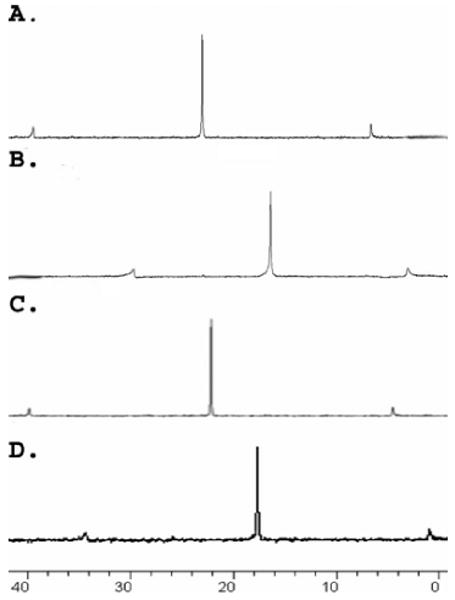
31P NMR spectra (300 MHz, CD2Cl2, 298 K) of (A) acceptor 1, (B) cuboctahedron 4, (C) acceptor 2, and (D) cuboctahedron 5.
The self-diffusion coefficient of a molecule is a measure of its mobility in solution and depends on the viscosity of the solvent and, more importantly, the “effective” size and shape of the molecule/supramolecule. Pulsed-gradient spin-echo (PGSE) experiments in conjunction with the Stokes–Einstein equation were used to determine approximate sizes of supramolecules 4 and 5. The measured values of the radius (R) are only approximate because of potential sources of intrinsic inaccuracy in the conversion of diffusion coefficient to radius.7 We carried out 1H PGSE experiments for both 4 and 5 in different solvents. In each case, the size of the supramolecule is comparable to that obtained by molecular force field modeling (Table 1). Both compounds' diffusion coefficients decrease with an increase in the viscosity coefficient of the solvent [see the Supporting Information (SI) for details of the PGSE experiments].
Table 1.
Diffusion Coefficients and Sizes of Cuboctahedral Compounds 4 and 5
| compound | molecular weight | diffusion coefficient (10−6 cm2/s) | diameter (nm) |
|---|---|---|---|
| 4 | 21208.46 | 2.13 ± 0.1b | 6.6 ± 0.2a |
| 2.2 ± 0.2c | 6.7 ± 0.3a | ||
| 6.9d | |||
| 5 | 24358.16 | 1.69 ± 0.2b | 8.1 ± 0.2a |
| 8.4d |
Calculated from the diffusion coefficient using the Stokes–Einstein equation.
Obtained from PGSE experiments (CD3COCD3, 298 K).
Obtained from electrochemical measurements.
Obtained from molecular modeling as the measured distance between the two most separated points of the functional units.
Elemental analysis of 4 and 5 and electrospray ionization (ESI) mass spectrometry provided further evidence for the formation of new cuboctahedral systems (see the SI). Because of the high molecular weight of 4, well-resolved isotopic distribution patterns were difficult to obtain. However, the ESI mass spectrum showed peaks at m/z 3383.8, 2879.0, and 2501.0 corresponding to [M – 6OTf]6+, [M – 7OTf]7+, and [M – 8OTf]8+, respectively.
Molecular force field simulations were used to gain further insight into the structural characteristics of cuboctahedral complexes 4 (Figure 2) and 5. A 1.0 ns molecular dynamics simulation (OPLS force field for 4, MMFF force field for 5) was used to equilibrate each supramolecule, and this was followed by energy minimization of the resulting structure to full convergence. The model structure of 4 features a well-defined cuboctahedron with an approximate diameter of 6.7 nm, and the distance between neighboring ferrocene units is 2.9 nm. Likewise, the simulations of 5 revealed a very similar cuboctahedral structure with flexible crown ether units and an overall size of 8 nm. Both cases reveal an internal cavity size of ∼5 nm, which is comparable to the size of some small globular proteins.
Figure 2.
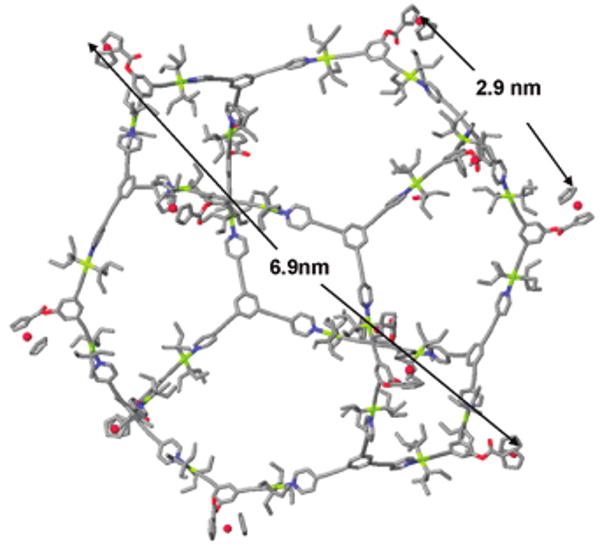
Molecular modeling of multiferrocenyl cuboctahedron 4.
The electrochemical properties of multiferrocene cuboctahedral complex 4 were also investigated. Cyclic voltammograms were recorded using a macroelectrode (∼1 mm2 Pt disk electrode) in acetone with 0.1 M n-Bu4NPF6 as the supporting electrolyte. Figure 3A shows that a single redox wave was observed for 4, suggesting that there are no interactions among the multiple redox centers at the periphery.6 Both the anodic and cathodic peak potentials were independent of the scan rate. Furthermore, the potential difference between the anodic and cathodic peak potentials (ΔEP) was 81 ± 8 mV, which is slightly larger than the theoretical value for an ideal reversible redox system (59 mV at 25 °C) as a result of the solution ohmic resistance. The statistical nature of the multielectron transfer process may cause the voltammetric peak to broaden as well.8
Figure 3.
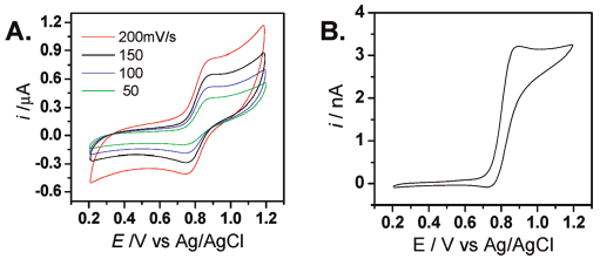
(A) Cyclic voltametry of compound 4 at different scan rates (50–200 mV/s) at a 1 mm2 Pt electrode. (B) Steady-state current response of compound 4 at 20 mV/s at a microsized (26 μm diameter) Pt disk electrode. Solution: 0.2 mM 4 in acetone containing 0.1 M n-Bu4NPF6.
No decomposition of the cuboctahedra during these redox processes was observed, as indicated by a thorough examination of the 1H NMR spectra of 4 before and after the redox studies. Electrochemical reversibility reveals fast electron transfer among the flexible peripheral redox sites. Such fast heterogeneous electron transfer can be explained by fast rotation or as the result of electrons hopping through space. Furthermore, the distance between two consecutive ferrocene units (2.9 nm) ensures that the structural rearrangement between 4 and 412+ is negligible.
In an attempt to obtain additional confirmation of the size of the ferrocenyl cuboctahedral assembly, 4 was subjected to steady-state electrochemical measurements by using a 26 μm diameter Pt disk electrode (Figure 3B). The microelectrode voltammetric response9 of 4 shows hysteresis in the forward and reverse scans, possibly indicating some adsorption of oxidized 4 at the Pt surface. Nevertheless, the voltammogram exhibits a reasonably well-defined limiting current and E1/2 values.
Plotting the voltammetric data as E versus log[(ilim − i)/i], where ilim is the limiting current determined by the mass transport of redox species, gives a linear relation with a slope of −61 mV/dec (see Figure S9 in the SI). This value is very close to the ideal value (−59 mV/n with n = 1), indicating that the redox species react one at a time, more or less independently of one another. In other words, there is an absence of strong intramolecular electronic-coupling bridges between the ferrocene moieties in 4.
Diffusion coefficients of electroactive compounds may also be calculated by electrochemical measurements. Here, the method of Denault et al.10 was employed. Potential-step chronoamperometry was performed with a 26 μm diameter Pt disk electrode. The current (it) was recorded as a function of time (t) while the potential was stepped from a value where no reaction happens to a value where the ferrocenyl groups are oxidized at the diffusion-controlled rate. In the present work, we stepped the potential from 0.2 to 1.0 V (vs Ag/AgCl). The slope of the linear region of the it/ilim versus t −1/2 plot was used to calculate the diffusion coefficient (D) of the redox complex (see Figure S10 in the SI). The obtained value of D (2.2 × 10−6 cm2/s) is very close to that measured by the PGSE method, and the calculated diameter of 3D cuboctahedron 4 (6.7 nm) obtained using the Stokes–Einstein equation is very similar to the simulated value (6.9 nm). The agreement between the diffusion coefficient and size values obtained from spectroscopic, electrochemical, and molecular modeling methods along with the NMR data firmly establish the structures of 4 and 5.
The D value obtained from the above chronoamerometric experiments was used to independently determine θsites, the number of electroactive sites (ferrocenyl groups), for compound 4 (see the SI). The experimentally measured value of θsites for compound 4, 11.8 ± 0.2, agrees with the expected value of 12, demonstrating that all of the ferrocenyl groups attached to the metallacycles are electroactive and further supporting the formation and structure of 4.
In conclusion, we have presented a general strategy for the synthesis of stable, multifunctional cuboctahedral complexes in which coordination-driven self-assembly allows for precise control over the positioning of either ferrocene or crown ether functionalities. Both functionalized supramolecular systems are obtained in high yield and are soluble in common organic solvents, facilitating structural characterization.11 All of the functional units retain their functional fidelity and surround a nanoscopic hollow “core” environment ideal for guest encapsulation. It is known that multiredox molecules that are active at one single potential over various scan rates, such as multiferrocenyl cuboctahedron 4, may find uses as multielectron catalysts and sensors. Also, the complementary recognition properties of the peripheral crown ether hosts and the interior cavity of the crown ether-functionalized cuboctahedron may be used to develop both the exo and endo receptor properties of 5. We are currently exploring the possibility of protein encapsulation in these multifunctional cuboctahedral structures, where functional groups are expected to provide an extra means of characterizing the guest-encapsulated species.
Supplementary Material
Acknowledgments
P.J.S. thanks the NIH (Grant GM-057052) for financial support. J.H. thanks the Pao Yu-Kong and Pao ZhaoLong Scholarship for financial support. H.S.W. thanks the NSF (Grant CHE-0616505) for financial support.
Footnotes
Supporting Information Available: Synthesis and analytical data of 4 and 5, PGSE measurements, cyclic and steady-state voltammetry, chronoamperometry, calculations of D and θsite, and molecular modeling procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) You CC, Würthner F. J Am Chem Soc. 2003;125:9716. doi: 10.1021/ja029648x. [DOI] [PubMed] [Google Scholar]; (b) Tominaga M, Suzuki K, Murase T, Fujita M. J Am Chem Soc. 2007;129:11950. doi: 10.1021/ja054069o. [DOI] [PubMed] [Google Scholar]; (c) Kamiya N, Tominaga M, Sato S, Fujita M. J Am Chem Soc. 2007;129:3816. doi: 10.1021/ja0693082. [DOI] [PubMed] [Google Scholar]; (d) Suzuki K, Kawano M, Sato S, Fujita M. J Am Chem Soc. 2007;129:10652. doi: 10.1021/ja073629b. [DOI] [PubMed] [Google Scholar]; (e) Suzuki K, Iida J, Sato S, Kawano M, Fujita M. Angew Chem, Int Ed. 2008;47:5780. doi: 10.1002/anie.200801700. [DOI] [PubMed] [Google Scholar]; (f) Sato S, Iida J, Suzuki K, Kawano M, Fujita M. Science. 2006;313:1273. doi: 10.1126/science.1129830. [DOI] [PubMed] [Google Scholar]
- 2.(a) Fréchet JM. Science. 1994;263:1710. doi: 10.1126/science.8134834. [DOI] [PubMed] [Google Scholar]; (b) Rulkens R, Lough AJ, Manners I, Lovelace SR, Grant C, Geiger WE. J Am Chem Soc. 1996;118:12683. [Google Scholar]; (c) Valerio C, Fillaut JL, Ruiz J, Guittard J, Blais JC, Astruc D. J Am Chem Soc. 1997;119:2588. [Google Scholar]; (d) Castro R, Cuadrado I, Alonso B, Casado CM, Moran M, Kaifer AE. J Am Chem Soc. 1997;119:5760. doi: 10.1021/ja970922a. [DOI] [PubMed] [Google Scholar]; (e) Anicet N, Anne A, Moiroux J, Savéant JM. J Am Chem Soc. 1998;120:7115. [Google Scholar]; (f) Nguyen P, Gomez-Elipe P, Manners I. Chem Rev. 1999;99:1515. doi: 10.1021/cr960113u. [DOI] [PubMed] [Google Scholar]; (g) Brinke G, Ikkala O. Science. 2002;295:2407. doi: 10.1126/science.1067794. [DOI] [PubMed] [Google Scholar]; (h) Stone DL, Smith DK, McGrail PT. J Am Chem Soc. 2002;124:856. doi: 10.1021/ja0117478. [DOI] [PubMed] [Google Scholar]; (i) Choi TL, Lee KH, Joo WJ, Lee S, Lee TW, Chae MY. J Am Chem Soc. 2007;129:9842. doi: 10.1021/ja0717459. [DOI] [PubMed] [Google Scholar]
- 3.(a) Stang PJ, Olenyuk B. Acc Chem Res. 1997;30:502. [Google Scholar]; (b) Leininger S, Olenyuk B, Stang PJ. Chem Rev. 2000;100:853. doi: 10.1021/cr9601324. [DOI] [PubMed] [Google Scholar]; (c) Seidel SR, Stang PJ. Acc Chem Res. 2002;35:972. doi: 10.1021/ar010142d. [DOI] [PubMed] [Google Scholar]; (d) Schwab PFH, Levin MD, Michl J. Chem Rev. 1999;99:1863. doi: 10.1021/cr970070x. [DOI] [PubMed] [Google Scholar]; (e) Gianneschi NC, Masar MS, III, Mirkin CA. Acc Chem Res. 2005;38:825. doi: 10.1021/ar980101q. [DOI] [PubMed] [Google Scholar]; (f) Cotton FA, Lin C, Murillo CA. Acc Chem Res. 2001;34:759. doi: 10.1021/ar010062+. [DOI] [PubMed] [Google Scholar]; (g) Fujita M, Tominaga M, Hori A, Therrien B. Acc Chem Res. 2005;38:369. doi: 10.1021/ar040153h. [DOI] [PubMed] [Google Scholar]; (h) Fiedler D, Leung DH, Bergman RG, Raymond KN. Acc Chem Res. 2005;38:349. doi: 10.1021/ar040152p. [DOI] [PubMed] [Google Scholar]; (i) Steel PJ. Acc Chem Res. 2005;38:243. doi: 10.1021/ar040166v. [DOI] [PubMed] [Google Scholar]
- 4.(a) Yang HB, Das N, Huang F, Hawkridge AM, Muddiman DC, Stang PJ. J Am Chem Soc. 2006;128:10014. doi: 10.1021/ja063377z. [DOI] [PubMed] [Google Scholar]; (b) Yang HB, Hawkridge AM, Huang SD, Das N, Bunge SD, Muddiman DC, Stang PJ. J Am Chem Soc. 2007;129:2120. doi: 10.1021/ja066804h. [DOI] [PubMed] [Google Scholar]; (c) Yang HB, Ghosh K, Northrop BH, Zheng YR, Lyndon MM, Muddiman DC, Stang PJ. J Am Chem Soc. 2007;129:14187. doi: 10.1021/ja073744m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ghosh K, Yang HB, Northrop BH, Lyndon MM, Zheng YR, Muddiman DC, Stang PJ. J Am Chem Soc. 2008;130:5320. doi: 10.1021/ja711502t. [DOI] [PubMed] [Google Scholar]; (e) Ghosh K, Zhao Y, Yang HB, Northrop BH, White HS, Stang PJ. J Org Chem. 2008;73:8553. doi: 10.1021/jo801692v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Northrop BH, Yang HB, Stang PJ. Chem Commun. 2008:5896. doi: 10.1039/b811712h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Lee SJ, Hupp JT. Coord Chem Rev. 2006;250:1710. [Google Scholar]; (h) Dinolfo PH, Hupp JT. J Am Chem Soc. 2004;126:16814. doi: 10.1021/ja045457d. [DOI] [PubMed] [Google Scholar]; (i) Würthner F, You CC, Saha-Möller C. Chem Soc Rev. 2004;33:133. doi: 10.1039/b300512g. [DOI] [PubMed] [Google Scholar]; (j) Merlau ML, del Pilar Mejia M, Nguyen ST, Hupp JT. Angew Chem, Int Ed. 2001;40:4239. doi: 10.1002/1521-3773(20011119)40:22<4239::AID-ANIE4239>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]; (k) Dinolfo PH, Williams ME, Stern CL, Hupp JT. J Am Chem Soc. 2004;126:12989. doi: 10.1021/ja0473182. [DOI] [PubMed] [Google Scholar]; (l) You CC, Hippius C, Grüne M, Würthner F. Chem—Eur J. 2006;12:7510. doi: 10.1002/chem.200600413. [DOI] [PubMed] [Google Scholar]
- 5.(a) Horne RW. Virus Structure. Academic; New York: 1974. [Google Scholar]; (b) Kushner DJ. Bacteriol Rev. 1969;33:302–345. doi: 10.1128/br.33.2.302-345.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olenyuk B, Whiteford JA, Fechtenkötter A, Stang PJ. Nature. 1999;398:796. doi: 10.1038/19740. [DOI] [PubMed] [Google Scholar]
- 7.Caskey DC, Yamamoto T, Addicott C, Shoemaker RK, Vacek J, Hawkridge AM, Muddiman DC, Kottas GS, Michl J, Stang PJ. The Stokes–Einstein equation was derived for a neutral spherical macroscopic object, whereas our cuboctahedral structures are charged and have molecular dimensions. J Am Chem Soc. 2008;130:7620. doi: 10.1021/ja710715e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flanagan JB, Margel S, Bard AJ, Anson FC. J Am Chem Soc. 1978;100:4248. [Google Scholar]
- 9.Bard AJ, Faulkener LR. Electrochemical Methods: Fundamentals and Applications. Wiley; New York: 1980. [Google Scholar]
- 10.(a) Denault G, Mirkin M, Bard AJ. J Electroanal Chem. 1991;308:27. [Google Scholar]; (b) Biondi C, Bellugi L. J Electroanal Chem. 1970;24:263. [Google Scholar]; (c) Amatore C, Azzali M, Calas P, Jutand A, Lefrou C, Rollin Y. J Electroanal Chem. 1990;288:45. [Google Scholar]; (d) Mirkin MV, Nilov AP. J Electroanal Chem. 1990;283:35. [Google Scholar]; (e) Nowinski SA, Anjo DM. J Chem Eng Data. 1989;34:265. [Google Scholar]
- 11.Both compounds are stable for days at room temperature in all common noncoordinating organic solvents (acetone, nitromethane, dichloromethane).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.