

Abstract
In Gram-negative bacteria, TonB-dependent outer membrane transporters (TBDTs) bind large, scarce organometallic substrates with high affinity preceding active transport. The cobalamin transporter BtuB requires the additional binding of two Ca2+ ions before substrate binding can occur, but the underlying molecular mechanism is unknown. Using the crystallographic structures available for different bound states of BtuB, we have carried out extended molecular dynamics simulations of multiple functional states of BtuB to address the role of Ca2+ in substrate recruitment. We find that Ca2+ binding both stabilizes and repositions key extracellular loops of BtuB, optimizing interactions with the substrate. Interestingly, replacement by Mg2+ abolishes this effect, in accordance with experiments. Using a set of new force field parameters developed for cyanocobalamin, we also simulated the substrate-bound form of BtuB, where we observed interactions not seen in the crystal structure between the substrate and loops previously found to be important for binding and transport. Based on our results, we suggest that the large size of cobalamin compared to other TBDT substrates explains the requirement of Ca2+ binding for high affinity substrate recruitment in BtuB.
Introduction
In order to import large yet essential nutrients (> 600 Daltons) such as iron-complexes and cobalamins, Gram-negative bacteria must actively transport them across both the outer membrane (OM) and the cytoplasmic membrane (CM)1. At the CM, these nutrients use ATP-driven ABC transporters to cross the membrane2,3. However, due to the high permeability of the OM to small molecules, chemical energy cannot be generated or stored in the periplasmic space; therefore, active transport at the OM has to rely on energy generated at the CM. To circumvent this limitation, one pathway bacteria have evolved is known as the TonB system. TonB interacts with proteins in the CM, where the proton-motive force is used to generate energy, and delivers that energy to TonB-dependent transporters (TBDTs) in the OM4,5. These transporters share a common structure, possessing 22-stranded β-barrels connected to a large, N-terminal “luminal domain” folded inside the barrel. Various substrates bind on the extracellular side to the luminal domain, interacting simultaneously with the extracellular loops of the β-barrel, preceding transport. These loops, 11 in total (see Fig. 1), have been implicated in multiple functions, including the binding of substrate as well as lethal phages and colicins6–12. They vary in length within a single TBDT and particularly between different TBDTs, inversely proportional to the size of the substrate, suggesting they might also function to occlude the binding site after substrate binding, thus preventing it from leaving during transport13.
Figure 1.
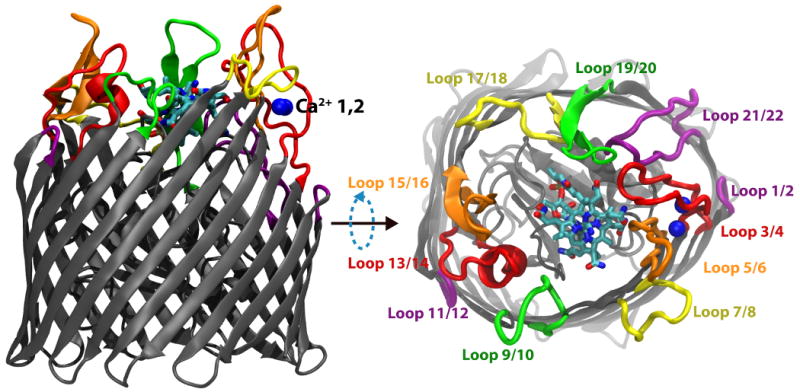
Membrane-plane and extracellular views of the Ca2+/CN-Cbl-bound structure of BtuB. The barrel and luminal domains are colored in grey, the two Ca2+ ions are in blue, and CN-Cbl is shown in a stick representation colored by atom type. The eleven extracellular loops are drawn using a thicker representation and labeled using different colors to distinguish them from their neighbors.
Once the substrate is bound, interaction between TonB and the TBDT triggers its transport, although the mechanism is still largely unknown. A short N-terminal sequence in the TBDT, known as the Ton-box, serves as the point of interaction with TonB14–18. The structures of TonB in complex with BtuB as well as in complex with FhuA, revealed that the Ton-box forms a β-strand which then joins the β-sheet in the C-terminal domain of TonB19,20. Recent evidence suggests a mechanical mode of interaction in which force applied to TonB is transmitted to the luminal domain of the TBDT, causing unfolding and/or additional structural deformation by which a substrate pathway can form5,13,21. In agreement with this model, experiments have demonstrated that residues in the luminal domain become accessible to fluorophores in the periplasm only during transport22,23, and that TonB is simultaneously bound to both the CM and OM24; however, alternative models of substrate path formation cannot yet be ruled out25–27.
One of the best structurally characterized TBDTs is the cobalamin (Cbl, e.g., vitamin B12) transporter BtuB. Crystallographic structures of BtuB have been determined in the apo, Ca2+-bound, substrate loaded, colicin-bound, and TonB-bound states8,9,19,28. These structures have revealed how all the components (i.e., substrate, Ca2+ ions, and TonB) come together preceding transport; however, they still leave a number of questions unanswered. For example, Ca2+ binding is known to increase BtuB's affinity for Cbl 1000-fold, while it does not appear to be necessary for substrate binding in any other TBDT27,29. In the Ca2+-bound structure of BtuB, the presence of Ca2+ ordered all or part of three extracellular loops not resolved in the apo structure (loops 3/4, 5/6, and 7/8, shown in Fig. 1) although how this ordering influences substrate binding is not clear9. Furthermore, Mg2+ can inhibit substrate binding to BtuB, suggesting that Mg2+ competes with Ca2+ for specific ion binding sites29. The structure of the Ca2+/cyanocobalamin (CN-Cbl)-bound BtuB exhibited only a few differences compared to the Ca2+-bound form. Loops 3/4, 5/6, and 7/8 become fully resolved in the Ca2+/CN-Cbl-bound structure, although loop 9/10 is no longer seen9.
In order to determine the role of the extracellular loops of BtuB in Cbl recruitment and binding, we have performed an extensive set (over 250 ns) of molecular dynamics (MD) simulations of nearly all resolved structures of BtuB in a lipid/water environment. MD simulations have been applied to the study of many outer membrane proteins, including the TBDTs BtuB and FhuA21,30–35. To include the substrate in our simulations, we first needed to parametrize CN-Cbl, missing in previous simulations21, for the CHARMM force field (see Methods). The subsequent simulations illustrate the dynamical differences between the extracellular loops in the apo, Ca2+-bound, and Mg2+-bound forms of BtuB. Furthermore, simulation of the Ca2+/CN-Cbl-bound state reveals multiple interactions between CN-Cbl and BtuB, including some not present in the original crystal structure. Simulations of two apo structures of BtuB obtained under different crystallization conditions reveals the fast, spontaneous binding of Ca2+ ions to the extracellular loops in both. Based on our results, we conclude that Ca2+ binding serves to both position the extracellular loops closer to the substrate binding site as well as stabilize them, permitting BtuB to bind Cbl with high affinity.
Results
The role of Ca2+ binding in BtuB
Prior to substrate binding, BtuB binds Ca2+, which is known to increase the affinity of BtuB for Cbl 1000-fold, through an unknown mechanism27,29. According to the Ca2+-bound and Ca2+/CN-Cbl-bound crystal structures9, two Ca2+ ions are ligated between extracellular loops 3/4 and 5/6 primarily by a number of aspartic acid residues. To investigate how Ca2+ affects the structure and dynamics of these loops and the binding of Cbl, we simulated both the apo and the Ca2+-bound states of BtuB for 30 ns each, comparing the resulting structural and dynamical differences.
As a measure of loop stability, we calculated both the root mean-square deviation (RMSD) and the root mean-square fluctuations (RMSF) over the course of both simulations. As shown in Fig. 2A, the RMSD for the extracellular loops in the apo structure is significantly higher compared to that for the loops in the Ca2+-bound structure, providing further evidence that the binding of Ca2+ stabilizes these loops. The RMSF per residue (Fig. 2B) presents a more detailed picture, giving information on individual residues instead of the loops as a whole. For loops 3/4 and 5/6, the difference at peak RMSF is negligible, but for those residues closer to the Ca2+ binding site, the difference is statistically significant (defined as greater than one standard deviation from the mean RMSF for the apo structure; see Supplementary Fig. S1F). We also observe an increase in the stability of substrate binding loop 1 (SB-1, residues 57-65 in the luminal domain), in the Ca2+-bound structure. Given that residues in loop 3/4 near the Ca2+ binding site interact with SB-1, the stabilizing effect of Ca2+ on loop 3/4 may then also stabilize SB-1. Interestingly, it has been reported that the insertion of an aspartate-proline dipeptide or 1-5 alanines between residues 50 and 51 decreases both Ca2+ and Cbl affinity27,36. This effect can be explained as the reverse of what we observe in our simulations; whereas we see a stabilizing effect of Ca2+ on SB-1, in the experiments, residue insertions near SB-1 likely destabilize the Ca2+ binding site.
Figure 2.
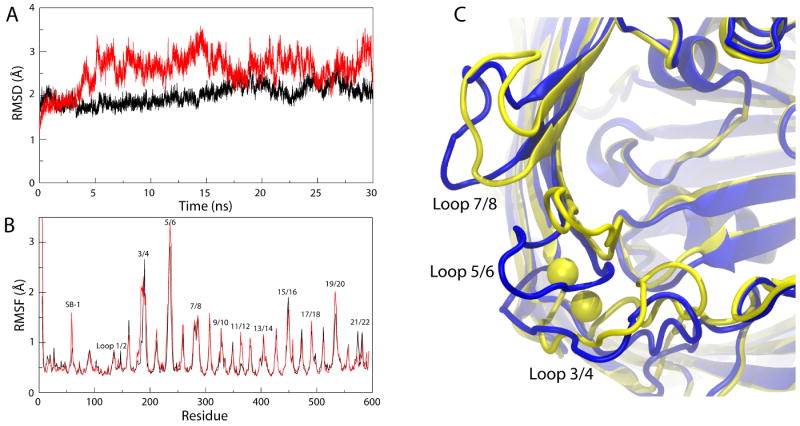
Differences between simulations of the Ca2+-bound and the apo structures of BtuB. (A) Root mean-square deviation (RMSD) of Cα atoms of BtuB's extracellular loops vs. time for the Ca2+-bound structure of BtuB (black) and the apo structure of BtuB (red), both in comparison to the CN-Cbl-bound structure of BtuB. (B) Root mean-square fluctuation (RMSF) vs. residue number for the two structures, colored as in A. (C) Comparison of the average structures, shown from the extracellular side, calculated from the last 20 ns out of the 30-ns simulations. The apo structure is shown in blue and the Ca2+-bound structure, with corresponding Ca2+ ions, in yellow.
The question still remains what purpose Ca2+ binding serves. It has been suggested that Ca2+ binding could restrain the loops and thus expose the Cbl binding site, increasing its binding affinity9. However, while we note a decrease in the loops' fluctuations, they still have the potential to occlude the Cbl binding site. In fact, we find that binding of Ca2+ shifts the average position of the loops closer to the Cbl binding site, up to 3.0 Å, 8.5 Å, and 3.4 Å for loops 3/4, 5/6, and 7/8, respectively (see Fig. 2C). Since all three loops contribute to CN-Cbl binding in the Ca2+/CN-Cbl-bound structure (5 out of 20 residues interacting with Cbl are found in these loops), bringing them closer to the binding site would position them for a closer interaction with the substrate, and, thus, could increase the affinity for Cbl9. The observed shift is in agreement with EPR data on the distance between certain residues in opposing loops (e.g., Thr188 in loop 3/4 and Gly399 in loop 13/14), which show a 2-3 Å decrease upon binding of Ca2+ (see comparison in Supplementary Table S1 and Supplementary Fig. S2)37. Thus Ca2+ binding can serve both to reposition the extracellular loops for optimal Cbl binding and then to further stabilize the binding site.
Effects of cyanocobalamin binding on BtuB
Binding of Cbl to BtuB occurs with very high affinity (sub-nanomolar) once BtuB is in its Ca2+-loaded state27,29. In the crystal structure, CN-Cbl interacts with 20 residues, both in the luminal domain and in extracellular loops 3/4, 5/6, 7/8 17/18, 19/20, and 21/22, divided approximately equally between hydrogen bonds and purely steric van der Waals contacts38. To examine these substrate-protein interactions, we simulated the Ca2+/CN-Cbl-bound BtuB for 30 ns, using the parameters we developed for CN-Cbl (see Methods).
We measured the RMSD of the extracellular loops as well as the RMSF for the entire protein. The RMSD of the loops in the Ca2+/CN-Cbl-bound state is comparable to that for the Ca2+-bound one (see Fig. 3B). The RMSF is also quite similar to that for the Ca2+-bound state, however with a few notable exceptions (see Fig. 3C). Extracellular loops 3/4 and 5/6 are significantly more stable due to their interactions with CN-Cbl; loops 7/8 and 19/20, which have few interactions with CN-Cbl, are instead less stable, and shift positions slightly (see also Supplementary Fig. S1C,D). The majority of hydrogen bonds between BtuB and CN-Cbl were the same as those observed in the crystal structure. However, an additional hydrogen bond between the side chain of Arg243 in loop 5/6 and the A-ring propionamide (the naming being defined in Chimento et al.38) formed, at the expense of one with the backbone of Leu63. The carbonyl of Gly92 in SB-3 formed a stable hydrogen bond with the hydroxide of CN-Cbl's ribose moiety as well.
Figure 3.
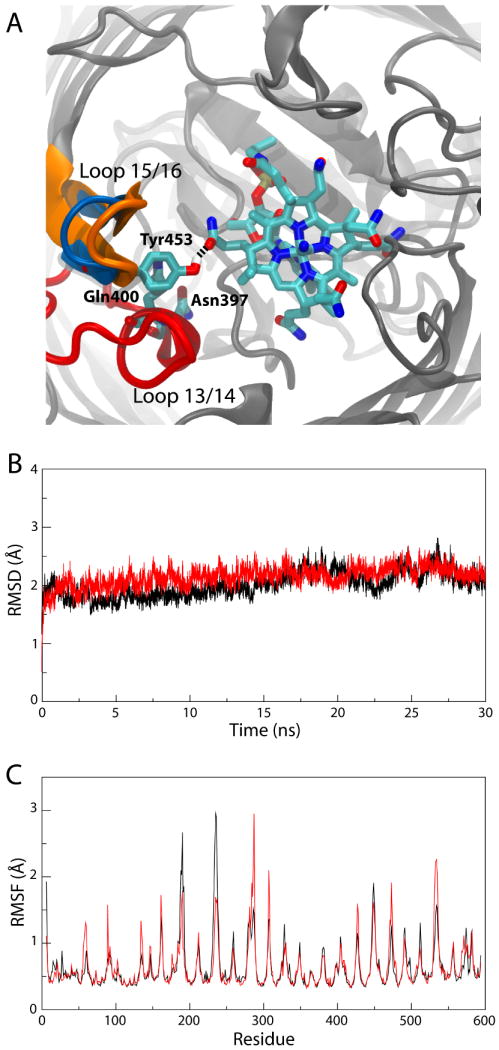
Simulation of the Ca2+/CN-Cbl-bound structure of BtuB. (A) Unique interactions observed during the simulation. The two loops involved, loops 13/14 and 15/16, are colored as in Fig. 1; the specific residues along with CN-Cbl are seen in a stick representation, colored by atom name. A snapshot of the simulation is shown in which Tyr453 hydrogen bonds to CN-Cbl. The crystallographic position of loop 15/16, farther away from CN-Cbl, is also indicated in blue. (B) RMSD of the extracellular loops vs. time and (C) RMSF vs. residue number for the Ca2+/CN-Cbl-bound structure of BtuB (red) and the Ca2+-bound structure (black).
We also observed interactions between loops 13/14 and 15/16 and CN-Cbl. These loops do not interact with CN-Cbl in the crystal structure, but experiments have indicated that they might be important for both substrate binding and transport in BtuB and in FepA and for transport only in FhuA7,9–11. Strands 13, 14, and their intervening loop form the “β-cantilever” motif, so-named due to its positioning inside the barrel, separated from adjacent β-strands, thus positioning it closer to the Cbl binding site than otherwise possible13. Hydrogen bonds between the C-ring propionamide of CN-Cbl and Tyr453 in loop 15/16 as well as Asn397 and Gln400 in loop 13/14 formed transiently over the course of the simulation (minimum hydrogen-bonding distances of 2.6 Å, 2.9 Å, and 2.8 Å, respectively); one example is shown in Fig. 3A. Loop 15/16 also moved toward CN-Cbl by 3 Å by the end of the 30-ns simulation. This movement is less than the 7-8 Å shift seen in the structure of BtuB with TonB when compared to the Ca2+/CN-Cbl-bound structure, although there, crystal contacts with an adjacent TonB may have influenced the loop's position19. The two different loop positions are also indicated in Fig. 3A.
The effect of Mg2+ in the Ca2+ binding site
Although the aspartate cage in BtuB is well suited for binding divalent ions, high affinity Cbl binding is dependent specifically on Ca2+29. The presence of Mg2+ in solution even inhibits high affinity binding at non-saturating concentrations of Ca2+, suggesting that Mg2+ ions can bind to BtuB, but do not confer the same increase in Cbl affinity as Ca2+29. The inhibitory effect of Mg2+ has been observed for other proteins with Ca2+ binding sites as well, including the C2 domains in eukaryotic signaling molecules and the tomato bushy stunt virus39,40; the reverse effect, Ca2+ inhibiting a Mg2+-dependent enzyme, has also been observed41. Additionally, the apo structure of BtuB was obtained in a high [Mg2+] solution, yet loops 3/4, 5/6, and 7/8 were not resolved9. Mg2+ has a smaller radius than Ca2+ (0.65 Å vs. 0.99 Å) and as a result, has a shorter optimal coordination distance (2.05 Å vs. 2.4 Å)42. Additionally, coordination of Ca2+ ions by carboxyl groups can be monodentate or bidentate, while coordination of Mg2+ ions is typically monodentate42.
In order to determine how the differences between Ca2+ and Mg2+ lead to distinct functional behaviors in BtuB, we simulated the Ca2+-bound crystal structure of BtuB with the Ca2+ ions replaced by Mg2+. We compared the RMSD of the Cα atoms of the extracellular loops of Ca2+-bound and Mg2+-bound BtuB, finding the loops in the Mg2+-bound state to exhibit much larger deviations over time than in the Ca2+-bound state (see Fig. 4C). In fact, the RMSD for the loops in the Mg2+-bound state has the same magnitude as that observed for the apo state, suggesting that the Mg2+ ions are unable to contain the loops' motions in the same way as Ca2+ ions. We also calculated the average structure of BtuB with Mg2+ bound, finding that the position of the loops is much closer to that in the apo state than in the Ca2+-bound state (see Supplementary Fig. S3).
Figure 4.
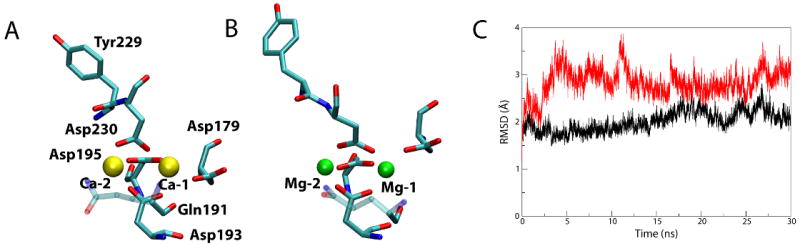
Comparison of Ca2+-bound and Mg2+-bound BtuB. (A) Ca2+ coordination after 30 ns. Residues interacting with the Ca2+ ions (yellow) are shown as sticks, colored by atom name. Gln191 is partially transparent so as not to obscure Asp193 behind it. (B) Mg2+ coordination after 30 ns. (C) RMSD of the extracellular loops vs. time for the simulation of the Mg2+-bound structure (red) compared to the simulation of the Ca2+-bound BtuB (black).
While the loops in the Mg2+-bound BtuB resemble those in the apo state, the Mg2+ ions do remain bound to the protein for the entire 30 ns of the simulation. To further distinguish between the binding of Mg2+ and Ca2+, we examined which residues coordinated the ions and with what coordination distances (Tables 1 and 2). Average coordination distances are approximately 0.3-0.5 Å shorter for Mg2+ compared to Ca2+, as expected from the ions' radii. The primary ligands for both Ca-1 and Mg-1 are the four aspartate residues, 179, 193, 195, and 230. The carbonyl of Gln191 also contributes to Ca-1 binding (average distance, davg=3.98 Å), but moves away from Mg-1 (davg=4.82 Å). Additionally, while coordination of Ca-1 by aspartates is largely bidentate, Asp193 and Asp195 switch to monodentate for Mg-1 (judged by the decrease in coordination distance for only one of the two carboxyl oxygens). The difference between Ca-2 and Mg-2 is similar; coordination by Asp230 is bidentate for Ca-2 but monodentate for Mg-2. Thus, the shorter coordination distance combined with changes in the ligands coordinating Mg2+ compared to Ca2+ prevent adequate stabilization and positioning of the extracellular loops for Cbl binding.
Table 1.
Ca/Mg-1 coordinationa.
| Atom name | occ. time (Ca) | occ. time (Mg) | avg. dist. (Ca) | avg. dist. (Mg) |
|---|---|---|---|---|
| Asp179 OD1 | 93.58 | 98.84 | 3.14 | 3.04 |
| Asp179 OD2 | 98.39 | 100.00 | 2.32 | 1.88 |
| Asp193 OD1 | 91.32 | 100.00 | 3.30 | 3.29 |
| Asp193 OD2 | 100.00 | 100.00 | 2.14 | 1.85 |
| Asp195 OD1 | 99.26 | 100.00 | 3.44 | 3.54 |
| Asp195 OD2 | 100.00 | 100.00 | 2.13 | 1.85 |
| Asp230 OD1 | 13.15 | 99.98 | 4.16 | 3.61 |
| Asp230 OD2 | 100.00 | 100.00 | 2.14 | 1.84 |
| Gln191 O | 54.88 | 1.61 | 3.98 | 4.82 |
| Asp193 O | 5.58 | 29.82 | 4.86 | 4.19 |
| Asp179 O | 2.92 | 1.71 | 6.11 | 5.87 |
Ca-1 and Mg-1 coordinating atoms are indicated by residue number and atom name. Occupancy (occ.) time is the fraction of time that each atom was within 4 Å of the Ca2+ or Mg2+ ion. The final two columns indicate the average distance (avg. dist.) between each atom and the corresponding ion over the course of the entire 30 ns of the simulation.
Table 2.
Ca/Mg-2 coordinationa.
| Atom name | occ. time (Ca) | occ. time (Mg) | avg. dist. (Ca) | avg. dist. (Mg) |
|---|---|---|---|---|
| Asp193 OD1 | 100.00 | 100.00 | 2.15 | 1.84 |
| Asp193 OD2 | 66.84 | 99.92 | 3.51 | 3.65 |
| Asp195 OD1 | 100.00 | 100.00 | 2.13 | 1.85 |
| Asp195 OD2 | 87.30 | 100.00 | 3.74 | 3.47 |
| Asp230 OD1 | 100.00 | 100.00 | 2.17 | 1.85 |
| Asp230 OD2 | 99.97 | 100.00 | 2.67 | 3.34 |
| Tyr229 O | 15.79 | 36.95 | 4.28 | 4.22 |
| Gln191 OE1 | 1.35 | 2.21 | 6.35 | 7.82 |
Ca-2 and Mg-2 coordinating atoms, along with their occupancy time and average distance from the ions, are given. See Table 1 for more details.
Spontaneous binding of Ca2+ and Mg2+ to the apo structures of BtuB
To determine how Ca2+ binds to BtuB, we simulated the apo state of BtuB with Ca2+ ions present in bulk solution at an effective concentration of 71 mM, i.e., much higher than that required for binding in vivo (KD = 30 nM), but within an order of magnitude of physiological extracellular Ca2+ concentration29. Binding of Ca2+ ions to the expected binding site between loops 3/4 and 5/6 occurred spontaneously in the simulation within nanoseconds. The Ca2+ ion designated Ca-1 by Chimento et al.9 binds first to Asp179 at t = 5 ns. A second Ca2+ ion, Ca-2, binds first to Asp193 at t =12 ns and then also Asp195 at t =13 ns. Additionally, Asp230 contacts Ca-1 at t =35 ns, helping to bring the ion much closer to its final location. The protein-Ca2+ interactions are strengthened over the course of the 50-ns simulation, resulting in Ca-1 being 4 Å from its crystallographic position and Ca-2 fluctuating between 6 and 9 Å away (see Fig. 5A). Interestingly, the order of binding of the Ca2+ ions is consistent with mutagenesis studies which suggest that Ca-1 is more important for Cbl binding than Ca-227. We also calculated the RMSD of extracellular loops 3/4, 5/6, and 7/8 in reference to the crystal structure of Ca2+-bound BtuB9 (Fig. 5B). While initially increasing, the RMSD peaked at 5.7 Å and then decreased over the latter half of the simulation to 2.6 Å, indicating that the apo structure, after binding of two Ca2+ ions, is approaching the Ca2+-bound structure.
Figure 5.
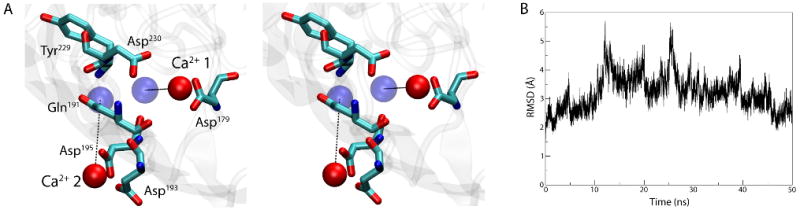
Binding of Ca2+ to the apo structure of BtuB. (A) Stereo view of the Ca2+ binding site after 50 ns of simulation. The Ca2+-binding residues are shown in a stick representation and the Ca2+ ions are drawn as red spheres. The positions of the ions in the crystal structure of the Ca2+-bound BtuB are indicated as blue spheres. (B) RMSD vs. time for loops 3/4, 5/6, and 7/8, compared to their positions in the crystal structure of the Ca2+-bound BtuB.
Simulation of the apo state of BtuB with Ca2+ ions in solution was repeated in order to ascertain the reproducibility of the results of the first simulation discussed above. The ions' initial positions were randomized before starting. In this simulation, only one Ca2+ ion was observed to bind between loops 3/4 and 5/6, contacting OD1 and OD2 of Asp230 as well as the backbone carbonyl of Asp193 and Gln191 at t =5.5 ns. As in the first simulation, this binding site is the same as that for Ca-1 in the crystallographic structure9. Although Ca2+ ions twice visited the region around the Ca-2 binding site during the 30-ns simulation, complete binding of an ion to this site was not achieved, an outcome which can be attributed to the short time scale of the simulation and the stochastic nature of the binding event.
Binding of Mg2+ ions from bulk solution to the apo state of BtuB was also observed in a separate 20-ns simulation in which all Ca2+ ions in solution were replaced by Mg2+ ions. After 6 ns, one Mg2+ ion became bound to Asp230 and Asp195, the two residues which ligate both Ca2+ ions in the crystallographic structure (see Fig. 4A). A second Mg2+ ion also interacts with Asp179, part of the binding site for Ca-1, from t =7 to t =12 ns. This interaction is, however, weak, with the ion-oxygen distance fluctuating between 4 Å to 13 Å before the ion completely leaves the binding site. A comparison of the ion-protein interaction energies reveals that the bound Mg2+ ion does not interact as strongly with BtuB as do Ca2+ ions in both spontaneous binding simulations; whereas the Mg2+ ion interacts with energy between -200 and -250 kcal/mol, the Ca2+ ions all reach energies of -350 to -500 kcal/mol (see Supplementary Fig. S4). Therefore, while Mg2+ ions can bind spontaneously to the same binding site as Ca2+, they do not do so in the same order or with the same strength.
An additional structure of the apo state of BtuB has been solved using different crystallization conditions. The majority of structures of BtuB are based on crystals in which the protein is surrounded by detergent micelles, referred to as the in surfo method9,19. Alternatively, in the in meso method, the protein rests in a lipid bilayer, thereby attempting to more closely replicate the native state28. Differences between the BtuB structures produced by the two methods include the resolution, or lack thereof, of certain regions. For example, the five N-terminal residues as well as loop 7/8 missing from the in surfo structure are resolved in the in meso structure, while SB-1 and loop 21/22 are present in the in surfo structure but not in the in meso one. Further differences include the positions of loops 9/10, 13/14, 15/16, and 19/20.
For comparison to the in surfo BtuB structure, we simulated the in meso structure for 30 ns. Similar to the in surfo structure, we observe two Ca2+ ions bind to loops 3/4 and 5/6 in the in meso BtuB structure. Ca-1 is 8.5 Å from its crystallographic position in the Ca2+-bound structure of BtuB while Ca-2 is 13.5 Å away, both moving closer over time. These distances are comparable to those for the simulation of the in surfo structure with Ca2+ ions present where, after 30 ns, Ca-1 and Ca-2 were both 8 Å away. Thus, with the exception of two extracellular loops whose positions may be crystallographic artifacts, the two structures do not display remarkably different behaviors. More details of the in meso simulation are provided in the Supplementary Data.
Discussion
By carrying out extended MD simulations of various bound states of BtuB, we have characterized the initial steps in the Cbl transport cycle. We have examined the role of Ca2+ in Cbl binding, finding Ca2+ ions to bind spontaneously to extracellular loops 3/4 and 5/6, stabilizing them and causing them to move closer to the Cbl binding site. Simulations of extracellular proteins, e.g., cadherin and other extracellular matrix proteins, have also demonstrated that the binding of Ca2+ ions plays a structural role by stabilizing and positioning key regions43–45. Additionally, we see a stabilizing effect of Ca2+ binding on SB-1, suggesting Ca2+ binding serves to position and stabilize multiple elements that contribute to the Cbl binding site. Destabilizing SB-1 may also affect the Ca2+ binding site, as suggested by the observation that amino acid insertions near SB-1 can diminish Ca2+ binding27,36; further insertions targeting SB-1 directly are necessary to test this suggestion. In contrast to the stabilizing effect of Ca2+, simulations of Mg2+ in the Ca2+ binding site demonstrated that while the Mg2+ ions remain bound, they significantly destabilize the binding site, explaining why experimentally, the presence of Mg2+ inhibits Cbl binding29.
We also examined the effect of Cbl binding, made possible by our development of simulation parameters for CN-Cbl in the CHARMM force field. When bound to BtuB, CN-Cbl exhibits a stabilizing effect on a number of extracellular loops of BtuB, as well as interactions with residues in loops 13/14 and 15/16 not present in the crystal structure. Deletion of each of these two loops is known to abrogate substrate binding and transport in FepA and BtuB, and transport only in FhuA, although it is unclear whether this is due to the loss of key interactions with the substrate or destabilization of the transporter's structure7,10,11. The role of loops 13/14 and 15/16 can now be tested by mutating the specific residues we observed to interact with CN-Cbl and then measuring the effect on its binding.
Based on these results, we can elaborate further on the transport cycle in TBDTs. One of the first steps, binding of Ca2+ ions, is only known to be necessary for BtuB, where we observe it to position loops closer to the Cbl binding site on the extracellular side, and to stabilize them in this position9,37. Since Ca2+ ions are likely always present at the outer membrane, this step does not seem to serve a regulatory role specific to Cbl transport38. Instead, we suspect that because Cbl is the largest substrate among the known TBDTs, it requires a larger number of interactions with the transporter than other substrates in order to achieve high affinity binding; the simultaneous arrangement of such interactions is made possible indirectly through the binding of Ca2+ ions. In the next step, the substrate binds to the transporter, interacting with certain extracellular and luminal protein loops. These interactions, through an allosteric mechanism, dislodge the Ton-box on the periplasmic side of the TBDT46,47. After binding of TonB to the Ton-box, energy generated at the inner membrane is transduced to the transporter. Although the details of the energy transduction step are still unknown, recent experiments indicate that TonB remains anchored to the CM during the process24. Disulfide crosslinking and residue accessibility experiments demonstrate that the luminal domain undergoes some unknown conformational change during transport, either within the barrel, wholly or partially48,49, or possibly being expelled from the barrel22. Previous simulations suggest a mechanical interaction in which the application of force partially unfolds the luminal domain is feasible21. However, it remains to be determined what is the specific balance between unfolding, removal as a single unit, and conformational change of the luminal domain within the barrel during transport5,22,50.
Methods
Modeling
DMPE lipid bilayer
The outer membrane of Gram-negative bacteria is composed primarily of a combination of lipopolysaccharides (LPS) in the outer leaflet and phosphatidylethanolamine (PE) lipids on the inner leaflet51,52. To approximate the outer membrane, we developed and used dimeristoyl phosphatidylethanolamine (DMPE) lipids. Previous simulations of outer membrane proteins have utilized various lipids including POPE21,51, POPC34, and DMPC30,53,54, for example. DMPE lipids were chosen for the current study because they have only 14 carbons in each tail (compared to 16 and 18 for POPE) and thus better match the hydrophobic thickness of BtuB55. A CHARMM topology file for DMPE is provided in the Supplementary Data.
Cyanocobalamin
Modeling of CN-Cbl for the CHARMM force field started from previously developed and tested AMBER parameters56. Since the potential energy functions in CHARMM and AMBER are nearly identical, transfer of bonded parameters, including equilibrium values and force constants for bonds, angles, and dihedrals, was straightforward for those atom types not already present in the CHARMM force field. For the charges in the AMBER force field, the electron density for CN-Cbl was calculated using semi-empirical methods and the resulting Mulliken charges were taken56. Although the accuracy of these charges has been questioned57, recent simulations have demonstrated they are functional in the CHARMM force field58. However, to avoid any potential discrepancies between the two different approaches, we chose to adopt CHARMM charges where possible from other compounds, and to calculate ab initio charges for the remaining atoms.
CN-Cbl can be broken down into a few separate chemical moieties, namely a corrin ring, associated methyl groups and acetamide side chains, a dimethylbenzimidazole (DMB), and a linker containing phosphate and ribose connecting the DMB to the corrin ring. To simplify calculations, ab initio charges were determined for only the corrin ring, along with the cobalt-bound cyano group and the DMB's cobalt-coordinating nitrogen, using the quantum chemistry package Gaussian 0359. The reduced system contained 51 atoms. The geometry was optimized and charges were calculated using density functional theory at the B3LYP/6-31+G* level. The normal procedure for developing CHARMM charges requires one to scale Mulliken charges resulting from the QM calculations in order to best reproduce the ab initio interaction of each atom with a TIP3P water molecule60. However, because the proper placement of such water molecules is no longer apparent for such a large compound, charges were instead determined from the electron density using Natural Population Analysis (NPA), which is known to be significantly more accurate than Mulliken charges and also was used in the development of CN-Cbl parameters for the GROMOS force field57,61. Charges on identical atom types were averaged and assigned, although variation was already small; these charges are given in Fig. 6A for all heavy atoms. Full topology and parameter sets are provided in the Supplementary Data.
Figure 6.

Parametrization of CN-Cbl. (A) Reduced model used for quantum chemical calculations in a ball and stick representation. The atoms are colored by type, with cobalt in green, and the final partial charges assigned to each heavy atom is given. (B) Simulation of CN-Cbl in a water box. The initial conformation is indicated as a thick stick representation colored by atom type. Snapshots of CN-Cbl taken every 4 ps from the 1-ns trajectory are overlaid as thin lines.
The parameters and charges were tested by simulating CN-Cbl in a water box for 1 ns. Fig. 6B shows 250 frames from the simulation (1 every 4 ps) overlaying the initial structure. A large flexibility is seen for the side chains and the linker, while the corrin ring remains stable, as expected. The cobalt-nitrogen distance for the axial nitrogen was 1.998 Å on average, in excellent agreement with the experimental value of 2.017 Å56.
Systems constructed
Multiple systems were built based on the available BtuB crystallographic structures. The apo state (PDB code 1NQE), the Ca2+-loaded state (1NQG), and the Ca2+/CN-Cbl-bound state (1NQH) were all simulated9. Additionally, we simulated the in meso structure of the apo state (2GUF)28. Missing extracellular loops in some of the structures were modeled based upon the TonB-bound BtuB structure which was complete19. Specifically, loops 3/4, 5/6, and 7/8 in the apo structure; loops 5/6 and 7/8 in the Ca2+-bound structure; loop 9/10 in the Ca2+/CN-Cbl-bound structure; and loops 3/4, 5/6, 21/22, and SB-1 in the in meso structure were all modeled.
Crystallographic ions and water molecules inside the barrel or near the loops were retained, along with CN-Cbl when present. Protonation states of titratable residues were assigned based on pKa calculations using propKa and an assumed pH of 762,63. The protein was placed in a DMPE lipid bilayer and then solvated. In nearly all simulations, Ca2+ and Cl− ions were added with a net concentration of 100 mM. For one simulation of the apo state (represented in Fig. 2), Na+ was used instead of Ca2+. Each system contained approximately 85,000 atoms.
Molecular dynamics
We used NAMD 2.664 for all molecular dynamics simulations as well as the CHARMM27 force field with the CMAP correction60,65. A multiple time-stepping algorithm was employed in which bonded interactions were calculated every 1 fs, short range non-bonded interactions every 2 fs, and long range interactions every 4 fs. Long range electrostatic interactions were calculated using the particle-mesh Ewald (PME) method with a grid density of more than 1/Å3.
Simulations were run at a constant temperature of 310K maintained using Langevin dynamics applied to all heavy atoms with a damping constant of 1 ps−1. A pressure of 1 atm was enforced along the z direction (normal to the membrane) with a Nosé-Hoover-Langevin piston; the membrane area was only allowed to fluctuate for the first 2 ns of equilibration, after which it was fixed (NPnTA ensemble). Periodic boundary conditions were used for all simulations.
Equilibration of each system was carried out in multiple stages. First, all atoms except the lipid tails were restrained for 250 ps of simulation, permitting the tails to “melt”. Next, protein backbone atoms were restrained for an additional 1.75 ns. Finally, all atoms were freed for the remainder of the simulation. We estimate the resulting area per lipid for our DMPE bilayer is 57-60 Å2.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (P41-RR005969, R01-GM067887 for J.G. and E.T., and R01-GM079800 for M.C.W. and E.T.). The authors gratefully acknowledge computer time provided by the TeraGrid resources (MCA06N060). J.G. also thanks Jan Saam, Chris Harrison, and Peter Freddolino for helpful discussions on force field parametrization.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nikaido H. Prevention of drug access to bacterial targets - permeability barriers and active efflux. Science. 1994;264:382–388. doi: 10.1126/science.8153625. [DOI] [PubMed] [Google Scholar]
- 2.Davidson AL, Chen J. ATP-binding cassette transporters in bacteria. Annu Rev Biochem. 2004;73:241–268. doi: 10.1146/annurev.biochem.73.011303.073626. [DOI] [PubMed] [Google Scholar]
- 3.Wen PC, Tajkhorshid E. Dimer opening of the nucleotide binding domains of ABC transporters after ATP hydrolysis. Biophys J. 2008;95:5100–5110. doi: 10.1529/biophysj.108.139444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Postle K, Kadner RJ. Touch and go: tying TonB to transport. Mol Microbiol. 2003;49:869–882. doi: 10.1046/j.1365-2958.2003.03629.x. [DOI] [PubMed] [Google Scholar]
- 5.Wiener MC. TonB-dependent outer membrane transport: going for Baroque? Curr Opin Struct Biol. 2005;15:394–400. doi: 10.1016/j.sbi.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Killmann H, Videnov G, Jung G, Schwarz H, Braun V. Identification of receptor binding sites by competitive peptide mapping: phages T1, T5, and phi 80 and colicin M bind to the gating loop of FhuA. J Bacteriol. 1995;177:694–698. doi: 10.1128/jb.177.3.694-698.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newton SMC, Igo JD, Scott DC, Klebba PE. Effect of loop deletions on the binding and transport of ferric enterobactin by FepA. Mol Microbiol. 1999;32:1153–1165. doi: 10.1046/j.1365-2958.1999.01424.x. [DOI] [PubMed] [Google Scholar]
- 8.Kurisu G, Zakharov SD, Zhalina MV, Bano S, Eroukova VY, Rokitskaya TI, et al. The structure of BtuB with bound colicin E3 R-domain implies a translocon. Nat Struct Biol. 2003;10:948–954. doi: 10.1038/nsb997. [DOI] [PubMed] [Google Scholar]
- 9.Chimento DP, Mohanty AK, Kadner RJ, Wiener MC. Substrate-induced transmembrane signaling in the cobalamin transporter BtuB. Nat Struct Biol. 2003;10:394–401. doi: 10.1038/nsb914. [DOI] [PubMed] [Google Scholar]
- 10.Endriβ F, Braun V. Loop deletions indicate regions important for FhuA transport and receptor functions in Escherichia coli. J Bacteriol. 2004;186:4818–4823. doi: 10.1128/JB.186.14.4818-4823.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuller-Schaefer CA, Kadner RJ. Multiple extracellular loops contribute to substrate binding and transport by the Escherichia coli cobalamin transporter BtuB. J Bacteriol. 2005;187:1732–1739. doi: 10.1128/JB.187.5.1732-1739.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cascales E, Buchanan SK, Duché D, Kleanthous C, Lloubés R, Postle K, et al. Colicin biology. Microbiol Mol Biol Rev. 2007;71:158–229. doi: 10.1128/MMBR.00036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chimento DP, Kadner RJ, Wiener MC. Comparative structural analysis of TonB-dependent outer membrane transporters: implications for the transport cycle. Proteins: Struct, Func, Bioinf. 2005;59:240–251. doi: 10.1002/prot.20416. [DOI] [PubMed] [Google Scholar]
- 14.Kadner RJ. Vitamin B12 transport in Escherichia coli: energy coupling between membranes. Mol Microbiol. 1990;4:2027–2033. doi: 10.1111/j.1365-2958.1990.tb00562.x. [DOI] [PubMed] [Google Scholar]
- 15.Lundrigan MD, Kadner RJ. Nucleotide sequence of the gene for the ferrienterochelin receptor FepA in Escherichia coli. homology among outer membrane receptors that interact with TonB. J Biol Chem. 1986;261:10797–10801. [PubMed] [Google Scholar]
- 16.Schramm E, Mende J, Braun V, Kamp RM. Nucleotide sequence of the colicin B activity gene cba: consensus pentapeptide among TonB-dependent colicins and receptors. J Bacteriol. 1987;169:3350–3357. doi: 10.1128/jb.169.7.3350-3357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cadieux N, Kadner RJ. Site-directed disulfide bonding reveals an interaction site between energy-coupling protein TonB and BtuB, the outer membrane cobalamin transporter. Proc Natl Acad Sci USA. 1999;96:10673–10678. doi: 10.1073/pnas.96.19.10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cadieux N, Bradbeer C, Kadner RJ. Sequence changes in the ton box region of BtuB affect its transport activities and interaction with TonB protein. J Bacteriol. 2000;182:5954–5961. doi: 10.1128/jb.182.21.5954-5961.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shultis DD, Purdy MD, Banchs CN, Wiener MC. Outer membrane active transport: structure of the BtuB:TonB complex. Science. 2006;312:1396–1399. doi: 10.1126/science.1127694. [DOI] [PubMed] [Google Scholar]
- 20.Pawelek PD, Croteau N, Ng-Thow-Hing C, Khursigara CM, Moiseeva N, Allaire M, et al. Structure of TonB in complex with FhuA, E. coli outer membrane receptor. Science. 2006;312:1399–1402. doi: 10.1126/science.1128057. [DOI] [PubMed] [Google Scholar]
- 21.Gumbart J, Wiener MC, Tajkhorshid E. Mechanics of force propagation in TonB-dependent outer membrane transport. Biophys J. 2007;93:496–504. doi: 10.1529/biophysj.107.104158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma L, Kaserer W, Annamalai R, Scott DC, Jin B, Jiang X, et al. Evidence of ball-and-chain transport of ferric enterobactin through FepA. J Biol Chem. 2007;282:397–406. doi: 10.1074/jbc.M605333200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devanathan S, Postle K. Studies on colicin B translocation: FepA is gated by TonB. Mol Microbiol. 2007;65:441–453. doi: 10.1111/j.1365-2958.2007.05808.x. [DOI] [PubMed] [Google Scholar]
- 24.Kaserer WA, Jiang X, Xiao Q, Scott DC, Bauler M, Copeland D, et al. Insight from TonB hybrid proteins into the mechanism of iron transport through the outer membrane. J Bacteriol. 2008;190:4001–4016. doi: 10.1128/JB.00135-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Letain TE, Postle K. TonB protein appears to transduce energy by shuttling between the cytoplasmic membrane and the outer membrane in Gram-negative bacteria. Mol Microbiol. 1997;24:271–283. doi: 10.1046/j.1365-2958.1997.3331703.x. [DOI] [PubMed] [Google Scholar]
- 26.Larsen RA, Postle K. In vivo evidence of TonB shuttling between the cytoplasmic and outer membrane in Escherichia coli. J Bacteriol. 2003;49:211–218. doi: 10.1046/j.1365-2958.2003.03579.x. [DOI] [PubMed] [Google Scholar]
- 27.Cadieux N, Barekzi N, Bradbeer C. Observations on the calcium dependence and reversibility of cobalamin transport across the outer membrane of Escherichia coli. J Biol Chem. 2007;282:34921–34928. doi: 10.1074/jbc.M707426200. [DOI] [PubMed] [Google Scholar]
- 28.Cherezov V, Yamashita E, Liu W, Zhalnina M, Cramer WA, Caffrey M. In meso structure of the cobalamin transporter, BtuB, at 1.95 Å resolution. J Mol Biol. 2006;364:716–734. doi: 10.1016/j.jmb.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradbeer C, Reynolds PR, Bauler GM, Fernandez MT. A requirement for calcium in the transport of cobalamin across the outer membrane of Escherichia coli. J Biol Chem. 1986;261:2520–2523. [PubMed] [Google Scholar]
- 30.Faraldo-Gómez JD, Smith GR, Sansom MSP. Molecular dynamics simulations of the bacterial outer membrane protein FhuA: A comparative study of the ferrichrome-free and bound states. Biophys J. 2003;85:1406–1420. doi: 10.1016/S0006-3495(03)74573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ash WL, Zlomislic MR, Oloo EO, Tieleman DP. Computer simulations of membrane proteins. Biochim Biophys Acta Biomembr. 2004;1666:158–189. doi: 10.1016/j.bbamem.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 32.Bond PJ, Sansom MSP. The simulation approach to bacterial outer membrane proteins (review) Molecular Membrane Biology. 2004;21:151–161. doi: 10.1080/0968760410001699169. [DOI] [PubMed] [Google Scholar]
- 33.Gumbart J, Wang Y, Aksimentiev A, Tajkhorshid E, Schulten K. Molecular dynamics simulations of proteins in lipid bilayers. Curr Opin Struct Biol. 2005;15:423–431. doi: 10.1016/j.sbi.2005.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luan B, Caffrey M, Aksimentiev A. Structure refinement of the OpcA adhesin using molecular dynamics. Biophys J. 2007;93:3058–3069. doi: 10.1529/biophysj.107.106724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bond PJ, Derrick JP, Sansom MSP. Membrane simulations of OpcA: gating in the loops? Biophys J. 2007;92:L23–L25. doi: 10.1529/biophysj.106.097311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bradbeer C, Gudmundsdottir A. Interdependence of calcium and cobalamin binding by wild-type and mutant BtuB protein in the outer-membrane of Escherichia coli. J Bacteriol. 1990;172:4919–4926. doi: 10.1128/jb.172.9.4919-4926.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim M, Xu Q, Murray D, Cafiso DS. Solutes alter the conformation of the ligand binding loops in outer membrane transporters. Biochemistry. 2008;47:670–679. doi: 10.1021/bi7016415. [DOI] [PubMed] [Google Scholar]
- 38.Chimento DP, Kadner RJ, Wiener MC. The Escherichia coli outer membrane cobalamin transporter BtuB: Structural analysis of calcium and substrate binding, and identification of orthologous transporters by sequence/structure conservation. J Mol Biol. 2003;332:999–1014. doi: 10.1016/j.jmb.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 39.Hogle J, Kirchhausen T, Harrison SC. Divalent cation sites in tomato bushy stunt virus. difference maps at 2.9 Å resolution. J Mol Biol. 1983;171:95–100. doi: 10.1016/s0022-2836(83)80315-5. [DOI] [PubMed] [Google Scholar]
- 40.Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: Structural and functional diversity. Prot Sci. 1996;5:2375–2390. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peeraer Y, Rabijns A, Collet JF, van Schaftingen E, de Ranter C. How calcium inhibits the magnesium-dependent enzyme human phosphoserine phosphatase. Eur J Biochem. 2004;271:3421–3427. doi: 10.1111/j.0014-2956.2004.04277.x. [DOI] [PubMed] [Google Scholar]
- 42.Katz AK, Glusker JP, Beebe SA, Bock CW. Calcium ion coordination: A comparison with that of beryllium, magnesium, and zinc. J Am Chem Soc. 1996;118:5752–5763. [Google Scholar]
- 43.Sotomayor M, Schulten K. The allosteric role of the Ca++ switch in adhesion and elasticity of C-cadherin. Biophys J. 2008;94:4621–4633. doi: 10.1529/biophysj.107.125591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohkubo YZ, Tajkhorshid E. Distinct structural and adhesive roles of Ca2+ in membrane binding of blood coagulation factors. Structure. 2008;16:72–81. doi: 10.1016/j.str.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 45.Diao J, Tajkhorshid E. Indirect role of Ca2+ in the assembly of extracelluar matrix proteins. Biophys J. 2008;95:120–127. doi: 10.1529/biophysj.108.129106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lukasik SM, Ho KWD, Cafiso DS. Molecular basis for substrate-dependent transmembrane signaling in an outer-membrane transporter. J Mol Biol. 2007;370:807–811. doi: 10.1016/j.jmb.2007.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferguson AD, Amezcua CA, Halabi NM, Chelliah Y, Rosen MK, Ranganathan R, et al. Signal transduction pathway of TonB-dependent transporters. Proc Natl Acad Sci USA. 2007;104:513–518. doi: 10.1073/pnas.0609887104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eisenhauer HA, Shames S, Pawelek PD, Coulton JW. Siderophore transport through Escherichia coli outer membrane receptor FhuA with disulfide-tethered cork and barrel domains. J Biol Chem. 2005;280:30574–30580. doi: 10.1074/jbc.M506708200. [DOI] [PubMed] [Google Scholar]
- 49.Chakraborty R, Storey E, van der Helm D. Molecular mechanisms of ferricsiderophore passage through the outer membrane receptor proteins of Escherichia coli. Biometals. 2007;20:263–274. doi: 10.1007/s10534-006-9060-9. [DOI] [PubMed] [Google Scholar]
- 50.Ferguson AD, Deisenhofer J. Metal import through microbial membranes. Cell. 2004;116:15–24. doi: 10.1016/s0092-8674(03)01030-4. [DOI] [PubMed] [Google Scholar]
- 51.Tieleman DP, Berendsen HJC. A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. Biophys J. 1998;74:2786–2801. doi: 10.1016/S0006-3495(98)77986-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Im W, Roux B. Ions and counterions in a biological channel: a molecular dynamics study of OmpF porin from Escherichia coli in an explicit membrane with 1 M KCl aqueous salt solution. J Mol Biol. 2002;319:1177–1197. doi: 10.1016/S0022-2836(02)00380-7. [DOI] [PubMed] [Google Scholar]
- 54.Robertson KM, Tieleman DP. Orientation and interactions of dipolar molecules during transport through OmpF porin. FEBS Lett. 2002;528:53–57. doi: 10.1016/s0014-5793(02)03173-3. [DOI] [PubMed] [Google Scholar]
- 55.Xu Q, Kim M, Ho KWD, Lachowicz P, Fanucci GE, Cafiso DS. Membrane hydrocarbon thickness modulates the dynamics of a membrane transport protein. Biophys J. 2008;95:2849–2858. doi: 10.1529/biophysj.108.133629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marques HM, Ngoma B, Egan TJ, Brown KL. Parameters for the AMBER force field for the molecular mechanics modeling of the cobalt corrinoids. J Mol Struc. 2001;561:71–91. [Google Scholar]
- 57.Kandt C, Xu ZT, Tieleman DP. Opening and closing motions in the periplasmic vitamin B-12 binding protein BtuF. Biochemistry. 2006;45:13284–13292. doi: 10.1021/bi061280j. [DOI] [PubMed] [Google Scholar]
- 58.Liu M, Sun T, Hu J, W WC, Wang C. Study on the mechanism of the BtuF periplasmic-binding protein for vitamin B-12. Biophys Chem. 2008;135:19–24. doi: 10.1016/j.bpc.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 59.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 03 (Revision B.05) Gaussian, Inc.; Pittsburgh, PA: 2003. [Google Scholar]
- 60.MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck J, Field MJ, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 61.Reed A, Weinstock RB, Weinhold F. Natural population analysis. J Chem Phys. 1985;83:735–746. [Google Scholar]
- 62.Li H, Robertson AD, Jensen JH. Very fast empirical prediction and interpretation of protein pKa values. Proteins: Struct, Func, Bioinf. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 63.Bas DC, Rogers DM, Jensen JH. Very fast prediction and rationalization of pK(a) values for protein-ligand complexes. Proteins: Struct, Func, Bioinf. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 64.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comp Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.MacKerell AD, Jr, Feig M, Brooks CL., III Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comp Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.