

Abstract
Classical swine fever (CSF) severity is dependent on the virulence of the CSF virus (CSFV) strain. The earliest event detected following CSFV infection is a decrease in lymphocytes number. With some CSFV strains this leads to lymphopenia, the severity varying according to strain virulence. This lymphocyte depletion is attributed to an induction of apoptosis in non-infected bystander cells. We collected peripheral blood mononuclear cells (PBMC) before and during 3 days post-infection with either a highly or moderately virulent CSFV strain and subjected them to comparative microarray analysis to decipher the transcriptomic modulations induced in these cells in relation to strain virulence. The results revealed that the main difference between strains resided in the kinetics of host response to the infection: strong and immediate with the highly virulent strain, progressive and delayed with the moderately virulent one. Also although cell death/apoptosis-related IFN stimulated genes (ISG) were strongly up-regulated by both strains, significant differences in their regulation were apparent from the observed differences in onset and extent of lymphopenia induced by the two strains. Furthermore, the death receptors apoptotic pathways (TRAIL-DR4, FASL-FAS and TNFa-TNFR1) were also differently regulated. Our results suggest that CSFV strains might exacerbate the interferon alpha response, leading to bystander killing of lymphocytes and lymphopenia, the severity of which might be due to the host’s loss of control of IFN production and downstream effectors regulation.
Keywords: classical swine fever virus, virulence, bystander apoptosis, type I interferon, microarray
1. INTRODUCTION
Classical swine fever virus (CSFV) is a Pestivirus, member of the Flaviviridae family. It is an enveloped virus with a 12.5-kb positive polarity single-stranded RNA genome. CSFV is the causal agent of a highly contagious disease in swine that leads to important economic losses worldwide. The severity of clinical signs varies according to host parameters such as age, breed or health status, but is also largely dependent on the virulence of the viral strains [29]. Highly virulent strains cause an acute haemorrhagic form of the disease that induces high mortality (more than 98% in piglets), whereas moderately virulent strains induce either a sub-acute or a chronic form of the disease giving pigs a chance to recover. Infection with low virulent strains results in either very mild or a total absence of clinical signs.
Complete genome sequences are available for a few CSFV strains and many studies have attempted to explain virulence in terms of genetic differences between CSFV strains: a poly-U insertion is only found in the 3′NTR region of avirulent strains [47], mutation in genes or their deletion (E1, E2, Erns or Npro) in highly virulent strains attenuates disease expression in swine [25, 32, 35]. However, back experiments failed to restore virulence in avirulent strains [31].
The earliest detectable event following CSFV infection, prior to viraemia has even been established, is a decrease in the number of circulating leukocytes [40]. This occurs irrespective of strain virulence whereas the time-course and intensity of leukocyte depletion vary according to strain virulence. The term leukopenia (white blood cell count less than 7 × 106 cells/mL) can be applied to infections with highly and moderately virulent CSFV strains. The mechanism of CSFV-induced immune cell depletion is still not fully understood but in peripheral blood and lymph nodes, it is attributed to cell death via the induction of apoptosis in the most depleted cells, i.e., the lymphocytes [6, 39, 40].
This apoptosis of the lymphocytes is not a direct consequence of the presence of virus or viral proteins in these cells as the main target cells to be infected by the virus in vivo are not lymphocytes but monocytes-macrophages [22]. This was clearly shown in an in situ double-staining experiment to detect CSFV and apoptosis which showed that most CSFV-positive cells were negative for apoptosis in the lymph nodes and vice versa [6]. In contrast, the in vitro infection of lymphocytes does not induce any cytopathogenic effects and therefore does not provoke any cell death [39]. Furthermore, in vitro studies showed that CSFV-infected cells prevent their type I interferon (IFN)-mediated suicide by interfering with IFN production [3]. In vivo, monocytes-macrophages, as well as plasmacytoid dendritic cells, are suspected to have a role in this indirect induction of apoptosis because of their enhanced release of proinflammatory cytokines, that unbalance the homeostasis of the cellular environment [13, 36, 41].
The interactions of two strains of CSFV with the immune cells were therefore studied in vivo in order to better understand the differential pathogenic effects observed with different CSFV strains and to try to identify some of the pathogenic mechanisms. We chose to work on peripheral blood mononuclear cells (PBMC) which give us access to the main infected cell population (monocytes) and the main affected cell population (lymphocytes) without killing the animals. This strategy allowed us to perform a 3 days kinetic study using a limited number of animals. Furthermore, blood sampling on the same animals at day 0 (prior infection) were used as controls to determine the baseline expression level for microarray analysis. The transcriptomic modulations induced in pigs in relation to the virulence of the CSFV strain were deciphered by comparative microarray analysis.
2. MATERIALS AND METHODS
2.1. Virus and animal infections
Two CSFV strains of different virulence were used in the experiments. The highly virulent Eystrup strain was kindly provided by Dr A. Summerfield (Institute of Virology and Immunoprophylaxis, Mittelhäusern, Switzerland) and the moderately virulent Paderborn strain was kindly provided by Prof. V. Moennig (EU CSF reference laboratory, Hanover, Germany).
Two groups of eight seven-week-old, specific-pathogen-free Large White pigs, from our protected facilities, were oronasally infected with 106 TCID50 of either the Eystrup or Paderborn strain. The dose administered was confirmed by back titration of the inocula by propagation on porcine kidney cell line PK15.
Experiments were performed in accordance with the animal welfare experimentation agreement granted by the Direction des Services Vétérinaires des Côtes d’Armor (AFSSA registration number B-22-745-1), under the responsibility of Marie-Frédérique Le Potier (agreement number 22-17).
2.2. Sample collection and treatments
Blood samples were collected on heparin and EDTA before infection on day 0 (D0) then on day 1 (D1), day 2 (D2) and day 3 (D3) post-infection (pi). PBMC were isolated from heparinized blood by Ficoll density gradient centrifugation (Eurobio, Les Ulis, France). Purified PBMC were then washed with PBS and kept frozen in Trizol reagent (Invitrogen Life Technologies Inc., Carlsbad, CA, USA) until RNA extraction.
Serum samples were purified from coagulated blood samples by centrifugation at 3 000 g for 5 min before being frozen at –20 °C.
2.3. CSFV infection assessment on whole blood
Lymphocyte counts were obtained from EDTA blood with a MS9 hematology analyzer (Melet Schloesing laboratoires, Osny, France).
The RNeasy mini kit (Qiagen, Courtaboeuf, France) was used for total RNA extraction from EDTA blood and CSFV genome detection was performed by real-time one-step RT-PCR using the Adiavet CSF kit (Adiagene, Saint-Brieuc, France) according to the manufacturer’s instructions. Viral genome quantifications were determined using a standardized RNA kindly provided by Béatrice Blanchard (Adiagene).
2.4. Microarray analysis
Total RNA was extracted by a standard Trizol RNA extraction protocol and assessed for quality and quantity with the Agilent 2100 Bioanalyzer. Half the RNA samples obtained from each pig on D0 were pooled to constitute the uninfected pig reference. RNA were converted to aminoallyl-RNA (aRNA) and either Cy3- or Cy5-labelled using the Amino Allyl Message Amp aRNA kit (Ambion) and CyDye (Cy3/Cy5) Reactive Dye Pack (Amersham). Each Cy3- (or Cy5-) coupled sample from infected pigs was then hybridized with a Cy5- (or Cy3-) coupled reference sample on a pig microarray slide containing 20 400 oligonucleotides from the US Pig Genome Coordination Program1. Hybridizations were performed overnight at 42 °C in ArrayIt hybridization chambers (Telechem, Sunnyvale, CA, USA). Microarrays were scanned using a Genepix 4000A scanner with the GenePix Pro 5.0 data acquisition and analysis software (Axon Instruments, Union City, CA, USA).
The GenePix output data were subjected to the Micro-Array Data Suite of Computed Analysis (MADSCAN) for normalization [19]. Variant genes were identified by applying the significance analysis of microarray (SAM) software with a false discovery rate set below 3% [45]. We first visualized genes that were differentially expressed in the infected in relation to the uninfected reference by SAM one-class analysis for each day in the infection time-course. We then chose a SAM two-class data method of analysis to pick out genes for which the mean expression levels in the Eystrup-infected pig samples were significantly different from those found in the Paderborn samples. The obtained sets of genes were characterized according to the Gene Ontology annotations performed with the Ingenuity Pathway Analysis 6 software2 (Ingenuity Systems Inc., Redwood City, CA, USA). Due to the incomplete annotation of the pig genome this final analysis was performed by homology with human, mouse or rat genes.
2.5. Real-time RT-PCR assays
Real-time RT-PCR (rRT-PCR) were performed on the same RNA samples as those used for microarray, to validate the microarray results on a limited number of selected genes (see Tab. I). rRT-PCR were run for each pig on each day before and after infection, using 5 ng of total RNA in a one-step Brilliant II SYBR GREEN qRT-PCR master mix (Agilent Technologies Inc., Palo Alto, CA, USA) with 0.2 μM of each primer. Each amplification reaction was performed at least in duplicate with the Chromo4 BIO-RAD real-time PCR system at the annealing temperature of 60 °C. Primer specificity was checked by melting curve analysis. Beta-Actin (ACTb) was used as endogenous control to normalize the quantification of target genes. The relative amounts of all assessed genes were calculated using the R = 2–ΔΔCT equation [23] with D0 as the relative reference. To compare microarray results with RT-PCR ones, we expressed data in Log2 ratio.
Table I.
Primer sequences used for real-time RT-PCR assays.
| Gene name | Forward primer sequence | Reverse primer sequence | Amplicon length (bp) | GenBank accession number |
|---|---|---|---|---|
| ACTb | CACGCCATCCTGCGTCTGGA | AGCACCGTGTTGGCGTAGAG | 380 | DQ845171 |
| CASP4 | CAGCAACCTTGGCAGACAGC | GAACCCTTTGTGACATCTCTCCAG | 145 | AK231138 |
| DR4 | TCGGTATGGACGCCTGAGTC | GATCGCCAGAAAAGGACCTTG | 56 | XM_001926723 |
| FAS | CATCGTGAGGGTCAATTCTGC | CATGTTTCCGTTTGCCAGG | 55 | NM_213839 |
| FASL | AAGAAGAAGAGGGACCACAATG | CTTTGGCTGGCAGACTCTCT | 149 | AY033634 |
| IFI16 | ACACTGACCGAAAGATGGAGATC | GCACCAGATACATCCCACTCAC | 122 | XM_001929212 |
| IFIH1 | GTAGAATTACCCATCACATTTCCC | CAAAATCCTATGAAAGGTCAAGTTG | 125 | NM_001100194 |
| IFNa | TCAGCACAGAGGGCTCGG | TGCATGACACAGGCTTCCAG | 103 | AY687280 |
| NMI | GGCCAAGCCGGTTTCATTAAAG | CCGGGCCTGCCTGTCATAC | 196 | AK234080 |
| OAS1 | TCCACTCCCCTCCCGACTC | GGCTTCCTTGACCTGTGTTCG | 133 | NM_214303 |
| TNFa | TGGCCCAAGGACTCAGATCAT | TCGGCTTTGACATTGGCTACA | 76 | EU682384 |
| TRAIL | GGAACGGTTTCTACAGAAGGGAAC | TCAGCAGTATAGGGTCAGGATAGC | 94 | NM_001024696 |
| XAF1 | AATGGATTTCCCCTTCCTTCGG | CGCCCTGAGCTTGCCATC | 102 | AK232641 |
2.6. Cytokines detection in serum
Interferon alpha (IFNa) was detected by ELISA as previously described [12], using a porcine recombinant IFNa with specific activity of 3.8 × 107 units (U)/mg (R&D Systems Inc., Minneapolis, MN, USA). Tumor necrosis factor alpha (TNFa) was detected using a commercial ELISA kit from Biosource (Camarillo, CA, USA). The results obtained with samples collected post-infection were expressed in relation to their respective D0 values.
2.7. Statistical analysis
The variations between strains for the lymphocyte counts, PCR and ELISA tests, were analyzed either by comparing data obtained with Eystrup infection, with data obtained with Paderborn infection using a Two-sample Student’s t-test, or by comparing the variations within each strain sample with their respective D0-value, using a Paired Two-sample Student’s t-test. These tests were performed using Systat 9 software (Systat Software, Inc., Point Richmond, CA, USA), with a limit of significance of p < 0.05 (indicated by *) or p < 0.01 (indicated by **). The error bars on the graphs are Standard Error (SE) bars.
3. RESULTS
3.1. Host response to the moderately virulent CSFV strain, in comparison to the highly virulent strain, is delayed
To correlate the physiologic and virologic events, transcriptomic modulations and virulence of the CSFV strains, the lymphocyte depletion was measured and the viral RNA was quantified in blood. A dramatic and regular decrease in the number of lymphocytes was observed from D1 pi with the Eystrup strain whereas it was delayed by one day with the Paderborn strain (Fig. 1A). The differences in the kinetics and severity of CSFV-induced lymphopenia between Eystrup- and Paderborn-infected pigs were statistically validated. The detection of viral genome started from D1 with Eystrup, from D2 with Paderborn (Fig. 1B). Viraemia was detected from D2 with Eystrup, from D3 with Paderborn (data not shown).
Figure 1.
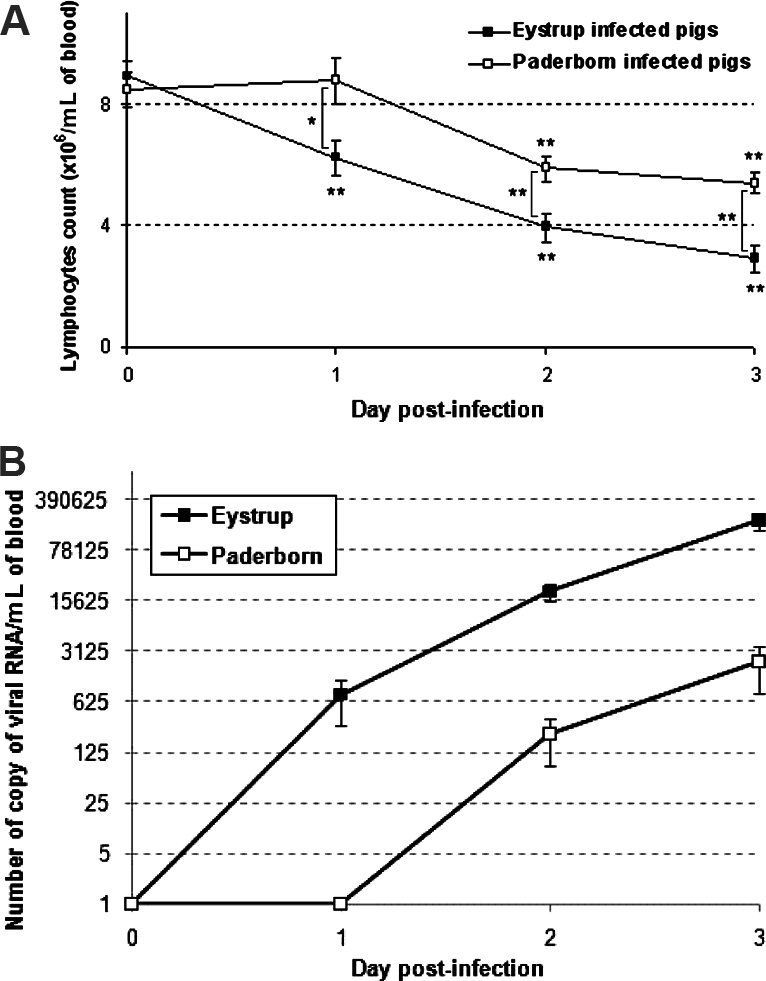
Blood lymphocyte counts (A) and virus genome quantification (B) for the time-course after CSFV infection. Results correspond to mean (± SE) results obtained for 8 pigs infected by Eystrup or 8 pigs infected by Paderborn. * p < 0.05 and ** p < 0.01 either compared each post-infection value with the respective D0 value using Paired Two-sample Student’s t-test, or the Eystrup values with Paderborn values using Two-sample Student’s t-test at each infection time-point.
Microarray data set was analyzed using first a one-class SAM analysis of gene expression in PBMC isolated from infected in relation to non-infected pigs revealed a mild and delayed response to Paderborn infection and limited homology with the response to the Eystrup strain (cf. online Supplementary Figs. S1 and S2, and Tabs. SI and SII available at www.vetres.org.).
A two-class SAM analysis was then performed to better assess the differences between our two infection models. A list of 292 genes for which the modulations in expression between Eystrup and Paderborn infections were different, was obtained from the 3 day-experiment (cf. online Supplementary Tab. SIII available at www.vetres.org). On day 1, 181 genes exhibited differential expression between the strains, 44 on D2 and 79 on D3 (Fig. 2). Among the genes modulated on D1, 169 were up-regulated by Eystrup in relation to Paderborn-induced expression and 12 down-regulated. Thus the modulation of response to infection was more immediate and dramatic with Eystrup than with Paderborn.
Figure 2.
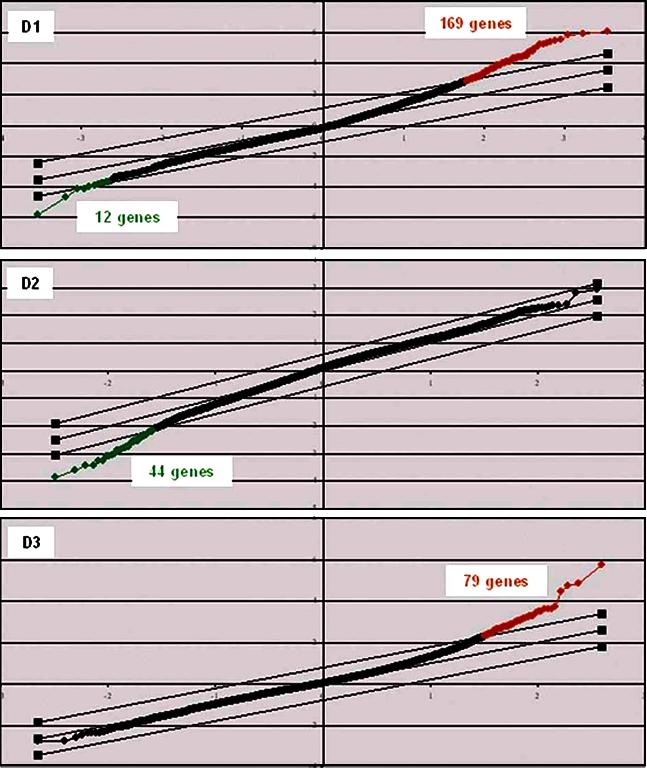
Two-class analysis SAM-plot of genes differently modulated by Eystrup in relation to Paderborn over 3 days post-infection. A two-class SAM analysis of microarray data was performed each day post-infection with the false discovery rate set at below 3%. In this analysis, gene expression in the 8 Eystrup samples was compared with gene expression in the 8 Paderborn samples. The x-axis values denote expected expression; y-axis values, observed expression; parallel transverse lines, threshold lines indicating a false discovery rate of < 3%. Red dots correspond to genes up-regulated by Eystrup in relation to Paderborn; black dots, invariant genes and green dots, down-regulated genes. (A color version of this figure is available online at www.vetres.org.)
3.2. Differential induction of IFN stimulated genes according to CSFV strains
The ontological analysis of the variant genes made apparent that “Cell Death” (16%) and “Immune Response” (12%) were the main two biological functions that were differently regulated between strains (Fig. 3).
Figure 3.

Distribution of genes differentially modulated by Eystrup in relation to Paderborn in biological function. Differently modulated genes between Eystrup and Paderborn infections obtained by the two-class SAM analysis over the 3 days post-infection were organized according to their biological processes annotation with the Ingenuity Analysis Pathway software. The 20 most important biological functions are shown. Percentages refer to the number of genes included in one function related to the remaining others. (A color version of this figure is available online at www.vetres.org.)
As given biological function can result from numerous types of cell signaling, we realized a second Ingenuity analysis that displayed that the main differentially modulated pathways were “Interferon Signaling” and the “Protein Ubiquitination Pathway”. For these two pathways, p-values were very significant (p < 0.0001) but the IFN pathway was the only one with both high significant p-value and high ratio (Fig. 4). Further investigation of these IFN signaling genes revealed that most of them were classified as “IFN stimulating genes” (ISG) and mainly corresponded to effectors of the IFN response. The variations of these ISG were highly pronounced with almost half displaying a Log2 ratio > 1.000 and 4 with a Log2 ratio > 2.000 (Tab. II). Up-regulation of these genes was immediate and massive for the 3 days following infection with the Eystrup strain, but was only apparent from the second day onwards for Paderborn.
Figure 4.

Signaling pathways distribution of genes differentially modulated by Eystrup in relation to Paderborn. Genes revealed by two-class SAM analysis over the 3 days post-infection were organized with the Ingenuity Pathway Analysis software according to the pathway in which they are involved in the current data base of this software, and classified according to the calculated –Log(p-values). The significance of the association between the data set and the signaling pathway was measured in two ways: ratio and p-value. Ratios refer to the proportion of variant genes from a pathway related to the number of genes included in the software database for this same pathway. The p-value, calculated using a right-tailed Fisher’s exact test, corresponds to the probability that the association between a set of focus genes and a given pathway is not due to random chance. The threshold corresponds to a limit of significance set by the software (p < 0.05). The first nine networks are shown.
Table II.
Changes in expression levels of ISG differently regulated after infection with the Eystrup strain of CSFV. The ISG listed were found differently regulated between the two CSFV strains. Eystrup (E) and Paderborn (P) gene expression values correspond to the mean Log2 ratio related to the non-infected reference. Bold data are two-class SAM significant.
| Gene name | GenBank accession number | Log2 ratio |
|||||
|---|---|---|---|---|---|---|---|
| D1 |
D2 |
D3 |
|||||
| E | P | E | P | E | P | ||
| Cell death/apotosis | |||||||
| IFIH1 | NM_001100194 | 1.630 | 0.126 | 2.304 | 1.658 | 1.993 | 1.545 |
| XAF1 | AK232641 | 1.058 | 0.244 | 2.183 | 1.315 | 1.696 | 1.485 |
| OAS1 | NM_214303 | 1.080 | 0.238 | 2.106 | 1.471 | 1.824 | 1.608 |
| IFI44 | AK233687 | 1.243 | 0.197 | 1.443 | 1.126 | 0.964 | 0.446 |
| NMI | AK234080 | 0.900 | −0.115 | 1.378 | 0.746 | 1.065 | 0.661 |
| CASP4 | AK231138 | 0.726 | −0.028 | 1.166 | 0.684 | 1.060 | 0.522 |
| CASP1 | NM_214162 | 0.948 | 0.206 | 0.804 | 0.915 | 1.020 | 0.678 |
| USP18 | NM_213826 | 0.851 | 0.021 | 0.649 | 0.712 | 0.781 | 0.535 |
| IFI16 | XM_001929212 | 0.530 | −0.052 | 0.632 | 0.451 | 0.481 | 0.228 |
| IRF7 | NM_001097428 | 0.477 | 0.081 | 0.478 | 0.503 | 0.572 | 0.305 |
| POMP | AK240274 | 0.178 | −0.301 | 0.393 | 0.188 | 0.343 | 0.170 |
| CD47 | NM_001025079 | 0.730 | −0.041 | 0.239 | 0.443 | 0.601 | 0.345 |
| ETS1 | NM_005238 | −0.181 | 0.344 | −0.306 | −0.063 | −0.079 | −0.254 |
| Antiviral response | |||||||
| ISG20 | NM_001005351 | 1.322 | 0.192 | 2.869 | 1.488 | 2.049 | 1.662 |
| GBP2 | NM_001128474 | 1.403 | 0.089 | 1.180 | 1.045 | 1.005 | 0.744 |
| GBP1 | NM_001128473 | 1.358 | 0.405 | 0.581 | 0.462 | 0.906 | 0.577 |
| CCL5 | NM_001129946 | −1.265 | −0.289 | −1.169 | −0.916 | −0.525 | −0.688 |
| Mobility/adhesion | |||||||
| SELL | NM_001112678 | 0.336 | 0.191 | 0.168 | 0.186 | 0.471 | 0.038 |
| CD9 | NM_214006 | −0.162 | 0.145 | −0.398 | 0.110 | −0.261 | −0.296 |
| VIM | AY368193 | −0.361 | 0.309 | −0.690 | −0.098 | −0.869 | −0.414 |
| Signaling | |||||||
| STAT2 | NM_213889 | 1.050 | 0.200 | 0.905 | 0.783 | 0.910 | 0.584 |
| JAK2 | NM_214113 | 0.553 | −0.018 | 0.039 | 0.092 | 0.481 | 0.161 |
| NCF2 | NM_001123142 | 0.359 | −0.107 | 0.211 | 0.291 | 0.495 | 0.128 |
| Antigen presentation | |||||||
| TAP2 | DQ227991 | 0.470 | 0.008 | 0.496 | 0.321 | 0.443 | 0.246 |
| SLA-DMb | NM_001113707 | 0.195 | 0.183 | −0.391 | 0.170 | −0.257 | −0.075 |
| SLA-DRa | NM_001113706 | −0.095 | 0.137 | −0.345 | 0.085 | −0.486 | −0.211 |
| Protein translation | |||||||
| WARS | EU715029 | 0.276 | 0.014 | 0.221 | 0.199 | 0.035 | 0.104 |
| EIF2S2 | XM_001929150 | 0.148 | −0.255 | 0.035 | −0.001 | 0.064 | −0.078 |
| Inflammatory response | |||||||
| AIF1 | XM_001928769 | 0.425 | −0.084 | 0.064 | 0.351 | 0.188 | 0.151 |
| Lymphocyte activation | |||||||
| CD69 | AF484233 | 0.710 | 0.279 | −0.030 | 0.012 | 0.605 | −0.423 |
| Unknown | |||||||
| IFIT3 | XM_001928703 | 1.663 | 0.370 | 2.723 | 2.045 | 2.459 | 1.779 |
3.3. The highly virulent CSFV strain induces immediate and strong overexpression of the ISG involved in cell death or apoptosis processes
Almost 50% of the ISG highlighted by the microarray were assigned a role in apoptosis or, more generally, in cell death induction and were qualified as “Cell death/apoptosis-related ISG” [5, 21]. We therefore focused on these genes to investigate if they might be responsible for the apoptosis induced in lymphocytes during CSFV infection. All 6 cell death/apoptosis-related ISG assessed by rRT-PCR were up-regulated from D1 to D3 pi with Eystrup and from D2 to D3 pi with Paderborn (Fig. 5). Significant differences between the two strains for all 6 genes were confirmed on D1 pi with higher Log2 ratios for Eystrup than for Paderborn. Therefore comparison of the microarray and rRT-PCR results confirmed the expression profiles. Thus, expression of the effector genes of the IFN response was considerably activated in pig PBMC after an in vivo CSFV infection. The level of activation was positively correlated with strain virulence.
Figure 5.

Real-time PCR validation of the microarray data, performed for 6 selected genes differently modulated by the Eystrup strain in relation to the Paderborn strain. The microarray results for Interferon induced with helicase C domain protein 1 (IFIH1) (A), Interferon gamma inducible protein 16 (IFI16) (B), X-linked inhibitor of apoptosis (XIAP) associated factor 1 (XAF1) (C), N-Myc interactor (NMI) (D), 2′,5′ oligoadenylate synthetase 1 (OAS1) (E) and Caspase-4 (CASP4) (F) are presented on A1-F1 whereas the RT-PCR results are presented on A2-F2. The microarray values are the mean (± SE) of relative gene expression (Log2 ratios) obtained for each post-infection value in relation to the non-infected reference. The presented PCR values are relative expressions calculated from the CT results on D1, D2 and D3 pi and related to D0, as described in Materials and methods. They correspond to the mean (± SE) of the results obtained for the 8 pigs infected by Eystrup or the 8 pigs infected by Paderborn. * p < 0.05 and ** p < 0.01 compared Eystrup values with Paderborn values using Two-sample Student’s t-test at each infection time-point.
3.4. ISG expression in PBMC is associated with a high serum level of IFNa
We then looked for an IFNa expression, the main antiviral IFN, in PBMC, that could be the origin of the cell death-related ISG up-regulations (Fig. 6A). A significant down-regulation was observed, compared to the basal expression level obtained in PBMC on D0, as early as D1 after the infection of pigs with the Eystrup strain, but only after D2 with the Paderborn strain. Down-regulation did not differ significantly between Eystrup and Paderborn during the 3 days post-infection.
Figure 6.
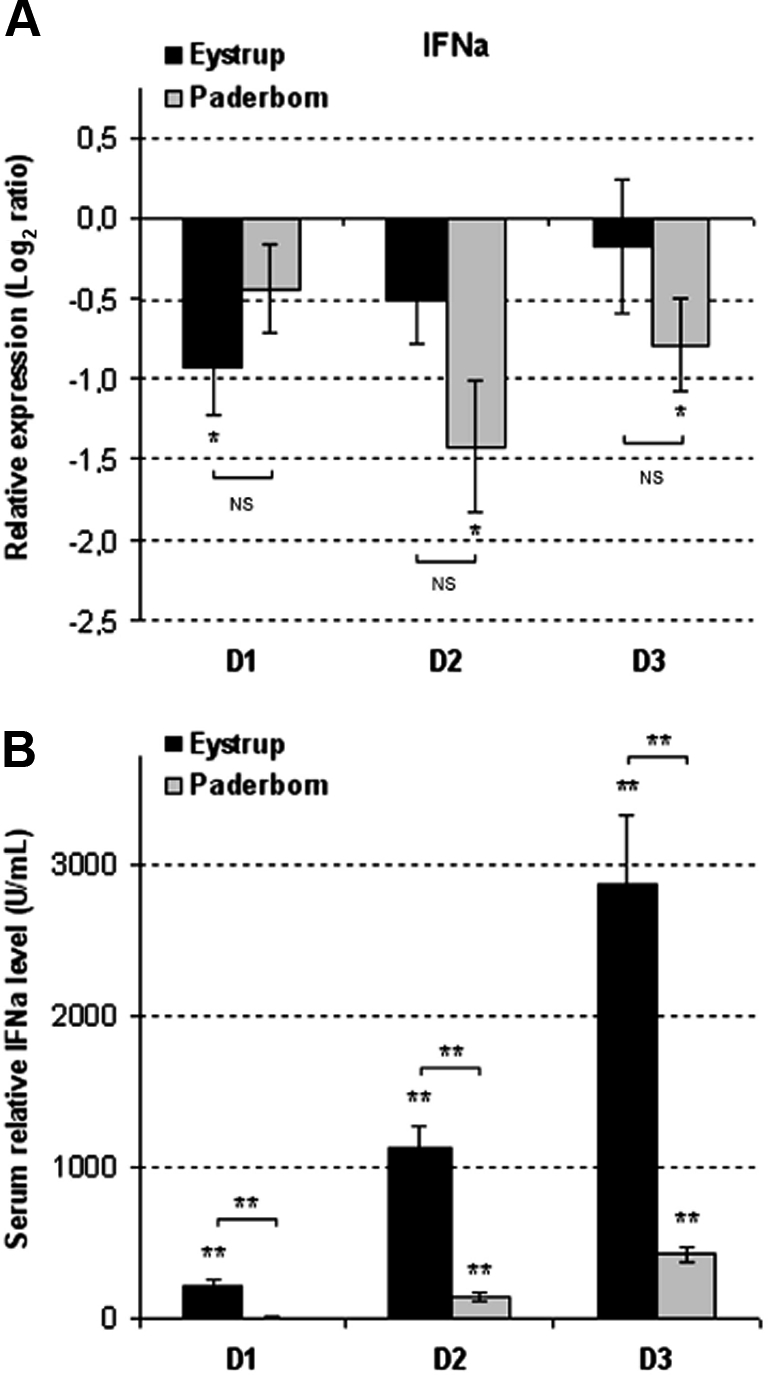
IFNa expression and production modulations in PBMC and sera induced by CSFV infection during the post-infection time-course. (A) IFNa expression assessed in PBMC by real-time RT-PCR. The presented values are the relative expression (Log2 Ratios) calculated from the CT results on D1, D2 and D3 pi in relation to D0 as described in Materials and methods. (B) IFNa levels in serum quantified by ELISA. The values are presented in relation to the levels quantified before infection. All data correspond to the mean (± SE) of results obtained for the 8 pigs infected by Eystrup or the 8 pigs infected by Paderborn. * p < 0.05 and ** p < 0.01 either compared each post-infection value with the respective D0 value using Paired Two-sample Student’s t-test, or Eystrup values with Paderborn values using Two-sample Student’s t-test at each infection time-point.
High levels of IFNa production during CSFV infection have however been previously described [13, 41], so we then looked for the secreted protein and its presence was confirmed in Eystrup-infected pig sera as early as day 1 post-infection (Fig. 6B). In pig sera infected with Paderborn, IFNa was detected only from D2, reaching on D3 a concentration seven times lower than with Eystrup (Fig. 6B). The serum IFNa concentrations for Eystrup and Paderborn differed significantly throughout the kinetics of infection and a one-day delay in IFNa detection was observed with Paderborn sera. Thus, the observed up-regulation of ISG could result from stimulation by the IFNa present in the environment of PBMC without an autocrine stimulation of these cells.
3.5. Different modulation of the death receptors-mediated apoptotic pathways according to CSFV virulence
An apoptotic pathway mediated by death receptors has already been suggested to induce apoptosis during CSFV infections [6, 39]. We therefore investigated the expression levels of the death receptors DR4 and FAS, and of their respective ligands Tumor necrosis factor related apoptosis inducing ligand (TRAIL) and Fas ligand (FASL). Although no significant variation for the TNFR1, the third type of death receptors (data not shown), was observed in our microarray results, we also included a detection of its ligand TNFa to complete the data.
The TRAIL-DR4 pathway showed a significant up-regulation of TRAIL by both CSFV strains over the 3 days of infection. Up-regulation with Eystrup was immediate and massive with the highest Log2 ratio obtained as early as D1. With Paderborn, the relative expression level rose slowly starting from a ratio of 0.89 on D1, reaching a ratio similar to that of Eystrup only on D3 (Fig. 7A). For DR4, the results showed an inverse regulation related to the CSFV strain with Eystrup displaying an immediate slight up-regulation on D1. Although non-significant, this up-regulation seems to try to be maintained on D2 and D3. Conversely Paderborn showed no variation in DR4 expression on D1 but a slight and progressive down-regulation from D2 to D3. Although this variation was small, the opposite regulation of DR4 by Eystrup and Paderborn strains was statistically significant (Fig. 7B).
Figure 7.

Variation in the expression level of death receptors and their ligands induced by CSFV infection. The expression of death receptors and their ligands was evaluated either at the RNA level (A–E) or at the protein level (F). (A–E) Expression modulations induced in PBMC by Eystrup and Paderborn strain infection were assessed by real-time RT-PCR. (A), (B) and (C) for the death receptor ligands: TRAIL, FASL and TNFa and (D) and (E) for their respective receptors: DR4 and FAS. The presented PCR values are the Log2 Ratios calculated from the CT results on D1, D2 and D3 pi and related to D0 as described in Materials and methods. (F) TNFa levels in serum were quantified by ELISA. Values are presented in relation to the levels quantified before infection. All data correspond to the mean (± SE) of results obtained for the 8 pigs infected by Eystrup or the 8 pigs infected by Paderborn. * p < 0.05 and ** p < 0.01 either compared each post-infection value with the respective D0 value using Paired Two-sample Student’s t-test, or Eystrup values with Paderborn values using Two-sample Student’s t-test at each infection time-point.
The ligand of the FASL-FAS pathway was also inversely regulated. This was only statistically validated on D2 post-infection although the trend was discernible on D1 (Fig. 7C). A similar modulation profile to that of TRAIL was observed for FAS, the up-regulation with Eystrup being observed as early as D1, but only significant from D2 onwards with Paderborn. However, the extent of modulation was much less pronounced for FAS than for TRAIL (FAS max Log2 ratio = 1.06 for Eystrup on D1 and 0.49 for Paderborn on D2) (Fig. 7D).
Concerning the TNFa-TNFR1 pathway, TNFa expression in the PBMC was enhanced higher and faster with Eystrup whereas no significant modulation of TNFa expression was observed over the 3 days of infection with Paderborn (Fig. 7E). These results were confirmed at the protein level (Fig. 7F).
Thus, TRAIL and FAS which are identified as ISG, displayed a similar modulation profile to the other cell death/apoptosis ISG revealed by microarray analysis. By contrast, the expression profile for non-ISG death receptors and ligands was different and the ISG and non-ISG could therefore be distinguished in relation to the modulations induced by the two CSFV strains, within the same route of death receptor-mediated apoptosis.
4. DISCUSSION
Due to the difficulties of reproducing in vitro some of the CSFV infection specificities observed in vivo such as the death of lymphocytes, the virus-host interactions in this study were investigated by transcriptomic analysis of PBMC isolated from pigs, infected either by the highly virulent Eystrup or the moderately virulent Paderborn strain of CSFV. The microarray results revealed that, despite a very strong host response toward Eystrup infection, with a large number of modulated genes and a limited number of genes in common with Paderborn infection, one of the main differences between the strains concerned the kinetics of the host response to the infection. The highly virulent Eystrup strain induced a strong and immediate host response whereas the moderately virulent Paderborn strain triggered a more progressive host response, one day later. Comparison of the host responses to Eystrup and Paderborn infections revealed that “immune response” and “cell death” were the main biological processes that were differently modulated between CSFV strains. Further analysis revealed that the IFN signaling was the main signaling pathway that differed between Eystrup and Paderborn. Several genes identified as IFN-stimulated genes and effectors of the IFN response were confirmed by rRT-PCR to be differently modulated between both strains.
The type I IFN response is usually associated with the host’s antiviral defenses but many viruses evade this defense by interfering either with its induction or its downstream effectors [8]. Silencing of type I IFN expression in in vitro CSFV-infected cells has been associated with the viral protein Npro which targets the proteasomal degradation of IRF3, an important inducer of type I IFN expression [2, 33]. However, a recent study has shown that impairment of the CSFV-induced IFN repression by Npro does not affect CSFV virulence in vivo, which indicates that the observed difference in virulence expression between CSFV strains is not dependent on their ability to use this virus protection mechanism [34]. No modulations of ISG, due to the silencing of type I IFN expression in infected cells, were revealed in vitro by either transcriptomic analyses in infected macrophages [4, 49] or proteomic analysis of infected PK15 [42]. However these ISG remained inducible in vitro following rIFNa treatment, demonstrating that CSFV does not target downstream effectors of the IFN response [37].
In our in vivo experiment, we demonstrated a strong up-regulation of ISG in PBMC and a down-regulation of IFNa in these same cells. However, very high levels of IFNa were detected in the sera from infected pigs, especially those infected with the highly virulent CSFV strain, as shown in previous studies [13, 41]. This IFNa serum production was correlated to viral RNA detection in blood and with ISG up-regulation (our results). In vivo, it has been shown that plasmacytoid dendritic cells (pDC) produce huge amounts of IFNa during CSFV infection [13] but that the number of circulating pDC decreases [41] and this could explain the IFNa reduced expression found in our PBMC. Summerfield and collaborators demonstrated a strong correlation between lymphopenia and the IFNa levels in sera from CSFV-infected pigs [41]. Our results confirm and extend this correlation to the up-regulation of ISG for both CSFV strains. Furthermore, the ISG involved in the cell death/apoptosis process are the ones most differently regulated between Eystrup and Paderborn, this difference in regulation matching the observed onset of lymphopenia with the two strains. Hence, the hypothesis established by Summerfield et al. in 2006 [41], then followed by Jamin et al. [13] and McCullough et al. [26], that type I IFN mediates CSFV-induced lymphopenia, is reinforced by our results and turned toward an apoptotic process as the most probable mechanism.
Type I IFN is known to promote the induction of apoptosis in infected cells [43], however IFNa has also been reported to induce death in various non-infected cells [14, 44]. For instance, the culture of a human lymphoma cell line with 250 U/mL of IFNa induces 40% of apoptosis after 48 h and 80% of apoptosis after 72 h [48]. During CSFV infection, it is well established that virus-induced apoptosis mainly occur in non-infected cells by an indirect mechanism also known as “bystander apoptosis” [6, 40].
This study sheds light on the CSFV-induced apoptosis in lymphocytes by identifying numerous up-regulated ISG, including the death ligand TRAIL and death receptor FAS, both involved in death receptors-mediated apoptosis pathways. An increase in FAS at the surface of lymphocytes isolated from CSFV-infected pigs has previously been described [39], suggesting that the FASL-FAS pathway is a mediator of CSFV-induced apoptosis and that this pathway could be a mechanism leading to bystander apoptosis [1]. In this study we confirmed the up-regulated expression of the ISG FAS in PBMC after CSFV infection of pigs and correlated this up-regulation at the onset of lymphopenia with the observed time lag between Eystrup and Paderborn. Interestingly, the expression of FASL also showed an opposite regulation between Eystrup and Paderborn, suggesting reduced possibilities of activation for this pathway during Paderborn’s infection. A type I IFN-mediated induction of apoptosis, independent of FASL-FAS, has already been described by McNally et al. in bystander CD8 T cells during T-cell response to some virus infections [27].
TRAIL-DR4 is another death receptor-mediated apoptotic pathway that has not been described with CSFV infection before. Our results suggest that due to the large and rapid up-regulation of TRAIL, this pathway could be of major significance in the induction of apoptosis. Several arguments corroborate this hypothesis. Some studies have shown that IFN-mediated apoptosis via TRAIL can occur in non-infected cells [30, 48]. During HIV-1 infection, the virus stimulates pDC to produce type I IFN and an up-regulation of TRAIL is observed in CD4+ T cells from infected patients, suggesting a cooperative role of pDC, IFNa and TRAIL in HIV-1 pathogenesis [10]. Similarly to CSFV, a study with the Ebola Virus, which also induces bystander killing lymphopenia, showed that a high proportion of cells expressed TRAIL rather than FAS and FASL suggesting that the TRAIL pathway could be preferentially involved during Ebola-induced lymphocyte cell death [9]. Lastly, X-linked inhibitor of apoptosis (XIAP) associated factor 1 (XAF1), shown by our results to be up-regulated during CSFV infection, is a crucial “cell death/apoptosis-related ISG” mediator of IFN-induced apoptosis via TRAIL [20, 28].
Interestingly, the expression of DR4, as for FASL, showed an opposite regulation between strains i.e., up-regulated by the highly virulent CSFV strain but down-regulated by the moderately virulent one. A resistance to TRAIL-induced apoptosis, due to a reduced expression of DR4, has been demonstrated in both normal and cancerous cells [17, 18]. The down-regulation of DR4 observed with the Paderborn infection, might result in a diminished sensitivity of PBMC to TRAIL-induced apoptosis, and might explain the reduced lymphopenia observed during Paderborn infection compared to that of Eystrup.
The induction of apoptosis can be mediated via three death receptor pathways. We have shown that the regulation of two of these pathways, both IFN-dependent, is altered during CSFV infection. TNFa-TNFR1, a third pathway reported to induce death receptors-mediated apoptosis, is not IFN-dependent. Given the high levels of TNFa produced in spleen macrophages [36], as well as dendritic cells and serum isolated from pigs infected with highly virulent CSFV strains [13], the TNFa-TNFR1 pathway has been suggested to be involved in the induction of CSFV apoptosis [6]. Our results cast some doubts on the relationship between TNFa production and lymphopenia. No significant induction of TNFa expression was observed on D2 with the moderately virulent strain Paderborn, whereas lymphopenia was clearly evident, and a low level of TNFa in the serum was found only on D3 pi and with no significant increase of lymphopenia compared to D2. Therefore TNFa is unlikely to be involved in the induction of apoptosis during Paderborn infection, although an effect of TNFa during the Eystrup infection, which is characterized by more severe lymphopenia, cannot be excluded and may be due to a synergic apoptotic effect of both IFNa and TNFa [24].
In conclusion, this comparison of two strains of different virulence, to analyze the modulations of gene expression induced by CSFV infection in vivo, reveals that type I IFN is the most probable inducer of lymphocyte depletion during CSFV infection. Thus, CSFV could subvert the host’s immune system, “turning the good IFN bad” as does HIV-1 in the bystander killing of non-infected lymphocytes [11]. This immunopathogenic mechanism also seems to be used by other leucopenic viruses, such as Dengue virus, African swine fever virus and Ebola virus but ISG induction is generally reduced in these viruses, unlike CSFV, by highly virulent strains compared to those of low virulence, suggesting that CSFV causes a variant IFN response subversion [7, 16, 46]. Some studies have shown that type I IFN can also induce lymphopenia by modifying the distribution of lymphocytes either by promoting their retention in lymphoid organs [38], or by increasing their adhesion and egress from the blood vessels [15]. Our microarray results do not rule out a possible involvement of additional processes, other than apoptosis, in CSFV-induced lymphopenia.
Online material
Supplementary online material provided by the author.
Acknowledgments
This work was supported by grants from the Conseil Régional de Bretagne including Fonds Social Européen, (211-B2-9/2005/ARED/PPCVIRUL) for the Patricia Renson PhD fellowship and by the EU Network of Excellence, EPIZONE (Contract No. FOOD-CT-2006-016236). We thank the Biogenouest transcriptomic platform for help with treatment of the microarray data. We are grateful to Bruno Jan and André Kéranflec’h for animal care and blood sampling, Beatrice Grasland for her advice on PCR assays and primer design, and Stephanie Bougeard for hers on statistical analysis of PCR data. We also thank Philippe Vannier for his support all along this study.
Footnotes
References
- 1.Badley A.D., McElhinny J.A., Leibson P.J., Lynch D.H., Alderson M.R., Paya C.V., Upregulation of Fas ligand expression by human immunodeficiency virus in human macrophages mediates apoptosis of uninfected T lymphocytes, J. Virol. (1996) 70:199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bauhofer O., Summerfield A., Sakoda Y., Tratschin J.D., Hofmann M.A., Ruggli N., Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation, J. Virol. (2007) 81:3087–3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bensaude E., Turner J.L., Wakeley P.R., Sweetman D.A., Pardieu C., Drew T.W., et al. , Classical swine fever virus induces proinflammatory cytokines and tissue factor expression and inhibits apoptosis and interferon synthesis during the establishment of long-term infection of porcine vascular endothelial cells, J. Gen. Virol. (2004) 85:1029–1037 [DOI] [PubMed] [Google Scholar]
- 4.Borca M.V., Gudmundsdottir I., Fernandez-Sainz I.J., Holinka L.G., Risatti G.R., Patterns of cellular gene expression in swine macrophages infected with highly virulent classical swine fever virus strain Brescia, Virus Res. (2008) 138:89–96 [DOI] [PubMed] [Google Scholar]
- 5.Chawla-Sarkar M., Lindner D.J., Liu Y.F., Williams B.R., Sen G.C., Silverman R.H., Borden E.C., Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis, Apoptosis (2003) 8:237–249 [DOI] [PubMed] [Google Scholar]
- 6.Choi C., Hwang K.K., Chae C., Classical swine fever virus induces tumor necrosis factor-alpha and lymphocyte apoptosis, Arch. Virol. (2004) 149:875–889 [DOI] [PubMed] [Google Scholar]
- 7.Gil S., Sepulveda N., Albina E., Leitao A., Martins C., The low-virulent African swine fever virus (ASFV/NH/P68) induces enhanced expression and production of relevant regulatory cytokines (IFNalpha, TNFalpha and IL12p40) on porcine macrophages in comparison to the highly virulent ASFV/L60, Arch. Virol. (2008) 153:1845–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haller O., Kochs G., Weber F., Interferon, Mx, and viral countermeasures, Cytokine Growth Factor Rev. (2007) 18:425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hensley L.E., Young H.A., Jahrling P.B., Geisbert T.W., Proinflammatory response during Ebola virus infection of primate models: possible involvement of the tumor necrosis factor receptor superfamily, Immunol. Lett. (2002) 80:169–179 [DOI] [PubMed] [Google Scholar]
- 10.Herbeuval J.P., Hardy A.W., Boasso A., Anderson S.A., Dolan M.J., Dy M., Shearer G.M., Regulation of TNF-related apoptosis-inducing ligand on primary CD4+ T cells by HIV-1: role of type I IFN-producing plasmacytoid dendritic cells, Proc. Natl. Acad. Sci. USA (2005) 102:13974–13979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herbeuval J.P., Shearer G.M., HIV-1 immunopathogenesis: how good interferon turns bad, Clin. Immunol. (2007) 123:121–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jamin A., Gorin S., Le Potier M.F., Kuntz-Simon G., Characterization of conventional and plasmacytoid dendritic cells in swine secondary lymphoid organs and blood, Vet. Immunol. Immunopathol. (2006) 114:224–237 [DOI] [PubMed] [Google Scholar]
- 13.Jamin A., Gorin S., Cariolet R., Le Potier M.F., Kuntz-Simon G., Classical swine fever virus induces activation of plasmacytoid and conventional dendritic cells in tonsil, blood, and spleen of infected pigs, Vet. Res. (2008) 39:7. [DOI] [PubMed] [Google Scholar]
- 14.Jiang J., Gross D., Nogusa S., Elbaum P., Murasko D.M., Depletion of T cells by type I interferon: differences between young and aged mice, J. Immunol. (2005) 175:1820–1826 [DOI] [PubMed] [Google Scholar]
- 15.Kamphuis E., Junt T., Waibler Z., Forster R., Kalinke U., Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia, Blood (2006) 108:3253–3261 [DOI] [PubMed] [Google Scholar]
- 16.Kash J.C., Muhlberger E., Carter V., Grosch M., Perwitasari O., Proll S.C., et al. , Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence, J. Virol. (2006) 80:3009–3020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim K., Fisher M.J., Xu S.Q., el-Deiry W.S., Molecular determinants of response to TRAIL in killing of normal and cancer cells, Clin. Cancer Res. (2000) 6:335–346 [PubMed] [Google Scholar]
- 18.Kurbanov B.M., Fecker L.F., Geilen C.C., Sterry W., Eberle J., Resistance of melanoma cells to TRAIL does not result from upregulation of antiapoptotic proteins by NF-kappaB but is related to downregulation of initiator caspases and DR4, Oncogene (2007) 26:3364–3377 [DOI] [PubMed] [Google Scholar]
- 19.Le Meur N., Lamirault G., Bihouee A., Steenman M., Bedrine-Ferran H., Teusan R., et al. , A dynamic, web-accessible resource to process raw microarray scan data into consolidated gene expression values: importance of replication, Nucleic Acids Res. (2004) 32:5349–5358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leaman D.W., Chawla-Sarkar M., Vyas K., Reheman M., Tamai K., Toji S., Borden E.C., Identification of X-linked inhibitor of apoptosis-associated factor-1 as an interferon-stimulated gene that augments TRAIL Apo2L-induced apoptosis, J. Biol. Chem. (2002) 277:28504–28511 [DOI] [PubMed] [Google Scholar]
- 21.Leaman D.W., Chawla-Sarkar M., Jacobs B., Vyas K., Sun Y., Ozdemir A., et al. , Novel growth and death related interferon-stimulated genes (ISGs) in melanoma: greater potency of IFN-beta compared with IFN-alpha2, J. Interferon Cytokine Res. (2003) 23:745–756 [DOI] [PubMed] [Google Scholar]
- 22.Lee W.C., Wang C.S., Chien M.S., Virus antigen expression and alterations in peripheral blood mononuclear cell subpopulations after classical swine fever virus infection, Vet. Microbiol. (1999) 67:17–29 [DOI] [PubMed] [Google Scholar]
- 23.Livak K.J., Schmittgen T.D., Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method, Methods (2001) 25:402–408 [DOI] [PubMed] [Google Scholar]
- 24.Manna S.K., Mukhopadhyay A., Aggarwal B.B., IFN-alpha suppresses activation of nuclear transcription factors NF-kappa B and activator protein 1 and potentiates TNF-induced apoptosis, J. Immunol. (2000) 165:4927–4934 [DOI] [PubMed] [Google Scholar]
- 25.Mayer D., Hofmann M.A., Tratschin J.D., Attenuation of classical swine fever virus by deletion of the viral N(pro) gene, Vaccine (2004) 22:317–328 [DOI] [PubMed] [Google Scholar]
- 26.McCullough K.C., Ruggli N., Summerfield A., Dendritic cells-At the front-line of pathogen attack, Vet. Immunol. Immunopathol. (2009) 128:7–15 [DOI] [PubMed] [Google Scholar]
- 27.McNally J.M., Zarozinski C.C., Lin M.Y., Brehm M.A., Chen H.D., Welsh R.M., Attrition of bystander CD8 T cells during virus-induced T-cell and interferon responses, J. Virol. (2001) 75:5965–5976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Micali O.C., Cheung H.H., Plenchette S., Hurley S.L., Liston P., LaCasse E.C., Korneluk R.G., Silencing of the XAF1 gene by promoter hypermethylation in cancer cells and reactivation to TRAIL-sensitization by IFN-beta, BMC Cancer (2007) 7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moennig V., Floegel-Niesmann G., Greiser-Wilke I., Clinical signs and epidemiology of classical swine fever: a review of new knowledge, Vet. J. (2003) 165:11–20 [DOI] [PubMed] [Google Scholar]
- 30.Oshima K., Yanase N., Ibukiyama C., Yamashina A., Kayagaki N., Yagita H., Mizuguchi J., Involvement of TRAIL/TRAIL-R interaction in IFN-alpha-induced apoptosis of Daudi B lymphoma cells, Cytokine (2001) 14:193–201 [DOI] [PubMed] [Google Scholar]
- 31.Risatti G.R., Borca M.V., Kutish G.F., Lu Z., Holinka L.G., French R.A., et al. , The E2 glycoprotein of classical swine fever virus is a virulence determinant in swine, J. Virol. (2005) 79:3787–3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Risatti G.R., Holinka L.G., Lu Z., Kutish G.F., Tulman E.R., French R.A., et al. , Mutation of E1 glycoprotein of classical swine fever virus affects viral virulence in swine, Virology (2005) 343:116–127 [DOI] [PubMed] [Google Scholar]
- 33.Ruggli N., Bird B.H., Liu L., Bauhofer O., Tratschin J.D., Hofmann M.A., N(pro) of classical swine fever virus is an antagonist of double-stranded RNA-mediated apoptosis and IFN-alpha/beta induction, Virology (2005) 340:265–276 [DOI] [PubMed] [Google Scholar]
- 34.Ruggli N., Summerfield A., Fiebach A.R., Guzylack-Piriou L., Bauhofer O., Lamm C.G., et al. , Classical swine fever virus can remain virulent after specific elimination of the interferon regulatory factor 3-degrading function of Npro, J. Virol. (2009) 83:817–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sainz I.F., Holinka L.G., Lu Z., Risatti G.R., Borca M.V., Removal of a N-linked glycosylation site of classical swine fever virus strain Brescia Erns glycoprotein affects virulence in swine, Virology (2008) 370:122–129 [DOI] [PubMed] [Google Scholar]
- 36.Sanchez-Cordon P.J., Nunez A., Salguero F.J., Pedrera M., Fernandez de Marco M., Gomez-Villamandos J.C., Lymphocyte apoptosis and thrombocytopenia in spleen during classical swine fever: role of macrophages and cytokines, Vet. Pathol. (2005) 42:477–488 [DOI] [PubMed] [Google Scholar]
- 37.Seago J., Hilton L., Reid E., Doceul V., Jeyatheesan J., Moganeradj K., et al. , The Npro product of classical swine fever virus and bovine viral diarrhea virus uses a conserved mechanism to target interferon regulatory factor-3, J. Gen. Virol. (2007) 88:3002–3006 [DOI] [PubMed] [Google Scholar]
- 38.Shiow L.R., Rosen D.B., Brdickova N., Xu Y., An J., Lanier L.L., et al. , CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs, Nature (2006) 440:540–544 [DOI] [PubMed] [Google Scholar]
- 39.Summerfield A., Knotig S.M., McCullough K.C., Lymphocyte apoptosis during classical swine fever: implication of activation-induced cell death, J. Virol. (1998) 72:1853–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Summerfield A., McNeilly F., Walker I., Allan G., Knoetig S.M., McCullough K.C., Depletion of CD4(+) and CD8(high+) T-cells before the onset of viraemia during classical swine fever, Vet. Immunol. Immunopathol. (2001) 78:3–19 [DOI] [PubMed] [Google Scholar]
- 41.Summerfield A., Alves M., Ruggli N., de Bruin M.G., McCullough K.C., High IFN-alpha responses associated with depletion of lymphocytes and natural IFN-producing cells during classical swine fever, J. Interferon Cytokine Res. (2006) 26:248–255 [DOI] [PubMed] [Google Scholar]
- 42.Sun J., Jiang Y., Shi Z., Yan Y., Guo H., He F., Tu C., Proteomic alteration of PK-15 cells after infection by classical swine fever virus, J. Proteome Res. (2008) 7:5263–5269 [DOI] [PubMed] [Google Scholar]
- 43.Tanaka N., Sato M., Lamphier M.S., Nozawa H., Oda E., Noguchi S., et al. , Type I interferons are essential mediators of apoptotic death in virally infected cells, Genes Cells (1998) 3:29–37 [DOI] [PubMed] [Google Scholar]
- 44.Thyrell L., Erickson S., Zhivotovsky B., Pokrovskaja K., Sangfelt O., Castro J., et al. , Mechanisms of Interferon-alpha induced apoptosis in malignant cells, Oncogene (2002) 21:1251–1262 [DOI] [PubMed] [Google Scholar]
- 45.Tusher V.G., Tibshirani R., Chu G., Significance analysis of microarrays applied to the ionizing radiation response, Proc. Natl. Acad. Sci. USA (2001) 98:5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ubol S., Masrinoul P., Chaijaruwanich J., Kalayanarooj S., Charoensirisuthikul T., Kasisith J., Differences in global gene expression in peripheral blood mononuclear cells indicate a significant role of the innate responses in progression of dengue fever but not dengue hemorrhagic fever, J. Infect. Dis. (2008) 197:1459–1467 [DOI] [PubMed] [Google Scholar]
- 47.Wang Y., Wang Q., Lu X., Zhang C., Fan X., Pan Z., et al. , 12-nt insertion in 3′ untranslated region leads to attenuation of classic swine fever virus and protects host against lethal challenge, Virology (2008) 374:390–398 [DOI] [PubMed] [Google Scholar]
- 48.Yanase N., Kanetaka Y., Mizuguchi J., Interferon-alpha-induced apoptosis via tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-dependent and -independent manner, Oncol. Rep. (2007) 18:1031–1038 [PubMed] [Google Scholar]
- 49.Zaffuto K.M., Piccone M.E., Burrage T.G., Balinsky C.A., Risatti G.R., Borca M.V., et al. , Classical swine fever virus inhibits nitric oxide production in infected macrophages, J. Gen. Virol. (2007) 88:3007–3012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary online material provided by the author.