

Abstract
Among the most exciting recent developments in structural biology is the structure determination of G-protein-coupled receptors (GPCRs), which comprise the largest class of membrane proteins in mammalian cells and have enormous importance for disease and drug development. The GPCR structures are perhaps the most visible examples of a nascent revolution in membrane protein structure determination. Like other major milestones in science, however, such as the sequencing of the human genome, these achievements were built on a hidden foundation of technological developments. Here, we describe some of the methods that are fueling the membrane protein structure revolution and have enabled the determination of the current GPCR structures, along with new techniques that may lead to future structures.
Keywords: membrane protein, crystallization, expression, bicells, lipid cubic phase
Introduction
Membrane protein structural biology is in the midst of a fledgling revolution reflected in the ∼180 solved membrane protein structures as of 2008—a number that is increasing exponentially (http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html).1 We now have more detailed views of remarkable molecular feats such as how a protein gets across the membrane,2–4 how a channel moves water with nary a proton escaping,5 and how a potassium ion passes across a membrane with amazing efficiency and selectivity6,7; to name just a few of the accomplishments in the past decade. Among the most important recent achievements is the determination of now four G-protein-coupled receptor (GPCR) structures. The first breakthrough was the structure of rhodopsin in 2000,8 followed 8 years later by opsin, our best model for the activated form of a GPCR.9,10 We had to wait until 2007 to see the first structure of a ligand-activated GPCR with the successful structure determination of a β2-adrenergic receptor structure.11,12 The β1-adrenergic receptor13 and an adenosine receptor14 structure followed in the next year.
Many different tricks were used to achieve the stability and conformational homogeneity required for GPCR crystallization including the use of antibody complexes,12 fusion proteins,11,14 tight binding ligands,10–14 stabilizing mutants,13 and special crystallization environments such as the lipid cubic phase11,14 and bicelles.12 The diversity of strategies used illustrates how the membrane protein structure revolution, like the soluble protein structure revolution before it, is being fed not by a transcendent breakthrough, but by the gradual accrual of new techniques that can be deployed where needed. Clearly, membrane protein structure determination is supported by all the developments in crystallography such as microfocus beamlines, sensitive detectors, automation, and a vast array of improved software, to name just a few, but here we focus on some of the tricks used specifically for the GPCR structures and some promising new protein engineering methods that may yield fruit in the future.
Three different crystallization methods were used for GPCR structures
Membrane proteins have evolved to function in a membrane environment, so it is not surprising that they are often not very stable when solubilized in detergents. Membrane proteins that can maintain structural integrity in micelles can be crystallized directly in detergent. As crystallization from detergent is straightforward using standard methods, it is by far the most popular way to crystallize membrane proteins. This method was used for rhodopsin, opsin, and for a stabilized variant of the β1-adrenergic receptor (see latter). It is nevertheless reasonable to assume that a more ideal medium from the membrane protein's perspective is a bilayer. It is also clear that structures can be altered by detergent extraction and by variations in bilayer properties so obtaining structures reflecting conformations in true membranes remains the ultimate goal.
Landau and Rosebusch15 achieved a major conceptual breakthrough by developing a method to crystallize from bilayers using the lipid cubic phase (see Martin Caffrey's comprehensive review16). In the lipid cubic phase, the bilayer is bent and organized into a lattice to make it continuous in three dimensions [Fig. 1(A)]. Thus, the formation of protein crystal nuclei can be fed by diffusion of protein from the rest of the lattice. The lipid cubic phase is extremely viscous and hard to manipulate, so special techniques are required to set up trials, but it has clearly proven worthy of the extra effort. The approach yielded the first high-resolution structure of bacteriorhodopsin17,18 and has been successfully used on ∼10 distinct proteins, culminating in one of the β2-adrenergic receptor structures11 and the adenosine receptor structure.14 Technology developments, largely from Martin Caffrey's group, were important contributors. In particular, additives were used to swell the lattice and facilitate the diffusion of larger proteins in the lipid mesophase,19 and a specialized robot was used for crystallization trial setups.20 Further developments, such as microfluidic devices, will continue contributing to the popularity and success of the method.
Figure 1.
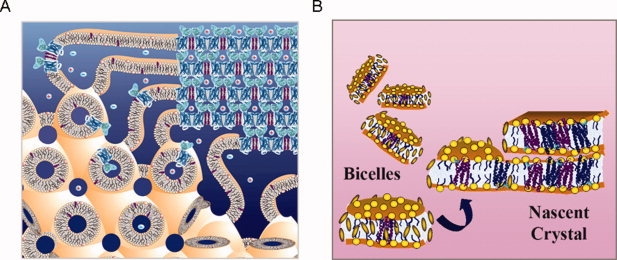
Alternative crystallization methods. (A) The in meso or lipid cubic phase method: The lipid cubic phase is illustrated toward the lower left and a possible mechanism of crystal formation is illustrated in the upper right. This figure was provided by Martin Caffrey. (B) The bicelle method: Bicelles are illustrated at the left of the figure. Lipid head groups are indicated by the yellow balls and the small brown oval disks are intended to represent small amphiphiles that protect the hydrophobic edges. A possible mechanism of crystal formation is illustrated at the right. For both methods, the mechanism of crystal formation is not known in detail, although both methods yield layered crystals reminiscent of a lamellar phase and both formulations can convert into a lamellar phase.
To avoid difficulties in working with the viscous lipid cubic phase, we tested whether bicelle formulations could be used to crystallize membrane proteins21,22 (reviewed in ref.23). Bicelles are bilayer discs that form in certain lipid amphiphile mixtures [Fig. 1(B)]. Protein can be readily incorporated into the bicelles where they will experience a bilayer environment.24,25 A major advantage of the bicelle method is that the solution is easily pipetted so that standard crystallization methods can be used. The phase behavior of bicelle mixtures can be complex.26–28 At higher temperatures, a perforated lamellar phase can form which may be an auspicious organization for crystal formation.29 Indeed, it is thought that lamellar phase formation occurs during lipid cubic phase crystallization.16 We successfully crystallized bacteriorhodopsin from a number of simple bicelle formulations21,22 and the method has now been used to determine the structures of three additional proteins, including one of the β2-adrenergic receptor structures.12,30,31 An aspect of both the lipid cubic phase method and the bicelle method that has yet to be fully explored is the range of lipid formulations that could be used. For example, cholesterol was an important additive used for the adenosine receptor structure. The bicelle method could be particularly flexible in this regard. Perhaps E. coli lipid compositions could be used for E. coli proteins and mammalian lipid compositions used for mammalian proteins. Whether bicelles could be formed with these alternative formulations needs investigation.
A variety of soluble domain additions
Crystal contacts in membrane protein crystals are often formed largely by soluble domains, particularly when crystallized from detergent. Thus, an early idea for improving the crystallizability of membrane proteins was to add additional soluble domains.32 A particularly effective, albeit difficult, way to do this is by developing monoclonal antibodies against the folded protein.33 In addition to adding a stable soluble domain to the complex, the antibody can stabilize and lock the protein in a unique conformation. Michel and coworkers33 first successfully used this approach to determine the structure of cytochrome c oxidase using a complex with an Fv fragment. Mackinnon's group then determined a high-resolution structure of the KcsA potassium channel using an Fab fragment,34 which is easier to obtain than the Fv fragment, an approach used for a number of subsequent structures including the β2-adrenergic receptor.12 While an ideal approach in principle, the effort required to obtain monoclonal antibodies is a major limitation. Methods to rapidly obtain antibodies using phage display approaches could greatly facilitate and broaden the use of the method.35,36 An additional concern is the possibility of locking the protein in a non-native conformation. Thus, Kobilka carefully selected an antibody that did not substantially alter antagonist binding for the crystallization of the β2-adrenergic receptor.37
Another key approach developed in the Kobilka laboratory was the introduction of T4 lysozyme into a loop of β2-adrenergic receptor (see Fig. 2),38 a method that was also used in the crystallization of the adenosine receptor.14 Fusion of a crystallizable fusion protein has assisted the crystallization of a number of small soluble proteins.39–41 A problem with the approach is that if the linkage between the two proteins is floppy, it can introduce additional flexibility which is deleterious to crystallization. A possible solution to this problem for membrane proteins is to insert the soluble domain between transmembrane loops, an idea first implemented by Prive et al.32 This is the approach used by the Kobilka group.38 They replaced the third intracellular loop on β2-adrenergic receptor with T4 lysozyme to diminish the conformational flexibility in the loop and also in an effort to reduce larger motions in the protein as a whole. With the success of the method on two GPCR structures,11,14 other similar attempts are sure to follow.
Figure 2.
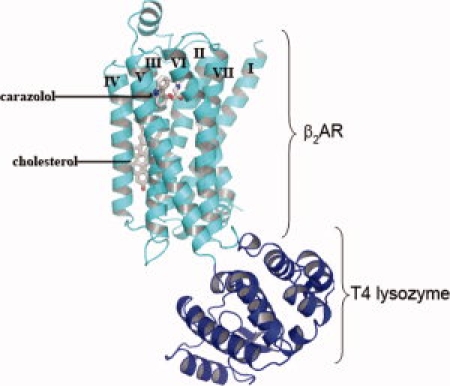
β2 adrenergic receptor structure with inserted T4 lysozyme domain. T4 lysozyme (dark blue) was inserted in the loop between TM segments 5 and 6 of the β2 adrenergic receptor (light blue) to aid crystallization by adding potential crystal contacts and reducing conformational heterogeneity. The bound antagonist, carazolol, and cholesterol are shown in stick representation. An interactive view is available in the electronic version of the article.
Stabilizing mutants
When stabilizing ligands or solution conditions are insufficient to produce a protein robust enough for crystallization, it may be possible to engineer the protein for increased stability. Indeed a number of GPCR structures utilized stabilizing mutations.13,42 The T4 lysozyme insertions discussed earlier improved the behavior of the receptors, but more subtle point mutants can also have a dramatic effect.
We discovered that stabilizing mutations can be quite common in membrane proteins, making it possible to create highly stable variants. In characterizing a library of cysteine substitutions in diacylglycerol kinase, we were surprised to find that about 1 in 10 greatly improved stability to thermal inactivation.43 We therefore screened a small library of random mutations and identified a collection of stabilizing mutations.44 Four mutations could be combined to yield a superstable variant that increased the half-life in octylglucoside from 6 min at 55°C to 35 min at 80°C. The stable mutants were useful for determining the NMR structure of the enzyme (Sanders, personal communication). A stable, conformationally locked Cys mutant was also important for the first crystals of lac permease.45,46
A similar approach was taken by Tate and coworkers47 to identify thermostable variants of the β1-adrenergic receptor. An impressive 318 positions were changed to alanine (or leucine if the wild-type residue was already alanine), expressed in E. coli and tested for thermal inactivation in detergent solution. E. coli expression was made possible by another technology development from Reinhardt Grisshammmer who showed that maltose binding protein fusions assist with GPCR expression.48 Of the 318 substitutions, 17 improved stability. Additional substitutions were tested at all 17 of these positions to look for more stable variants and 5 positions yielded better substitutions. The single mutants were combined ultimately into an active receptor variant with dramatically improved stability containing six mutations. The engineered thermostable protein was used in structure determination. The positions of the stabilizing point mutations are shown on the structure in Figure 3.
Figure 3.
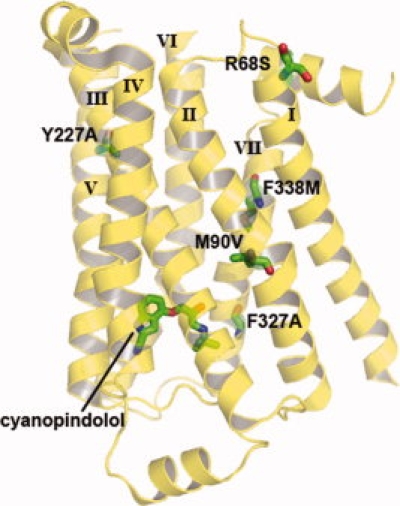
Stabilizing mutations introduced into the β1 adrenergic receptor. Tate and coworkers screened 318 positions of the β1 receptor for stabilizing mutations. Single mutants were combined to give an active receptor with six mutations: R68S, M90V, Y227A, A282L, F327A, and F338M. The hextuple mutant increased the Tm by 21° and ultimately enabled the crystallization and structure determination of the receptor. The bound antagonist, cyanopindolol, and the stabilizing mutations are shown in stick representation, with the exception of A282L because it is in a loop region that was unresolved in the crystal structure. An interactive view is available in the electronic version of the article.
The Tate group extended this work to the adenosine A2a receptor, but in this case they have stabilized both the antagonist and agonist binding conformations.49 The latter holds promise for obtaining crystals of the agonist bound form, which would finally provide detailed information on at least one of the major conformational changes that occur in GPCR signaling.
Not all targets are amenable to large scale activity screening so it would be useful to have stability screens that are not activity dependent and can be performed in crude extract. No techniques applicable to membrane proteins exist to our knowledge.
More rational protein engineering approaches
Another way to obviate the need to screen hundreds of mutants would be to develop more targeted approaches to stabilization. The structural information now available for GPCRs can facilitate designed stabilization of other members of the family.
The Oprian group has successfully engineered stabilizing disulfide bonds into rhodopsin and opsin, greatly improving stability without large alterations in their conformations.50,51 A disulfide crosslinked rhodopsin was sufficiently stable that the protein could be heterologously expressed and purified and yielded a crystal structure.42 There is no reason a similar approach could not be used for other unstable GPCRs (and other proteins), to the extent that reasonable homology models can be built for disulfide design. A clear advantage of this strategy is that the protein can be conformationally locked, which should improve crystal formation.
The Stevens group has utilized a more targeted mutant screen to stabilize the β2-adrenergic receptor.52 They recognized that a glutamate residue in TM3 of the β2-adrenergic receptor, which projected into the interface of TM3 with TM4 and TM5, was most commonly a large hydrophobic in other family members. They therefore reasoned correctly that changing the Glu to a large hydrophobic might stabilize the protein. The concept of changing non-consensus residues to consensus residues has been successful in stabilizing soluble proteins53 and should perhaps be tested more extensively for membrane proteins, given the success with the β2-adrenergic receptor.
Recently, Baneres and coworkers54 presented an entirely new way to engineer stability and conformational rigidity into a GPCR. In this work, a Zn2+ binding site was introduced into the leukotriene receptor, BLT1, by the introduction of three histidine residues predicted to be close in space based on a homology model (see Fig. 4). The Zn2+ binding site was placed so that it would bridge the likely flexible interface between TM3 and TM6. The engineered receptor bound Zn2+ with 8 μM affinity. The presence of Zn2+ dramatically stabilized the receptor and locked it in the ground state conformation. The potential of this technique for stabilizing GPCRs for crystallization and locking them in different functional conformations appears great.
Figure 4.
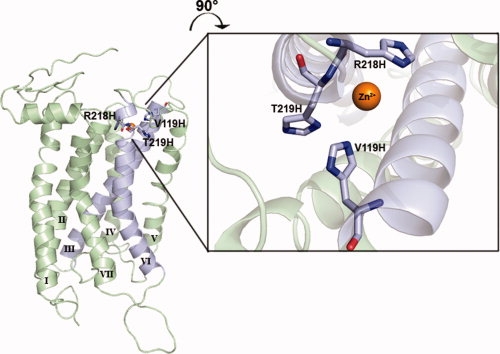
Stabilization of the leukotriene receptor, BLT1, using an engineered metal binding site. To restrain transmembrane helices 3 and 6 (light blue), Baneres and coworkers engineered a Zn2+ binding site by introducing three His residues at positions predicted to be close in space.54 One residue in TM3, Val119, and two residues in TM6, Arg218, and Thr219 were selected from the BLT1 model to mutate to histidine. The figure shows a model with the three side chains replaced with histidines for illustration only. It is not intended to be an accurate representation of the binding site as no energy optimization was performed. Baneres generously provided the BLT1 homology model coordinates used for this figure. An interactive view is available in the electronic version of the article.
Expression
High-level expression is an extremely important factor in structure determination. In this regard, the current GPCR structures did not make use of any particular innovations. Rhodopsin was obtained from a naturally abundant source and the rest were expressed using baculovirus systems. Many GPCRs, like other membrane proteins, are not easily expressed, however, so deeper penetration into the family will require developments in this area.55
Although eukaryotic expression systems will continue to be utilized as they can provide a more natural milieu for GPCRs, E. coli seems particularly promising as many GPCRs can already be expressed in their active form in E. coli.48,56–59 Expression of unmodified GPCRs in E. coli is generally very low, so various modifications are often used. For example, Grishammer showed greatly improved expression of the neurotensin receptor by fusion to periplasmically expressed maltose binding protein. This approach was used by the Tate group in screening for GPCRs with improved stability. A mistic fusion was also found to help GPCR expression.60
In an exciting development, the Pluckthun group found that point mutants within the GPCR itself can greatly improve expression and can simultaneously improve stability.59 They developed a screen for functional expression of the neurotensin receptor-1 by using a tight-binding fluorescently labeled ligand so that E. coli cells expressing functional receptor could be selected by fluorescence activated cell sorting. They then used rounds of enrichment of randomly mutagenized receptors, retaining cells with the highest levels of fluorescence. Remarkably, the best sequence not only improved expression in E. coli ∼10-fold, but also improved expression ∼3-fold in mammalian cells. The evolved receptor also had improved stability in detergent. These results suggest that low expression may be at least partially a result of poor stability or folding. Consistent with this view, stabilized rhodopsin could also be heterologously expressed.42 It remains to be seen whether this is a general phenomenon, however, and whether it translates to other membrane proteins. A great advantage of this technique is that functionality can be retained and optimized, albeit only the function selected for. For example, one residue in neurotensin receptor-1 had to be restored to maintain full signaling.59 This should be a powerful approach for improving the properties of GPCRs and other membrane proteins, as long as a rapid screen can be developed.
The E. coli host can also be engineered to improve expression of particular proteins.57,58,61,62 Recently, Georgiou and coworkers57,58 were able to improve expression of a canabanoid receptor by overexpression of a protease FtsH and by a Tn5 insertion into DnaJ. Perhaps additional engineering can yield further E. coli strains optimized for GPCR expression and folding.
Another promising development is cell free expression, which can obviate such problems as mRNA degradation, toxicity of high-level protein production or proteolysis of the expressed protein. There are now numerous demonstrations of high-level cell free expression of membrane proteins, including GPCRs, and refolding and reconstitution of activity for many.63–65 A key question for structural studies is what fraction of the protein remains in an altered or inactive conformation and how much conformational impurity can be tolerated for crystallization trials. There is now one case of a cell free expressed membrane protein, EmrE, that was sufficiently homogenous to yield diffraction quality crystals.66 Techniques for efficient refolding could ultimately make this approach the method of choice for membrane protein production.
Conclusion
While many tricks have been developed to tackle difficult structural targets like GPCRs, membrane protein crystallography is still not a job for the faint of heart. There is no sure-fire, or even likely, road to success and few of the options are easy. Nevertheless, it is always nice to have validated choices. Continued method development must therefore be a priority. So far all the GPCR structures have been obtained by crystallography. Solution and solid state NMR is starting to make a significant dent, however, and it appears that GPCR-sized proteins may become accessible to these techniques.67–69 For both crystallography and NMR, the key elements that can pave the way to a structure are high-level expression, protein stability, and conformational homogeneity. New methods that can attack these fundamental issues more reliably, more generally and with greater ease of implementation will continue to build the membrane protein structure revolution.
References
- 1.White SH. The progress of membrane protein structure determination. Protein Sci. 2004;13:1948–1949. doi: 10.1110/ps.04712004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van den Berg B, Clemons WM, Jr, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA. X-ray structure of a protein-conducting channel. Nature. 2004;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- 3.White SH, von Heijne G. How translocons select transmembrane helices. Annu Rev Biophys. 2008;37:23–42. doi: 10.1146/annurev.biophys.37.032807.125904. [DOI] [PubMed] [Google Scholar]
- 4.Zimmer J, Nam Y, Rapoport TA. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature. 2008;455:936–943. doi: 10.1038/nature07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tajkhorshid E, Nollert P, Jensen MO, Miercke LJ, O'Connell J, Stroud RM, Schulten K. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science. 2002;296:525–530. doi: 10.1126/science.1067778. [DOI] [PubMed] [Google Scholar]
- 6.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 7.Gouaux E, Mackinnon R. Principles of selective ion transport in channels and pumps. Science. 2005;310:1461–1465. doi: 10.1126/science.1113666. [DOI] [PubMed] [Google Scholar]
- 8.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsa G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 9.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 10.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 11.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 13.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landau E, Rosenbusch J. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc Natl Acad Sci USA. 1996;93:14532–14535. doi: 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caffrey M. Crystallizing membrane proteins for structure determination: use of lipidic mesophases. Annu Rev Biophys. 2008 doi: 10.1146/annurev.biophys.050708.133655. in press. [DOI] [PubMed] [Google Scholar]
- 17.Pebay-Peyroula E, Rummel G, Rosenbusch J, Landau E. X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science. 1997;277:1676–1681. doi: 10.1126/science.277.5332.1676. [DOI] [PubMed] [Google Scholar]
- 18.Luecke H, Richter H, Lanyi J. Proton transfer pathways in bacteriorhodopsin at 2.3 angstrom resolution. Science. 1998;280:1934–1937. doi: 10.1126/science.280.5371.1934. [DOI] [PubMed] [Google Scholar]
- 19.Cherezov V, Clogston J, Papiz MZ, Caffrey M. Room to move: crystallizing membrane proteins in swollen lipidic mesophases. J Mol Biol. 2006;357:1605–1618. doi: 10.1016/j.jmb.2006.01.049. [DOI] [PubMed] [Google Scholar]
- 20.Cherezov V, Peddi A, Muthusubramaniam L, Zheng YF, Caffrey M. A robotic system for crystallizing membrane and soluble proteins in lipidic mesophases. Acta Crystallogr D Biol Crystallogr. 2004;60:1795–1807. doi: 10.1107/S0907444904019109. [DOI] [PubMed] [Google Scholar]
- 21.Faham S, Boulting GL, Massey EA, Yohannan S, Yang D, Bowie JU. Crystallization of bacteriorhodopsin from bicelle formulations at room temperature. Protein Sci. 2005;14:836–840. doi: 10.1110/ps.041167605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faham S, Bowie JU. Bicelle crystallization: a new method for crystallizing membrane proteins yields a monomeric bacteriorhodopsin structure. J Mol Biol. 2002;316:1–6. doi: 10.1006/jmbi.2001.5295. [DOI] [PubMed] [Google Scholar]
- 23.Faham S, Rachna U, Abramson J, Bowie J. In: Current topics in membranes. Delucas L, editor. Elsevier Press; Atlanta GA. (in press) [Google Scholar]
- 24.Sanders CR, II, Landis GC. Reconstitution of membrane proteins into lipid-rich bilayered mixed micelles for NMR studies. Biochemistry. 1995;34:4030–4040. doi: 10.1021/bi00012a022. [DOI] [PubMed] [Google Scholar]
- 25.Sanders CR, Prosser RS. Bicelles: a model membrane system for all seasons? Structure. 1998;6:1227–1234. doi: 10.1016/s0969-2126(98)00123-3. [DOI] [PubMed] [Google Scholar]
- 26.Nieh MP, Raghunathan VA, Glinka CJ, Harroun TA, Pabst G, Katsaras J. Magnetically alignable phase of phospholipid “bicelle” mixtures is a chiral nematic made up of wormlike micelles. Langmuir. 2004;20:7893–7897. doi: 10.1021/la048641l. [DOI] [PubMed] [Google Scholar]
- 27.Triba MN, Warschawski DE, Devaux PF. Reinvestigation by phosphorus NMR of lipid distribution in bicelles. Biophys J. 2005;88:1887–1901. doi: 10.1529/biophysj.104.055061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Dam L, Karlsson G, Edwards K. Direct observation and characterization of DMPC/DHPC aggregates under conditions relevant for biological solution NMR. Biochim Biophys Acta. 2004;1664:241–256. doi: 10.1016/j.bbamem.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Prosser RS, Hwang JS, Vold RR. Magnetically aligned phospholipid bilayers with positive ordering: a new model membrane system. Biophys J. 1998;74:2405–2418. doi: 10.1016/S0006-3495(98)77949-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luecke H, Schobert B, Stagno J, Imasheva ES, Wang JM, Balashov SP, Lanyi JK. Crystallographic structure of xanthorhodopsin, the light-driven proton pump with a dual chromophore. Proc Natl Acad Sci USA. 2008;105:16561–16565. doi: 10.1073/pnas.0807162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ujwal R, Cascio D, Colletier JP, Faham S, Zhang J, Toro L, Ping P, Abramson J. The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc Natl Acad Sci USA. 2008;105:17742–17747. doi: 10.1073/pnas.0809634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prive GG, Verner GE, Weitzman C, Zen KH, Eisenberg D, Kaback HR. Fusion proteins as tools for crystallization: the lactose permease from Escherichia coli. Acta Crystallogr D Biol Crystallogr. 1994;50:375–379. doi: 10.1107/S0907444993014301. [DOI] [PubMed] [Google Scholar]
- 33.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 35.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, Koide S, Sidhu SS. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J Mol Biol. 2007;373:924–940. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 36.Tereshko V, Uysal S, Koide A, Margalef K, Koide S, Kossiakoff AA. Toward chaperone-assisted crystallography: protein engineering enhancement of crystal packing and X-ray phasing capabilities of a camelid single-domain antibody (VHH) scaffold. Protein Sci. 2008;17:1175–1187. doi: 10.1110/ps.034892.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Day PW, Rasmussen SG, Parnot C, Fung JJ, Masood A, Kobilka TS, Yao XJ, Choi HJ, Weis WI, Rohrer DK, Kobilka BK. A monoclonal antibody for G protein-coupled receptor crystallography. Nat Methods. 2007;4:927–929. doi: 10.1038/nmeth1112. [DOI] [PubMed] [Google Scholar]
- 38.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 39.Donahue JP, Patel H, Anderson WF, Hawiger J. Three-dimensional structure of the platelet integrin recognition segment of the fibrinogen gamma chain obtained by carrier protein-driven crystallization. Proc Natl Acad Sci USA. 1994;91:12178–12182. doi: 10.1073/pnas.91.25.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smyth DR, Mrozkiewicz MK, McGrath WJ, Listwan P, Kobe B. Crystal structures of fusion proteins with large-affinity tags. Protein Sci. 2003;12:1313–1322. doi: 10.1110/ps.0243403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhan Y, Song X, Zhou GW. Structural analysis of regulatory protein domains using GST-fusion proteins. Gene. 2001;281:1–9. doi: 10.1016/s0378-1119(01)00797-1. [DOI] [PubMed] [Google Scholar]
- 42.Standfuss J, Xie G, Edwards PC, Burghammer M, Oprian DD, Schertler GF. Crystal structure of a thermally stable rhodopsin mutant. J Mol Biol. 2007;372:1179–1188. doi: 10.1016/j.jmb.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lau FW, Nauli S, Zhou Y, Bowie JU. Changing single side-chains can greatly enhance the resistance of a membrane protein to irreversible inactivation. J Mol Biol. 1999;290:559–564. doi: 10.1006/jmbi.1999.2905. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Y, Bowie JU. Building a thermostable membrane protein. J Biol Chem. 2000;275:6975–6979. doi: 10.1074/jbc.275.10.6975. [DOI] [PubMed] [Google Scholar]
- 45.Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301:610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 46.Smirnova IN, Kaback HR. A mutation in the lactose permease of Escherichia coli that decreases conformational flexibility and increases protein stability. Biochemistry. 2003;42:3025–3031. doi: 10.1021/bi027329c. [DOI] [PubMed] [Google Scholar]
- 47.Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. Conformational thermostabilization of the beta1-adrenergic receptor in a detergent-resistant form. Proc Natl Acad Sci USA. 2008;105:877–882. doi: 10.1073/pnas.0711253105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grisshammer R, Duckworth R, Henderson R. Expression of a rat neurotensin receptor in Escherichia coli. Biochem J. 1993;295(Pt 2):571–576. doi: 10.1042/bj2950571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Magnani F, Shibata Y, Serrano-Vega MJ, Tate CG. Co-evolving stability and conformational homogeneity of the human adenosine A2a receptor. Proc Natl Acad Sci USA. 2008;105:10744–10749. doi: 10.1073/pnas.0804396105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Struthers M, Yu H, Oprian DD. G protein-coupled receptor activation: analysis of a highly constrained, “straitjacketed” rhodopsin. Biochemistry. 2000;39:7938–7942. doi: 10.1021/bi000771f. [DOI] [PubMed] [Google Scholar]
- 51.Xie G, Gross AK, Oprian DD. An opsin mutant with increased thermal stability. Biochemistry. 2003;42:1995–2001. doi: 10.1021/bi020611z. [DOI] [PubMed] [Google Scholar]
- 52.Roth CB, Hanson MA, Stevens RC. Stabilization of the human beta2-adrenergic receptor TM4-TM3-TM5 helix interface by mutagenesis of Glu122(3.41), a critical residue in GPCR structure. J Mol Biol. 2008;376:1305–1319. doi: 10.1016/j.jmb.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davidson AR. Multiple sequence alignment as a guideline for protein engineering strategies. Methods Mol Biol. 2006;340:171–181. doi: 10.1385/1-59745-116-9:171. [DOI] [PubMed] [Google Scholar]
- 54.Martin A, Damian M, Laguerre M, Parello J, Pucci B, Serre L, Mary S, Marie J, Baneres JL. Engineering a G protein-coupled receptor for structural studies: stabilization of the BLT1 receptor ground state. Protein Sci. 2009;18:727–734. doi: 10.1002/pro.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCusker EC, Bane SE, O'Malley MA, Robinson AS. Heterologous GPCR expression: a bottleneck to obtaining crystal structures. Biotechnol Prog. 2007;23:540–547. doi: 10.1021/bp060349b. [DOI] [PubMed] [Google Scholar]
- 56.Breyer RM, Strosberg AD, Guillet JG. Mutational analysis of ligand binding activity of beta 2 adrenergic receptor expressed in Escherichia coli. EMBO J. 1990;9:2679–2684. doi: 10.1002/j.1460-2075.1990.tb07453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Skretas G, Georgiou G. Engineering G protein-coupled receptor expression in bacteria. Proc Natl Acad Sci USA. 2008;105:14747–14748. doi: 10.1073/pnas.0807741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Skretas G, Georgiou G. Genetic analysis of G protein-coupled receptor expression in Escherichia coli: inhibitory role of DnaJ on the membrane integration of the human central cannabinoid receptor. Biotechnol Bioeng. 2009;102:357–367. doi: 10.1002/bit.22097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sarkar CA, Dodevski I, Kenig M, Dudli S, Mohr A, Hermans E, Pluckthun A. Directed evolution of a G protein-coupled receptor for expression, stability, and binding selectivity. Proc Natl Acad Sci USA. 2008;105:14808–14813. doi: 10.1073/pnas.0803103105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roosild TP, Greenwald J, Vega M, Castronovo S, Riek R, Choe S. NMR structure of Mistic, a membrane-integrating protein for membrane protein expression. Science. 2005;307:1317–1321. doi: 10.1126/science.1106392. [DOI] [PubMed] [Google Scholar]
- 61.Massey-Gendel E, Zhao A, Boulting G, Kim HY, Balamotis MA, Seligman LM, Nakamoto RK, Bowie JU. Genetic selection system for improving recombinant membrane protein expression in E. coli. Protein Sci. 2009;18:372–383. doi: 10.1002/pro.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 63.Klammt C, Schwarz D, Dotsch V, Bernhard F. Cell-free production of integral membrane proteins on a preparative scale. Methods Mol Biol. 2007;375:57–78. doi: 10.1007/978-1-59745-388-2_3. [DOI] [PubMed] [Google Scholar]
- 64.Klammt C, Schwarz D, Eifler N, Engel A, Piehler J, Haase W, Hahn S, Dotsch V, Bernhard F. Cell-free production of G protein-coupled receptors for functional and structural studies. J Struct Biol. 2007;158:482–493. doi: 10.1016/j.jsb.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 65.Klammt C, Schwarz D, Lohr F, Schneider B, Dotsch V, Bernhard F. Cell-free expression as an emerging technique for the large scale production of integral membrane protein. FEBS J. 2006;273:4141–4153. doi: 10.1111/j.1742-4658.2006.05432.x. [DOI] [PubMed] [Google Scholar]
- 66.Chen YJ, Pornillos O, Lieu S, Ma C, Chen AP, Chang G. X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci USA. 2007;104:18999–19004. doi: 10.1073/pnas.0709387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang G. NMR of membrane-associated peptides and proteins. Curr Protein Pept Sci. 2008;9:50–69. doi: 10.2174/138920308783565714. [DOI] [PubMed] [Google Scholar]
- 68.McDermott AE. Structure and dynamics of membrane proteins by magic angle spinning solid-state NMR. Annu Rev Biophys. 2009 doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- 69.Gong XM, Franzin CM, Thai K, Yu J, Marassi FM. Nuclear magnetic resonance structural studies of membrane proteins in micelles and bilayers. Methods Mol Biol. 2007;400:515–529. doi: 10.1007/978-1-59745-519-0_35. [DOI] [PMC free article] [PubMed] [Google Scholar]