

Abstract
The protein scaffold is a peptide framework with a high tolerance of residue modifications. The cysteine-stabilized αβ motif (CSαβ) consists of an α-helix and an antiparallel triple-stranded β-sheet connected by two disulfide bridges. Proteins containing this motif share low sequence identity but high structural similarity and has been suggested as a good scaffold for protein engineering. The Vigna radiate defensin 1 (VrD1), a plant defensin, serves here as a model protein to probe the amino acid tolerance of CSαβ motif. A systematic alanine substitution is performed on the VrD1. The key residues governing the inhibitory function and structure stability are monitored. Thirty-two of 46 residue positions of VrD1 are altered by site-directed mutagenesis techniques. The circular dichroism spectrum, intrinsic fluorescence spectrum, and chemical denaturation are used to analyze the conformation and structural stability of proteins. The secondary structures were highly tolerant to the amino acid substitutions; however, the protein stabilities were varied for each mutant. Many mutants, although they maintained their conformations, altered their inhibitory function significantly. In this study, we reported the first alanine scan on the plant defensin containing the CSαβ motif. The information is valuable to the scaffold with the CSαβ motif and protein engineering.
Keywords: plant defensin, α-amylase, alanine scan, circular dichroism spectroscopy, fluorescence spectroscopy, protein engineering
Introduction
The protein scaffold is a peptide framework that exhibits a high tolerance of its residue modifications.1 To introduce tailored functions into protein scaffolds is a major challenge and has a unique significance in protein design.1,2 The structural core of suitable protein scaffolds should be compact and rigid, and its folding properties should tolerate the amino acid alteration in a contiguous structure or by changing the length and sequence of loops.3 Protein design requires detailed knowledge of protein folding structure, function, and dynamics, but it also challenges our understanding of the principles underlying protein structure.4,5 The cysteine-stabilized αβ (CSαβ) motif consists of an α-helix and an antiparallel triple-stranded β-sheet. The α-helix and the β-sheet are connected by two disulfide bridges.6–10 Proteins with the CSαβ motif are widely distributed among plants, insects, arachnidia, and mollusca.8,10 They exhibit a wide spectrum of biological activities, including antimicrobial activity, enzyme inhibitory function, inhibition of protein translation, and sweet taste.6,11–16 Proteins with the CSαβ motif share low sequence identity but high structural similarity.17 The few conserved residues are restricted to the eight cysteines, a glycine on the β2 strand and an aromatic residue on the Loop 1.11 The relationship between the conserved simple three-dimensional structure and high diversity of biological function is especially interesting.
For their low sequence identity, multiple biological function, and high structure similarity, proteins with the CSαβ motif are thought as good candidates for protein engineering.18 Some proteins containing this motif had been engineered to exhibit new functions either by minimal residue substitution or loop exchange.18–20 Some researches focused on medical applications of the scaffold with the CSαβ motif.21–23 Protein engineering approach and drug screening strategy have been utilized to generate new proteinous antibiotics with the motif.24 It is thought that the proteins with the CSαβ motif have high potential in drug development.25 Furthermore, sequences with low identity fold to a specific three-dimensional structure, which is particularly curious. It is worth to probe the residue governing the structural stability, biological function, and interaction with its counterparts. The information is not only useful in understanding the properties of the CSαβ motif but also can be applied to protein engineering.26
The Vigna radiate defensin 1 (VrD1), a 46-residue basic peptide containing a CSαβ motif, belongs to the plant defensin family. It has been reported as the first defensin with insecticidal activity against the larvae of the Callosobruchus chinensis. The three-dimensional structure of VrD1 has been determined by nuclear magnetic resonance.27 In this study, VrD1 is served as a model, and a systematic alanine substitution was performed to investigate the amino acid tolerance of the CSαβ motif. The goal of this study is to probe the availability for protein engineering of residues in the CSαβ motif relating to their structural stability and biochemical function. We reported the first complete alanine scan on the CSαβ motif. Our results showed that the secondary structures of all mutants were well preserved. The secondary structure of VrD1 and its mutants can tolerate temperatures over 95°C and high concentration of chaotropic agents although they exhibited various enzyme inhibitory abilities. Several positions on the CSαβ motif were crucial to the enzyme inhibitory function through direct or indirect interactions to regulate the inhibitory function.
Results and Discussion
In this study, 32 of 38 noncysteine and alanine positions were probed. Five mutants, T2A, E8A, G27A, G31A, and N45A, could not be obtained during the molecular cloning or protein purification processes. The G31 is conserved in all described proteins containing CSαβ motif. The G31 is related to stereo constraints between the side chains of the nearby aromatic residues, and a tiny residue is required by the close contacts between the helix and the β2 strand.28,29 Replacing charged or neutral residue by alanine sometimes destabilizes the mutated protein.30,31 It is likely that mutation of these residues (E8, G27, and G31) contributed to the instability of VrD1. The amino acid tolerance of these five positions remained to be tested. The first disulfide bridge, formed by C3 and C46, was not involved in the CSαβ motif. To investigate the role of the first disulfide bridge, the alanine substations of cysteines on positions 3 and 46 (C3AC46A) were generated to remove the disulfide bridge. The molecular weights of purified recombinant proteins were verified with mass spectrometry (Table I). The observed molecular weights of all purified proteins met their oxidized theoretical values. The verified proteins were used in further experiments. A multiple sequence alignment was performed to analyze the conserved positions of plant defensins containing CSαβ motif (see Fig. 1). The highly conserved positions are labeled in red, and the positions dominated by certain amino acids are labeled in blue. The highly conserved positions included the eight cysteines and a glycine on position 31 (related to the positions on VrD1). The residue positions 1, 10, 26, and 38 are dominated by certain types of amino acids. A positively charged amino acid is usually on position 1. There is always a residue with aromatic side chain on position 10 and followed by a glycine. On position 26, there is usually a charged residue, either positive or negative. The position 38 is dominated by a positively charged amino acid. These conserved sites have a possibility of being important to the structural stability and/or biochemical functions.
Table I.
Data Summaries of VrD1 and Its Mutants
| Group | Residue no. | Protein | Theoretical M.W. (Da)a | Observed M.W. (Da) | TMA inhibition (%)b | Cm (M)c | [θ]215 (nm)d |
|---|---|---|---|---|---|---|---|
| rVrD1 | 5211.2 | 5210 | 87 ± 18 | 4.17 | −2990 ± 37 | ||
| I | 4 | M4A | 5150.8 | 5151 | 15 ± 6 | 3.66 | −2974 ± 82 |
| 6 | K6A | 5154.1 | 5155 | 6 ± 2 | 2.57 | −2382 ± 158 | |
| 9 | G9A | 5224.9 | 5226 | 7 ± 1 | 3.27 | −3774 ± 37 | |
| 10 | W10A | 5096.1 | 5097 | 4 ± 1 | N/D | −3305 ± 79 | |
| 14 | L14A | 5168.8 | 5167 | 27 ± 5 | 4.51 | −3388 ± 107 | |
| 36 | M36A | 5150.8 | 5152 | 34 ± 10 | 3.88 | −2766 ± 38 | |
| 38 | R38A | 5126.1 | 5128 | 25 ± 3 | 2.97 | −2927 ± 34 | |
| 44 | V44A | 5182.8 | 5183 | 29 ± 16 | 3.14 | −2591 ± 20 | |
| II | 1 | R1A | 5125.8 | 5126 | 39 ± 9 | 3.29 | −3171 ± 183 |
| 17 | T17A | 5180.8 | 5181 | 50 ± 10 | 3.32 | −3056 ± 216 | |
| 18 | T18A | 5180.8 | 5181 | 57 ± 12 | 4.50 | −3181 ± 59 | |
| 21 | H21A | 5144.8 | 5145 | 46 ± 14 | 4.33 | −2686 ± 162 | |
| 22 | S22A | 5194.9 | 5195 | 67 ± 14 | 2.84 | −3371 ± 96 | |
| 24 | K24A | 5153.8 | 5154 | 48 ± 10 | 2.81 | −3084 ± 145 | |
| 25 | N25A | 5167.8 | 5198 | 44 ± 9 | 3.49 | −2405 ± 123 | |
| 26 | R26A | 5126.1 | 5126 | 52 ± 6 | 2.76 | −2706 ± 199 | |
| 29 | I29A | 5168.8 | 5167 | 41 ± 12 | 3.32 | −3298 ± 52 | |
| 37 | T37A | 5180.8 | 5181 | 63 ± 8 | 2.98 | −2840 ± 57 | |
| 39 | T39A | 5180.8 | 5181 | 45 ± 6 | 3.32 | −2999 ± 54 | |
| 43 | L43A | 5168.8 | 5167 | 40 ± 6 | 4.69 | −3111 ± 45 | |
| III | 5 | I5A | 5168.8 | 5169 | 89 ± 18 | 2.93 | −3654 ± 130 |
| 7 | K7A | 5154.1 | 5153 | 83 ± 9 | 2.53 | −3077 ± 49 | |
| 11 | G11A | 5224.9 | 5225 | 90 ± 17 | 4.66 | −2897 ± 83 | |
| 12 | K12A | 5154.1 | 5154 | 81 ± 9 | 2.48 | −2847 ± 160 | |
| 15 | I15A | 5168.8 | 5168 | 96 ± 13 | 3.87 | −2637 ± 145 | |
| 16 | D16A | 5166.9 | 5167 | 83 ± 9 | 2.96 | −2963 ± 96 | |
| 28 | Y28A | 5118.9 | 5119 | 100 ± 20 | 3.10 | −3311 ± 67 | |
| 30 | G30A | 5224.9 | 5225 | 72 ± 20 | 3.26 | −2695 ± 125 | |
| 32 | N32A | 5168.2 | 5167 | 71 ± 9 | 2.64 | −2775 ± 116 | |
| 34 | K34A | 5154.1 | 5153 | 79 ± 8 | 3.31 | −2762 ± 135 | |
| 35 | G35A | 5224.9 | 5225 | 78 ± 16 | 3.44 | −2541 ± 109 | |
| 41 | Y41A | 5118.9 | 5119 | 100± 21 | 3.73 | −3299 ± 93 | |
| 46 | C3AC46A | 5148.7 | 5150 | 95 ± 21 | 2.69 | −2808 ± 231 |
All mutants are classified into three groups according to their TMA inhibition abilities. Within each group, the data are sorted by the position of mutated point. Group I: TMA inhibition ability < 35%; Group II: 35% < TMA inhibition ability < 70%; Group III: 70% < TMA inhibition ability.
N/D: undetectable.
Oxidized molecular weight.
TMA inhibition ability of 20 μM VrD1.
Chemical melting point (Cm).
Mean-residue ellipticity at 215 nm.
Figure 1.
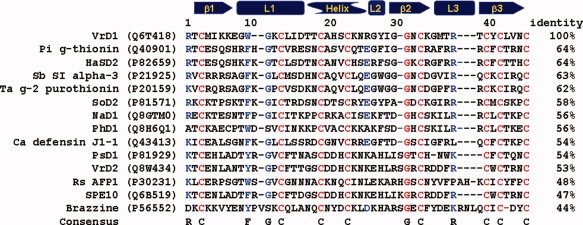
Sequence alignment of various plant defensins. The sequences were retrieved from the Swiss-Prot database, and the code in the quotation marker is the entry code of each sequence. The secondary structure regions are displayed on the top of the alignment. The alignment is performed with Vector NTI 10.0. The highly conserved positions are labeled in red. The dominated amino acids on certain positions are labeled in blue.
Secondary structure tolerance
To investigate the secondary structure stability of each mutant, the CD spectra were scanned and analyzed. The mean-residue ellipticity [θ] at 215 nm ([θ]215) of the wild-type VrD1 and mutants is listed in Table I. The example CD spectra of several mutants and wild-type VrD1 are shown in Figure 2. The CD spectra of all the mutants were not significantly different from the spectrum of wild-type VrD1. These results indicated that the secondary structures were well preserved for all mutants even in the absence of the first disulfide bridge formed by C3 and C46. It implied that the secondary structure of VrD1 could tolerate the amino acid substitutions in most positions. The thermostability to temperature and denaturants was also monitored by circular dichroism spectroscopy. It was found that the secondary structure of VrD1 and its mutants could be maintained even at temperatures more than 95°C (Fig. 2 inset). The secondary structure of VrD1 also tolerated to 6M guanidine HCl (Gdn-HCl) and 7M urea. The structure was completed destroyed only in the presence of dithiothreitol (data not shown). These results indicated that the disulfide bridges were extremely crucial in maintaining the structure of the protein containing CSαβ motif.
Figure 2.
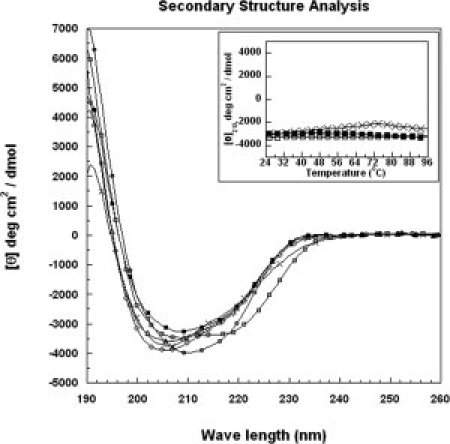
CD spectra of VrD1 and its mutants. The secondary structure was analyzed by circular dichroism spectroscopy. The recombinant VrD1 (50 μM) was dissolved in 20 mM phosphate buffer, and the CD spectrum was scanned from 190 to 260 nm. The CD signals in millidegree were converted to mean-residue ellipticity [θ]. The inset shows temperature melting curves of the VrD1 and VrD1 mutants. Closed quadrate: wild type, open circle: G9A, open quadrate: W10A, open diamond: T17A, open triangle: K24A, and cross: C3AC46A double mutant.
Intrinsic fluorescence and chemical denaturation
The intrinsic fluorescence spectra of wild-type VrD1 and mutants were analyzed. The wild-type VrD1 and mutants, except for the W10A, could be excited at 280 nm (Ex λ280) and exhibited an emission λmax at 348 nm (Em λ348) (see Fig. 3). The W10A exhibited an emission λmax at 303 nm and this indicated that the fluorescence profile was altered when the single tryptophan residue was mutated. This intrinsic fluorescence of VrD1 wild type and the other mutants was produced by the tryptophan. The intensity of emission λmax of tryptophan was solvent dependent and related to the protein conformation.32 Variations of the intensity of tryptophan fluorescence can be utilized to measure the alteration of protein conformation and stability by chemical denaturation with Gdn-HCl.33,34 To further investigate the chemical stabilities of VrD1 and its derivatives, the protein unfolding in the presence of Gdn-HCl was measured via intrinsic tryptophan fluorescence. The fluorescence spectra of wild-type VrD1 in different concentrations of Gdn-HCl are shown in Figure 4(A). The Cm values (midpoint of the unfolding transition) of wild-type VrD1 and its derivatives were determined at the Em λ348 in the presence of Gdn-HCl. The Cm values of all proteins were calculated and listed in Table I. The Gdn-HCl denaturation curves of some mutants were shown, and a native-like two-state transition curve could be observed among these mutants [Fig. 4(B)]. The calculated Cm value of wild-type VrD1 was 4.17M, and the Cm values of mutants were varied from 4.69M (L43A) to 2.48M (K12A). The Cm values of five mutants, L14A, L43A, G11A, T18A, and H21A, were higher than that of wild-type VrD1. The other mutants showed lower Cm values than that of wild-type VrD1. Although the chemical stabilities monitored by intrinsic tryptophan fluorescence of these alanine-substitution mutants were varied, the globular fold of these proteins did not change significantly [Figs. 2 and 3].
Figure 3.
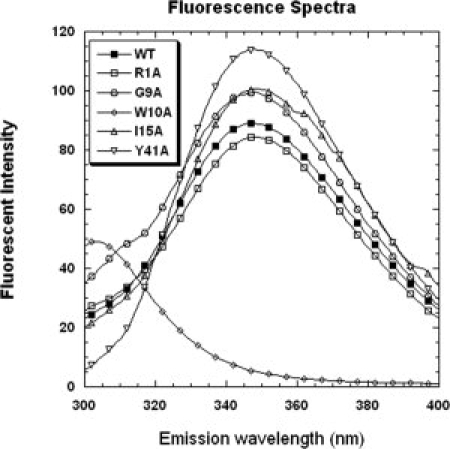
Intrinsic fluorescence spectra of VrD1 and its mutants. The wild-type VrD1 and mutants were dissolved in 10 mM Tris buffer pH 7.5 containing 100 mM NaCl and 1 mM EDTA at a final concentration of 7.5 μM. The excitation λ was set at 280 nm and the emission λ from 295 to 400 nm was detected. The Em λmax of wild-type VrD1 and mutants, except for the W10A, was detected at 348 nm. The Em λmax of W10A was detected at 303 nm.
Figure 4.
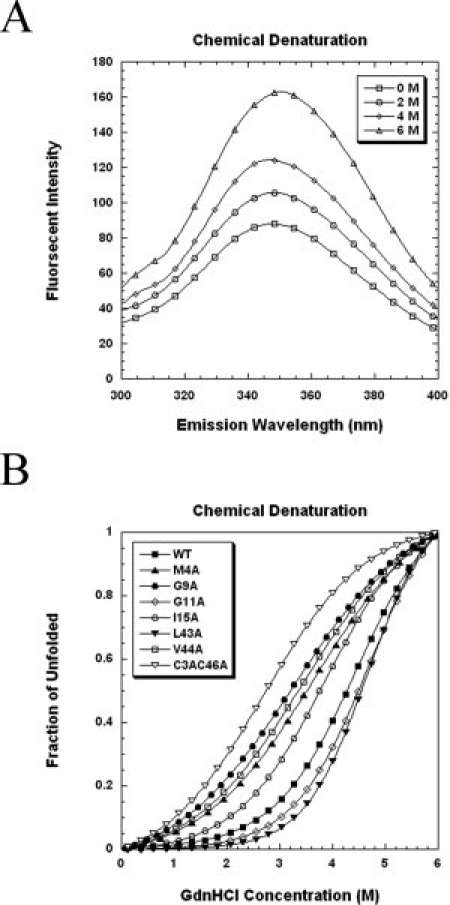
Denaturant induced unfolding of VrD1 and its mutants. The measurement of chemical denaturation was based on the intrinsic fluorescence intensity change at emission λ348. The proteins were exposed to different concentrations of Gdn-HCl for at least 16 h at room temperature and the fluorescence spectra were scanned. After substrate the spectrum without Gdn-HCl, the chemical melting point was calculated by fitting the fraction of the unfolding curve to a two-step unfolding model. Some curves are presented. (A) Effect of increasing Gdn-HCl concentration on intrinsic fluorescence of VrD1. (B) Comparison of the Gdn-HCl induced unfolding of VrD1 and its mutants.
Enzyme inhibitory function
To test the function of mutated VrD1, the Tenebrio molitor α-amylase (TMA) inhibition assay was used to evaluate the activity of these mutants as described in previous studies.18,27 The wild-type VrD1 displayed a maximal TMA inhibition (about 87% inhibition) at 20 μM, and this was consistent with the TMA inhibitory ability of the native VrD1 [Fig. 5(A)].27 The TMA-inhibitory abilities at 20 μM of wild-type VrD1 and mutants were compared [Table I and Fig. 5(B)]. According to these maximal inhibitory abilities, all mutants were classified into three groups and their locations were also displayed in the VrD1 structure [Table I and Fig. 6(A,B)]. The Group I exhibited the weak TMA inhibitory ability (<35%), the Group II exhibited the medium TMA inhibitory ability (35–70%), and the Group III exhibited the strong TMA inhibitory ability (>70%) (Table I).
Figure 5.
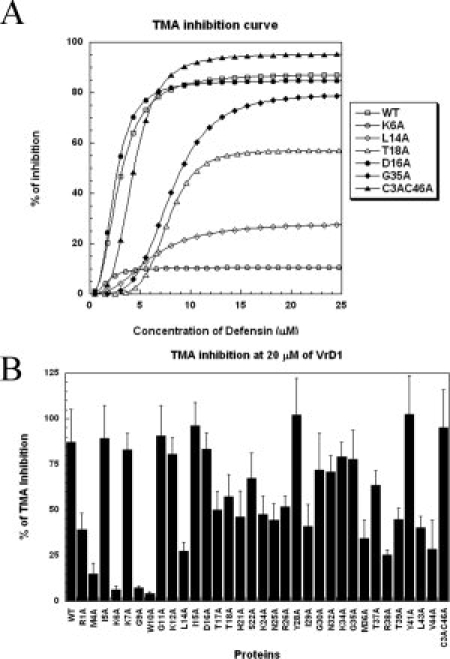
Inhibitory activities of VrD1 and its mutants against TMA. (A) Comparison of the TMA inhibitory activities of VrD1 and its mutants at difference concentration. The TMA inhibitory abilities of VrD1 and its mutants were titrated in the presence of different concentration of defensins. The maximal inhibitory effect can be achieved at 13 μM of wild-type VrD1. (B) Comparison of the maximal TMA inhibitory activities of VrD1 and its mutants. The TMA inhibition ability of VrD1 and its mutants was compared in the presence of 20 μM of proteins. The assays were performed as described in the Materials and Methods section. The inhibitory abilities of mutants and wild type were assayed in duplicate and repeated. The data are presented as mean ± SD.
Figure 6.
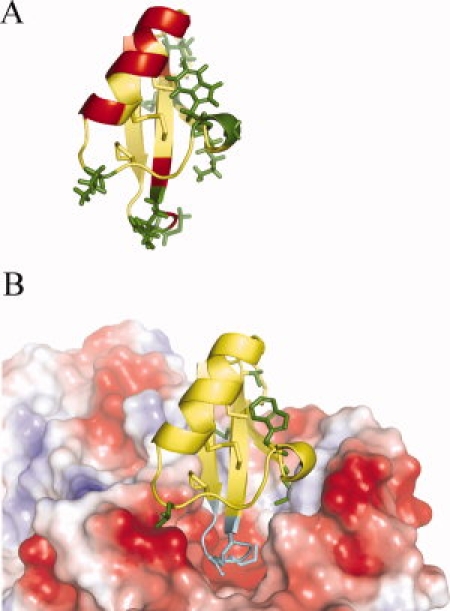
Mutated position mapping and molecular docking. (A) Mapping of the mutated positions on the VrD1 structure. The structure of VrD1 (1TI5) is display in ribbon with PyMol v0.99. The residues of Group I, II, and III are labeled in green, red, and yellow; respectively. The side chains of the Group I residues are displayed in sticks. (B) Molecular docking simulation of VrD1 and TMA. The molecular docking was simulated with PatchDock and the result was visualized with PyMol v0.99. The VrD1 is displayed in ribbon and TMA is displayed in electrostatic surface potential. The positively and negatively charged areas are represented in blue and red, respectively. The side chains of the Group I residues are displayed in green sticks. The VrD1 loop 3 can insert to the catalytic pocket of TMA and is displayed in teal. The side chains of M36 and R38 are also displayed in sticks. The Group I residues are located on the binding surface of VrD1.
There were eight mutants in Group I and the mutated positions were located within the β1 strand (M4A, K6A), β3 strand (V44A), the 310 helix (G9A, W10A), the Loop 3 (M36A, R38A), and the Loop 1 (L14A). These residues are mapped in the VrD1 structure in Figure 6(A). Although they were scattered on the different segments of VrD1, these residues all projected from the same side of the protein. The molecular docking simulation also revealed that these residues could form an interface between VrD1 and TMA [Fig. 6(B)]. Most of the Group II residues, labeled in red in Figure 6(A), were located in different region from the Group I, especially for the helix segment. There were seven mutated positions (T17A, T18A, H21A, S22A, K24A, N25A, and R26A) sat on the one side of the helix, which was the noncysteine side. Although the helical conformation and protein stability seemed not to be affected by the alanine substitution on these positions, it is possible that they may remotely affect the inhibitory function by the disulfide linkages between the α-helix and β-sheet of this CSαβ motif.
The Loop 3 of VrD1 was proposed as a functional region for TMA inhibitory activity, and the molecular docking model also showed that it can be inserted into the catalytic pocket of TMA [Fig. 6(B)].18 In the catalytic pocket of TMA, three acidic residues, D185, E222, and D287, were directly involved in the degradation of starch.35,36 Four mutated position (M36A, R38A, T37A, and T39A) in the Loop 3 showed weak or medium inhibition confirming the important role of this loop in the TMA inhibitory function of VrD1. Interestingly, alanine-substitution of two residues located on the 310 helix, G9A and W10A, almost abolished the TMA inhibitory function [Table I and Fig. 5(B)]. These results confirmed that the both residues are critical in TMA inhibitory function. The relationship between the unique 310 helix and the TMA inhibitory function still needs further investigation. In the Group III mutants, alanine-substitution for these residues caused changes in TMA inhibitory abilities ranging from 70 to 100% [Table I and Fig. 5(B)]. The contributions to TMA inhibition of these residues in Group III appear less critical than Group I and II mutants. In addition, the double mutant, C3AC46A, exhibited similar inhibitory ability as did the wild-type VrD1.
Materials and Methods
Plasmid construction, polymerase chain reaction, and site-directed mutagenesis
The synthesized VrD1 gene was constructed into the expression vector pET32a(+) between BamHI and XhoI. An acidic hydrolysis site, -Asp-Pro-, was inserted into the intergenic region between the His-tag and VrD1 to generate pET32a(+)-VrD1 (Supporting Information).37,38 The point mutation on VrD1 was performed with polymerase chain reaction (PCR)-based site-directed mutagenesis. The plasmids of wild-type pET32a(+)-VrD1 were utilized as templates, and the synthesized mutated oligo nucleotides were applied as primers. A proof reading DNA polymerase, Phusion Ultra II PFU (Merck, USA), was used in the PCR. After being digested by DpnI to remove the templates, the PCR products were heat-shock transformed to E. coli Top10. The transformed bacterial cells were plated out on a LB agar plate containing ampicillin and incubated at 37°C for 18 h. The plasmids were prepared from the positive clones and sequenced.
Protein purification, identification, purity, and protein assay
The plasmid was heat-shock transformed to E. coli BL21 (DE3) for producing recombinant proteins. The E coli cells were grown in 1 L of LB broth containing ampicillin to OD600 0.6 at 37°C, and 1 mM isopropyl-β-d-thiogalactopyranoside was added to induce the production of recombinant VrD1. The cells were harvested by centrifugation and the wet pastes were resuspended in 10 mM Tris-HCl (pH 8.0) containing 100 mM NaCl. After occasionally being inverted at 4°C for 30 min, the cells were lysed with sonication, centrifuged, and then the supernatants were collected. The His-tagged recombinant proteins were isolated with immobilized metal affinity chromatography. To cut-off the tags from the recombinant proteins, the purified recombinant proteins were diluted, and the pH was adjusted to 1.4 with HCl. After being incubated at 55°C for 16 h, the protein solution was neutralized to pH 7, and the positive charged proteins were purified by a weak cation exchange column (CM Hitrap 5 mL, GE Life Science USA) with a gradient from 50 to 400 mM NaCl in 30 mL of 20 mM Tris (pH 7.0). The recombinant VrD1 was further purified through a preparation-grade HPLC C18 column with a gradient of acetonitrile. The final purified proteins were dried in a spin vacuum and dissolved in 10 mM phosphate buffer (pH 8). The protein amounts were measured with a BAC protein assay kit (Pierce, CA). The molecular weight of purified protein was verified with mass spectrometry. The verified protein should have an observed value meeting its oxidized theoretical value. The protein purity was examined in an analysis-grade HPLC C18 column.18,27
Tenebrio molitor α-amylase inhibition assay
The α-amylase was purified from the extracts of T. molitor larvae as described.27 The fractions were assayed for α-amylase activity, and the fractions with α-amylase activity were pooled, desalted, and concentrated. To test the inhibition ability of recombinant VrD1 and its mutants, the purified recombinant VrD1 and mutants were dissolved in 10 mM phosphate buffer pH 7.0 at a final concentration of 100 μM as a stock solution. Different amounts of defensin stock solution were mixed with 0.06 μM TMA in 10 mM phosphate buffer (pH 7.0) containing 1 mM CaCl2. The final volume of mixture was 200 μL. The mixtures were incubated at 30°C for 30 min, then 250 μL 0.4% (w/v) Zulkowsky starch (Merck) in reaction buffer was added and vortex. After incubation at 30°C for 10 min, the reaction was stopped by adding 250 μL 1% (w/v) dinitrosalicylic acid (Sigma, USA) in 0.4N NaOH and boiled in a boiling water bath for 5 min. After cooling in ice, the enzyme activity was monitored by the absorbance at 546 nm.
Circular dichroism, fluorescence spectroscopy, and chemical denaturation experiments
The secondary structure analysis was performed in an AVIV 62A DS circular dichroism spectrometer. The wild-type VrD1 and its mutants were prepared in 20 mM sodium phosphate buffer (pH 7.0) at a concentration of 50 μM. To reduce the disulfide bridges, the VrD1 was exposed to 1 mM dithiothreitol at 37°C for 30 min. The spectra of protein samples were converted to mean-residue ellipticity [θ].39 To measure the intrinsic fluorescence, the samples were dissolved in 10 mM Tris-HCl (pH 7.0) containing 100 mM NaCl and 1 mM EDTA (fluorescence buffer) at a final concentration of 7.5 μM. The excitation wavelength was set at 280 nm and the emission wavelengths from 300 to 400 nm were detected in a Hitachi F-7000 fluorescence spectrophotometer. The chemical denaturation experiment was based on the change of the fluorescence intensity at emission λ348. In brief, the samples were prepared in the fluorescence buffer containing Gdn-HCl at a range from 0 to 6M and set on bench for 16 h before assay. The fluorescence intensity of the emission λ348 was observed, and the following equation was applied to calculate the fraction of denaturation.
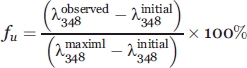 |
(1) |
In the Eq. (1), the fu represents the fraction of denaturation and the 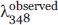 refers to the fluorescence intensity observed in the presence of different concentration of Gdn-HCl. The
refers to the fluorescence intensity observed in the presence of different concentration of Gdn-HCl. The 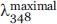 and
and 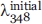 are the fluorescence intensities observed in the presence of 6 and 0M of Gdn-HCl, respectively.
are the fluorescence intensities observed in the presence of 6 and 0M of Gdn-HCl, respectively.
The chemical melting point (Cm) was calculated by fitting the fraction of denaturation data from Eq. (1) to nonlinear least-squares regression based on the two-state unfolding model as below:
| (2) |
In the Eq. (2), the Y represents the value of the spectroscopic property of the protein at a given Gdn-HCl concentration [D]. The YF and YU are the denoted intercepts. The mF and mU are the slopes of the baseline of the native state and the unfold protein state, respectively. The mG is a measure of the dependence of ΔGo on [D] and ΔGo(H2O) is the free energy change in the absence of denaturants.40,41
Molecular docking and multiple sequence alignment
The Modeller 9v3 was utilized to construct the three-dimensional structures of the mutants, and the wild-type VrD1 structure (PDB code 1TI5) was used as the template.18,27 The docking of VrD1 to TMA was performed with Patch Dock Beta 1.3 (http://bioinfo3d.cs.tau.ac.il/PatchDock/) and the default setting was used.42 To analyze the interactions of VrD1 and TMA, the result of the molecular docking was analyzed with the Ligplot.43 The docking results were visualized with PyMol v0.99. Multiple sequence alignment was performed with Vector NTI 10.0.44
Conclusions
The elucidation of the amino acid substitution tolerance in the model protein represents a starting point for understanding the interaction with targets and protein engineering of CSαβ motif. We undertake a comprehensive alanine substitution study to elucidate the amino acid tolerance of the protein containing CSαβ motif. Our results show that most of the noncysteine positions of the CSαβ motif can tolerate the amino acid substitution. The alanine substitution of each position does not affect the protein structure, although the inhibitory effect of some mutants is altered even abolished. The enzyme inhibitory function is significantly decreased when the residues located on binding surface are mutated. The CSαβ motif can serve as a suitable scaffold for protein engineering, and the information provided in this study will facilitate the development of protein designs utilizing the CSαβ motif.
Acknowledgments
The authors thank Ms. Fen-Shung Wu for her assistance with the mass spectrometry.
References
- 1.Hey T, Fiedler E, Rudolph R, Fiedler M. Artificial, non-antibody binding proteins for pharmaceutical and industrial applications. Trends Biotechnol. 2005;23:514–522. doi: 10.1016/j.tibtech.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 2.Pessi A, Bianchi E, Crameri A, Venturini S, Tramontano A, Sollazzo M. A designed metal-binding protein with a novel fold. Nature. 1993;362:367–369. doi: 10.1038/362367a0. [DOI] [PubMed] [Google Scholar]
- 3.Skerra A. Alternative non-antibody scaffolds for molecular recognition. Curr Opin Biotechnol. 2007;18:295–304. doi: 10.1016/j.copbio.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Nikkhah M, Jawad-Alami Z, Demydchuk M, Ribbons D, Paoli M. Engineering of β-propeller protein scaffolds by multiple gene duplication and fusion of an idealized WD repeat. Biomol Eng. 2006;23:185–194. doi: 10.1016/j.bioeng.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Chen Z, Zhao H. Rapid creation of a novel protein function by in vitro coevolution. J Mol Biol. 2005;348:1273–1282. doi: 10.1016/j.jmb.2005.02.070. [DOI] [PubMed] [Google Scholar]
- 6.Assadi-Porter FM, Aceti DJ, Markley JL. Sweetness determinant sites of brazzein, a small, heat-stable, sweet-tasting protein. Arch Biochem Biophys. 2000;376:259–265. doi: 10.1006/abbi.2000.1726. [DOI] [PubMed] [Google Scholar]
- 7.Fant F, Vranken WF, Borremans FAM. The three-dimensional solution structure of Aesculus hippocastanum antimicrobial protein 1 determined by 1H nuclear magnetic resonance. Proteins. 1999;37:388–403. doi: 10.1002/(sici)1097-0134(19991115)37:3<388::aid-prot7>3.3.co;2-6. [DOI] [PubMed] [Google Scholar]
- 8.Sun YM, Liu W, Zhu RH, Wang DC, Goudet C, Tytgat J. Roles of disulfide bridges in scorpion toxin BmK M1 analyzed by mutagenesis. J Pept Res. 2002;60:247–256. doi: 10.1034/j.1399-3011.2002.21021.x. [DOI] [PubMed] [Google Scholar]
- 9.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 10.Zhu S, Gao B, Tytgat J. Phylogenetic distribution, functional epitopes and evolution of the CSαβ superfamily. Cell Mol Life Sci. 2005;62:2257–2269. doi: 10.1007/s00018-005-5200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen G-H, Hsu M-P, Tan C-H, Sung H-Y, Kuo CG, Fan M-J, Chen H-M, Chen S, Chen C-S. Cloning and characterization of a plant fefensin VaD1 from Azuki bean. J Agric Food Chem. 2005;53:982–988. doi: 10.1021/jf0402227. [DOI] [PubMed] [Google Scholar]
- 12.Clauss MJ, Mitchell-Olds T. Functional divergence in tandemly duplicated Arabidopsis thaliana trypsin inhibitor genes. Genetics. 2004;166:1419–1436. doi: 10.1534/genetics.166.3.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong JH, Ng TB. Sesquin, a potent defensin-like antimicrobial peptide from ground beans with inhibitory activities toward tumor cells and HIV-1 reverse transcriptase. Peptides. 2005;26:1120–1126. doi: 10.1016/j.peptides.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Spelbrink RG, Dilmac N, Allen A, Smith TJ, Shah DM, Hockerman GH. Differential antifungal and calcium channel-blocking activity among structurally related plant defensins. Plant Physiol. 2004;135:2055–2067. doi: 10.1104/pp.104.040873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song X, Wang J, Wu F, Li X, Teng M, Gong W. cDNA cloning, functional expression and antifungal activities of a dimeric plant defensin SPE10 from Pachyrrhizus erosus seeds. Plant Mol Biol. 2005;57:13–20. doi: 10.1007/s11103-004-6637-y. [DOI] [PubMed] [Google Scholar]
- 16.Lobo DS, Pereira IB, Fragel-Madeira L, Medeiros LN, Cabral LM, Faria J, Bellio M, Campos RC, Linden R, Kurtenbach E. Antifungal Pisum sativum defensin 1 interacts with Neurospora crassa cyclin F related to the cell cycle. Biochemistry. 2007;46:987–996. doi: 10.1021/bi061441j. [DOI] [PubMed] [Google Scholar]
- 17.Stec B. Plant thionins—the structural perspective. Cell Mol Life Sci. 2006;63:1370–1385. doi: 10.1007/s00018-005-5574-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin KF, Lee TR, Tsai PH, Hsu MP, Chen CS, Lyu PC. Structure-based protein engineering for α-amylase inhibitory activity of plant defensin. Proteins. 2007;68:530–540. doi: 10.1002/prot.21378. [DOI] [PubMed] [Google Scholar]
- 19.Vita C, Roumestand C, Toma F, Menez A. Scorpion toxins as natural scaffolds for protein engineering. Proc Natl Acad Sci USA. 1995;92:6404–6408. doi: 10.1073/pnas.92.14.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vita C, Drakopoulou E, Vizzavona J, Rochette S, Martin L, Menez A, Roumestand C, Yang Y-S, Ylisastigui L, Benjouad A, Gluckman JC. Rational engineering of a miniprotein that reproduces the core of the CD4 site interacting with HIV-1 envelope glycoprotein. Proc Natl Acad Sci USA. 1999;96:13091–13096. doi: 10.1073/pnas.96.23.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Gaal L, Mertens I, Ballaux D, Verkade HJ. Modern, new pharmacotherapy for obesity. A gastrointestinal approach. Best Pract Res Clin Gastroenterol. 2004;18:1049–1072. doi: 10.1016/j.bpg.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 22.Thevissen K, Kristensen H-H, Thomma BPHJ, Cammue BPA, Francois IEJA. Therapeutic potential of antifungal plant and insect defensins. Drug Discov Today. 2007;12:966–971. doi: 10.1016/j.drudis.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 23.Yang Y-F, Lyu P-C. The proteins of plant defensin family and their application beyond plant disease control. Recent Pat DNA Gene Seq. 2008;2:214–218. doi: 10.2174/187221508786241684. [DOI] [PubMed] [Google Scholar]
- 24.Thomma BPHJ, Cammue BPA, Thevissen K. Mode of action of plant defensins suggests therapeutic potential. Curr Drug Targets Infect Disord. 2003;3:1–8. doi: 10.2174/1568005033342000. [DOI] [PubMed] [Google Scholar]
- 25.Vila-Perello M, Tognon S, Sanchez-Vallet A, Garcia-Olmedo F, Molina A, Andreu D. A minimalist design approach to antimicrobial agents based on a thionin template. J Med Chem. 2006;49:448–451. doi: 10.1021/jm050882i. [DOI] [PubMed] [Google Scholar]
- 26.Boman HG. Antibacterial peptides: basic facts and emerging concepts. J Intern Med. 2003;254:197–215. doi: 10.1046/j.1365-2796.2003.01228.x. [DOI] [PubMed] [Google Scholar]
- 27.Liu YJ, Cheng CS, Lai SM, Hsu MP, Chen CS, Lyu PC. Solution structure of the plant defensin VrD1 from mung bean and its possible role in insecticidal activity against bruchids. Proteins. 2006;63:777–786. doi: 10.1002/prot.20962. [DOI] [PubMed] [Google Scholar]
- 28.Lay FT, Schirra HJ, Scanlon MJ, Anderson MA, Craik J. The three-dimensional solution structure of NaD1, a new floral defensin from Nicotiana alata and its application to a homology model of the crop defense protein alfAFP. J Mol Biol. 2003;325:175–188. doi: 10.1016/s0022-2836(02)01103-8. [DOI] [PubMed] [Google Scholar]
- 29.Landon C, Vovelle F, Sodano P, Pajon A. The active site of drosomycin, a small insect antifungal protein, delineated by comparison with the modeled structure of Rs-AFP2, a plant antifungal protein. J Pept Res. 2000;56:231–238. doi: 10.1034/j.1399-3011.2000.00757.x. [DOI] [PubMed] [Google Scholar]
- 30.Cunningham B, Wells J. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 31.Pakula AA, Young VB, Sauer RT. Bacteriophage lambda cro mutations: effects on activity and intracellular degradation. Proc Natl Acad Sci USA. 1986;83:8829–8833. doi: 10.1073/pnas.83.23.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alston R, Lasagna M, Grimsley G, Scholtz J, Reinhart G, Pace C. Peptide sequence and conformation strongly influence tryptophan fluorescence. Biophys J. 2008;94:2280–2287. doi: 10.1529/biophysj.107.116921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennion BJ, Daggett V. The molecular basis for the chemical denaturation of proteins by urea. Proc Natl Acad Sci USA. 2003;100:5142–5147. doi: 10.1073/pnas.0930122100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pace CN, Treviño S, Prabhakaran E, Scholtz JM. Protein structure, stability and solubility in water and other solvents. Philos Trans R Soc Lond B Biol Sci. 2004;359:1225–1235. doi: 10.1098/rstb.2004.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Payan F. Structural basis for the inhibition of mammalian and insect α-amylases by plant protein inhibitors. Biochim Biophys Acta. 2004;1696:171–180. doi: 10.1016/j.bbapap.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 36.Franco OL, Rigden DJ, Melo FR, Grossi-de-Sa MF. Plant α-amylase inhibitors and their interaction with insect α-amylases: structure, function and potential for crop protection. Eur J Biochem. 2002;269:397–412. doi: 10.1046/j.0014-2956.2001.02656.x. [DOI] [PubMed] [Google Scholar]
- 37.Vance JE, LeBlanc DA, London RE. Cleavage of the X-Pro peptide bond by pepsin is specific for the trans isomer. Biochemistry. 1997;36:13232–13240. doi: 10.1021/bi970918b. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Jing L, Xu K. A unique approach for high level expression and production of a recombinant cobra neurotoxin in Escherichia coli. J Biotechnol. 2002;94:235–244. doi: 10.1016/s0168-1656(01)00429-1. [DOI] [PubMed] [Google Scholar]
- 39.Cheng C-S, Chen M-N, Lai Y-T, Chen T, Lin K-F, Liu Y-J, Lyu P-C. Mutagenesis study of rice nonspecific lipid transfer protein 2 reveals residues that contribute to structure and ligand binding. Proteins. 2008;70:695–706. doi: 10.1002/prot.21520. [DOI] [PubMed] [Google Scholar]
- 40.Samuel D, Liu Y-J, Cheng C-S, Lyu P-C. Solution structure of plant nonspecific lipid transfer protein-2 from rice (Oryza sativa) J Biol Chem. 2002;277:35267–35273. doi: 10.1074/jbc.M203113200. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y-J, Samuel D, Lin C-H, Lyu P-C. Purification and characterization of a novel 7-kDa non-specific lipid transfer protein-2 from rice (Oryza sativa) Biochem Biophys Res Commun. 2002;294:535–540. doi: 10.1016/S0006-291X(02)00509-0. [DOI] [PubMed] [Google Scholar]
- 42.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 44.Lu G, Moriyama EN. Vector NTI, a balanced all-in-one sequence analysis suite. Brief Bioinform. 2004;5:378–388. doi: 10.1093/bib/5.4.378. [DOI] [PubMed] [Google Scholar]