

Abstract
Hair morphology is highly differentiated between populations and among people of European ancestry. Whereas hair morphology in East Asian populations has been studied extensively, relatively little is known about the genetics of this trait in Europeans. We performed a genome-wide association scan for hair morphology (straight, wavy, curly) in three Australian samples of European descent. All three samples showed evidence of association implicating the Trichohyalin gene (TCHH), which is expressed in the developing inner root sheath of the hair follicle, and explaining ∼6% of variance (p = 1.5 × 10−31). These variants are at their highest frequency in Northern Europeans, paralleling the distribution of the straight-hair EDAR variant in Asian populations.
Main Text
Hair morphology is one of the more conspicuous features of human variation and is particularly diverse among people of European ancestry, for which around 45% of individuals have straight hair, 40% have wavy hair, and 15% have curly hair.1 The degree of curliness is correlated with the distribution of hair keratins and cell type within the hair fiber, with the number of mesocortical cells decreasing as the curl intensifies.2 Recent studies have identified Asian-specific alleles of the EDAR and FGFR2 genes that are associated with thick, straight hair, suggesting that these variants arose after the divergence of Asians and Europeans.3,4 However, the genetic variants influencing hair curliness in Europeans (which has been shown to be highly heritable5) are unknown.
We conducted genome-wide association analyses in three Australian family samples: one sample of adolescent twins and their siblings (1649 individuals from 837 families) and two samples of adult twin pairs (S1, 1945 individuals from 1210 families; S2, 1251 individuals from 845 families) ascertained from the general population (Table 1).5 In the adolescent sample, hair curliness was rated on a three-point scale (Straight, Wavy, or Curly). In the adult samples, participants reported whether their hair was Straight or Curly (S1) or Straight, Wavy, or Curly (S2). To account for the differences in phenotype collection and age across the samples, each sample was analyzed independently and meta-analysis was used for combining the three sets of results. These studies were performed with the approval of the appropriate ethics committees and the informed consent of all participants.
Table 1.
Characteristics of the Three Cohorts
| Cohort 1 | Cohort 2 | Cohort 3 | |
|---|---|---|---|
| Birth years | 1982–1996 | 1903–1964 | 1965–1972 |
| Age mean (range) | 14 (9–24) | 47 (29–94) | 39 (30–42) |
| No. of females | 830 | 1483 | 697 |
| No. of males | 819 | 462 | 554 |
| Percentage of females with straight hair | 63.5% | 72.6% | 49.1% |
| Percentage of males with straight hair | 71.5% | 69.3% | 56.5% |
| Percentage of females with wavy hair | 27.9% | - | 35.7% |
| Percentage of males with wavy hair | 21.6% | - | 30.9% |
| Percentage of females with curly hair | 8.6% | 27.4% | 15.2% |
| Percentage of males with curly hair | 6.9% | 30.7% | 12.6% |
The genotypic data used in the current study derives from a larger genotyping project involving seven waves of genotyping that drew participants from our 1988 and 1990 adult health and lifestyle studies6 and adolescent melanoma risk factors study.7,8 The genotypic data from each project are described in Table 2. Standard quality-control filters were applied to the genotyping from each project, restricting the imputation to samples and SNPs with high data quality (Table 2). Individuals were screened for non-European ancestry, resulting in a sample of 16,140 genotyped individuals (Figure S2, available online). So that bias was not introduced to the imputed data, a set of SNPs common to the seven subsamples was used for imputation (n = 274,604). Imputation was undertaken with the use of the phased data from the HapMap samples of European ancestry (CEU; build 36, release 22) and MACH.9
Table 2.
Summary Information for the Seven Waves of Genotyping and the Quality Control Undertaken
| Project 1: ALCO CIDR | Project 2: ALCO deCODE | Project 3: MIG deCODE | Project 4: EUTWIN | Project 5: ADOL deCODE | Project 6: GL_CIDR | Project 7: WH deCODE | |
|---|---|---|---|---|---|---|---|
| Primary phenotype | Alcohol use (population sample) | Alcohol use (population sample) | Migraine (case/control sample) | Lipid levels (population sample) | Melanoma risk factors (population sample) | Glaucoma (population sample) | Womens' health (case/control sample) |
| Genotyping lab | CIDR | deCODE | deCODE | University of Helsinki | deCODE | CIDR | deCODE |
| Illuminia SNP platform | HumanCNV370-Quadv3 | HumanCNV370-Quadv3 | Human610-Quad | Human 317K | Human610-Quad | Human610-Quad | Human610-Quad |
| No. of genotyped samples | 4241 | 2611 | 999 | 462 | 4391 | 657 | 2360 |
| No. of genotyped SNPs | 343,955 | 344,962 | 592,385 | 318,210 | 592,392 | 589,296 | 562,193 |
| BeadStudio GenCall score < 0.7 | 24,494 | 27,459 | 46,931 | NAa | 47,418 | 36,877 | 57,589 |
| SNPs with call rate < 0.95 | 11,584 | 7537 | 8038 | 5021 | 8447 | 12,455 | 33,459 |
| SNPs with HWE failure p < 10−6 | 4318 | 1194 | 1221 | 67 | 2841 | 15,474 | 1763 |
| SNPs with MAF < 0.01/ only 1 observed allele | 7874 | 8976 | 33,347 | 264 | 33,347 | 28,607 | 24,509 |
| No. of SNPs after QC | 323093 | 321,267 | 530,922 | 312,937 | 529,379 | 531,042 | 518,948 |
| Percentage of genotyped SNPs | 93.93% | 93.13% | 89.62% | 98.34% | 89.36% | 90.11% | 92.31% |
For each project, DNA was extracted in accordance with standard protocols. Across projects, participants were genotyped on the Illumina 317K, 370K, or 610K SNP platforms, and genotypes were called with the Illumina BeadStudio software. After the quality control (QC) of the individual projects, the data from the seven waves of genotyping were integrated. As shown in Figure S1, a number of samples were duplicated among the various genotyping projects, allowing for cross-project QC. After integration of the data sets, the data were screened for missingness within individuals (>5%, taking into account the number of SNPs that were genotyped for each individual), pedigree and sex errors, and Mendelian errors (genotypes for all family members for a given SNP were removed upon detection of errors). After QC, in cases where one individual from a monozygotic twin pair had been genotyped, duplicate genotypes were assigned to the ungenotyped cotwin, resulting in a sample of 16,507 individuals. After screening for non-European ancestry (Figure S2), this resulted in a final sample of 16,140 individuals. HWE denotes Hardy-Weinburg equilibrium.
GenCall data were not available for this sample.
So that we could take full advantage of the information available in the ordinal scale, the data were analyzed via a multifactorial threshold model that describes discrete traits as reflecting an underlying normal distribution of liability (or predisposition). Liability, which represents the sum of all the multifactorial effects, is assumed to reflect the combined additive effects of a large number of genes and environmental factors, each of small effect, and is characterized by phenotypic discontinuities that occur when the liability reaches a given threshold.10 A total test of association was used, in which the dosage (MACH mldose) data for each SNP in turn were included within the threshold model, resulting in an additive test of association. In addition, fixed effects of sex and age (both linear and quadratic effects) and age-by-sex interactions were included with the threshold models in all data analyses, such that the trait value for individual j from family i was parameterized as: . The relatedness between the participants was explicitly modeled, accounting for the sex of relative pairs, and the phenotypic variances were constrained to unity. The association test statistic was computed by comparing the fit (minus twice log-likelihood) of the full model, which included the effect of the given SNP, to that of a nested model, in which the SNP effect had been dropped from the model. The difference in log-likelihoods follows an asymptotic chi-square distribution with the degrees of freedom equal to the difference in estimated parameters between the two models (in this case one). The genomic inflation factors of the three samples ranged from 0.98 to 1.02 (Figure S3), indicating that the test correctly controlled for the relatedness of the participants and that potential technical and stratification artifacts had a negligible impact on the results.
Four highly correlated single-nucleotide polymorphisms (SNPs) (rs17646946, rs11803731, rs4845418, rs12130862; r2 > 0.8, D′ > 0.95 within the HapMap CEU sample) on chromosome 1q21.3 (Figure 1B) reached our genome-wide significance threshold of 5 × 10−8, which corrects for ∼1 million independent common variants in the genome11 (Table 3, Figure S4). The association was found in all three samples, suggesting that the effect is robust to the age differences between the samples and that the liability threshold model accounted for the differences in the phenotypic definition across the samples. Meta-analysis of the three samples using an N (individuals)-weighted analysis in Metal (see Web Resources) resulted in highly significant p values for SNPs that are within this region and fall on a haplotype tagged by the directly genotyped SNP rs17646946 (p = 1.5 × 10−31) (Figures 1A and 1B, Table 3). The association in the 1q21.3 region is centered on the Trichohyalin gene TCHH and accounted for ∼6% of the variance (Figure 1E, Table 3). Further analysis showed that association at the haplotype level did not offer additional predictive power. Including the best SNPs as covariates in the analyses yielded no further evidence of association, completely accounting for the signal at this locus (Figure S5). We found neither evidence for epistasis between these SNPs and any other SNP across the genome nor any heterogeneity between sexes (Figures S5 and S6). Finally, although analysis of copy-number variation (CNV) across the region in the adolescent sample found evidence for CNV in 18 individuals, it was too infrequent to explain the observed effect (Table S2). Table S3 lists all SNPs with a combined p value of less than 1 × 10−5 for the meta-analysis. A second region of suggestive association was observed on chromosome 4q21.21 (rs1268789; p = 6.58 × 10−8), centered on the Fraser syndrome 1 gene FRAS1. We also examined evidence for association within the meta-analysis for the list of 170 candidate genes published by Fujimoto et al.4 (Table S4). In addition to the association observed in the TCHH region, strong association signals were observed in WNT10A, associated with odonto-onycho-dermal dysplasia, which is characterised by dry hair and a broad range of ectodermal phenotypes12 (2q35; rs7349332; p = 1.36 × 10−6).
Figure 1.
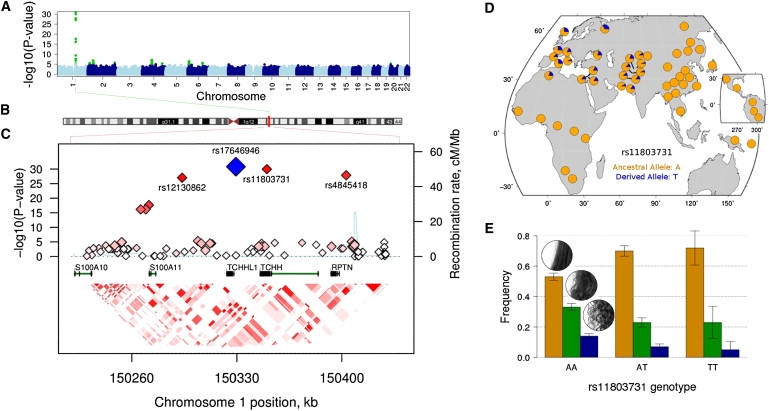
Genome-wide Association Results
(A) Manhattan plot showing the results for the genome-wide meta-analysis of hair morphology across three independent samples. SNPs with a p < 10−5 are highlighted in green.
(B) Karyotype of chromosome 1 highlighting the 1q21 region.
(C) Regional association and linkage disequilibrium plot for the 1q21 region. The most-associated genotyped SNP is shown in blue, and the color of the remaining markers reflects the linkage disequilibrium (r2) with the top SNP in each panel (increasing red hue associated with increasing r2). The recombination rate (right-hand y axis) is plotted in light blue and is based on the CEU HapMap population. Exons for each gene are represented by vertical bars, based on all isoforms available from the March 2006 UCSC Genome Browser assembly.
(D) Minor allele frequency for the TCHH SNP rs11803731, based on the Human Genome Diversity Project.29
(E) Frequency of straight (orange bars), wavy (green bars), and curly (blue bars) hair as a function of the rs11803731 genotype in a sample of unrelated individuals (n[AA] = 43; n[AT] = 493; n[TT] = 1132). With more T alleles, the proportion of straight hair increases. Vertical bars correspond to the 95% confidence intervals on the prevalence.
Table 3.
Details of the Genome-wide Significant SNPs in the 1q21.3 Region across the Three Samples
| rs17646946 | rs11803731 | rs4845418 | rs12130862 | |
|---|---|---|---|---|
| Position (bp) | 150,329,391 | 150,349,949 | 150,402,854 | 150,293,639 |
| Genotyped or imputed | genotyped | imputed | imputed | imputed |
| Minor (reference) allele | A | T | C | T |
| Major allele | G | A | G | A |
| Minor allele frequency | 18.3% | 18.4% | 17.3% | 18.1% |
| Hardy-Weinburg equilibrium p value | 0.73 | 0.75 | 0.70 | 0.79 |
| Rsq (imputation accuracy metric) | - | 0.92 | 0.96 | 0.98 |
| Adolescent sample (n = 1649) | ||||
| Allelic effect (β)a | 0.41 | 0.42 | 0.42 | 0.39 |
| P-value | 1.24 × 10−11 | 1.76 × 10−11 | 3.68 × 10−11 | 7.59 × 10−11 |
| Adult S1 (n = 1945) | ||||
| Allelic effect (β)a | 0.50 | 0.50 | 0.49 | 0.45 |
| P-value | 8.18 × 10−13 | 2.51 × 10−12 | 2.22 × 10−11 | 9.69 × 10−11 |
| Adult S2 (n = 1251) | ||||
| Allelic effect (β)a | 0.44 | 0.44 | 0.43 | 0.42 |
| P-value | 7.91 × 10−11 | 1.37 × 10−10 | 1.15 × 10−9 | 8.16 × 10−10 |
| Cross-Sample Calculations | ||||
| Explained varianceb averaged across samples | 6.11% | 6.11% | 5.79% | 5.22% |
| Meta analysis (p value) | 1.50 × 10−31 | 3.18 × 10−31 | 4.43 × 10−29 | 3.12 × 10−28 |
The allelic β reported here should be interpreted with reference to the liability threshold model, which maps the data onto a standard normal distribution in which the cutpoints between categories are mapped against the z distribution. For example, a β of .41 indicates that the threshold dividing the straight from wavy categories is moved 0.41 z units to the right for each risk allele that an individual possesses.
Calculated as [2p(1 − p)]β2, in which p is the minor allele frequency and β is the additive allelic effect.
Of the four most-associated SNPs in the 1q21 region, we focused on rs11803731 (p = 3.2 × 10−31), because this is a coding, nonsynonymous variant located in the third exon of TCHH (although more work is required for confirmation that this is the causal variant). The T allele at rs11803731 is the derived state and shows a striking geographic specificity to Europe and western-central Asia, reaching its highest frequency in Northern Europeans (Figure 1E), suggesting that the variant arose somewhere in this broad region. The modern frequency and distribution of de novo mutations will generally be determined by random genetic drift and migration. However, because rs11803731 influences a highly visible phenotype, it is an intuitively obvious target for natural or sexual selection.13 The EDAR gene that controls hair thickness shows one of the most convincing signatures of positive selection in the East Asian genome.4 rs11803731 is among the top 2.5% most-differentiated SNPs across the genome between Europeans and other Hapmap II populations (gauged by the FST-based locus-specific branch length test14). Although previous analysis of extended haplotype homozygosity patterns in the Human Genome Diversity Project (HGDP) cohort also shows tentative evidence of genetic hitchhiking for the 1q21.3 region in some European populations (ref. 15 and Figure S7), the overall evidence of selection is ambiguous. However, the genetic signatures of positive selection at individual loci, as detected with current tests, will vary depending on the timing, the strength of the selective event, the genomic characteristic of the region, and the genetic architecture (number, frequency, and effect size of causal loci) of the phenotype in question16 and thus may not be as obvious as those associated with other superficial traits. For example, the OCA2 gene region is a well-established target of selection17 and is known to influence human pigmentation traits, especially eye color.18,19
The effect of the rs11803731 variant, replacement of a leucine by a methionine at position 790 of the TCHH protein, was predicted by in silico analyses with the programs PolyPhen20 and PMut.21 PolyPhen predicted the L790M change to be “benign,” whereas PMut predicted this change to be “neutral.” No results were returned for other prediction programs, including SIFT22 (see Web Resources) (with either the SNP ID or the protein sequence used) and SNPs3D23 (see Web Resources) which contained no record of the rs11803731 SNP. Such predictions do not preclude a functional role, given that the effect of the SNP may be regulatory rather than structural, particularly as the amino acid replacement falls outside of α-helical regions.23 Surface-exposed methionines can be oxidized by reactive oxygen species posttranslationally, which if left unrepaired can result in changes in protein structure and activity and can lead to altered protein regulation.24
Alternatively, rs11803731 may be associated with structural variation. TCHH is a single-stranded α-helical protein with two or three highly repetitive regions, depending on the species (Figure S8). In sheep, the reference protein (CAA79165.1) is 1549 amino acids long, but variation in the number of complete and partial repeats in the C-terminal repeat region is seen across different strains.25,26 In the human TCHH protein, repeat lengths range from approximately 6 to 30 amino acids, corresponding to 18 to 90 bp of DNA sequence. A number of SNPs and insertion or deletion polymorphisms are present, particularly in the first and third repeat regions (dbSNP; see Web Resources), and this gene might harbor allelic length variants, as seen in sheep and in another highly repetitive gene in the human chromosome 1p21 region, involucrin (IVL), where alleles differ across human populations in the number of both short tandem repeats and single base changes within repeated sequence.27,28 Such length variation has not been reported for TCHH, and it remains to be determined experimentally whether such variation is common, affects the structure or length of the protein, and/or is tagged by SNPs flanking the repeated regions.
In conclusion, we report a quantitative trait locus that affects hair form in Europeans. The association accounts for ∼6% of the variance in hair morphology in this group and falls within the Trichohyalin gene, which has a known role in hair formation. The patterns of allele frequencies are striking, with the highest frequency of these variants observed in Northern Europeans (Figure 1), paralleling the observation of the straight-hair EDAR variant in Asian populations (Figure S9).
Acknowledgments
We thank the twins and their families for their participation. We also thank Dixie Statham, Ann Eldridge, Marlene Grace, Kerrie McAloney (sample collection); Lisa Bowdler, Steven Crooks (DNA processing); David Smyth, Harry Beeby, and Daniel Park (IT support). Funding was provided by the Australian National Health and Medical Research Council (241944, 339462, 389927, 389875, 389891, 389892, 389938, 442915, 442981, 496739, 552485, 552498), the Australian Research Council (A7960034, A79906588, A79801419, DP0770096, DP0212016, DP0343921), the FP-5 GenomEUtwin Project (QLG2-CT-2002-01254), and the U.S. National Institutes of Health (NIH grants AA07535, AA10248, AA13320, AA13321, AA13326, AA14041, MH66206). A portion of the genotyping on which this study was based (Illumina 370K scans on 4300 individuals) was carried out at the Center for Inherited Disease Research, Baltimore (CIDR), through an access award to our late colleague Dr. Richard Todd (Psychiatry, Washington University School of Medicine, St Louis). Statistical analyses were carried out on the Genetic Cluster Computer, which is financially supported by the Netherlands Scientific Organization (NWO 480-05-003). S.E.M., D.R.N., A.F.M., M.A.R.F., S.M., D.L.D., and G.W.M. are supported by the National Health and Medical Research Council (NHMRC) Fellowship Scheme.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes browser, http://www.1000genomes.org
Genetic Cluster Computer, http://www.geneticcluster.org
Metal, http://www.sph.umich.edu/csg/abecasis/Metal/index.html
PolyPhen, http://genetics.bwh.harvard.edu/pph/
SNPs3D, http://www.snps3d.org/
Sorting Intolerant from Tolerant (SIFT), http://sift.jcvi.org/
References
- 1.Loussouarn G., Garcel A.L., Lozano I., Collaudin C., Porter C., Panhard S., Saint-Leger D., de La Mettrie R. Worldwide diversity of hair curliness: a new method of assessment. Int. J. Dermatol. 2007;46(Suppl 1):2–6. doi: 10.1111/j.1365-4632.2007.03453.x. [DOI] [PubMed] [Google Scholar]
- 2.Thibaut S., Barbarat P., Leroy F., Bernard B.A. Human hair keratin network and curvature. Int. J. Dermatol. 2007;46(Suppl 1):7–10. doi: 10.1111/j.1365-4632.2007.03454.x. [DOI] [PubMed] [Google Scholar]
- 3.Fujimoto A., Nishida N., Kimura R., Miyagawa T., Yuliwulandari R., Batubara L., Mustofa M.S., Samakkarn U., Settheetham-Ishida W., Ishida T. FGFR2 is associated with hair thickness in Asian populations. J. Hum. Genet. 2009;54:461–465. doi: 10.1038/jhg.2009.61. [DOI] [PubMed] [Google Scholar]
- 4.Fujimoto A., Kimura R., Ohashi J., Omi K., Yuliwulandari R., Batubara L., Mustofa M.S., Samakkarn U., Settheetham-Ishida W., Ishida T. A scan for genetic determinants of human hair morphology: EDAR is associated with Asian hair thickness. Hum. Mol. Genet. 2008;17:835–843. doi: 10.1093/hmg/ddm355. [DOI] [PubMed] [Google Scholar]
- 5.Medland S.E., Zhu G., Martin N. Estimating the heritability of hair curliness in twins of European ancestry. TRHG. 2009;12:514–518. doi: 10.1375/twin.12.5.514. [DOI] [PubMed] [Google Scholar]
- 6.Hansell N.K., Agrawal A., Whitfield J.B., Morley K.I., Zhu G., Lind P.A., Pergadia M.L., Madden P.A., Todd R.D., Heath A.C. Long-term stability and heritability of telephone interview measures of alcohol consumption and dependence. Twin Res. Hum. Genet. 2008;11:287–305. doi: 10.1375/twin.11.3.287. [DOI] [PubMed] [Google Scholar]
- 7.Zhu G., Duffy D.L., Eldridge A., Grace M., Mayne C., O'Gorman L., Aitken J.F., Neale M.C., Hayward N.K., Green A.C. A major quantitative-trait locus for mole density is linked to the familial melanoma gene CDKN2A: a maximum-likelihood combined linkage and association analysis in twins and their sibs. Am. J. Hum. Genet. 1999;65:483–492. doi: 10.1086/302494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu G., Montgomery G.W., James M.R., Trent J.M., Hayward N.K., Martin N.G., Duffy D.L. A genome-wide scan for naevus count: linkage to CDKN2A and to other chromosome regions. Eur. J. Hum. Genet. 2007;15:94–102. doi: 10.1038/sj.ejhg.5201729. [DOI] [PubMed] [Google Scholar]
- 9.Li Y., Abecasis G. Mach 1.0: Rapid haplotype reconstruction and missing genotype inference. Am. J. Hum. Genet. 2006;S79:2290. [Google Scholar]
- 10.Neale M.C., Cardon L.R. Kluwer Academic Publishers; Dordrecht, the Netherlands: 1992. Methodology for Genetic Studies of Twins and Families. [Google Scholar]
- 11.International Hap Map Consortium The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 12.Adaimy L., Chouery E., Megarbane H., Mroueh S., Delague V., Nicolas E., Belguith H., de Mazancourt P., Megarbane A. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: the odonto-onycho-dermal dysplasia. Am. J. Hum. Genet. 2007;81:821–828. doi: 10.1086/520064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chernova O. Evolutionary aspects of hair polymorphism. Biol. Bull. 2006;33:43–52. [PubMed] [Google Scholar]
- 14.Shriver M.D., Kennedy G.C., Parra E.J., Lawson H.A., Sonpar V., Huang J., Akey J.M., Jones K.W. The genomic distribution of population substructure in four populations using 8,525 autosomal SNPs. Hum. Genomics. 2004;1:274–286. doi: 10.1186/1479-7364-1-4-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pickrell J.K., Coop G., Novembre J., Kudaravalli S., Li J.Z., Absher D., Srinivasan B.S., Barsh G.S., Myers R.M., Feldman M.W. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sabeti P.C., Schaffner S.F., Fry B., Lohmueller J., Varilly P., Shamovsky O., Palma A., Mikkelsen T.S., Altshuler D., Lander E.S. Positive natural selection in the human lineage. Science. 2006;312:1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- 17.Voight B.F., Kudaravalli S., Wen X., Pritchard J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duffy D.L., Montgomery G.W., Chen W., Zhao Z.Z., Le L., James M.R., Hayward N.K., Martin N.G., Sturm R.A. A three-single-nucleotide polymorphism haplotype in intron 1 of OCA2 explains most human eye-color variation. Am. J. Hum. Genet. 2007;80:241–252. doi: 10.1086/510885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sturm R.A., Duffy D.L., Zhao Z.Z., Leite F.P., Stark M.S., Hayward N.K., Martin N.G., Montgomery G.W. A single SNP in an evolutionary conserved region within intron 86 of the HERC2 gene determines human blue-brown eye color. Am. J. Hum. Genet. 2008;82:424–431. doi: 10.1016/j.ajhg.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrer-Costa C., Gelpí J., Zamakola L., Parraga I., de la Crux X., Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- 22.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee S.C., Kim I., Marekov L., O'Keefe E., Parry D., Steiner P. The structure of human trichohyalin. J. Biol. Chem. 1993;268:12164–12174. [PubMed] [Google Scholar]
- 24.Stadtman E.R., Moskovitz J., Levine R. Oxidation of methionine residues of proteins: biological consequences. Antioxid. Redox Signal. 2003;5:577–582. doi: 10.1089/152308603770310239. [DOI] [PubMed] [Google Scholar]
- 25.Fietz M.J., McLaughlan C.J., Campbell M.T., Rogers G.E. Analysis of the sheep trichohyalin gene: potential structural and calcium-binding roles of trichohyalin in the hair follicle. J. Cell Biol. 1993;121:855–865. doi: 10.1083/jcb.121.4.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLaren R.J., Rogers G.R., Davies K.P., Maddox J.F., Montgomery G.W. Linkage mapping of wool keratin and keratin-associated protein genes in sheep. Mamm. Genome. 1997;8:938–940. doi: 10.1007/s003359900616. [DOI] [PubMed] [Google Scholar]
- 27.Djian P., Delhomme B., Green H. Origin of the polymorphism of the involucrin gene in Asians. Am. J. Hum. Genet. 1995;56:1367–1372. [PMC free article] [PubMed] [Google Scholar]
- 28.Urquhart A., Gill P. Tandem-repeat internal mapping (TRIM) of the involucrin gene: repeat number and repeat-pattern polymorphism within a coding region in human populations. Am. J. Hum. Genet. 1993;53:279–286. [PMC free article] [PubMed] [Google Scholar]
- 29.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.