

Abstract
Human immunodefi ciency virus type-1 integrase (IN) is a new and novel target for inhibitors. Strand transfer inhibitors effectively prevent concerted integration of viral DNA by IN into the host chromosomes. Raltegravir is the fi rst approved strand transfer inhibitor for the treatment of HIV-1/AIDS. We propose a mechanistic hypothesis as to “when and where” these inhibitors are active in virus-infected cells. Using native agarose gel electrophoresis, we identified a transient synaptic complex (SC) wherein IN non-covalently juxtaposes two viral DNA ends. SC possesses many properties associated with the cytoplasmic preintegration complex (PIC) in infected cells, including concerted integration. Our results show that the strand transfer inhibitors effectively “trap” or inactivate the SC preventing concerted integration. It follows that the IN-viral DNA complex is “trapped” by the inhibitors via a transient intermediate within the cytosolic PIC before entry into the nucleus.
Introduction
Acquired immunodeficiency syndrome (AIDS), caused by human immunodeficiency virus type-1 (HIV-1), has been a serious public health problem for approximately 25 years. There are about 34 million people infected with HIV-1 worldwide. Despite the significant advances in understanding of the biology and pathogenesis of HIV-1, efforts have not been universally successful to prevent its spread. Educational programs on how to prevent new infections have had limited success on a global basis, although there are numerous local successful programs. However, the spread can also be attributed to unique characteristics of the virus and its high mutation rate which has hindered the development of an effective vaccine. HAART (highly active antiretroviral therapy) has been very successful in the treatment of infected individuals and preventing infection of new born children. Our review will focus on the development and mode of action of inhibitors to a novel target, the HIV-1 integrase (IN).
The HIV-1 polymerase gene encodes a polyprotein which upon proteolytic processing yields three viral enzymes; reverse transcriptase, protease and IN, all of which are essential for viral replication. Integration of HIV-1 DNA by IN into the cellular genome is an obligatory step in the retrovirus life cycle resulting in permanent infection and establishment of a latent proviral reservoir for virus production. Integration mediated by IN requires two independent steps. In the cytoplasm, IN binds to the ends of reverse transcribed blunt-ended DNA and processes the 3′ends by removing two nucleotides (GT) adjacent to conserved CA motif, exposing the active 3′-hydroxyl group. In the nucleus, the second reaction occurs when IN covalently joins the processed viral DNA ends by a concerted integration mechanism into the cellular genome [1]. Due to the unique nature of IN and the absence of a cellular counterpart, IN is an obvious target for drug development. Intense efforts by Merck and Co resulted in development of first FDA approved drug Raltegravir (marketed as “Isentress”) targeting HIV-1 IN (http://www.fda.gov/bbs/topics/news/2007/new01726.html). Raltegravir is a strand transfer inhibitor which blocks the joining of the processed viral DNA ends into the host chromosome. Raltegravir has been successful in naïve as well as patients who have undergone HAART and harbor strains resistant to combinations of drugs targeting reverse transcriptase and protease [2, 3]. This article discusses our recent results to understand the mechanisms of strand transfer inhibitors that prevent the formation of concerted integration products using a reconstituted system employing purified HIV-1 IN and viral DNA substrates. Based on our results, we propose a hypothesis as to “when and where” these inhibitors are active in infected cells.
HIV-1 IN contains 288 amino acids (aa) and is composed of three structural domains. The N-terminal domain (1–50 aa) contains a histidine-histidine-cysteine-cysteine (HHCC) motif involved in zinc binding [4]. This domain is associated with multimerization of IN and enhances its catalytic activity. The catalytic core domain (CCD) (50–212 aa) contains the catalytic D, D-35-E motif located at positions 64, 116 and 152, respectively. The D, D-35-E motif is conserved in all retrovirus and retrotransposons INs [5]. Mutations in this motif abolish the activity of IN and viral replication [6]. The C-terminal domain (213–288 aa) makes specific contacts with viral DNA in a IN-DNA complex but also binds non-specific DNA [7]. There are no crystal structures available for full-length IN or IN complexed with DNA substrates presumably due to a flexible loop in CCD and low solubility of the protein. Individual as well as two domain structures consisting of N-terminal domain with CCD and CCD with C-terminal domain have been solved [8–10].
The first major advancement in the development of IN inhibitors came from Merck and Co in the form of diketo acid inhibitors L-731,988 and L-708,906 [11]. These compounds inhibited the strand transfer reaction catalyzed by recombinant IN using blunt-ended oligonucleotides at IC50s of 80 and 150 nM for L-731,988 and L-708,906, respectively, while the IC50s were 1 to 2 μM for inhibition of HIV-1 replication in single-cycle assays. These inhibitors specifically inhibited the strand transfer reaction catalyzed by IN. The IC50 for inhibition of 3′OH processing by recombinant IN was ∼70 times (∼6000 nM) higher for L-731,988. The next generation of successful inhibitors were the naphthyridine carboxamide inhibitors that employed a similar active diketo acid moiety (Fig. 1). The diketo acid group chelates Mg++ in the active site of IN required for the catalysis [12]. L-870,810 and L-870,812 were the two prominent successful members of the naphthyridine carboxamide family for inhibiting viral integration in infected cells [13] and in infected rhesus macaques [14], respectively. L-870,810 and L-870,812 also had low IC50s of 8 nM and 40 nM, respectively, for inhibiting strand transfer using oligonucleotides substrates with recombinant IN [13, 14].
Figure 1.

Chemical structures of IN strand transfer inhibitors used in this study. L-841,411 belongs to the diketo acid family. L-870,810 and L-870,812 are the members of naphthyridine carboxamide family. MK-0518, the first FDA approved inhibitor targeting IN is also shown and belongs to the hydroxypyrimidinone carboxamide group. The diverse class of IN strand transfer inhibitors contain the diketo acid moiety (shown in blue) which interacts with Mg++ in the IN active site.
Discussion
Our studies with the diketo acid and naphthyridine carboxamide strand transfer inhibitors have possibly provided a new understanding of their mode of action. We investigated how these inhibitors selectively inhibit concerted integration by the use of the synaptic complex (SC) on native agarose gels [15]. IN juxtaposes two viral blunt-ended DNA molecules in a non-covalent manner in the SC (Fig. 2) which mimics properties associated with the cytoplasmic preintegration complex (PIC) found in HIV-1 infected cells. SC is the transient intermediate in the concerted integration pathway in vitro similar to the cytosolic PIC, which eventually migrates to the nucleus and preferentially integrates into active transcription units [16]. Within SC and cytosolic PIC, the viral DNA ends are slowly processed by IN while strand transfer activity is constrained within both complexes. SC is converted to the strand transfer complex (STC) upon binding to supercoiled DNA target [15, 17]. The STC contains the concerted integration product (Fig. 2), the result of concerted insertion of two donor molecules in the target DNA [17, 18] which mimics the same integration reaction in vivo. Integration of viral DNA into the target DNA results in the characteristic HIV-1 5 bp host-site duplication at the sites of insertion [17, 19–21]. IN binds to the terminal 32 nucleotides at the viral ends in SC and STC as revealed by DNaseI footprinting of the complexes suggesting a specific interaction of IN with the long terminal repeat (LTR) ends (manuscript in preparation).
Figure 2.
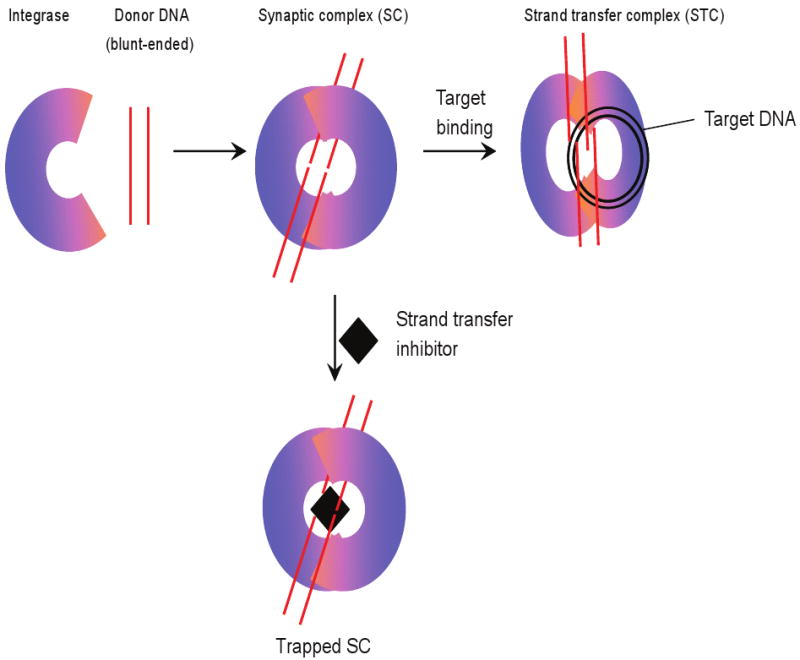
Mechanism of IN strand transfer inhibitors. IN juxtaposes two viral DNA ends forming the synaptic complex (SC). The viral DNA is processed by IN cleaving the terminal dinucleotide (GT) from their 3′ OH ends. Processing of the viral DNA induces a conformational change in SC which facilitates the binding of target DNA, forming the strand transfer complex. However, when SC is formed in presence of a strand transfer inhibitor, the inhibitor binds near the catalytic site which prevents target binding. The 3′ OH processing of the viral DNA ends is not affected by inhibitor at low nM concentrations. The inhibitor alters the structure of SC rendering it catalytically inactive and results in the accumulation of the “trapped” SC, analogous to the inhibitor inactivated preintegration complex in vivo.
With blunt-ended DNA substrates, concerted integration is inhibited by L-870,810 and L-870,812 with IC50s of 55 and 102 nM, respectively [15]. The IC50 value for concerted integration is different from the IC95 values obtained in vivo for L-870,810. L-870,810 has an IC95 of 15 nM in presence of 10% FBS. The reason for this discrepancy in the potency of inhibition in vivo and in vitro could be due to several possibilities. The simplest explanation would be that concerted integration in vitro may not recapitulate the in vivo integration process in some aspects, particularly the number of active complexes in both cases. We have observed that the IC50 of L-870,810 is dependent on the IN concentration which is presumably related to the number of active complexes at a given concentration [15]. Secondly, concerted integration in the reconstituted system involves concerted insertion of two separate DNA molecules (1.6 kbp in length) into target DNA while in vivo, it is a unimolecular reaction where the PIC inserts two ends of a single viral DNA molecule prepackaged within this nucleoprotein complex into chromosomes. However, the IC50 value obtained with L-870,812 (102 nM) matches reasonably well with the IC95 value of 250 nM in vivo [14].
These strand transfer inhibitors are significantly less effective with the use of 3′OH pre-processed substrates, which are effectively recruited by IN for near immediate strand transfer reactions since no 3′OH processing is necessary with this pre-processed substrate for concerted integration [15, 22]. The IC50s for inhibiting the concerted integration product formed with pre-processed substrates are higher; 1400 nM and 9600 nM for L-870,810 and L-870,812, respectively. Similar results were obtained with diketo acid L-841,411; IC50s for inhibition of concerted integration were 110 nM and 4100 nM with blunt and pre-processed substrates, respectively [15].
The reason(s) for paradoxical nature of preferential inhibition of concerted integration using blunt-ended DNA as substrate is not clear. However, it suggests that inhibitors interact significantly differently within SC formed with blunt and pre-processed substrates in vitro. Here, it is important to point out again that after viral entry into the cell and reverse transcription of the viral RNA genome in the cytoplasm, the resulting DNA in the PIC is blunt-ended. The 3′OH processing of the viral DNA is a slow and time-dependent process and by the time the PIC enters the nucleus, approximately 70%–90% of the viral DNA molecules have processed ends [23, 24]. Hence, in cells infected with HIV-1, the inhibitor first encounters PICs that contain blunt-ended DNA which is undergoing 3′OH processing. Similar IC50s obtained in inhibiting concerted integration in vitro as well as inhibiting the HIV-1 replication in vivo suggest that IN complexed with blunt-ended DNA undergoing 3′OH processing is the preferred nucleoprotein complex for inhibitor binding to IN.
In support of our data, cytoplasmic PIC isolated from HIV-1 infected cells grown for 5 h in presence of L-731,988 was found to be catalytically inactive [11]. The results suggested that the inhibitor had modified the PIC before entry into the nucleus and rendered the cytoplasmic PIC inactive. As stated previously, these inhibitors do not affect 3′OH processing of the viral DNA ends either in vivo or within the SC at low nM concentrations in vitro.
The partially ineffective strand transfer inhibition with 3′OH pre-processed oligonucleotides substrates in comparison to blunt-ended substrates by inhibitors has been observed by other groups [25, 26]. The reactions containing blunt-ended DNA oligonucleotides are inhibited with an IC50s two to 10-fold lower than that observed in the reactions with pre-processed DNA ends. The inhibitors need to be in close proximity to the IN active-site within the SC for effective inhibition. Our assay does not allow us to determine exactly when the inhibitor interacts with IN to block strand transfer activity and yet not affecting 3′OH processing. However, it appears that the inhibitor may interact with a transient structural IN-DNA intermediate arising prior to or immediately after the 3′OH processing event. Inhibitor may bind to IN-DNA complex before processing but most likely would not yet be in right orientation to block the active site and hence does not inhibit 3′OH processing. Conformational change in IN-DNA complex caused by 3′OH processing gives inhibitor an access to the active site and block the strand transfer reaction [27, 28]. In any case, the inhibitors appear to have the best opportunity to “trap” the IN-DNA complex in the cytosolic PIC before it encounters the chromosomal DNA for integration.
Summarizing the above observations, we speculate that inhibitors likely bind to a transient IN-DNA structure formed prior to or upon 3′OH processing within SC or the cytosolic PIC. We observed an accumulation of this trapped transient structure (“trapped” SC) (Fig. 2) in our inhibitor titration experiments which supports this possibility [15]. A similar interfacial inhibition mechanism was proposed for HIV-1 IN strand transfer inhibitors [29]. In our assays with pre-processed substrate, the transient conformation in IN observed during 3′OH processing does not exist and hence, refractive to effective inhibition in comparison to blunt-end substrate. To further support our hypothesis, we formed the SC containing one blunt-ended and one pre-processed DNA to determine if only one blunt-end is sufficient for effective inhibition at low nM concentrations of strand transfer inhibitors. L-870,810 inhibited the formation of concerted integration products with this SC containing one blunt-ended and one pre-processed DNA ends with an IC50 of 60 nM [22]. This IC50 value with L-870,810 was nearly double than what we observed with the SC formed with two blunt-ended DNA molecules (IC50 of 30 nM) under identical experimental conditions [22]. Similar results were obtained with L-870,812. The data suggest a single blunt-end in SC is sufficient to effectively inhibit concerted integration, a possibility which also occurs within the PIC as it transverses the cytoplasm in the presence of inhibitors.
Conclusions
L-870,810, L-870,812 and L-841,411 are the earlier generation inhibitors of Raltegravir (Fig. 1). However, the mechanism of the inhibition caused by Raltegravir is similar [26, 30]. Surely new insights into the mechanism associated with the strand transfer inhibitors will be coming from different avenues of research. It is essential that ongoing efforts along multiple lines of research be continued because IN drug-resistant mutants are cross-resistant to a variety of structurally distinct strand transfer inhibitors [22, 26, 31]. For patients, it will be of great significance that resistant virus mutants against an IN inhibitor and mutants resistant to HAART remain sensitive to other structurally distinct inhibitors targeting IN.
Acknowledgments
This work was supported by NIH AI31334 and CA16312 to DPG.
Footnotes
Copyright in this article, its metadata, and any supplementary data is held by its author or authors. It is published under the Creative Commons Attribution By licence. For further information go to: http://creativecommons.org/licenses/by/3.0/.
Competing Interests: Authors declare no competing interests.
Author's Contributions: KKP and DPG helped design the research and wrote the manuscript. KKP performed some of the experiments.
References
- 1.Bushman FD, Craigie R. Activities of human immunodeficiency virus (HIV) integration protein in vitro: specific cleavage and integration of HIV DNA. Proc Natl Acad Sci USA. 1991;88:1339–43. doi: 10.1073/pnas.88.4.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Markowitz M, Morales-Ramirez JO, Nguyen BY, Kovacs CM, Steigbigel RT, Cooper DA, Liporace R, Schwartz R, Isaacs R, Gilde LR, et al. Antiretroviral activity, pharmacokinetics, and tolerability of MK-0518, a novel inhibitor of HIV-1 integrase, dosed as monotherapy for 10 days in treatment-naive HIV-1-infected individuals. J Acquir Immune Defic Syndr. 2006;43:509–15. doi: 10.1097/QAI.0b013e31802b4956. [DOI] [PubMed] [Google Scholar]
- 3.Steigbigel RT, Cooper DA, Kumar PN, Eron JE, Schechter M, Markowitz M, Loutfy MR, Lennox JL, Gatell JM, Rockstroh JK, et al. Raltegravir with optimized background therapy for resistant HIV-1 infection. N Engl J Med. 2008;359:339–54. doi: 10.1056/NEJMoa0708975. [DOI] [PubMed] [Google Scholar]
- 4.Zheng R, Jenkins TM, Craigie R. Zinc folds the N-terminal domain of HIV-1 integrase, promotes multimerization, and enhances catalytic activity. Proc Natl Acad Sci USA. 1996;93:13659–64. doi: 10.1073/pnas.93.24.13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kulkosky J, Jones KS, Katz RA, Mack JP, Skalka AM. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol Cell Biol. 1992;12:2331–8. doi: 10.1128/mcb.12.5.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engelman A, Craigie R. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J Virol. 1992;66:6361–9. doi: 10.1128/jvi.66.11.6361-6369.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lutzke RA, Plasterk RH. Structure-based mutational analysis of the C-terminal DNA-binding domain of human immunodeficiency virus type 1 integrase: critical residues for protein oligomerization and DNA binding. J Virol. 1998;72:4841–8. doi: 10.1128/jvi.72.6.4841-4848.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science. 1994;266:1981–6. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- 9.Wang JY, Ling H, Yang W, Craigie R. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. Embo J. 2001;20:7333–43. doi: 10.1093/emboj/20.24.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen JC, Krucinski J, Miercke LJ, Finer-Moore JS, Tang AH, Leavitt AD, Stroud RM. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc Natl Acad Sci USA. 2000;97:8233–8. doi: 10.1073/pnas.150220297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science. 2000;287:646–50. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- 12.Grobler JA, Stillmock K, Hu B, Witmer M, Felock P, Espeseth AS, Wolfe A, Egbertson M, Bourgeois M, Melamed J, et al. Diketo acid inhibitor mechanism and HIV-1 integrase: implications for metal binding in the active site of phosphotransferase enzymes. Proc Natl Acad Sci USA. 2002;99:6661–6. doi: 10.1073/pnas.092056199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hazuda DJ, Anthony NJ, Gomez RP, Jolly SM, Wai JS, Zhuang L, Fisher TE, Embrey M, Guare JP, Jr, Egbertson MS, et al. A naphthyridine carboxamide provides evidence for discordant resistance between mechanistically identical inhibitors of HIV-1 integrase. Proc Natl Acad Sci USA. 2004;101:11233–8. doi: 10.1073/pnas.0402357101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hazuda DJ, Young SD, Guare JP, Anthony NJ, Gomez RP, Wai JS, Vacca JP, Handt L, Motzel SL, Klein HJ, et al. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science. 2004;305:528–32. doi: 10.1126/science.1098632. [DOI] [PubMed] [Google Scholar]
- 15.Pandey KK, Bera S, Zahm J, Vora A, Stillmock K, Hazuda D, Grandgenett DP. Inhibition of human immunodeficiency virus type 1 concerted integration by strand transfer inhibitors which recognize a transient structural intermediate. J Virol. 2007;81:12189–99. doi: 10.1128/JVI.02863-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–9. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 17.Pandey KK, Sinha S, Grandgenett DP. Transcriptional coactivator LEDGF/p75 modulates human immunodeficiency virus type 1 integrase-mediated concerted integration. J Virol. 2007;81:3969–79. doi: 10.1128/JVI.02322-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M, Mizuuchi M, Burke TR, Jr, Craigie R. Retroviral DNA integration: reaction pathway and critical intermediates. EMBO J. 2006;25:1295–304. doi: 10.1038/sj.emboj.7601005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinha S, Grandgenett D. Recombinant HIV-1 integrase exhibits a capacity for full-site integration in vitro that is comparable to that of purified preintegraton complexes from virus-infected cells. J Virol. 2005;79:8208–16. doi: 10.1128/JVI.79.13.8208-8216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinha S, Pursley MH, Grandgenett DP. Efficient concerted integration by recombinant human immunodeficiency virus type 1 integrase without cellular or viral cofactors. J Virol. 2002;76:3105–13. doi: 10.1128/JVI.76.7.3105-3113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li M, Craigie R. Processing of viral DNA ends channels the HIV-1 integration reaction to concerted integration. J Biol Chem. 2005;280:29334–9. doi: 10.1074/jbc.M505367200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zahm JA, Bera S, Pandey KK, Vora A, Stillmock K, Hazuda D, Grandgenett DP. Mechanisms of human immunodeficiency virus type-1 concerted integration as related to strand transfer inhibition and drug resistance. Antimicrob Agents Chemother. 2008;52:3358–68. doi: 10.1128/AAC.00271-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H, Engelman A. Asymmetric processing of human immunodeficiency virus type 1 cDNA in vivo: implications for functional end coupling during the chemical steps of DNA transposition. Mol Cell Biol. 2001;21:6758–67. doi: 10.1128/MCB.21.20.6758-6767.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller MD, Farnet CM, Bushman FD. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J Virol. 1997;71:5382–90. doi: 10.1128/jvi.71.7.5382-5390.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchand C, Zhang X, Pais GC, Cowansage K, Neamati N, Burke TR, Jr, Pommier Y. Structural determinants for HIV-1 integrase inhibition by beta-diketo acids. J Biol Chem. 2002;277:12596–603. doi: 10.1074/jbc.M110758200. [DOI] [PubMed] [Google Scholar]
- 26.Marinello J, Marchand C, Mott BT, Bain A, Thomas CJ, Pommier Y. Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochemistry. 2008;47:9345–54. doi: 10.1021/bi800791q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Tsiang M, Yu F, Hung M, Jones GS, Zeynalzadegan A, Qi X, Jin H, Kim CU, Swaminathan S, Chen JM. Modeling, analysis, and validation of a novel HIV integrase structure provide insights into the binding modes of potent integrase inhibitors. J Mol Biol. 2008;380:504–19. doi: 10.1016/j.jmb.2008.04.054. [DOI] [PubMed] [Google Scholar]
- 28.Dicker IB, Samanta HK, Li Z, Hong Y, Tian Y, Banville J, Remillard RR, Walker MA, Langley DR, Krystal M. Changes to the HIV long terminal repeat and to HIV integrase differentially impact HIV integrase assembly, activity, and the binding of strand transfer inhibitors. J Biol Chem. 2007;282:31186–96. doi: 10.1074/jbc.M704935200. [DOI] [PubMed] [Google Scholar]
- 29.Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discov. 2005;4:236–48. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 30.Miller M, Witmer M, Stillmock K, Felock P, Ecto L, Flynn J, Schleif W, Dornadula G, Danovich R, Hazuda D. Biochemical and antiviral activity of MK-058, a potent HIV integrase inhibitor. AIDS 2006—XVI International AIDS Conference; Toronto. 2006. Oral abstract session Abstract no. THAA0302. [Google Scholar]
- 31.Dayam R, Al-Mawsawi LQ, Neamati N. HIV-1 integrase inhibitors : An emerging clinical reality. Drugs R D. 2007;8:155–68. doi: 10.2165/00126839-200708030-00003. [DOI] [PubMed] [Google Scholar]