


Abstract
Adenosine-to-inosine (A-to-I) editing has been shown to be an important mechanism that increases protein diversity in the brain of organisms from human to fly. The family of ADAR enzymes converts some adenosines of RNA duplexes to inosines through hydrolytic deamination. The adenosine recognition mechanism is still largely unknown. Here, to investigate it, we analyzed a set of selectively edited substrates with a cluster of edited sites. We used a large set of individual transcripts sequenced by the 454 sequencing technique. On average, we analyzed 570 single transcripts per edited region at four different developmental stages from embryogenesis to adulthood. To our knowledge, this is the first time, large-scale sequencing has been used to determine synchronous editing events. We demonstrate that edited sites are only coupled within specific distances from each other. Furthermore, our results show that the coupled sites of editing are positioned on the same side of a helix, indicating that the three-dimensional structure is key in ADAR enzyme substrate recognition. Finally, we propose that editing by the ADAR enzymes is initiated by their attraction to one principal site in the substrate.
INTRODUCTION
Adenosine-to-inosine (A-to-I) RNA editing is catalyzed by a family of enzymes called adenosine deaminases that act on RNA (ADARs) (1,2). Two enzymes, ADAR1 and ADAR2, have been shown to have catalytic activity on substrates in the mammalian brain (3). These enzymes convert A-to-I within structured RNA that is largely double stranded (ds). There are three dsRNA binding domains (dsRBDs) in ADAR1, while ADAR2 has two dsRBDs. Both enzymes have one catalytic deamination domain in the C-terminal.
In vivo, A-to-I editing has been categorized into two types: (i) hyper-editing of multiple adenosines in longer and almost completely duplexed structures. Hyper-editing has been found almost unanimously within untranslated regions and the functional consequences still remain unclear (4–6) and (ii) site selectively edited substrates, where a few adenosines are targeted within an imperfect RNA fold-back structure. The properties that make an RNA prone for site selective editing are still not fully understood, but the assumption is that internal mismatches and bulges within an RNA duplex are important for ADAR selectivity (7–9). Site selectively edited substrates have mainly been found in transcripts coding for proteins involved in neurotransmission (3). In most of these substrates, the editing events have been shown to have functional consequences on neural ion channels. Inosine is interpreted as a guanosine (G) by the translational machinery. Thus, if the editing event occurs within the encoded sequence of an mRNA, then it can give rise to amino acid changes and variant functional properties of the final protein. Besides, intronic editing has the potential to produce multiple isoforms by inducing alternative splicing. ADAR2 has been shown to auto-edit its own pre-mRNA but only −1 out of the several edited sites within intron 4 creates a new splice site with the possible consequence of a truncated protein if translated (10).
In this study, we chose three substrates that are selectively edited at several sites to determine the mechanism of target recognition. The selected transcripts code for the ADAR2 protein, the serotonin receptor 5-HT2C and the kainate glutamate receptor subunit GluR-6. ADAR1 and ADAR2 have specific but overlapping specificities for site selective editing. Of the five sites that are edited in the 5-HT2C transcript (A, B, C′, C and D), the A-site is prone to ADAR1 editing while the D-site is mostly edited by ADAR2 (11,12). The other sites have the potential to be edited by both ADAR1 and ADAR2. The glutamate receptor subunit GluR-6 is edited at four sites. Two of these sites (I/V and Y/C) are located in transmembrane segment 1 (TM1) of the receptor, affecting the Ca2+ permeability of the channel (13). These are both edited by ADAR2 (12).
Recently using the 454 amplicon sequencing technique, we showed that editing of most selectively edited substrates are regulated during development (14). We took advantage of this knowledge to study coupling of edited sites in individual transcripts at different levels of editing efficiency. The high number of sequenced transcripts allowed us to develop a statistical basis to deduce coupling of nearby editing events in a substrate also at low levels of editing efficiency. Here, we introduce new data indicating that there is a coupling between edited sites at semi-fixed distances from each other. Furthermore, we show that the ADAR enzymes preferentially recognize adenosines located in the major grooves on the same side of an A-form double-helix structure.
MATERIALS AND METHODS
454 AMPLICON SEQUENCING FOR EDITING ANALYSES
RNA was isolated from mouse brains at embryonic day 15 and 19 and postnatal day 2 and 21 using TRIzol (Invitrogen). The preparation of the sample for sequencing was as described in ref. (14). The products were sequenced using the 454 amplicon sequencing technique (15) according to the instructions by the manufacturer (Roche).
Identification of edited transcripts from the 454 sequencing
Identification of edited transcripts was done by categorization of transcript through the recognition of the sequencing primer. The editing pattern for each of the three transcripts was directly compiled from string matching in Perl. The string was the nucleotide sequence of the target region with the editing site(s) which were matched to A or G, i.e. with the instruction ‘OR(A OR G)’. All A:s and G:s were directly calculated through the matching.
Determination of editing frequencies and coupling
To determine coupled editing for a given transcript, we used a χ2-test and cluster analyses. The null-hypothesis for the χ2-test is independent editing for a given pair of targeted sites. The result of the χ2-test is either rejection or acceptance. Here, rejection of the null-hypothesis indicates the pair as mutually edited with a significance set by the p-value. Since we perform multiple measurements of χ2, we also make a Bonferroni correction where an adjusted p-value p′ is used: p′ = p/n, where n is the number of measurements and p = 0.05 (16). Each developmental stage is separated in the calculations and we also apply the Bonferroni correction accordingly.
For the cluster analyses we used XLSTAT, an Excel add-in (Microsoft). Since a target position is either edited or not edited, the data is binary and can be set to (1 or 0). In the cluster analysis, we use a similarity matrix where each element is calculated according to Dice coefficients, Dij = 2a/(2a + b + c). Dice coefficients are used in cluster analyses when the data is binary and twice the weight are put on agreements. This is in contrast to the χ2 analyses where 0 (not edited) and 0 (not edited) is weighted equally to 1 (edited) and 1 (edited). Here, ‘a’ is the total number of transcripts having 1 and 1 for a position pair. Furthermore, ‘b’ and ‘c’ are 1 and 0 or 0 and 1, while ‘d’ is 0 and 0. Given the Dice values XLSTAT groups each position into classes and also produce dendrograms that show the distances between classes (Figure 2). Here the distance to the splitting (root) between classes corresponds to the level of similarity, i.e. the longer distance the more dissimilar, which in this case indicates less mutual editing.
Figure 2.

The resulting dendrograms from the cluster analyses of Adar2 edited positions at the four developing stages with the corresponding clustering of positions (classes) below. An annotation of the same class denotes positive coupling properties. The dendrograms have been made using XLSTAT (see ‘Materials and Methods’ section).
Molecular modeling
RNA secondary structures were predicted by MC-Fold (17) from sequence alone using the publicly available Web site (http://www.major.iric.ca/MC-Fold) with the following parameters: MaxBulge 2; UseBulge 2; MeanNrgy −1.0; CapNrgy: +0.5; Explore 21%; and Branches 9. The lowest-energy structures were then submitted to MC-Sym from script automatically generated from the MC-Fold output using the default parameters: method ‘exhaustive’; model_limit 100; rmsd 3.0. The tertiary models were scored, relieved and energy minimized using the options offered by the MC-Sym working directories (see the Web site for details).
RESULTS
Regulation of editing during development depends on site location
To investigate the recognition mechanism for selective editing, we chose three transcripts known to be subject to multiple A-to-I editing events in the mammalian brain: GluR-6, 5-HT2C and Adar2. Total RNA from mouse brain was extracted and amplified by reverse transcriptase followed by a polymerase chain reaction (RT-PCR) using primers specific for amplification of regions including site −28 to +28 in Adar2 (nine sites); the A, B, C′, C and D site in 5-HT2C and the I/V and Y/C sites in GluR-6 (Figure 1). The products were subsequently sequenced according to the 454 amplicon sequencing protocol (14). The advantages with this technique for editing analyses is that even low levels of editing efficiency can be detected. Also, this sequencing method produces a large number of sequenced transcripts (reads) giving a high statistical significance when modification by RNA editing is determined. All sequences corresponding to the individual A-to-I editing substrates were analyzed for A to G changes. The extent of editing was determined for different developmental stages of the mouse brain: embryonic day 15 and 19, (E15 and E19), as well as post-natal day 2 and 21, (P2 and P21). In all three substrates, an increase in editing efficiency during development could be observed for several sites (Figure 1). However, in the transcripts of the serotonin receptor 5-HT2C, the nature of the five edited sites can be divided into two groups: (i) increase in editing during development and (ii) close to a constant level of editing through development. The efficiency of editing at site A and B increase rapidly from E15 to E19, but show only a moderate increase after birth (Figure 1A). The D site in this transcript have no pre-natal increase in editing but a high level of close to 50% editing already at E15. After birth, the increase is similar to what was observed for the A and B sites. Dissimilar from the other sites, C′ and C, located next to each other, have a low level of editing that is constant through development.
Figure 1.

Developmental regulation of editing in the Adar2 (A), 5-HT2C (B) and GluR-6 (C) transcripts. The efficiency of editing is plotted against the age of the mouse. On average 600 sequences were analyzed. Detailed information on number of edited transcripts can be found in ref. (14) and at the NCBI Short Read Archive (SRA), accession number SRA008179. The sequential context in which the edited position (in bold) reside for the Adar2, 5-HT2C and GluR-6 transcripts are shown on the top of graphs in A, B and C.
The Adar2 pre-mRNA is edited at many sites within intron 4 (7). We analyzed the efficiency of editing at nine of these sites located in the proximity of each other. Also in this substrate, the developmental regulation of editing can be divided into two groups. Editing of sites (+24, +23, +10 and −1) increases during development, whereas for the other sites (+28, −2, −4, −27 and −28) it increases only moderately or not at all, ending with a low efficiency of editing (under 20%) in the adult animal (Figure 1B). The most efficiently edited site is at +24, where more than 80% of the transcripts are edited in the adult brain.
The I/V and Y/C sites edited in the kainate receptor transcript GluR-6 were analyzed. Editing of these two sites is regulated during development in a similar way as the efficiently edited sites in the 5-HT2C and Adar2 transcripts. A dramatic increase in editing efficiency is observed from the embryonic stages to P2, and at a slower rate of increase up to P21, where 74 and 80% of the transcripts are edited at the I/V and Y/C sites, respectively (Figure 1C).
Taken together these results indicate that edited sites within a substrate are regulated differently and the most efficiently edited sites follow a similar trend of increased editing during development.
Is the editing enzyme attracted to specific sites?
Using the 454 amplicon sequencing method, editing in a large number of individual transcripts can be analyzed. We wanted to see if there is a preference in the affinity for different sites in the editing substrates. For each developmental time point, 129–1329 transcripts were analyzed from the three different substrates (Figure 1). Since the efficiency of editing is low during embryogenesis, we assumed that the amount of active editing enzyme is limited at this stage. If certain sites have higher affinity for the enzyme, then this should be reflected in the pattern of edited sites. At E15, the majority of the edited 5-HT2C transcripts (26%) were singularly edited at the D site (Supplementary Table S1). After further development, both A and D are seen as singularly edited sites in the transcripts analyzed at E19, P2 and P21. The fact that we only detect singularly edited sites at A and D suggests that these are sites of the highest affinity for editing, and we thus consider them to be principal editing sites. Interestingly, these sites are preferentially edited by different ADAR enzymes, the A site by ADAR1 and the D site by ADAR2 (11,12). This might explain why this transcript has dual principal sites. Only one principal editing site can be identified in the Adar2 transcript. Through development the +24 site is clearly the dominant site in singularly edited transcripts (Supplementary Table S1). At day P2, 247 of the 781 transcripts analyzed are edited only at the +24 site. Hence, our interpretation is that the +24 site has the highest affinity for editing in the Adar2 transcript. This is also the most efficiently edited site at P21 with more than 80% of the transcripts edited. Even though there are only two sites of editing in proximity to each other in the GluR-6 transcript, one of them (Y/C) is found to be the dominant site for singularly edited transcripts with the peak at E19 with 25% of the transcripts edited only at the Y/C site (6% for I/V). Following the same pattern as for the other transcripts, Y/C is also the most efficiently edited site in the transcript.
Taken together, the frequency of singularly edited transcripts in all substrates analyzed peak when the activity of the editing enzyme is limited and decrease during late development with an increased editing activity (Supplementary Figure S1). At the same time, the number of multiple edited transcripts increases with development. We propose that the singularly edited transcripts indicate principal edited sites with high affinity for the editing enzyme, although we cannot exclude an initial binding of the enzyme at other sites.
Coupling occurs between edited sites
We wanted to know if editing at the principal editing site is coupled to other sites that show a similar trend of increased editing efficiency during development. The 454 sequencing method allowed us to statistically analyze the editing events in each target transcript individually. We can therefore determine if distinct editing events show any combinatorial behavior, such as interdependence for instance; i.e. determining if a position, N, in a target transcript is edited, then is there another position, N′, also edited? Alternatively, if position N is not edited, then is N′ edited or not? To answer this type of questions, we used a χ2-test with significance p = 0.05 and 1 degree of freedom. The null hypothesis is that the positions are independently edited (see also ‘Materials and Methods’ section). In Supplementary Figure S1, the sequential context of the sites and a schematic χ2 matrix are shown. Along the solid diagonal arrow, there is a pattern where either the two positions are edited (G and G) or not edited (A and A). The dotted diagonal arrow follows entries that correspond to the reverse, A and G, or G and A. If a and/or d in the solid diagonal is significantly favored, i.e. if a + d ≫ c + b, they are considered to be positively coupled. A negative coupled pair of positions would significantly favor the elements along the dotted diagonal. Since we performed multiple statistical measurements of coupled sites, for example 36 different χ2-values were calculated from 36 2 × 2 matrices in the Adar2 transcript, we made a Bonferroni correction to the p-value per target region and developmental stage. The motivation for this was that if p is assigned to 0.05, we statistically anticipate to falsely reject the null-hypothesis one time if we perform the measurement 20 times (1/20 = 0.05). It should be noted that the usage of a Bonferroni correction of the p-value is considered to be very strict by its implementation, and thus we increase the quality level of the results. After the Bonferroni correction, we have significances of p = 0.0014 and 0.005 for Adar2 and 5-HT2C, respectively. The corresponding critical χ2-values are 10.22 and 7.88 and any pair having a higher χ2-value is considered to be significantly coupled (either negative or positive).
In addition to the χ2-test, we made cluster analyses on the substrates with more than two edited sites. Mutual editing patterns were compiled into classes where members in a class share a synchronous editing pattern within the same target transcript (Figures 2 and 3). An intrinsic feature of the cluster analyses, not seen from the χ2-test, is that more than two (a pair) positions can be categorized into the same class. Hence, we detect coupling between more than two edited positions directly. The cluster analyses also show how closely related editing at different sites are.
Figure 3.

The resulting dendrograms from the cluster analyses of 5-HT2C edited positions at the four developing stages with the corresponding clustering of positions (classes).
Overall, there are several sites that are positively coupled (Table 1 and Supplementary Table S2). We consider two positions to be coupled (+) if only one of the tests show coupling, or strongly coupled (++) if both the χ2-test and the cluster analysis show coupled properties. To analyze coupling of edited sites at different levels of editing, we applied the coupling analysis on the four different developmental time points.
Table 1.
Coupling between edited sites in mouse brain at developmental day P2
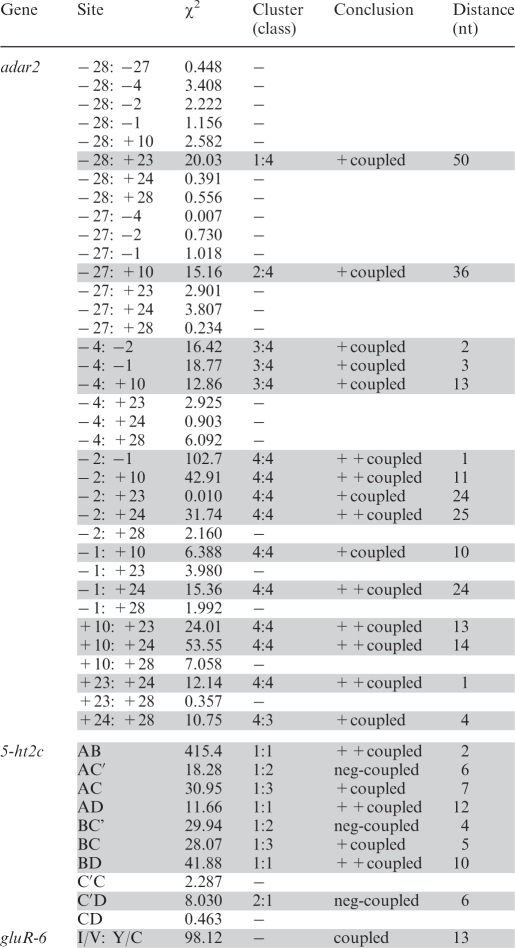 |
If both the χ2-test and the cluster analyses show coupling it is assign (++). If one test show coupling it is indicated as (+). The indication ‘neg’ means negatively coupled sites. After the Bonferroni correction, column values should be compared to >10.22 for the Adar2 sites and >7.88 for the 5-HT2c sites. For developmental day E15, E19 and P21 see Supplementary Table X. n.d. = not determined.
In the Adar2 transcript, there is a strong coupling between the +24, +10 and −1 site. According to the cluster analysis these sites are coupled at all developmental stages indicating that coupling occurs regardless of editing efficiency (Figure 2). Another site that is strongly coupled to these three sites at E15, E19 and P2, is +23. This coupling is also confirmed by the χ2-test. Both analyses indicate that even if the strongest coupling is between sites +24, +10 and −1, coupling is also seen between other sites.
In the 5-HT2C transcript, the D site is strongly coupled to A and B, while the C site is more weakly coupled to this group (Figure 3). Strikingly, the C′ site is consistently detached from the other classes in the cluster analyses. At day P2, it is even clearly negatively coupled to the A, B and D sites and show no coupled properties at the other three stages. Coupling between sites A and B has previously been stated (18). The A and B sites are only separated by one nucleotide; the coupling can therefore be explained by a slipping of the enzyme between the sites. However, the coupling between sites A and D was harder to foresee since they are preferentially edited by different ADAR enzymes. Indeed, in several cases, coupling occurs between sites edited by different enzymes. Therefore, coupling does not discriminate between sites that are edited by ADAR1 or ADAR2, indicating that there is a communication between the two proteins on the same transcript.
Coupling of edited sites occurs at defined distances from each other
An interesting question based on the coupling results is whether there is any consensus between the edited sites that are coupled. To visualize such a pattern, we calculated all distances between sites that showed coupling, xsd (see distance, Table 1). The edited positions in the Adar2 transcript revealed a pattern of ‘hot-spot’ editing at a semi-fixed distance from each other. There are four hot-spots: [− 28, −27]; [− 4, −2, −1]; [+10] and [+23, +24, +28]. Hot-spot [+10] has a spacer distance of 11–14 nucleotides (nt) from the nearby sites. We decided to call this semi-fixed spacer distance asd ≃ 12 nt. The distance between the hot-spot at −2 and −27 is 2 × asd and between +10 and −27, there is 3 × asd = 36 nt. For every possible xsd, 1–55, we counted the number of coupled sites in the Adar2 transcript with that spacer distance (Figure 4). There is a clear pattern of preferred spacer distances of coupled editing events. Pairs of coupled edited sites have xsd in the intervals: 1–4, 10–14, 23–26, 36–37 and 50–51. The peaks in the graph are centered on the average distance between coupled pairs in each interval and the height reflects the number of coupled pairs in the corresponding interval. The most common xsd is in the range 23–26 nts with seven coupled edited pairs in this region. Noteworthy, is that the interval peaks are multiples of asd and that there are adenosines in between these hot-spots that are not edited.
Figure 4.

The result from the compilation of clustering of spacer distances of coupled positions in Adar2. The x-axis show the discontinuous available spacer distances (1–55) in nucleotides (nts). Couplings indicate the number of coupled position pairs having the corresponding distance. The peaks are at the spacer distances that tend to group: 1–4, 10–14, 23–26, 36–37 and 50–51. The height of a peak indicates to the total number of coupled positions in the group to the right.
The two edited sites I/V and Y/C in GluR-6 are separated by 13 nt (1 × asd). Both sites in GluR-6 are efficiently edited but could in theory show either mutual editing or detached editing on the same transcript. Strikingly and in convincing correlation with our results from the coupling analyses of Adar2, we found strong coupled properties between the I/V and Y/C sites consistently through development (Table 1 and Supplementary Table S2).
In the 5-HT2C transcript, the space between edited positions differs from Adar2 and GluR-6. The A to D and B to D sites are separated by 12 and 10 nts, respectively. These are coupled following the pattern seen in Adar2 and GluR-6. The A and B sites, separated by 1 nt, are also strongly positively coupled, hence following the pattern seen in Adar2. Interestingly, a negative coupling can be seen between A and C′, B and C′ as well as D and C′, which are separated by 5, 3 and 6 nts, respectively. This result indicates that other sites are rarely edited when editing at the C′ site occurs.
Based on observations made in these three transcripts, we conclude that distances between coupled sites (xsd) cluster at distances that are multiples of asd. Hence, there is a clear pattern of synchronous editing of sites separated by n × asd, where n = (0), 1, 2, 3, 4.
Coupling of edited sites is directional initiating at the principal site
Our model of consecutive binding of ADARs would suggest that editing within a transcript is directional from the principal edited site. To analyze this, we used the Adar2 transcript and the +24 principal site of editing. If we denote edited or not edited at a site with 1 or 0, then for the four consecutive sites, starting with the +24 site, we expect patterns like ‘1 1 1 1’, ‘1 1 0 0’ or ‘1 1 1 0’, but more rarely ‘1 0 0 1’. When the sequences were analyzed, we saw that our model is supported by 98.3, 94.3, 92.1 and 73.6% of the transcripts for the different developmental stages. Therefore, the data strongly support the model of directional editing initiated at the principal site.
The tertiary structure determines coupling
To better understand why coupling between edited sites are at certain distances to each other, we predicted the tertiary structure of Adar2 and the 5-HT2C from their sequence information using the MC-Fold and MC-Sym algorithms (17); http://www.major.iric.ca/MC-Pipeline; see Methods section). These assign and score small RNA building blocks observed in NMR and crystallographic data that best accommodate the sequence, which are then assembled into secondary and tertiary structures. All adenosines at the sites of editing are involved in the formation of a base pair with either U and score small RNA building blocks observed in NMR and crystallographic data that best accommodate the sequence, which are then assembled into secondary and tertiary structures. All adenosines at the sites of editing are involved in the formation of a base pair with either U (e.g. +23, −1, −2 and −4 in Adar2) or C (e.g. +24 and +10 in Adar2). In all the models we built, the AU base pairs adopt the Watson–Crick geometry. The AC pairs are less stable but in all cases they include at least one H-bond involving the NH2 donor group or N1 acceptor of the A, and are thus isosteric to the Watson-Crick geometries. Both AU and AC base pairs expose the sugar and Hoogsteen edges of the adenosines. The details of the interaction between the Adar2 and 5-HT2C RNA with the editing enzyme are not known. However, it is tempting to speculate from the invariant base pairing geometry that the editing mechanism is the same in both cases and at all sites.
Strikingly, the predicted structures revealed that the Adar2 sites +24, +23, +10, −1, −2 and −4, which were shown to be coupled in at least two developmental stages, are all located on the same side of the helix (Figure 5A). Apart from the edited sites that have been analyzed experimentally in this study, the Adar2 transcript is edited at several other sites on the opposite strand of the predicted hairpin structure (7). These sites had to be excluded since the maximum length of the RNA we isolated was set to ∼200 nt and the upstream edited sites are located close to 1500 nt from the region we chose to include in our analysis. However, superimposition of these sites onto our MC-Sym structure reveals that the most extensively edited sites (− 1476, −1500) are located on the same side of the helix as the ones we show are coupled (data not shown). Worth mentioning is also that the 17 other adenosines in this region show no or marginal signs of editing. These results indicate that editing preferentially occurs at sites on the same side of the helix and that editing of these sites, are strongly coupled.
Figure 5.

Predicted tertiary structure of the edited RNA substrates Adar2 and 5-HT2C. (A) Right, the Adar2 transcript seen from the front as indicated by the arrow in the secondary RNA structure to the left. Edited nucleotides are indicated in red. (B) To the right is the 5-HT2C transcripts seen from the front as indicated by the arrow in the secondary RNA structure to the left. Edited nucleotides are indicated in red.
The serotonin receptor transcript 5-HT2C differs in where the edited sites are located in the tertiary structure (Figure 5B). Out of the five edited sites, three are located on the same side of the helix structure. These three sites (A, B and D) are strongly coupled to each other in all combinations. Interestingly, the other two sites (C′ and C) are located on the opposing side of the helix. As previously mentioned these sites show low efficiency of editing without a strong coupling to other sites. The C′ site is even negatively coupled to the sites on the other side of the helix (Table 1). This result gives further evidence that efficiently edited sites are located on the same side of the RNA helix and that editing of these sites is positively coupled.
DISCUSSION
The mechanism behind the recognition of ADAR editing sites has not been fully elucidated. From previous work we know that the ADAR enzymes recognize specific adenosines within dsRNA interrupted by bulges and loops (19). We have previously shown that bulges and internal loops are important for editing specificity in a natural substrate, but not for binding (8,20). Furthermore, ADAR2 associates preferentially with an imperfect RNA fold-back structure over a perfect RNA duplex within the same molecule (21). By foot-printing analysis from our work and that of others, it is also known that the ADAR2 protein covers a region of 11–16 nucleotides on the natural GluR-B substrate at the R/G site (20,22). Consistent with the known length requirement for dsRNA binding domains (dsRBDs), a minimal duplex length of 15–20 nucleotides is required for deamination (23). The binding by both dsRBDs (I and II) in ADAR2 has been confirmed by NMR tertiary structure analysis of the GluR-B R/G substrate (24). Even though dsRBDs are not highly sequence specific, it has been shown that the dsRBDs of ADAR2 binds selectively to a natural substrate and that this binding is different from the binding of dsRBDs in the dsRNA binding protein kinase (PKR) (9). Also the deamination domain contributes to the recognition of a substrate. Although the details for this recognition is less clear, it has been shown that the specificity for a substrate can be changed by exchanging the ADAR1 and ADAR2 deamination domains (25). The specificity of ADAR1 and ADAR2 overlaps, although some sites are edited entirely by one enzyme or the other. There is a bias in the nearest neighbor preference to an edited site where the 5′ upstream nearest neighbor rarely constitutes a G (26). ADAR2 also shows a 3′ nearest neighbor preference where U = G > C = A (27). Furthermore, there is a preference for a cytosine opposing the targeted adenosine. This preference seems more pronounced for ADAR1 than for ADAR2 (25).
Still, it is largely unknown why only certain adenosines in a duplex are subjected to editing while others that appear to be in the right context in the secondary structure are not. Here, we analyzed the initial recognition of an edited substrate and the coupling between edited sites within transcripts edited at several sites. We used the 454 amplicon sequencing protocol to evaluate single transcript A-to-I editing in mouse brain. All transcripts analyzed in this study have edited sites with a certain distance from each other, consistent with n × 12 nt, where n = (0), 1, 2, 3, 4. With two sets of statistical tools, compiling data from four different time points during development, we show that editing at the distance of 12 nts (asd) is strongly coupled. Coupled editing at multiples of asd also leaves room for more than two enzymes interacting with the target RNA. In addition, positively coupled positions at a distance of 1–2 nucleotides were found (Table 1). Coupling of the adjacent edited sites are probably a result of an ADAR slipping to neighboring adenosines. As previously suggested, upon ADAR/RNA binding, adjacent adenosines could be sequentially edited before disassociation (19). We also observed coupling of sites at a distance of ∼24, ∼36 and ∼50 nucleotides in the Adar2 transcript. This observation suggests that one edited site can influence editing at other sites even if they are at high intramolecular distances from each other, either by direct contact or through binding of several enzymes along the helix structure. Furthermore, through development there are multiple transcripts with one single editing event. In Adar2, 27% of the transcripts are singularly edited at the +24 site, indicating that this is the principal site to attract the ADAR enzyme.
The editing pattern seen in the 5-HT2C receptor transcript is both coherent with and different from what we observed in the other transcripts. Also in this transcript there is a strong coupling between edited sites at a distance of 12 nucleotides (A and D). However, there are also edited sites at a distance of ∼6 nucleotides. Interestingly, these sites show: no, weak, or even negative coupling to the other sites (Table 1). Coupling and recognition of edited sites in the substrates can be explained by analyzing the predicted tertiary structure of these RNAs. All edited sites that show a strong coupling are located on the same side of the RNA helix while the negatively coupled sites are on opposite sides in the structure (Figure 5). During embryogenesis, the principal editing site in the 5-HT2C substrate is the D site. At embryonic day 15 (E15), 27% of the transcripts are edited only at the D site (14). An interesting observation is that from day E19 to P21 transcripts with singularly edited sites at the A and the D site are found at the same levels. We interpret this as a possibility to initiate editing of the transcript at either the A or the D site, but that ADAR2 editing the D site initiate editing earlier than ADAR1 since it is the only site edited during early development. Furthermore, previous results from our laboratory show that the editing pattern ABD is considerably more frequent than only AD (14). This is confirmed in our cluster analyses showing a stronger coupling between A and B than with D (Figure 3). This is somewhat surprising since A is edited by ADAR1 and B mainly by ADAR2, just like the D site.
In conclusion, our current model is that a principal editing site will initially attract the editing complex. Subsequently, other adenosines will be deaminated through two different processes: (i) deamination of adjacent adenosines by the same ADAR complex; (ii) deamination of adenosines located at distances that are multiples of asd by consecutive complexes binding to the same stem target (Figure 6). We further hypothesize that the less efficiently edited adenosines that are not principal sites have to fulfill the following criteria to be edited: (i) be located in a stem loop structure; (ii) have preferred nearest neighbor nucleotides; and (iii) be located on the same side of the helix as the principal editing site. Thus, adenosines that appear to be in the right context, fulfilling the first two criteria, will remain non-edited if they are not in a structurally favorable position with respect to the site of editing initiation. A model where one ADAR enzyme is processive, editing several consecutive sites is less likely than multiple enzymes binding to the same transcript since there would be no reason then to find editing only at certain distances (n × asd) from each other. We therefore propose that consecutive enzymes bind in register to deaminate subsequent adenosines after the first attraction to the principal site (Figure 6). One explanation why we find editing initiating at specific sites might be that editing at the principal site leads to a conformational change of the RNA molecule that facilitates further editing at other sites. A substrate conformational change upon editing has previously been suggested at the R/G site of GluR-B by the Beal laboratory (22). In future analyses, we would like to determine if the editing pattern is different in absence of the principal editing site.
Figure 6.

Model for multiple A-to-I editing of site selectively edited RNA. Editing is initiated at a principal editing site, possibly during transcription. A second site is edited at the distance of asd from the initiation site. This site is situated one helical turn from the first site. Consecutive editing can occur at the distance of approximately n × asd, but also immediately adjacent to edited hot-spots.
It has previously been proposed that the ADAR enzymes interact with their substrate as a dimer, where the dsRBDs have been shown to be important for the dimer formation of ADAR2 (28,29). The dimer interface to the RNA has been suggested to be similar to the 2-fold (‘yin-yang’) symmetry adopted by cytidine deaminases (30,31). Here, both monomers interact symmetrically with the helical structure of the RNA. In these homodimers only one catalytic center is active, whereas the other center binds a downstream U residue. Our data support this model, since we only find coupled positions located on the same side of the helix. It is therefore unlikely that two active catalytic centers bind symmetrically. Nonetheless, we cannot rule out the possibility of other dimer interfaces interacting with the RNA such as a ‘head-to-tail’ binding or that one monomer is not facing the RNA. However, given the homology between the cytidine and adenosine deaminases this is unlikely.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Swedish Research Council [NT2006-5415 to M.Ö.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Jan-Olov Persson for his help on the statistical analysis. They thank Lars Wieslander for fruitful discussions and for critically reading the paper. They would also like to thank Christina Holmberg and the 454 sequence facility at the KTH Genome Center, Stockholm.
REFERENCES
- 1.Bass BL, Nishikura K, Keller W, Seeburg PH, Emeson RB, O'C;onnell MA, Samuel CE, Herbert A. A standardized nomenclature for adenosine deaminases that act on RNA. RNA. 1997;3:947–949. [PMC free article] [PubMed] [Google Scholar]
- 2.Polson AG, Crain PF, Pomerantz SC, McCloskey JA, Bass BL. The mechanism of adenosine to inosine conversion by the double-stranded RNA unwinding/modifying activity: a high-performance liquid chromatography-mass spectrometry analysis. Biochemistry. 1991;30:11507–11514. doi: 10.1021/bi00113a004. [DOI] [PubMed] [Google Scholar]
- 3.Gommans WM, Dupuis DE, McCane JE, Tatalias NE, Maas S. In: RNA and DNA Editing. Smith HC, editor. New Jersey: John Wiley & Sons, Inc.; 2008. pp. 3–30. [Google Scholar]
- 4.Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2:e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blow M, Futreal PA, Wooster R, Stratton MR. A survey of RNA editing in human brain. Genome Res. 2004;14:2379–2387. doi: 10.1101/gr.2951204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004;22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 7.Dawson TR, Sansam CL, Emeson RB. Structure and sequence determinants required for the RNA editing of ADAR2 substrates. J. Biol. Chem. 2004;279:4941–4951. doi: 10.1074/jbc.M310068200. [DOI] [PubMed] [Google Scholar]
- 8.Källman AM, Sahlin M, Öhman M. ADAR2 A–> I editing: site selectivity and editing efficiency are separate events. Nucleic Acids Res. 2003;31:4874–4881. doi: 10.1093/nar/gkg681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stephens OM, Haudenschild BL, Beal PA. The binding selectivity of ADAR2's; dsRBMs contributes to RNA-editing selectivity. Chem. Biol. 2004;11:1239–1250. doi: 10.1016/j.chembiol.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 10.Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999;399:75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- 11.Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- 12.Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 13.Kohler M, Burnashev N, Sakmann B, Seeburg PH. Determinants of Ca2 +permeability in both TM1 and TM2 of high affinity kainate receptor channels: diversity by RNA editing. Neuron. 1993;10:491–500. doi: 10.1016/0896-6273(93)90336-p. [DOI] [PubMed] [Google Scholar]
- 14.Wahlstedt H, Daniel C, Ensterö M, Öhman M. Large-scale mRNA sequencingdetermines global regulation of RNA editing during brain development. Genome Res. 2009;19:978–986. doi: 10.1101/gr.089409.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonferroni CE. Studi in Onore del Professore Salvatore Ortu Carboni. Italy: Rome; 1935. pp. 13–60. [Google Scholar]
- 17.Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Emeson RB, Samuel CE. Serotonin-2C receptor pre-mRNA editing in rat brain and in vitro by splice site variants of the interferon-inducible double-stranded RNA-specific adenosine deaminase ADAR1. J. Biol. Chem. 1999;274:18351–18358. doi: 10.1074/jbc.274.26.18351. [DOI] [PubMed] [Google Scholar]
- 19.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Öhman M, Källman AM, Bass BL. In vitro analysis of the binding of ADAR2 to the pre-mRNA encoding the GluR-B R/G site. RNA. 2000;6:687–697. doi: 10.1017/s1355838200000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klaue Y, Källman AM, Bonin M, Nellen W, Öhman M. Biochemical analysis and scanning force microscopy reveal productive and nonproductive ADAR2 binding to RNA substrates. RNA. 2003;9:839–846. doi: 10.1261/rna.2167603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yi-Brunozzi HY, Stephens OM, Beal PA. Conformational changes that occur during an RNA-editing adenosine deamination reaction. J. Biol. Chem. 2001;276:37827–37833. doi: 10.1074/jbc.M106299200. [DOI] [PubMed] [Google Scholar]
- 23.Doyle M, Jantsch MF. New and old roles of the double-stranded RNA-binding domain. J. Struct. Biol. 2002;140:147–153. doi: 10.1016/s1047-8477(02)00544-0. [DOI] [PubMed] [Google Scholar]
- 24.Stefl R, Xu M, Skrisovska L, Emeson RB, Allain FH. Structure and specific RNA binding of ADAR2 double-stranded RNA binding motifs. Structure. 2006;14:345–355. doi: 10.1016/j.str.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 25.Wong SK, Sato S, Lazinski DW. Substrate recognition by ADAR1 and ADAR2. RNA. 2001;7:846–858. doi: 10.1017/s135583820101007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polson AG, Bass BL. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J. 1994;13:5701–5711. doi: 10.1002/j.1460-2075.1994.tb06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lehmann KA, Bass BL. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 2000;39:12875–12884. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- 28.Cho DS, Yang W, Lee JT, Shiekhattar R, Murray JM, Nishikura K. Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. J. Biol. Chem. 2003;278:17093–17102. doi: 10.1074/jbc.M213127200. [DOI] [PubMed] [Google Scholar]
- 29.Gallo A, Keegan LP, Ring GM, O'C;onnell MA. An ADAR that edits transcripts encoding ion channel subunits functions as a dimer. EMBO J. 2003;22:3421–3430. doi: 10.1093/emboj/cdg327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerber AP, Keller W. RNA editing by base deamination: more enzymes, more targets, new mysteries. Trends Biochem. Sci. 2001;26:376–384. doi: 10.1016/s0968-0004(01)01827-8. [DOI] [PubMed] [Google Scholar]
- 31.Navaratnam N, Fujino T, Bayliss J, Jarmuz A, How A, Richardson N, Somasekaram A, Bhattacharya S, Carter C, Scott J. Escherichia coli cytidine deaminase provides a molecular model for ApoB RNA editing and a mechanism for RNA substrate recognition. J. Mol. Biol. 1998;275:695–714. doi: 10.1006/jmbi.1997.1506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.