

Abstract
Promyelocytic leukemia oncogenic domains (PODs), also called nuclear domain 10 (ND10), are subnuclear structures that have been implicated in a variety of cellular processes as well as the life cycle of DNA viruses including papillomaviruses. In order to investigate the interplay between papillomaviruses and PODs, we analyzed the status of PODs in organotypic raft cultures of human keratinocytes harboring HPV genome that support the differentiation-dependent HPV life cycle. The number of PODs per nucleus was increased in the presence of HPV genomes selectively within the poorly differentiated layers, but was absent in the terminally differentiated layers of the stratified epithelium. This increase in PODs was correlated with an increase in abundance of post-translationally modified PML protein. Neither the E2 dependent transcription nor viral DNA replication was reliant upon the presence of PML. Implications of these findings in terms of HPV's interaction with its host are discussed.
Keywords: PML, papillomavirus, raft cultures, viral DNA replication, viral transcription
Introduction
Promyelocytic leukemia (PML) oncogenic domains (PODs), also called PML nuclear bodies (PML-NBs) or nuclear domain 10 (ND10), are subnuclear structures with size ranging from 0.1 to 1.0 μm that are attached to the nuclear matrix. They are characterized by the presence of PML protein, which is essential for POD assembly. A number of other nuclear proteins localize to PODs either constitutively such as Daxx, Sp100 and CREB binding protein (CBP) or in response to certain stimuli. PODs have been implicated in a wide variety of cellular processes including transcriptional regulation, protein modification, DNA repair, chromatin metabolism, apoptosis, p53 pathways, senescence and interferon response. Mammalian cells commonly contain 5-30 PODs, depending on the cell type, and the number, size and the composition of PODs can be altered in response to stimuli including heat shock, genotoxic insults, interferon treatment and viral infection (Borden, 2002; Dellaire and Bazett-Jones, 2004; Eskiw et al., 2003; Everett et al., 1999; Maul et al., 2000; Pearson and Pelicci, 2001; Regad and Chelbi-Alix, 2001; Zhong et al., 2000b). The number of PODs is increased upon interferon treatment due to transcriptional upregulation of PML and Sp100 (Chelbi-Alix et al., 1995; Lavau et al., 1995; Stadler et al., 1995). Therefore, PODs have been hypothesized to participate in cellular defense mechanisms against incoming pathogens; though, the function of PODs (or PML, Sp100) in the innate immune response remains poorly understood. Many viruses, particularly DNA viruses, have been shown to cause disruption of PODs and mutant viruses that lack the ability to disrupt PODs replicate inefficiently, providing further support that PODs mediate cellular defense (Ahn and Hayward, 1997; Chee et al., 2003; Everett, 2001; Korioth et al., 1996; Leppard and Everett, 1999; Muller and Dejean, 1999; Puvion-Dutilleul et al., 1995a; Puvion-Dutilleul et al., 1995b). For instance, an immediate early gene product of herpes simplex virus-1 (HSV-1), ICP-0, targets PML for the proteosome dependent degradation (Everett et al., 1998; Maul et al., 1993). Consistent with the hypothesis that HSV-1 counteracts cellular defenses mediated by PML or PODs, the replication defective phenotype of an ICP-0 mutant HSV-1 can be rescued in human fibroblasts in which PML expression is stably knocked down, (Everett et al., 2006).
PODs or constituents of PODs have also been physically linked to distinct steps or processes in the life cycle of many DNA viruses, such as HSV-1, cytomegalovirus (CMV), adenovirus and simian virus 40 (SV40) (Everett, 2001; Ishov and Maul, 1996). The genomes of these DNA viruses can be found in close association with PODs during infection and proteins encoded by these viruses that regulate transcription and/or replication of the genomes can associate with components of PODs, such as PML, Daxx or Sp100. These observations have led to the hypothesis that these viruses associate with PODs in order to support viral transcription and/or replication of the genome. For instance, plasmids containing the SV40 origin of DNA replication have been shown to localize to or nearby to PODs in the presence of SV40 large T antigen, which is essential for SV40 DNA replication (Tang et al., 2000). The genome of HSV-1 associates with PODs upon infection in the time frame when transcription of immediate early genes takes place; and at later stages of the life cycle, PML along with the DNA repair machinery is recruited to globular nuclear domains that are sites of HSV-1 DNA replication and contain essential viral DNA replication proteins such as the DNA binding proteins (ICP8 or UL29) and the viral polymerase (UL30) (Burkham et al., 1998; Everett and Murray, 2005; Everett et al., 2003; Wilkinson and Weller, 2004).
Papillomaviruses are a group of small nonenveloped viruses with circular double stranded DNA genomes approximately 8 kbs long that infect the squamous stratified epithelia in cutaneous or mucosal sites and usually cause hyperproliferative lesions in a variety of higher vertebrates including humans. A subset of mucosotropic human papillomaviruses (HPVs), called high risk HPVs, are frequently associated with human cancers including cervical cancer, other anogenital cancers and a subset of head and neck cancers (zur Hausen, 2002). The life cycle of papillomavirus is tightly linked to the differentiation process of the host squamous epithelia. HPVs gain access to and infect basal cells within the squamous epithelia presumably at sites of wounding and establish their DNA genome as extrachromosomal replicons at low copies in the host nucleus. In these undifferentiated, mitotically active cells, a subset of viral genes called the early genes are expressed and the viral DNA is replicated on average once per cell cycle and efficiently segregated to daughter cells, thereby leading to the stable maintenance of the viral genome within the infected tissue. As no progeny virus is made in basal cells, this is referred to as the nonproductive stage of the life cycle. Once daughter cells leave the basal compartment and initiate terminal differentiation, the productive stage of the viral life cycle begins. Here, viral DNA is amplified, structural or capsid proteins expressed and progeny viruses assembled (Howley, 2001). The virus encodes at least five early genes (E1, E2, E5, E6 and E7) that are involved in viral DNA replication and transcription (E1, E2) (Lambert, 1991) and in creating a cellular environment that is supportive of the viral replicative life cycle (E5, E6 and E7) (Fehrmann et al., 2003; Flores et al., 2000; Genther et al., 2003; Thomas et al., 1999). Three ‘late’ genes (E1ˆE4, L1, L2) are selectively expressed in the terminally differentiated cells. They are involved in viral DNA amplification (E1ˆE4) (Nakahara et al., 2005; Wilson et al., 2005) or form the protein capsid (L1 and L2 are the major and minor capsid protein, respectively) of the progeny virions (Kirnbauer et al., 1993). Several reports have demonstrated that early proteins (E1, E2, E6 and E7) as well as late proteins (E1ˆE4 and L2) associate with PODs or protein constituents of PODs, suggesting that PODs play an important role in the papillomavirus life cycle. Based on these studies, it has been hypothesized that PODs are sites for the initiation of viral infection, DNA replication and DNA encapsidation (Becker et al., 2003; Bischof et al., 2005; Day et al., 2004; Day et al., 1998; Florin et al., 2002; Guccione et al., 2004; Heino et al., 2000; Roberts et al., 2003; Swindle et al., 1999).
In this study, we analyzed the status of PODs in cells that harbor papillomavirus genome extrachromosomally and determined whether PODs are required for discrete processes the papillomaviral life cycle. We found that the number of PODs was increased in cells that harbor papillomaviral genomes, and that this increase in human keratinocytes was observed predominantly within undifferentiated cells. Analysis of PML proteins indicated that the increase in the number of PODs correlated with increased post-translational modification of PML. Nevertheless, while we were able to recapitulate the effect of PML on early steps in infection by papillomavirus, we found that replication and transcription of transfected papillomaviral genomes were not dependent upon PML.
Results
PODs are increased within poorly differentiated cells in the organotypic raft cultures of NIKS cells harboring HPV genome
In order to examine the effect of papillomaviruses on PODs in the context of the viral life cycle, we analyzed the status of PODs in the organotypic (raft) cultures harboring HPV genome extrachromosomally that support the differentiation-dependent life cycle of papillomaviruses. Sections from paraffin-embedded rafts of NIKS (Normal Immortalized Keratinocytes, previously referred to as BC-1-EP/SL), a spontaneously immortalized line of human foreskin keratinocytes known to support the HPV life cycle, that do or do not harbor the HPV16, HPV18 or HPV31 genome were subjected to indirect immunofluorescence (IF) staining using anti-human PML antibody. Fig. 1 shows representative IF images obtained using confocal microscopy. PML was detected as nuclear speckles consistent with the pattern of PODs described in the literature (Maul et al., 2000). The intensity and the number of PODs was increased in rafts of NIKS harboring HPV16 genome when compared to rafts of NIKS not harboring HPV genomes (Fig. 1). This increase was observed predominantly within the poorly differentiated basal and parabasal layers of the stratified epithelia (white boxes in Fig. 1A and B, enlarge images are shown in Fig. 1C and D) in which the early, nonproductive stage of the viral life cycle takes place. This same result was observed in rafts of NIKS harboring HPV31 genomes using an alternative PML antibody (supplemental Fig.1) and in rafts of NIKS harboring HPV18 genomes (data not shown).
Figure 1. Immunofluorescence analysis of PODs in rafts that do or do not harbor HPV16 genome.
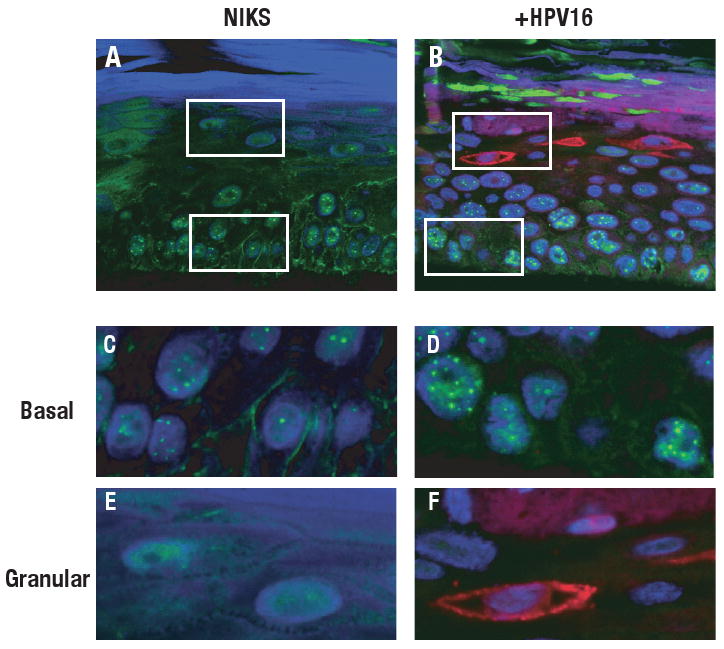
Cross sections from paraffin-embedded rafts of NIKS cells (A, C, E) or NIKS cells harboring HPV16 genome (B, D, F) were double stained with anti-human PML rabbit polyclonal antibody and anti-E1ˆE4 mouse monoclonal antibody as described in Materials and Methods. PML was detected using Alexa 488-conjugated goat anti-rabbit antibody (green) and E1ˆE4 was detected using Alexa 598-conjugated goat anti-mouse antibody (red). Nuclei were counterstained with TOTO-3 (blue). All images were analyzed using confocal microscopy (MRC1024, Bio-Rad/Zeiss). PML was detected as nuclear speckles in nucleus. Enlarged images from the area in basal cell layers (C, D) or granular layers (E, F) are shown. White boxes in panel A and B indicate the areas shown in the enlarged images (panels C-F).
Interestingly, we failed to find PML-positive nuclear speckles in the granular layers of rafts of NIKS not harboring HPV genomes (a white box in Fig.1A, an enlarged image is shown in Fig. 1E). This suggests that PODs disappear in human keratinocytes as they terminally differentiate. An alternative explanation might be that the epitope recognized by the PML ab used in our immunofluorescence staining shown in Figure 1 was masked somehow in the terminally differentiated cells. However, a similar, selective loss of detection of PML in the terminally differentiating compartment was observed with a second monoclonal antibody for PML that recognizes a different epitope (supplemental Fig. 1).
The granular layer is the cellular compartment that, in tissues harboring HPV genomes, supports expression of late viral proteins and assembly of progeny virus. It has been shown previously that viral late proteins, E1ˆE4 and L2 protein, when overexpressed in heterologous cell types, associate with PML or PODs (Day et al., 1998; Florin et al., 2002; Roberts et al., 2003). Therefore, we were interested in knowing if PODs were specifically retained in those terminally differentiating epithelial cells supporting the productive stage of the viral life cycle. In order to analyze the status of PODs specifically in those terminally differentiating keratinocytes in which the productive stage of the viral life cycle had been initiated, we performed double immunofluorescence staining with antibodies to PML and the late viral protein E1ˆE4. E1ˆE4 is known to be expressed in all cells in which viral DNA amplification, an initial aspect of the late stage of the viral life cycle, arises (Middleton et al., 2003; Nakahara et al., 2005). As shown in Fig. 1B and 1F, we were not able to detect PODs in E1ˆE4 positive-cells, indicating that PODs are lost in the differentiation-dependent manner even in the presence of HPV genome.
The viral minor capsid protein L2 protein forms aggregates in rafts harboring HPV genome that is independent of PODs
PODs have been hypothesized to be sites of papillomaviral DNA encapsidation (Becker et al., 2003; Florin et al., 2002); which is a step in the late stage of the viral life cycle the efficiency of which depends on L2 (Holmgren et al., 2005). However, we found that PODs are absent in terminally differentiating epithelial cells including those that express E1ˆE4 and therefore have undergone progeny viral DNA amplification, a step that occurs prior to virus assembly and DNA encapsidation (Fig. 1). This predicts that the subsequent steps of viral assembly including DNA encapsidation does not require PODs. In agreement with our observations, Becker et al. found that the assembly of virus like particles, VLPs, occurs in PML-null MEFs. Interestingly, they also found that L2 still accumulated in nuclear dots in PML-null MEFs, and they observed that L1 was recruited into these nuclear dots (Becker et al., 2004). To examine the sub-cellular localization of L2 in the the natural host cells, i.e. terminally differentiating human keratinocytes, we performed immunofluorescence for L2 and PML on sections from rafts of NIKS harboring HPV31 genome (available L2 antibodies for HPV16 do not efficiently detect L2 in rafts by IF). As shown in Figure 2, L2 was detected in small to large nuclear aggregates in the granular layers and none of these L2-positive cells were positive for PML. These results indicate that in the natural host cell in which virus assembly occurs, as in PML-null MEFs, L2 forms aggregates in the absence of PML or PODs.
Figure 2. Immunofluorescence analysis of PODs and L2 in rafts harboring HPV31 genome.
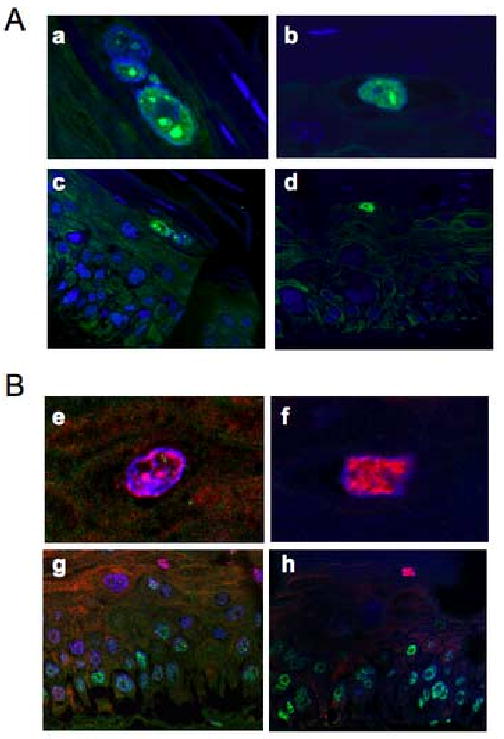
Cross sections from paraffin-embedded rafts of NIKS cells harboring HPV31 genome were stained with anti-L2 rabbit polyclonal antibody (A) or double stained with anti-L2 antibody and anti-human PML mouse monoclonal antibody (B). L2 was detected with goat anti-rabbit antibody conjugated with Alexa 488 (green in A) or conjugated with Alexa 546 (red in B). PML was detected with goat anti-mouse antibody conjugated with Alexa 488 (green in B). Nuclei were counterstained with TOTO-3 (blue). All images were analyzed using confocal microscopy (MRC1024, Bio-Rad/Zeiss). Enlarged images for L2 positive cells are shown in top (a, b, e, f) and overall views of rafts are shown in bottom (c,d,g,h).
The number of PODs is increased two-fold in NIKS cells harboring extrachromosomally replicating HPV genome
To evaluate more quantitatively whether PODs are increased within poorly differentiated human keratinocytes cells of rafts harboring HPV genomes (Fig.1), we examined the status of PODs in NIKS grown on feeders in monolayer, a condition that maintains keratinocytes in the poorly differentiated state. NIKS were grown on coverslips with feeder cells for 2 days and subjected to IF staining using antibodies against PML or Daxx, both components of PODs. Feeder cells were removed prior to fixation as described in Materials and Methods. Fig. 3A shows representative IF images of NIKS that do or do not harbor HPV genomes. Consistent with the observations made in raft cultures, the intensity and the number of nuclear speckles detected with antibodies to PML and Daxx was increased in monolayer cultures of NIKS harboring HPV16, HPV18 or HPV31 genomes, as compared to monolayer cultures of NIKS not harboring HPV genomes. We quantified the number of nuclear speckles positive for PML or Daxx in each population (Fig. 3B). NIKS harboring HPV genomes contained approximately two fold more nuclear speckles than the parental, HPV-negative NIKS. These differences were statistically significant. We concluded that the number of PODs was increased in NIKS harboring extrachromosomal HPV genomes.
Figure 3. Immunofluorescence analysis of monolayer of NIKS that do or do not harbor HPV genome.
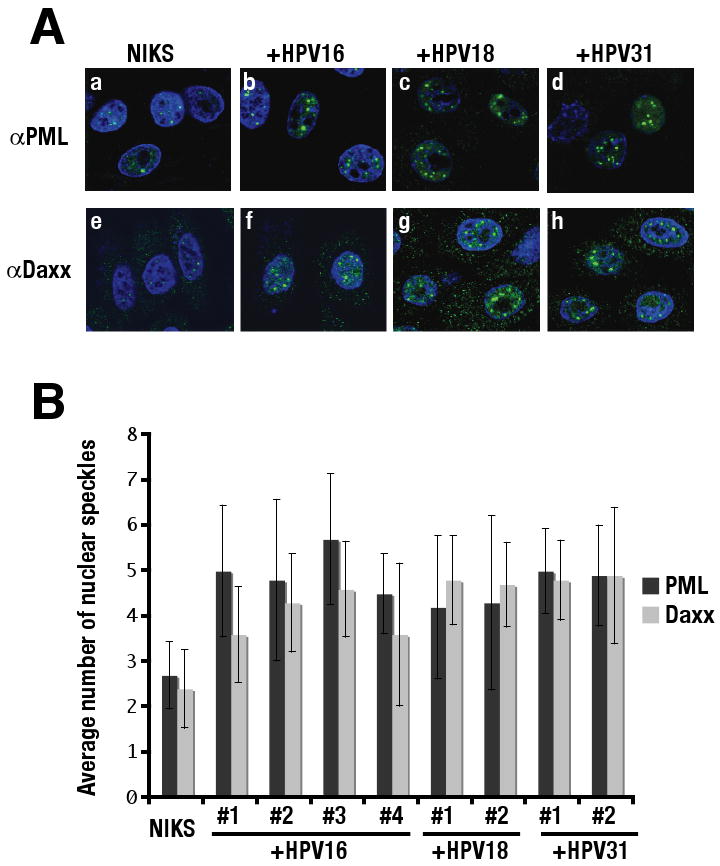
(A) Monolayer cultures of NIKS cells or NIKS cells harboring HPV16 genome, HPV18, HPV31 genomes were subjected to indirect immunofluorescent analysis using anti-human PML antibody (a-d) or anti-human Daxx antibody (e-h) as described in Materials and Methods. PML as well as Daxx was detected using Alexa 488-conjugated goat anti-rabbit antibody (green) and nuclei were counterstained with TOTO-3 (blue). (B) Quantification of the number of nuclear speckles in NIKS cells that do or do not harbor HPV genome. The numbers of nuclear speckles positive for PML or Daxx were counted in NIKS cells, four independently generated NIKS cell populations harboring HPV16 genome, and two independent NIKS cell populations each harboring either HPV18 or HPV31 as indicated. The number of nuclear speckles was significantly increased in all NIKS cell populations harboring HPV genome, compared to parental NIKS cells (p < 0.05). P value was evaluated using Wilcoxon rank sum test (two sided).
We also tested whether the number of PODs was increased in mouse C127 fibroblasts harboring bovine papillomavirus type 1 (BPV-1) genomes. The number of PML-positive nuclear speckles was increased in two clones of C127 cells harboring BPV-1 genome (clone B and ID13 (Law et al., 1981; Lowy et al., 1980)), compared to parental, BPV-1 negative C127 cells (data not shown). Thus, the increased frequency of PODs is a feature conferred by both animal and human papillomaviruses.
In addition, we compared the number of nuclear speckles positive for PML or Daxx in clonal populations of cervical epithelial cells derived from a patient infected with HPV16 in which the viral genomes exist either as extrachromosomal replicons (designated as W12E) or integrated into the host genome (designated as W12I) (Jeon and Lambert, 1995). W12E cells contained a statistically significant, approximately two-fold higher number of PML-positive PODs (6.6±1.2 and 6.4±1.8 in clones 20863 and 20850, respectively) compared to W12I (3.7±0.6, 3.6±0.5 and 2.9±0.7 for clones 20861, 21402 and 20822, respectively) cells. A similar two-fold difference was observed with Daxx antibody (data not shown). These data indicated that the extrachromosomal state of the papillomaviral genome correlates with an increased number of PODs. Taken together, these IF data demonstrate that papillomaviruses cause an increase in the number of PODs in the non-productive stage of the viral life cycle, possibly in association with extrachromosomal viral DNA replication.
Slower migrating forms of PML are increased in the cells harboring papillomavirus genome
Since PML protein is known to be required for POD formation, we tested whether the levels of PML protein were increased in human and mouse cells harboring papillomavirus genomes. Total cellular protein was isolated in the cell lysis buffer containing 20mM NEM which inhibits desumoylation and an equal amount of the protein lysate was subjected to Western blot analysis (Fig. 4A). As a control, the protein lysate was isolated from NIKS infected with 10 MOI of HSV-1 at 6 hrs post-infection, because HSV-1 is known to cause degradation of PML upon infection (lane 1 in Fig. 4A). Protein lysate was also isolated from mouse embryonic fibroblasts (MEFs) generated from either PML-null or PML-sufficient mice (generous gifts from Dr.Gerd G Maul at The Wistar Institute). In lysates from NIKS, a major band specific for PML migrating at around 105 KDa as well as several minor bands was detected (left panel lane 1 to 6 in Fig. 4A). All of these bands were detected with a level significantly reduced in NIKS infected with HSV-1, confirming that these protein species are PML specific. In mouse fibroblasts, the major PML-specific band had an apparent molecular weight slightly larger than 105 KDa. It and some minor bands were not detected in PML-null MEFs (lane 7 in Fig. 4A). As shown in Fig. 4A, the patterns of migration of PML proteins were affected by the presence of papillomavirus genomes (Lane 3 to 6, compared to 2, lane 10 and 11, compared to 8 and 9). In all cases we saw an increased abundance particularly of slower migrating forms of PML (brackets, Fig. 4A) in the human keratinocytes as well as the mouse fibroblasts harboring papillomavirus genomes. These slower migrating forms of PML could represent altered post-translational modification states of PML (Boddy et al., 1996; Chang et al., 1995) or alternative isoforms that arise from differential splicing transcripts. PML encodes three lysines that are conjugated with small ubiquitin modifier -1 (SUMO-1) and the conjugation of a SUMO-1 molecule adds the size of the conjugated proteins each approximately 12 KDa (Dohmen, 2004). Sumoylation of PML is required for proper POD formation and for recruiting proteins such as Daxx into PODs (Ishov et al., 1999; Zhong et al., 2000a). This and the facts that we observed a correlation between the increased number of PODs and the accumulation of slower migrating forms of PML led us to suspect that the latter represent sumoylated forms of PML.
Figure 4. Western blot analysis for PML in NIKS and C127 cells that do or do not harbor papillomaviral genomes.
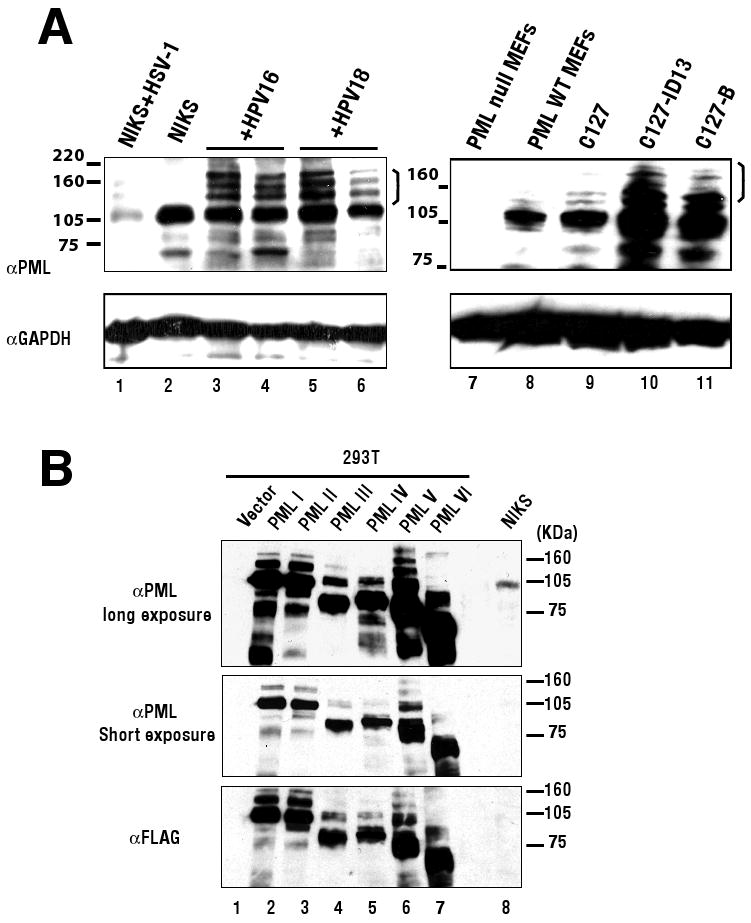
(A) The protein lysate was isolated from cells in the lysis buffer containing 20mM of NEM. 100 μg of protein from NIKS cells (lane 1 -6) or 20 μg of mouse cells (lane 7–11) was electrophoresed through a 7.5% SDS-polyacrylamide gel and transferred to PVDF membrane. Human PML protein was detected using anti-human PML mouse monoclonal antibody (Top panel: lane 1-6) and mouse PML protein was detected using anti-mouse PML mouse monoclonal antibody (Top panel: lane 7-11). GAPDH was detected as a loading control (bottom panels). NIKS cells were infected with HSV-1 at 10 MOI and harvested at 6 hrs post-infection (lane 1). Two independent NIKS cells populations harboring HPV16 (lane 3,4) or HPV18 (lane 5,6) were shown for comparison to parental NIKS cells (lane 2). C127 cells clone B or ID13 (lane 10 or 11, respectively), were compared to parental C127 cells (lane 9). Mouse embryonic fibroblasts (MEFs) generated from a PML knock out mouse (lane 7; PML null MEFs) or from its wildtype counterpart (lane 8; PML WT MEFs) were included as a negative or positive control for mouse cells. Molecular weight markers are depicted on left of top panels. A bracket on the right of top panels indicates slower migrating forms of PML described in the text. (B) The expression vector encoding each isoform of human PML cDNA (PMLI to PML VI) that were FLAG tagged at N-terminus or the empty vector was transfected into 293T cells and the protein lysate was harvested at 48hrs post-transfection. 5 μg of protein lysate from 293T cells and 100 μg of protein lysate from NIKS cells were electrophoresed. Exogenously expressed PMLs in 293T cells (lane 1 to 7) and endogenous PMLs in NIKS cells (lane 8) were detected using anti-human PML antibody (top two panels). After stripping, the membrane was subjected to immnodetection using anti-FLAG antibody (bottom panel). Molecular weight markers are depicted on right of each panel.
To investigate further the nature of the slower migrating forms of PMLs, we compared the electrophoretic mobility of PML in NIKS to that in 293T cells transfected with vectors expressing individual PML isoforms (Fig. 4B). The expression vectors encoding each of six isoforms of human PML cDNA, PML I to VI (Beech et al., 2005), or the empty vector was transfected into 293T cells and the protein lysate was isolated. All PMLs were epitope tagged at N-terminus with FLAG. 5 μg of the protein lysate from 293T cells transfected were electrophoresed together with 100 μg of the protein lysate from NIKS cells and PML proteins were detected by Western blot using anti-human PML antibody or anti-FLAG antibody. As shown in Fig. 4B, the major bands of PML isoform I and II (the largest predicted isoforms) were detected at size around 105KDa (lane 2 and 3 in Fig. 4B). This size was consistent with the size of the major endogenous PML detected in NIKS cells (lane 8 in Fig. 4B: long exposure). As predicted, the major bands of all other PML isoforms were smaller than 105KDa (lane 4 to 7). Minor bands that migrated slower than the major bands for each PML isoform likely reflect post-translationally modified derivatives of the individual PML isoform exogenously expressed. These slower migrating forms (Fig. 4B) were in the same apparent size range as the slower migrating forms of PML detected in NIKS harboring HPV genomes (bracket, Fig. 4A). We interpreted these results to indicate that the slower migrating forms of PML that accumulated in HPV-positive NIKS cells were post-translationally modified PML. Western blot analysis for Daxx protein revealed no significant change in levels of Daxx in NIKS and C127 cells harboring papillomavirus genome (data not shown). Thus, the presence of papillomavirus genome specifically affects PML protein.
PML is not required for the E2 dependent transcription in BPV-1 transfected cells
Day et al. recently investigated the trafficking of papillomavirus particles in infected cells (Day et al., 2004). They found that DNA encapsidated in papillomavirus pseudovirions together with the minor capsid protein L2 co-localized to PODs by 24 hrs post-infection. They also found an increased expression of either encapsidated reporter gene or encapsidated viral genome in PML-null MEFs that had been complimented with a human PML cDNA. This led the authors to hypothesize that PODs are destinations for papillomavirus-encapsidated DNAs and contribute to the establishment of viral infections. Using the same cells Day et al. used, we recapitulated their results using BPV-1 particles (data not shown). Thus the influence of PML on papillomavirus infection, and the observed increase in PODs in papillomavirus positive cells led us to question what specific viral processes were influenced by PML and PODs.
To investigate in more detail what viral processes might be dependent upon PODs and specifically PML, we monitored the efficiency of viral transcription and DNA replication in these same pair of PML-null MEFs that are or are not complemented with the human PML cDNA. These cells are called HP3 (PML-null MEFs transduced with a retrovirus carrying a human PML cDNA) and Hemp (PML-null MEFs transduced with the empty retroviral vector) cells. Papillomaviral transcription relies upon a family of gene products encoded by the viral E2 translational open reading frame (ORF). The full-length BPV-1 E2 ORF gene product is called E2 transcriptional activator (E2TA) because it activates transcription of viral promoters through its ability to bind the viral genome at multiple copies of a partially palindromic DNA binding site called E2BS. Two shorter E2 ORF gene products that are generated through initiation of translation at an internal ATG (E2TR) or through mRNA splicing (E8E2TR) are transcriptional repressors that can inhibit the function of E2TA via their ability to bind E2BSs and/or to form heterodimers with E2TA. We tested the efficiency of transcription supported by E2TA using a luciferease reporter assay. The reporter plasmid, pBS1073, encodes four E2BSs in the upstream of the minimal tymidine kinase promoter which drives expression of firefly luciferase. pBS1073 was used to monitor E2TA activity. pBS1013, a parental plasmid to pBS1073 that does not contain any E2BSs, was used as a negative control. To provide further verification that we were monitoring E2TA function in these experiments, we co-transfected the reporter plasmids not only with a recombinant plasmid containing the wild type BPV-1 viral genome but also with ones carrying mutant BPV-1 genomes disrupted in their expression of either E2TA or E2TR expression (Lambert et al., 1988; Lambert et al., 1990). To monitor efficiency for the transfections, a GFP expressing plasmid was included in the transfections. All transfections were done in triplicate and three independent sets of transfections were performed to account for experimental variation. The luciferase activities from cells transfected with BPV-1 genomes were normalized to that from cells transfected with an empty vector. As shown in Figure 5, the luciferase activity from pBS1073 was increased when the recombinant plasmid containing the wild type BPV-1 was co-transfected. As expected, a minimal luciferase activity was detected from pBS1013. The BPV-1-dependent induction in luciferase expressed from pBS1073 in the PML-positive HP-3 cells or the PML-negative Hemp cells was 8 or 12 fold, respectively. These increases were not significantly different from each other (p=0.12), indicating that PML is not required for the E2-dependent transcription. As predicted, the mutant BPV-1 genome inactivated for E2TA did not support increased transcription of luciferase from pBS1073 as compared to that from pBS1013; whereas, the E2TR-defective BPV-1 genome led to heightened E2TA activity. In the later case the similar level of E2TA-dependent activity was evident in the HP-3 cell and Hemp cell lines (18 and 21 fold increase, respectively). Similar results were obtained whether cells were isolated at 48 (data not shown) or 72 hours (Fig. 4) post-transfection. The efficiency of the transfection in Hemp and HP3 cells was similar based upon quantification of the frequency of GFP-positive cells (data not shown). Thus, we concluded that the presence of PML or PODs does not influence the transcriptional activity of the E2 in transfected cells.
Figure 5. E2 dependent transcription assay in HP-3 cells or Hemp cells.
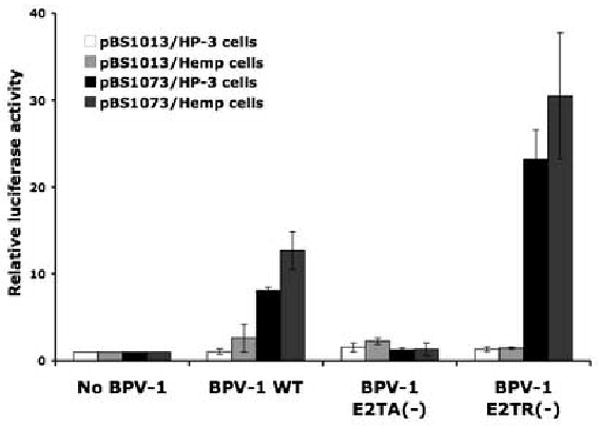
A plasmid encoding full sequence of BPV-1 wild type genome or mutant BPV-1 genomes defective for E2TA (BPV-1 E2TA(-)) or E2TR (BPV-1 E2TR(-)), was co-transfected a reporter plasmid, pBS1073 or pBS1013 into HP-3 cells or Hemp cells. An empty vector was transfected as a control (no BPV-1). pBS1073 contains four E2 binding sites (E2BS) located upstream of the minimal thymidine kinase promoter, which drives expression of firefly luciferease; therefore, luciferase expression from pBS1073 is E2-dependent. pBS1013, a parental plasmid of pBS1073, which does not contain E2BSs, was used as a negative control. At 72 hrs post-transfection, cell lysates were harvested and luciferase activity was measured as described in the Materials and Methods. All transfections were done in triplicate. Average luciferase activity from cells transfected with the empty vector (no BPV-1) was set at 1. Y-axis indicates fold increase of a luciferase activity from cells transfected with the indicated BPV-1 DNA. Shown is a representative experiment; this experiment was repeated three times.
PML is not required for the viral DNA replication of BPV-1 in transfected cells
Swindle et al. reported that, in the presence of HPV E1 and E2, a plasmid containing an HPV origin of DNA replication co-localized to or close by nuclear foci containing PML protein in transiently transfected cells (Swindle et al., 1999). This finding led the authors to conclude that papillomaviral DNA replication occurs at or near these nuclear foci. To investigate whether PML is required for papillomaviral DNA replication, a recombinant plasmid carrying either the wild type BPV-1 genome or a mutant BPV-1 genome defective in expression of E2 and the viral DNA helicase E1 (both E1 and E2 are required for initial BPV-1 DNA replication) (Lambert et al., 1988), was transfected into HP3 and Hemp cells. A GFP expression vector was included in the transfections and its expression was monitored at 24 hours post-transfection, allowing us to confirm that the efficiency of the transfection was comparable amongst all samples. Low molecular weight “Hirt” DNA was isolated at day 7 post-transfection. Hirt DNA from an equal number of cells was digested with BamHI, which linearizes the BPV-1 plasmid, in the presence or the absence of Dpn I, which can digest only the input, bacterially synthesized DAM-methylated DNA. Thus the Dpn I-resistant DNA represents DNA that has replicated in the transfected mammalian cells. Bacterially synthesized DNA, pGEM-3Z, was added at the time of cell lysis as an internal control to ensure that the efficiency of DNA isolation was comparable for individual samples and that Dpn I digestion was complete (data not shown for no Dpn I digestion control). These digested DNAs were subjected to BPV-1 specific Southern analysis. Three independent transfections were performed to account for experimental variation. The results from a representative experiment are shown in Figure 6. Similar levels of Dpn I-resistant wild type BPV-1 DNA accumulated in Hemp and HP3 cells. As predicted, no Dpn I-resistant BPV-1 was observed using the E1/E2 mutant viral genome, confirming that we were monitoring E1 and E2-dependent replication. These results indicated that PML and PODs are not required for viral DNA replication in transfected cells.
Figure 6. Short-term replication of BPV-1 DNA in HP-3 cells or Hemp cells.
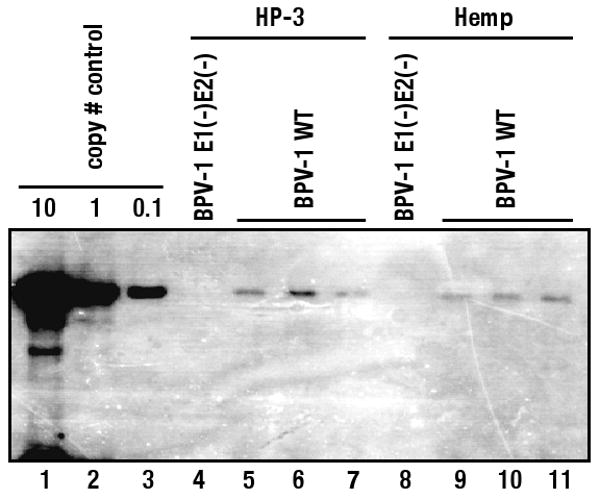
A plasmid encoding full sequence of BPV-1 wild type genome or mutant BPV-1 genome, defective for both E1 and E2 (BPV-1E1(-)E2(-)), was co-transfected with pGFP-N1 into HP-3 cells (lane 4-7) or Hemp cells (lane 8-11). Hirt DNA was isolated from cells at 7 days after transfection and subjected to BamHI and Dpn I digestion. Hirt DNA from equal number of cells was electrophoresed though 0.8% agarose gels and Dpn I resistant BPV-1 DNA was detected by Southern blot. A plasmid encoding wild type BPV-1 linearized by BamHI digestion was loaded for copy number controls (lane 1-3). No Dpn I resistant BPV-1 DNA was detected in Hirt DNA isolated from HP-3 cells and Hemp cells transfected with E1 and E2 defective BPV-1 transfection (lane 4, 8), validating that BPV-1 DNA replication was E1 and E2 dependent. Dpn I-resistant DNA was detected in all three independent transfections of wild type BPV-1 genome into either HP-3 (lane 5-7) or Hemp (lanes 9-11) cells.
Discussion
The association of several viral proteins of papillomaviruses with PODs or PML has been reported, the best-studied being L2, the minor capsid protein that is implicated viral DNA encapsidation. Several laboratories have reported that L2 is co-localized with PML in the nucleus and giving the classical punctate nuclear pattern seen for PODs. Our previous work as well as work done by Day et al. have shown that L1, a major capsid protein, as well as E2 protein co-localizes with PML in the presence of L2, but not in the absence of L2. These results led to the hypothesis that PODs may provide sites for viral DNA encapsidation (Becker et al., 2003; Day et al., 1998; Florin et al., 2002; Heino et al., 2000). These prior studies were not carried out in the natural host cells in which progeny virus assembly normally occurs. In the current study, we found that terminally differentiating keratinocytes, the host cells in which progeny virus assembly does occur, did not contain PML positive nuclear speckles even in the presence of HPV genomes and that L2 forms aggregates in the absence of PODs in those terminally differentiating keratinocytes (Fig.1 and Fig. 2). Thus we must conclude that viral DNA encapsidation occurs independent of PODs or PML. Interestingly, Becker et.al found that Daxx, which is detected in a diffuse or occasionally microdispersed pattern in the nucleus of PML-null cells or in human fibroblasts in which the expression of PML is knock down by shRNA (Everett et al., 2006; Zhong et al., 2000a), was attracted to nuclear aggregates of L2 in the PML-null MEFs (Becker et al., 2004). Recently, it was reported that the localization of L2 with PODs was dependent on the expression level of L2 as high but not low levels of L2 resulted in the localization to PODs. Furthermore, the localization of L2 to PODs was not affected by the treatment of interferon β that increases PODs via transcriptional upregulation of PML and Sp100 (Kieback and Muller, 2006). Taken together with our findings illustrated in Figure 2, we hypothesize that L2 protein may self-aggregate and attract POD-associated proteins into nuclear foci that resemble POD structures when it's expressed at high level. The direct target of L2 in forming these POD-like nuclear bodies may be Daxx protein or other POD-associated proteins but not PML per se. Since L2 has been indicated to play a key role for viral DNA encapsidation, this de novo formation of POD-like structures may be a part of viral assembly processes in the terminally differentiating cells where L2 is expressed at a high level. It would be interesting, therefore, to analyze the localization of Daxx in the terminally differentiating cells in the presence or the absence of HPV genome to study POD-like structures (unfortunately, antibodies for Daxx that we tested did not work on paraffin embedded sections of rafts). An example of de novo formation of POD-like structures has also been described for HSV-1 infected cells immediately upon infection (Everett et al., 2004).
Although we did not observe the detectable influence of HPV genome on the absence of PODs in terminally differentiating cells, we did observe that the frequency of PODs was increased in the basal to intermediate layers of rafts cultures and in monolayer cultures of NIKS harboring HPV genomes (Figs.1 and 3) as well as C127 cells harboring BPV-1 genome (data not shown). Quantification revealed that the number of PODs, using either abs to PML or Daxx, was approximately two fold increased in NIKS harboring HPV genomes. Furthermore, W12E cells contained two-fold greater numbers of PODs than W12I cells and the number of PODs in W12I cells was similar to that of HPV-negative NIKS, indicating that the presence of extrachromosomally replicating papillomavirus genomes correlates with the increase in numbers of PODs. Both E6 and E7 oncoproteins of HPV16 are selectively expressed in the W12I cells (Jeon and Lambert, 1995). Therefore we conclude that expression of the E6 and the E7 oncogenes is not sufficient to account for the increased number of PODs.
Western blot analysis showed that slower migrating forms of PMLs were concomitantly increased in NIKS cells as well as C127 cells harboring papillomavirus genomes and that the slower migrating forms of PML were likely post-translationally modified forms of PMLs (Fig. 4). Sumoylation of PML is required for proper POD formation. In PML-null MEFs, POD related proteins such as Daxx and Sp100 fail to localize to similar nuclear structures. Introduction into these cells of the mutant PML that lacks all three sumoylation sites does not rescue POD formation whereas the introduction of the wild type PML does (Zhong et al., 2000a). Given this requirement of sumoylation for POD formation we raise the possibility that sumoylation of PML is enhanced and/or desumoylation of the PML is suppressed in the presence of extrachromosomal papillomavirus genomes.
Little is known about what controls sumoylation of PML. The number of PODs can change during the cell cycle, increasing in S phase and decreasing during mitosis, and in response to cellular stresses. Although the mechanisms underlining these processes has not been clearly elucidated, they appear to be in part mediated by phosphorylation and subsequent sumoylation or desumoylation of PML. Everett et al. have shown that sumoylated forms of PML are absent and instead a phosphatase sensitive form of PML newly appears in mitotic cells, indicating that PML is desumoylated and phosphorylated prior to or during mitosis. They also showed that the treatment of a phosphatase inhibitor to interphase cells facilitated desumoylation of PML, suggesting that PML is phosphorylated and subsequently desumoylated in mitosis (Everett et al., 1999). In contrast, treatment with arsenic trioxide (AS2O3) induces sumoylation of PML in a variety of cells, and in APL cells, in which PODs are normally disrupted, AS2O3 restores PODs (Zhu et al., 1997)((Bischof et al., 2005). Hayakawa and Privalsky reported that AS2O3 induced phosphorylation of PML through mitogen activated kinase (MAPK) pathways and increased phoshorylation of PML was associated with increased sumoylation of PML (Hayakawa and Privalsky, 2004). Viral E5 protein of HPV16 as well as BPV-1 has shown to activate a growth factor receptor, epidermal growth factor receptor (EGFR) or platelet-derived growth receptor (PDGF-R) respectively, resulting in the enhancement of MAPK-dependent signal transduction pathways (Crusius et al., 1997; Crusius et al., 2000; Petti et al., 1991). Therefore, it is possible that the E5 induces sumoylation of PML and subsequent formation of PODs by activating the MAPK pathways. This is consistent with the observation that W12E cells contain greater numbers of PODs than W12I cells, in which the E5 expression is lost due to integration (Jeon and Lambert, 1995). However, it is also relevant to note that long-term treatment with AS2O3 eventually results in the proteosome-mediated degradation of PML. Therefore, if the constitutive activation of MAPK pathways by E5 induces sumoylation and subsequent increase of PODs in cells harboring papillomavirus genome, there must be other mechanisms provided by the virus to suppress subsequent degradation of the PML.
Direct interaction of HPV E6 and/or E7 with several isoforms of PML has been reported. Bischof et al. have shown that HPV16, HPV11 or HPV6b E7 co-immunoprecipitates with all isoforms of the PML tested and that these E7 proteins, but not the E6 proteins from these same HPVs, can inhibit PML IV-induced senescence (Bischof et al., 2005). In contrast, Guccione et al. have reported that HPV11 as well as HPV18 E6 protein co-immunoprecipitates with PML I to IV and can inhibit PML IV-induced senescence (Guccione et al., 2004). In those studies, the consequence of the E7 or the E6 interaction with PML isoforms, other than in the context of PML IV-induced senescence, was not addressed. It is possible that the interaction of the E6 and/or the E7 protein with some isoforms of PML may provide an additional mechanism that facilitates the increase in the number of PODs in the presence of E5, but clearly from our PML IF results with W12I cells, the expression of E6 and E7 alone is not sufficient.
Whereas the above discussion considers the potential role of viral oncoproteins in causing the increased frequency of PODs, our data is also consistent with the alternative hypothesis that some intrinsic cellular responses to extrachromosomal viral DNA is responsible for the observed increase in PODs. Jul-Larsen et al. found that newly synthesized viral DNA in cells infected by SV40 or polyomavirus BK (BKV) accumulated at or near to PODs and that this sequestration of the DNA to PODs was dependent on active viral DNA replication. A significant portion of the accumulated DNA was single stranded (ssDNA) leading these investigators to hypothesize that the viral genomes are recruited to PODs as a part of a DNA damage response (Jul-Larsen et al., 2004). Indeed, ssDNA is also recruited to PODs upon UV exposure and depletion of PML sensitizes cells to UV-induced apoptosis, suggesting that PODs are processing sites for single stranded, damaged DNA (Boe et al., 2006). Therefore, it is tempting to speculate that extrachromosomally replicating papillomavirus DNA also is associated with PODs and this is driven by DNA damage responses. Consistent with this hypothesis, Swindle et al. observed that E1 and E2 proteins of HPV11 co-localize with a plasmid containing the replication origin of HPV11 in or juxtaposed to nuclear foci containing PML in transient replication experiments and that this colocalization was not observed in the absence of the ori sequence (Swindle et al., 1999). The number of PODs increases upon DNA damage. This increase has been attributed in part to p53-dependent transcriptional upregulation of PML (Borden, 2002; Dellaire and Bazett-Jones, 2004; Guo et al., 2000). It has also been shown that PML is phosphorylated by DNA damage checkpoint kinase Chk2 within 1 hr of irradiation (although protein levels of PML are not changed) and that phosphorylation of PML by Chk2 is abrogated in the absence of ataxia telangiectasia-mutated (ATM) (Yang et al., 2002). Indeed, Dellaire et al. reported that the initial increase of PODs upon irradiation was independent of p53 as well as protein synthesis and new PODs were formed from preexisting PODs by fission. Furthermore, they showed that this increase of PODs was delayed or inhibited by a loss of function of Niimegen breakage syndrome (NBS) -1, ATM, Chk2 and ATR (ATM- and Rad3 related), all of which have been detected in PODs constitutively or induced upon DNA damage (Dellaire et al., 2006). Other DNA repair proteins such as BLM, a RecQ DNA helicase, have been detected in PODs (Sanz et al., 2000; Yankiwski et al., 2001). It is therefore interesting to consider the possibility that extrachromosomally replicating papillomaviral DNA molecules, because they may accumulate single stranded regions of DNA as does polyomavirus DNA, or single or double stranded breaks (Taylor et al., 2003), could elicit a cellular DNA damage response that in turn induces the number of PODs. Such a DNA damage response would ultimately be over-ridden by the action of the viral oncoproteins E6 and E7, or in the case of polyomaviruses, large T antigen, which can inhibit DNA damage responses (Ahuja et al., 2005; Helt and Galloway, 2003; Munger et al., 2004). Thus, while initial DNA damage responses that can lead to the induction of PODs may be intact in cells harboring extrachromosomal viral genomes, the intended consequence of this response in terms of eliciting a functional DNA damage response is over-ridden by the viral oncoproteins. Thus, it is perhaps not surprising that neither we (Fig. 6), nor Jul-Larsen et al, in their studies on polyomavirus (Jul-Larsen et al., 2004), found any influence of PML-status on the efficiency of viral DNA replication.
A number of reports have indicated that the PML can influence transcription in a positive or a negative manner. For instance, the overexpression of PML resulted in transcriptional activation of major histocompatibility complex (MHC) class I transporter TAP-1 (Zheng et al., 1998) and the glucocorticoid receptor (Lin et al., 2003), whereas it caused the transcriptional repression of EGFR (Vallian et al., 1998). This transcriptional regulation is mediated by the interaction of PML with transcriptional factors such as Daxx, a repressor for the glucocorticoid receptor, and Sp-1, an activator for the EGFR. Block et al. have recently shown that the subnuclear targeting of reporter plasmids to PODs results in increased or reduced expression of reporters in a promoter dependent manner (Block et al., 2006). The results of Day et al. and our confirmatory experiments (data not shown) indicate that targeting the BPV-1 genome to PODs upon infection enhances initial transcription of viral early genes, which is mediated by enhancer elements found in the long control region (LCR) (Day et al., 2004). However we also observed that PML does not have a direct role on the activity of the viral E2 transactivator (Fig. 5), which is presumably expressed at times post-initial infection. Thus, the importance of PML and PODs in papillomavirus transcription may be transient in nature.
Materials and Methods
Tissue culture and virus infection
NIKS were maintained at subconfluence onto mitomycin C treated m1 3T3 feeder cells in F medium with all supplements as previously described (Lambert et al., 2005). C127 cells were cultured in Dulbecco's Modified Eagle Media (D-MEM, Invitrogen Co., Carlsbad, CA) containing 10% fetal bovine serum (FBS), Penicillin and Streptomycin (100 μg/ml). HP-3 cells and Hemp cells were cultured in media containing G418 (500 μg/ml) and Hygromycin B (400μg/ml) as previously reported (Day et al., 2004). NIKS were plated at 1X106 cells in 10-cm tissue culture plate without feeder cells in low calcium F medium with all supplements (Lambert et al., 2005) on the day before HSV-1 infection. NIKS were incubated in 2 ml of low calcium F medium containing HSV-1 KOS strain (a kind gift from Dr. Curtis R. Brandt at University of Wisconsin-Madison) at a multiplicity of infection (MOI) of 10 for two hours at 37 °C to allow the viruses to attach, briefly washed and incubated in 5 ml of freshly replaced low calcium F medium with all supplements for subsequent 6 hours until harvesting.
Indirect immunofluorescence
NIKS were plated on coverslips with feeder cells and cultured in monolayer for 2 days before the staining. Feeder cells were removed by incubating cells in 0.02% EDTA/Phosphate buffered saline (PBS) prior to fixation. NIKS were fixed in 4% paraformaldehyde (PFA) in PBS for 15 mins at room temperature and washed with PBS three times for 5 mins. Subsequently, cells were incubated in 0.01% Triton-X 100 in PBS for 15mins at room temperature and washed with PBS three times for 5 mins before proceeding with immunostaining. Cross sections of rafts on plus coated slides were deparaffinized in xylene, rehydrated using a serial grade of ethanol and then microwaved in 10 mM citrate buffer pH 6.0 for 20 mins for antigen unmasking before proceeding immunostaining as previously described (Nakahara et al., 2005). The indirect immunofluorescence staining was done as follows. Samples were incubated in a blocking solution (3% bovine serum albumin in PBS) for 30 mins at room temperature and then incubated with a primary antibody diluted in the blocking solution for over night at 4 °C. Samples were washed with PBS three times for 5 mins at room temperature and incubated with a fluorophore conjugated secondary antibody diluted in PBS for 30 mins at room temperature. NIKS grown on coverslips were mounted onto slides with the fluorescence mounting medium (Vector Lab., Burlingame, CA) added 2μM of TOTO-3 (Invitrogen) and analyzed using confocal microscopy (MRC-1024, Bio-rad/Zeiss, Thornwood, NY). Rafts were sealed in the mounting media added 2μM of TOTO-3 with coverslips and analyzed. Antibodies used for indirect immunofluorescence were followings. Anti-human PML rabbit polyclonal antibody (H-238, Santa Cruz Biotechnology Inc., Santa Cruz, CA) at 1:100 dilution, anti-human PML monoclonal mouse antibody (PG-M3, Santa Cruz) at 1:200 dilution, anti-human Daxx rabbit polyclonal antibody (M-112, Santa Cruz) at 1:100 dilution, anti-E1ˆE4 monoclonal antibody at 1:20 dilution, anti-HPV31 L2 rabbit polyclonal antibody at 1:500 dilution. Alexa Fluor 488 goat anti–rabbit antibody (Invitrogen) as a secondary antibody to detect hPML or Daxx at 1:500 dilution and Alexa Fluor 594 goat anti–mouse antibody as a secondary antibody detect E1ˆE4 at 1:500 dilution. To quantify the number of PML or Daxx positive nuclear speckles, the number of nuclear speckles was counted over 200 cells from 10-20 images that were randomly chosen for individual NIKS populations. The total number of nuclear speckles per image was divided by the total number of cells per image to derive the average number of the nuclear speckles per cell. The statistical analysis was performed using the Wilcoxon rank sum test (two sided).
Western blotting
Cells were washed in ice-cold PBS twice and scraped off in ice-cold lysis buffer (50 mM Tris pH7.4, 150 mL NaCl, 1 mM EDTA, 1mM phenylmethysulfonyl, 1 μg/ml leupeptin, 1 μg/ ml pepstain, 1 mM NaF, 1% Nonidet-40, 0.5% deoxycholate, 0.5% Sodium dodecyl sulfate, SDS) containing 20mM N-ethylmaleimide (Sigma-Aldrich., St. Louis, MO) and then incubated on ice for 30 mins. The detergent soluble fraction was separated from the insoluble material by centrifugation at 14,000 rpm for 15 min, 4°C (Eppendorf Model 5415C Microcentrifuge). Total protein concentration of the soluble fraction was measured using DC protein assay kit instructed as manufacture's protocol (Bio-Rad, Hercules, CA). Equal amount of protein was mixed with an appropriate volume of 6X SDS loading buffer (1X; 62.5 mM Tris-HCl pH6.8, 10% glycerol, 2%SDS, 0.02% bromophenol blue) containing 100 mM DTT and boiled for 5 min. Protein samples were electrophoresed through an SDS-7.5 % polyacrylamide gel (ready-made gel, Bio-Rad) and transferred onto PVDF membrane (Milipore, Billerica, MA). The membrane was blocked with 5% skimmed milk in PBS-T (PBS containing 0.1% Tween-20) for 1 hr at room temperature and incubated overnight at 4°C with a primary antibody diluted in the blocking solution. The membrane was washed with PBS-T three times for 10 min at room temperature and incubated for 1hr at room temperature with a secondary antibody (Peroxidase conjugated anti mouse IgG, Jackson ImmunoResearch, cat.#715-035-150, West Grove, PA) diluted 1:10,000 in the blocking solution. The membrane was again washed with PBS-T three times for 10 min each at room temperature and antibody conjugates were detected using ECL plus kit (GE healthcare, Piscataway, NJ) according to the manufacture's instruction. To reuse membranes, they were stripped by agitating in the stripping buffer (62.5mM Tris-HCl pH 6.7, 2%SDS, 100mM β-mercaptoethanol) for 25 mins at room temperature and washed with PBS-T three times for each 5 mins. After stripping, membranes were blocked in the blocking buffer for overnight at 4°C, then incubated with the alternative primary antibody. Primary antibodies used included: anti-human PML mouse monoclonal antibody (clone IB9, MBL, Woburn, MA) at 1:500 dilution, anti- mouse PML mouse monoclonal antibody (clone 36.1-104, Upstate/Milipore, Billerica, MA) at 1:500 dilution, anti- GAPDH mouse monoclonal antibody (6C5, Chemion/Milipore) at 1:50,000 dilution, and anti-FLAG mouse monoclonal antibody (M2, Sigma-Aldrich) at 1:500. To compare the migration pattern of each PML isoform, the expression vector encoding the FLAG tagged human PML or the empty vector control (provided by Dr.Keith N Leppard (Beech et al., 2005)) was transfected into 293T cells using lipofectamine 2000 (Invitrogen) according to the manufacture's instruction. 48 hours after trasfection, the protein lysate was isolated as described above.
Luciferase assay
The luciferase reporter plasmid, pBS1013 or pBS1073 (Kim et al., 2003), and the recombinant plasmids encoding the wild type BPV-1 (p146-2 (Yang et al., 1985)) or mutant BPV-1 (p771-2 (Lambert et al., 1988) or p1472-1 (Lambert et al., 1990)) genome were cotransfected into the mouse fibroblast cell lines, Hemp and HP3, to monitor the levels of E2 dependent transcription in the absence or the presence of PML, respectively. Cells were plated on 6 well- tissue culture plates in 10% FBS in D-MEM media without antibiotics at 2×105 cells/well the day before transfection. 1 μg of pBS1013 or pBS1073, 1 μg of pEGFP-N1 (Clontech, Mountain View, CA) and 4 μg of a BPV-1 encoding plasmid or an empty vector were mixed with 30 μl of Superfect transfection reagent (Qiagen, Valencia, CA) in 100 μl of Opti-MEM (Invitrogen) and incubated for 15 mins at room temperature. The cell culture media was removed and replaced with the DNA-Superfect in 500 μl of 10%FBS in D-MEM. After incubation for 4 hrs, the media containing the transfection complex was removed and replaced with the fresh cell culture media (10% FBS in D-MEM containing G418 and Hygromycin B). 72 hrs after the transfection, cells were washed twice with PBS and lysed in 200 μl of a luciferase cell culture lysis buffer (Promega, Madison, WI). 20 μl of cell lysate was mixed with 100 μl of a luciferase assay substrate (Promega, Madison, WI) and the relative light unit (RLU) was measured by a luminometer (BD biosciences, San Jose, CA). Protein concentration in cell lysate was measured using DC protein assay kit to normalize RLU per μg protein for each sample. All transfections were done triplicate and three independent sets of experiments were performed. Statistical analysis was done using the two-sided Wilcoxon ran sum test.
Transient replication assay
Transfection was done using a Nucleofector as per manufacture's instruction using the MEF 2 Nucleofector kit, (Amaxa Inc., Gaithersburg, MD). HP-3 cells and Hemp cells were plated on 15 cm tissue culture plates at 5×105 cells/plate 2 days before transfection. Cells were removed by trypsin treatment and the number of cells was counted. 1×106 cells were mixed with 100 μl of a transfection solution and 4 μg of the recombinant plasmid encoding the wild type BPV-1 or a mutant BPV-1 deficient for E1 and E2 (p1308-1 (Lambert et al., 1988)) together with 1 μg of pEGFP-N1.. Transfection was done using the program T-20. Cells were placed back on 10 cm tissue culture plates and cultured for 7 days. Low molecular weight Hirt DNA, was extracted from cells as described previously (Hirt, 1967). 2 ng of bacterially synthesized pGEM-3Z (Promega) was added to lysis solution to compare efficiency of DNA isolation amongst samples and to monitor completion of Dpn I digestion. Hirt DNA isolated from an equal number of cells was digested with BamHI with or without Dpn I for 8 hrs at 37°C and electrophoresed through an 0.8% agarose gel. The gel was transferred to a nylon membrane (Hybond-N, GE healthcare) and subjected to BPV-specific Southern blot as previously described (Lambert et al., 1990).
Supplementary Material
Raft cultures harboring the wild type HPV31 or a L2 mutant of HPV31 were previously described (Holmgren et al., 2005). Cross sections of rafts were double stained with anti-human PML monoclonal antibody and anti-L2 rabbit polyclonal antibody as described in Fig.2. Consistent with Fig.1, the intensity as well as the number of PODs detected by the PML monoclonal antibody was increased in basal to intermediate layers of rafts harboring wild type HPV31 as well as a L2 mutant of HPV31 genome compared to rafts of NIKS not harboring HPV31 genome. PODs were not detected in the granular layers of rafts regardless of the presence of HPV31 genome, indicating that the failure of detecting PODs by anti-PML rabbit polyclonal antibody (Fig.1) is not due to the lack of reactivity of the antibody.
Acknowledgments
We thank all members of Lambert lab and Dr. Bill Sugden for helpful discussion. We especially thank David Vereide for assistance in establishing the IF conditions for detecting PML. We thank W.M.Keck Laboratory for Biological imaging at UW-Madison for technical assistance with performing confocal microscopy. We also thank Dr. Gerd G Maul (The Wistar Institute), Dr. Curtis R Brandt (UW-Madison), Dr. Patricia M. Day (National Cancer Research) and Dr.Keith N Leppard (The University of Warwick) for kindly providing cells, viruses and plamsids. This study was supported by a grant from the NIH (CA022443).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn JH, Hayward GS. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J Virol. 1997;71(6):4599–613. doi: 10.1128/jvi.71.6.4599-4613.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja D, Saenz-Robles MT, Pipas JM. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005;24(52):7729–45. doi: 10.1038/sj.onc.1209046. [DOI] [PubMed] [Google Scholar]
- Becker KA, Florin L, Sapp C, Maul GG, Sapp M. Nuclear localization but not PML protein is required for incorporation of the papillomavirus minor capsid protein L2 into virus-like particles. J Virol. 2004;78(3):1121–8. doi: 10.1128/JVI.78.3.1121-1128.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KA, Florin L, Sapp C, Sapp M. Dissection of human papillomavirus type 33 L2 domains involved in nuclear domains (ND) 10 homing and reorganization. Virology. 2003;314(1):161–7. doi: 10.1016/s0042-6822(03)00447-1. [DOI] [PubMed] [Google Scholar]
- Beech SJ, Lethbridge KJ, Killick N, McGlincy N, Leppard KN. Isoforms of the promyelocytic leukemia protein differ in their effects on ND10 organization. Exp Cell Res. 2005;307(1):109–17. doi: 10.1016/j.yexcr.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Bischof O, Nacerddine K, Dejean A. Human papillomavirus oncoprotein E7 targets the promyelocytic leukemia protein and circumvents cellular senescence via the Rb and p53 tumor suppressor pathways. Mol Cell Biol. 2005;25(3):1013–24. doi: 10.1128/MCB.25.3.1013-1024.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Block GJ, Eskiw CH, Dellaire G, Bazett-Jones DP. Transcriptional regulation is affected by subnuclear targeting of reporter plasmids to PML nuclear bodies. Mol Cell Biol. 2006;26(23):8814–25. doi: 10.1128/MCB.00636-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy MN, Howe K, Etkin LD, Solomon E, Freemont PS. PIC 1, a novel ubiquitin-like protein which interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene. 1996;13(5):971–82. [PubMed] [Google Scholar]
- Boe SO, Haave M, Jul-Larsen A, Grudic A, Bjerkvig R, Lonning PE. Promyelocytic leukemia nuclear bodies are predetermined processing sites for damaged DNA. J Cell Sci. 2006;119(Pt 16):3284–95. doi: 10.1242/jcs.03068. [DOI] [PubMed] [Google Scholar]
- Borden KL. Pondering the promyelocytic leukemia protein (PML) puzzle: possible functions for PML nuclear bodies. Mol Cell Biol. 2002;22(15):5259–69. doi: 10.1128/MCB.22.15.5259-5269.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkham J, Coen DM, Weller SK. ND10 protein PML is recruited to herpes simplex virus type 1 prereplicative sites and replication compartments in the presence of viral DNA polymerase. J Virol. 1998;72(12):10100–7. doi: 10.1128/jvi.72.12.10100-10107.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KS, Fan YH, Andreeff M, Liu J, Mu ZM. The PML gene encodes a phosphoprotein associated with the nuclear matrix. Blood. 1995;85(12):3646–53. [PubMed] [Google Scholar]
- Chee AV, Lopez P, Pandolfi PP, Roizman B. Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J Virol. 2003;77(12):7101–5. doi: 10.1128/JVI.77.12.7101-7105.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelbi-Alix MK, Pelicano L, Quignon F, Koken MH, Venturini L, Stadler M, Pavlovic J, Degos L, de The H. Induction of the PML protein by interferons in normal and APL cells. Leukemia. 1995;9(12):2027–33. [PubMed] [Google Scholar]
- Crusius K, Auvinen E, Alonso A. Enhancement of EGF- and PMA-mediated MAP kinase activation in cells expressing the human papillomavirus type 16 E5 protein. Oncogene. 1997;15(12):1437–44. doi: 10.1038/sj.onc.1201312. [DOI] [PubMed] [Google Scholar]
- Crusius K, Rodriguez I, Alonso A. The human papillomavirus type 16 E5 protein modulates ERK1/2 and p38 MAP kinase activation by an EGFR-independent process in stressed human keratinocytes. Virus Genes. 2000;20(1):65–9. doi: 10.1023/a:1008112207824. [DOI] [PubMed] [Google Scholar]
- Day PM, Baker CC, Lowy DR, Schiller JT. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc Natl Acad Sci U S A. 2004;101(39):14252–7. doi: 10.1073/pnas.0404229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day PM, Roden RB, Lowy DR, Schiller JT. The papillomavirus minor capsid protein, L2, induces localization of the major capsid protein, L1, and the viral transcription/replication protein, E2, to PML oncogenic domains. J Virol. 1998;72(1):142–50. doi: 10.1128/jvi.72.1.142-150.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellaire G, Bazett-Jones DP. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays. 2004;26(9):963–77. doi: 10.1002/bies.20089. [DOI] [PubMed] [Google Scholar]
- Dellaire G, Ching RW, Ahmed K, Jalali F, Tse KC, Bristow RG, Bazett-Jones DP. Promyelocytic leukemia nuclear bodies behave as DNA damage sensors whose response to DNA double-strand breaks is regulated by NBS1 and the kinases ATM, Chk2, and ATR. J Cell Biol. 2006;175(1):55–66. doi: 10.1083/jcb.200604009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohmen RJ. SUMO protein modification. Biochim Biophys Acta. 2004;1695(13):113–31. doi: 10.1016/j.bbamcr.2004.09.021. [DOI] [PubMed] [Google Scholar]
- Eskiw CH, Dellaire G, Mymryk JS, Bazett-Jones DP. Size, position and dynamic behavior of PML nuclear bodies following cell stress as a paradigm for supramolecular trafficking and assembly. J Cell Sci. 2003;116(Pt 21):4455–66. doi: 10.1242/jcs.00758. [DOI] [PubMed] [Google Scholar]
- Everett RD. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene. 2001;20(49):7266–73. doi: 10.1038/sj.onc.1204759. [DOI] [PubMed] [Google Scholar]
- Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J Virol. 1998;72(8):6581–91. doi: 10.1128/jvi.72.8.6581-6591.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Lomonte P, Sternsdorf T, van Driel R, Orr A. Cell cycle regulation of PML modification and ND10 composition. J Cell Sci. 1999;112(Pt 24):4581–8. doi: 10.1242/jcs.112.24.4581. [DOI] [PubMed] [Google Scholar]
- Everett RD, Murray J. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J Virol. 2005;79(8):5078–89. doi: 10.1128/JVI.79.8.5078-5089.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol. 2006;80(16):7995–8005. doi: 10.1128/JVI.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Sourvinos G, Leiper C, Clements JB, Orr A. Formation of nuclear foci of the herpes simplex virus type 1 regulatory protein ICP4 at early times of infection: localization, dynamics, recruitment of ICP27, and evidence for the de novo induction of ND10-like complexes. J Virol. 2004;78(4):1903–17. doi: 10.1128/JVI.78.4.1903-1917.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Sourvinos G, Orr A. Recruitment of herpes simplex virus type 1 transcriptional regulatory protein ICP4 into foci juxtaposed to ND10 in live, infected cells. J Virol. 2003;77(6):3680–9. doi: 10.1128/JVI.77.6.3680-3689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrmann F, Klumpp DJ, Laimins LA. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J Virol. 2003;77(5):2819–31. doi: 10.1128/JVI.77.5.2819-2831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores ER, Allen-Hoffmann BL, Lee D, Lambert PF. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J Virol. 2000;74(14):6622–31. doi: 10.1128/jvi.74.14.6622-6631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin L, Sapp C, Streeck RE, Sapp M. Assembly and translocation of papillomavirus capsid proteins. J Virol. 2002;76(19):10009–14. doi: 10.1128/JVI.76.19.10009-10014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genther SM, Sterling S, Duensing S, Munger K, Sattler C, Lambert PF. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J Virol. 2003;77(5):2832–42. doi: 10.1128/JVI.77.5.2832-2842.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guccione E, Lethbridge KJ, Killick N, Leppard KN, Banks L. HPV E6 proteins interact with specific PML isoforms and allow distinctions to be made between different POD structures. Oncogene. 2004;23(27):4662–72. doi: 10.1038/sj.onc.1207631. [DOI] [PubMed] [Google Scholar]
- Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, Pandolfi PP. The function of PML in p53-dependent apoptosis. Nat Cell Biol. 2000;2(10):730–6. doi: 10.1038/35036365. [DOI] [PubMed] [Google Scholar]
- Hayakawa F, Privalsky ML. Phosphorylation of PML by mitogen-activated protein kinases plays a key role in arsenic trioxide-mediated apoptosis. Cancer Cell. 2004;5(4):389–401. doi: 10.1016/s1535-6108(04)00082-0. [DOI] [PubMed] [Google Scholar]
- Heino P, Zhou J, Lambert PF. Interaction of the papillomavirus transcription/replication factor, E2, and the viral capsid protein, L2. Virology. 2000;276(2):304–14. doi: 10.1006/viro.2000.0342. [DOI] [PubMed] [Google Scholar]
- Helt AM, Galloway DA. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis. 2003;24(2):159–69. doi: 10.1093/carcin/24.2.159. [DOI] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26(2):365–9. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Holmgren SC, Patterson NA, Ozbun MA, Lambert PF. The minor capsid protein L2 contributes to two steps in the human papillomavirus type 31 life cycle. J Virol. 2005;79(7):3938–48. doi: 10.1128/JVI.79.7.3938-3948.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howley PM, Lowy DR. Papillomaviruses and their replication Fields virology. 4th. Lippincott Williams; WilkinsPhiladelphia, PA: 2001. pp. 2197–2229. [Google Scholar]
- Ishov AM, Maul GG. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J Cell Biol. 1996;134(4):815–26. doi: 10.1083/jcb.134.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss JF, 3rd, Maul GG. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J Cell Biol. 1999;147(2):221–34. doi: 10.1083/jcb.147.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S, Lambert PF. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc Natl Acad Sci U S A. 1995;92(5):1654–8. doi: 10.1073/pnas.92.5.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jul-Larsen A, Visted T, Karlsen BO, Rinaldo CH, Bjerkvig R, Lonning PE, Boe SO. PML-nuclear bodies accumulate DNA in response to polyomavirus BK and simian virus 40 replication. Exp Cell Res. 2004;298(1):58–73. doi: 10.1016/j.yexcr.2004.03.045. [DOI] [PubMed] [Google Scholar]
- Kieback E, Muller M. Factors influencing subcellular localization of the human papillomavirus L2 minor structural protein. Virology. 2006;345(1):199–208. doi: 10.1016/j.virol.2005.09.047. [DOI] [PubMed] [Google Scholar]
- Kim K, Garner-Hamrick PA, Fisher C, Lee D, Lambert PF. Methylation patterns of papillomavirus DNA, its influence on E2 function, and implications in viral infection. J Virol. 2003;77(23):12450–9. doi: 10.1128/JVI.77.23.12450-12459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirnbauer R, Taub J, Greenstone H, Roden R, Durst M, Gissmann L, Lowy DR, Schiller JT. Efficient self-assembly of human papillomavirus type 16 L1 and L1-L2 into virus-like particles. J Virol. 1993;67(12):6929–36. doi: 10.1128/jvi.67.12.6929-6936.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korioth F, Maul GG, Plachter B, Stamminger T, Frey J. The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp Cell Res. 1996;229(1):155–8. doi: 10.1006/excr.1996.0353. [DOI] [PubMed] [Google Scholar]
- Lambert PF. Papillomavirus DNA replication. J Virol. 1991;65(7):3417–20. doi: 10.1128/jvi.65.7.3417-3420.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert PF, Baker CC, Howley PM. The genetics of bovine papillomavirus type 1. Annu Rev Genet. 1988;22:235–58. doi: 10.1146/annurev.ge.22.120188.001315. [DOI] [PubMed] [Google Scholar]
- Lambert PF, Monk BC, Howley PM. Phenotypic analysis of bovine papillomavirus type 1 E2 repressor mutants. J Virol. 1990;64(2):950–6. doi: 10.1128/jvi.64.2.950-956.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert PF, Ozbun MA, Collins A, Holmgren S, Lee D, Nakahara T. Using an immortalized cell line to study the HPV life cycle in organotypic “raft” cultures. Methods Mol Med. 2005;119:141–55. doi: 10.1385/1-59259-982-6:141. [DOI] [PubMed] [Google Scholar]
- Lavau C, Marchio A, Fagioli M, Jansen J, Falini B, Lebon P, Grosveld F, Pandolfi PP, Pelicci PG, Dejean A. The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene. 1995;11(5):871–6. [PubMed] [Google Scholar]
- Law MF, Lowy DR, Dvoretzky I, Howley PM. Mouse cells transformed by bovine papillomavirus contain only extrachromosomal viral DNA sequences. Proc Natl Acad Sci U S A. 1981;78(5):2727–31. doi: 10.1073/pnas.78.5.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppard KN, Everett RD. The adenovirus type 5 E1b 55K and E4 Orf3 proteins associate in infected cells and affect ND10 components. J Gen Virol. 1999;80(Pt 4):997–1008. doi: 10.1099/0022-1317-80-4-997. [DOI] [PubMed] [Google Scholar]
- Lin DY, Lai MZ, Ann DK, Shih HM. Promyelocytic leukemia protein (PML) functions as a glucocorticoid receptor co-activator by sequestering Daxx to the PML oncogenic domains (PODs) to enhance its transactivation potential. J Biol Chem. 2003;278(18):15958–65. doi: 10.1074/jbc.M300387200. [DOI] [PubMed] [Google Scholar]
- Lowy DR, Dvoretzky I, Shober R, Law MF, Engel L, Howley PM. In vitro tumorigenic transformation by a defined sub-genomic fragment of bovine papilloma virus DNA. Nature. 1980;287(5777):72–4. doi: 10.1038/287072a0. [DOI] [PubMed] [Google Scholar]
- Maul GG, Guldner HH, Spivack JG. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0) J Gen Virol. 1993;74(Pt 12):2679–90. doi: 10.1099/0022-1317-74-12-2679. [DOI] [PubMed] [Google Scholar]
- Maul GG, Negorev D, Bell P, Ishov AM. Review: properties and assembly mechanisms of ND10, PML bodies, or PODs. J Struct Biol. 2000;129(23):278–87. doi: 10.1006/jsbi.2000.4239. [DOI] [PubMed] [Google Scholar]
- Middleton K, Peh W, Southern S, Griffin H, Sotlar K, Nakahara T, El-Sherif A, Morris L, Seth R, Hibma M, Jenkins D, Lambert P, Coleman N, Doorbar J. Organization of human papillomavirus productive cycle during neoplastic progression provides a basis for selection of diagnostic markers. J Virol. 2003;77(19):10186–201. doi: 10.1128/JVI.77.19.10186-10201.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Dejean A. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J Virol. 1999;73(6):5137–43. doi: 10.1128/jvi.73.6.5137-5143.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78(21):11451–60. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara T, Peh WL, Doorbar J, Lee D, Lambert PF. Human papillomavirus type 16 E1circumflexE4 contributes to multiple facets of the papillomavirus life cycle. J Virol. 2005;79(20):13150–65. doi: 10.1128/JVI.79.20.13150-13165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson M, Pelicci PG. PML interaction with p53 and its role in apoptosis and replicative senescence. Oncogene. 2001;20(49):7250–6. doi: 10.1038/sj.onc.1204856. [DOI] [PubMed] [Google Scholar]
- Petti L, Nilson LA, DiMaio D. Activation of the platelet-derived growth factor receptor by the bovine papillomavirus E5 transforming protein. Embo J. 1991;10(4):845–55. doi: 10.1002/j.1460-2075.1991.tb08017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puvion-Dutilleul F, Chelbi-Alix MK, Koken M, Quignon F, Puvion E, de The H. Adenovirus infection induces rearrangements in the intranuclear distribution of the nuclear body-associated PML protein. Exp Cell Res. 1995a;218(1):9–16. doi: 10.1006/excr.1995.1125. [DOI] [PubMed] [Google Scholar]
- Puvion-Dutilleul F, Venturini L, Guillemin MC, de The H, Puvion E. Sequestration of PML and Sp100 proteins in an intranuclear viral structure during herpes simplex virus type 1 infection. Exp Cell Res. 1995b;221(2):448–61. doi: 10.1006/excr.1995.1396. [DOI] [PubMed] [Google Scholar]
- Regad T, Chelbi-Alix MK. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene. 2001;20(49):7274–86. doi: 10.1038/sj.onc.1204854. [DOI] [PubMed] [Google Scholar]
- Roberts S, Hillman ML, Knight GL, Gallimore PH. The ND10 component promyelocytic leukemia protein relocates to human papillomavirus type 1 E4 intranuclear inclusion bodies in cultured keratinocytes and in warts. J Virol. 2003;77(1):673–84. doi: 10.1128/JVI.77.1.673-684.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz MM, Proytcheva M, Ellis NA, Holloman WK, German J. BLM, the Bloom's syndrome protein, varies during the cell cycle in its amount, distribution, and co-localization with other nuclear proteins. Cytogenet Cell Genet. 2000;91(14):217–23. doi: 10.1159/000056848. [DOI] [PubMed] [Google Scholar]
- Stadler M, Chelbi-Alix MK, Koken MH, Venturini L, Lee C, Saib A, Quignon F, Pelicano L, Guillemin MC, Schindler C, et al. Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene. 1995;11(12):2565–73. [PubMed] [Google Scholar]
- Swindle CS, Zou N, Van Tine BA, Shaw GM, Engler JA, Chow LT. Human papillomavirus DNA replication compartments in a transient DNA replication system. J Virol. 1999;73(2):1001–9. doi: 10.1128/jvi.73.2.1001-1009.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Bell P, Tegtmeyer P, Maul GG. Replication but not transcription of simian virus 40 DNA is dependent on nuclear domain 10. J Virol. 2000;74(20):9694–700. doi: 10.1128/jvi.74.20.9694-9700.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor ER, Dornan ES, Boner W, Connolly JA, McNair S, Kannouche P, Lehmann AR, Morgan IM. The fidelity of HPV16 E1/E2-mediated DNA replication. J Biol Chem. 2003;278(52):52223–30. doi: 10.1074/jbc.M308779200. [DOI] [PubMed] [Google Scholar]
- Thomas JT, Hubert WG, Ruesch MN, Laimins LA. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc Natl Acad Sci U S A. 1999;96(15):8449–54. doi: 10.1073/pnas.96.15.8449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallian S, Gaken JA, Gingold EB, Kouzarides T, Chang KS, Farzaneh F. Modulation of Fos-mediated AP-1 transcription by the promyelocytic leukemia protein. Oncogene. 1998;16(22):2843–53. doi: 10.1038/sj.onc.1201837. [DOI] [PubMed] [Google Scholar]
- Wilkinson DE, Weller SK. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J Virol. 2004;78(9):4783–96. doi: 10.1128/JVI.78.9.4783-4796.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Fehrmann F, Laimins LA. Role of the E1--E4 protein in the differentiation-dependent life cycle of human papillomavirus type 31. J Virol. 2005;79(11):6732–40. doi: 10.1128/JVI.79.11.6732-6740.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HY, Fratta W, Majane EA, Costa E. Isolation, sequencing, synthesis, and pharmacological characterization of two brain neuropeptides that modulate the action of morphine. Proc Natl Acad Sci U S A. 1985;82(22):7757–61. doi: 10.1073/pnas.82.22.7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Kuo C, Bisi JE, Kim MK. PML-dependent apoptosis after DNA damage is regulated by the checkpoint kinase hCds1/Chk2. Nat Cell Biol. 2002;4(11):865–70. doi: 10.1038/ncb869. [DOI] [PubMed] [Google Scholar]
- Yankiwski V, Noonan JP, Neff NF. The C-terminal domain of the Bloom syndrome DNA helicase is essential for genomic stability. BMC Cell Biol. 2001;2:11. doi: 10.1186/1471-2121-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P, Guo Y, Niu Q, Levy DE, Dyck JA, Lu S, Sheiman LA, Liu Y. Proto-oncogene PML controls genes devoted to MHC class I antigen presentation. Nature. 1998;396(6709):373–6. doi: 10.1038/24628. [DOI] [PubMed] [Google Scholar]
- Zhong S, Muller S, Ronchetti S, Freemont PS, Dejean A, Pandolfi PP. Role of SUMO-1-modified PML in nuclear body formation. Blood. 2000a;95(9):2748–52. [PubMed] [Google Scholar]
- Zhong S, Salomoni P, Pandolfi PP. The transcriptional role of PML and the nuclear body. Nat Cell Biol. 2000b;2(5):E85–90. doi: 10.1038/35010583. [DOI] [PubMed] [Google Scholar]
- Zhu J, Koken MH, Quignon F, Chelbi-Alix MK, Degos L, Wang ZY, Chen Z, de The H. Arsenic-induced PML targeting onto nuclear bodies: implications for the treatment of acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 1997;94(8):3978–83. doi: 10.1073/pnas.94.8.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2(5):342–50. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Raft cultures harboring the wild type HPV31 or a L2 mutant of HPV31 were previously described (Holmgren et al., 2005). Cross sections of rafts were double stained with anti-human PML monoclonal antibody and anti-L2 rabbit polyclonal antibody as described in Fig.2. Consistent with Fig.1, the intensity as well as the number of PODs detected by the PML monoclonal antibody was increased in basal to intermediate layers of rafts harboring wild type HPV31 as well as a L2 mutant of HPV31 genome compared to rafts of NIKS not harboring HPV31 genome. PODs were not detected in the granular layers of rafts regardless of the presence of HPV31 genome, indicating that the failure of detecting PODs by anti-PML rabbit polyclonal antibody (Fig.1) is not due to the lack of reactivity of the antibody.