

Abstract
Reduced food intake brings about an adaptive decrease in energy expenditure that contributes to the recidivism of obesity following weight loss. Insulin and leptin inhibit food intake through actions in the central nervous system that are partly mediated by FoxO1. We show that FoxO1 ablation in pro–opiomelanocortin (Pomc) neurons (Pomc–Foxo1−/−) reduces food intake without affecting energy expenditure. Analyses of hypothalamic neuropeptides in Pomc–Foxo1−/− mice reveal selective increases of α–Msh and COOH–cleaved β–endorphin, the products of Carboxypeptidase E (Cpe)–dependent processing of Pomc. We show that Cpe is decreased in diet–induced obesity, and that FoxO1 deletion offsets the decrease, protecting against weight gain. Moreover, moderate Cpe overexpression in the arcuate nucleus phenocopies features of the FoxO1 mutation. The dissociation of food intake from energy expenditure in Pomc–Foxo1−/− mice represents a model for therapeutic intervention in obesity, and raises the possibility of targeting Cpe to develop weight loss medications.
Obesity and its associated co–morbidities pose a growing threat to public health 1. The outlook for their pharmacological treatment remains dim 2. Thus, identification of molecular pathways that can be effectively and safely targeted for therapy has acquired new urgency. A rational approach involves interrogation of signaling mechanisms that elicit satiety, such as those activated by insulin and leptin in hypothalamic neurons 3,4, with the goal of identifying tractable candidates for therapeutic intervention.
When insulin is delivered to the brain 5,6, its anorexigenic effects require activation of signaling by α–melanocyte–stimulating hormone (α–Msh) produced by hypothalamic Pomc neurons 7. Hypothalamic insulin receptor signaling results in nuclear exclusion and inactivation of transcription factor FoxO1 8,9. This in turn inhibits food intake through Agouti–related protein (Agrp) repression 8,10. Moreover, fatty acid–induced insulin resistance is associated with impaired FoxO1 nuclear export in Pomc neurons 11, implicating FoxO1 and its targets in this cell type in the pathogenesis of metabolic diseases.
Appetite–controlling neuropeptides are synthesized as pro–hormones that undergo limited posttranslational modifications to generate mature peptides with distinct biological functions. Unlike Agrp, whose anorexigenic actions are modulated by, but independent of posttranslational cleavage 12, Pomc must be processed to inhibit food intake. Pomc is cleaved into several biologically active peptides, including α–Msh and β–endorphin (β–Ep). This process is mediated by prohormone convertases (Pc1 and Pc2), Cpe, and peptidyl α–amidating monooxygenase (Pam) 13,14 (Fig. 1a). Mice with disrupted Pc1 or Pc2 (encoded by Pcsk1 and Pcsk2, respectively) show distinct patterns of impaired pro–peptide cleavage in different organs, including hypothalamic Pomc 15-17. Mice expressing a catalytically inactive Pc1 mutant are obese 18, as is a compound heterozygote for Pc1 mutations 19. A naturally occurring loss–of–function mutation of Cpe (Cpefat/fat) is associated with obesity, infertility and diabetes in mice 20. But owing to Cpe's pleiotropic actions, the mechanisms linking loss of Cpe function with the phenotype is unclear 20-22. Cpe variants resulting in altered activity have been reported in patients with early–onset type 2 diabetes 23.
Figure 1.
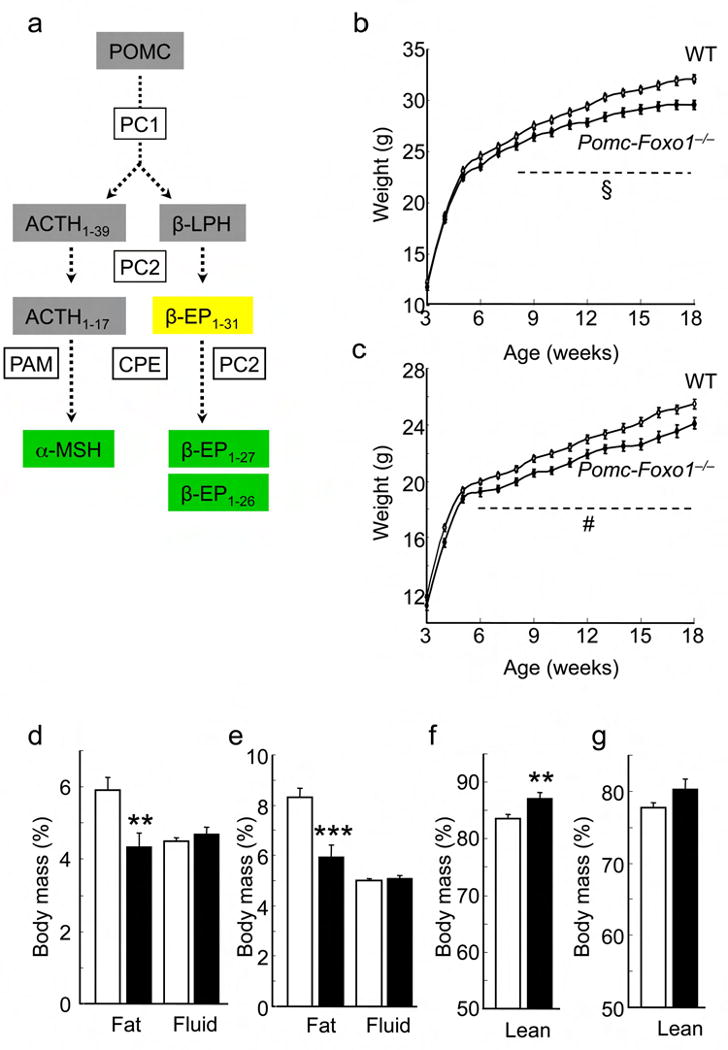
Body weight and composition. (a) Hypothalamic Pomc processing: Prohormone convertase1/2: Pc1/2; carboxypeptidase E: Cpe; peptidyl α–amidating monooxygenase: Pam; adrenocorticotrophic hormone: Acth, beta–lipotropin: β–Lph; alpha–melanocyte stimulating hormone: α–Msh; beta–endorphin: β–Ep. Orexigenic peptides are shaded in yellow, and anorexigenic peptides in green. (b) Growth curves of male (n = 38–82) and (c) female (n = 53–88) mice on normal chow (NCD). § = P ≤0.0001; # = P <0.05 (ANOVA). (d) Body fat and fluid content of 14–week–old male (n = 31–72) and (e) female (n = 23–53) mice on NCD. (f, g) Lean body mass of mice in (d) and (e), respectively. Data represent mean ± SEM. * = P <0.05; ** = P ≤0.01; *** = P ≤0.001 by Student's t–test for unpaired data.
Pc1 and Pc2 substrates include anorexigenic (Pomc 24, pro–corticotropin–releasing hormone 25) and orexigenic (pro–neuropeptide Y 26, Agrp 12, pro–melanin–concentrating hormone 27) hypothalamic pro–peptides. Pc1 and Pc2 activities are differentially regulated in different cell types in response to nutritional and hormonal cues 13,28. Leptin reverses the inhibition of hypothalamic Pcsk1 and Pcsk2 in response to fasting 29, increasing production of anorexigenic α–Msh 13,29 that acts on melanocortin 4 receptors (Mc4r) in target areas such as the paraventricular nucleus of the hypothalamus (PVN) 4. In contrast, there remain yawning gaps in our understanding of Cpe's role in this system, despite its documented role in rodent obesity 20.
In this study, we provide genetic evidence of a key role of the FoxO1–Cpe regulatory axis in the integration of food intake and energy expenditure.
Results
Somatic deletion of FoxO1 in Pomc neurons
In view of FoxO1's role in hypothalamic neuropeptide signaling 8,10, and of lingering uncertainties as to its primary target genes in this cell type 8, we generated Pomc–Foxo1−/− mice. Four lines of evidence document somatic deletion of the gene: (i) mating of Gt(ROSA)26Sortm2Sho (Rosa–Gfp) reporter mice with Pomc(cre) transgenics 30 resulted in GFP immunoreactivity that largely overlapped with endogenous Pomc expression patterns (Supplementary Fig. 1a); (ii) allele–specific PCR showed Foxo1 recombination in the pituitary–where ∼40% of cells express Pomc–but not in other brain regions or peripheral tissues (Supplementary Fig. 1b); (iii) pituitary Foxo1 mRNA was reduced by ∼40% in Pomc–Foxo1−/−mice (Supplementary Fig. 1c); and (iv) immunoprecipitation of chromatin (ChIP) isolated from mediobasal hypothalami (MBH) showed FoxO1 bound to the Pomc promoter 8 in WT, but not in Pomc–Foxo1−/− mice (Supplementary Fig. 1d).
Pomc–Foxo1−/− mice were born at Mendelian frequency with normal appearance. Their basal and stress–induced serum corticosterone, as well as pituitary Pomc levels were comparable to WT mice (Supplementary Fig. 1e,f), demonstrating that FoxO1 ablation doesn't affect corticotroph function 31.
Reduced body weight and fat content in Pomc–Foxo1−/− mice
Body weight of Pomc–Foxo1−/− mice was similar to WT littermates until ∼six weeks of age, but became ∼15% lower thereafter (Fig. 1b,c). Reduced body weight was associated with lower body mass index (BMI), despite shorter naso–anal length (Supplementary Fig. 2a–d), indicating that it is not solely an effect of reduced body size of the mutant animals. The change in body weight was accounted for by ∼27 % decrease of fat content (Fig. 1d,e), and was associated with a relative (%), but not absolute (g per mouse) increase of lean mass (Fig. 1f,g) 32.
Dissociation of food intake from energy expenditure in Pomc–Foxo1−/− mice
To evaluate the leanness of Pomc–Foxo1−/− mice, we measured food intake and energy expenditure. Cumulative ad libitum food intake was significantly reduced in mutant mice (Fig. 2a,b), more so in females when normalized by lean mass (Fig. 2c,d). In contrast, energy expenditure (Supplementary Fig. 3a,b), locomotor activity (Supplementary Fig. 3c), and respiratory quotient (Supplementary Fig. 3d) were unchanged. The decrease in food intake was independent of changes in circadian feeding patterns (Supplementary Fig. 3e). Insulin and glucose levels in the fasting, fed and refed states, as well as insulin sensitivity and tolerance to a glucose load, were also normal (data not shown).
Figure 2.
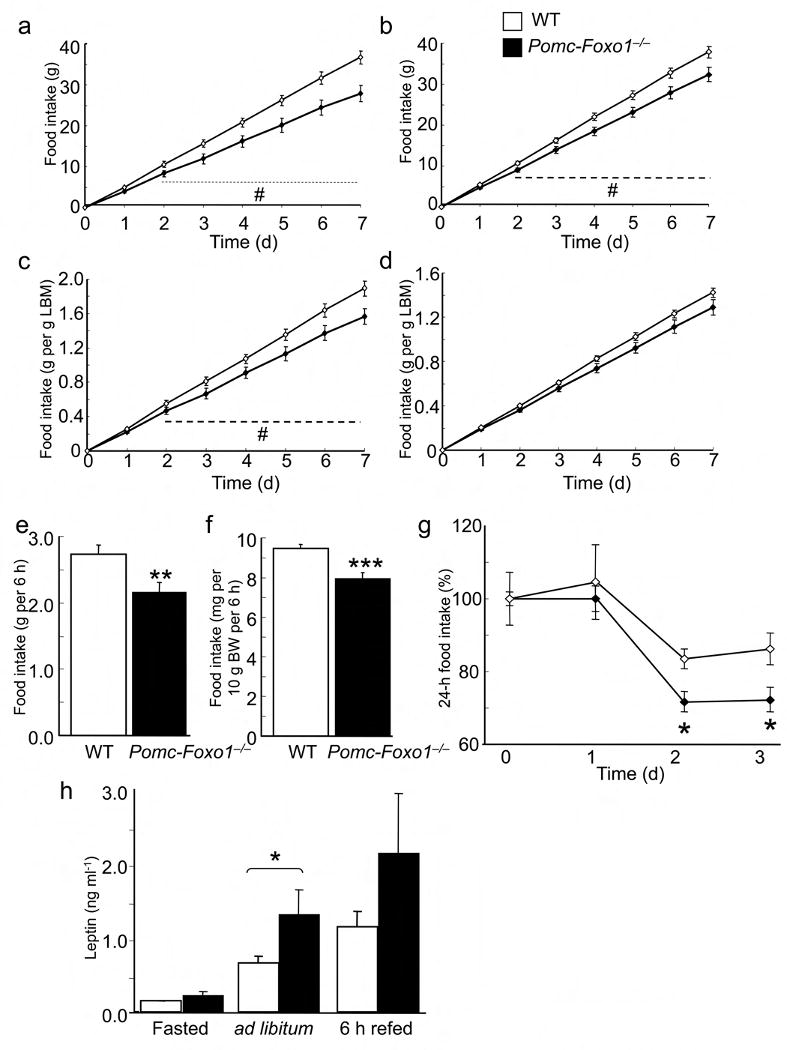
Food intake and leptin sensitivity. (a) Ad libitum intake of NCD in 14–week–old female (n = 6–17) and (b) male (n = 9–14) mice. (c, d) Ad libitum NCD intake corrected for average lean body mass (LBM) of mice in (a) and (b), respectively. (e) Absolute refeeding intake in 18–week–old male mice (n = 10–11). (f) Refeeding response normalized by body weight of mice in (e). (g) 24–h NCD intake in 15–week–old, body weight–matched male mice after intraperitoneal leptin injection. PBS was injected at day 0 (n = 4). (h) Serum leptin levels in male mice after 16–h fast, ad libitum feeding, and 24–h fast followed by 6–h refeeding (n = 16–37). Data represent mean ± SEM. * = P <0.05; ** = P ≤0.01; *** = P ≤0.001 by unpaired t–test. # = P ≤0.05 by ANOVA.
To characterize the hypophagia of Pomc–Foxo1−/− mice, we measured food intake following an 18–h fast. Mutant mice exhibited an ∼18% decrease in “rebound” food intake compared to WT littermates both in absolute values (Fig. 2e) and when corrected for body weight (Fig. 2f). These data indicate that leanness of Pomc–Foxo1−/− mice results from an unusual dissociation of hypophagia from reduced energy expenditure 33.
Given the interaction between insulin/FoxO1 and leptin/Stat3 signaling 8, we tested leptin sensitivity in Pomc–Foxo1−/− mice. Leptin curtailed food intake more markedly in Pomc–Foxo1−/−than in WT mice (Fig. 2g), consistent with increased leptin sensitivity. Surprisingly, Pomc–Foxo1−/− mice showed a near doubling of leptin levels in all conditions tested (Fig. 2h).
Pomc and Npy/Agrp neuron number in Pomc–Foxo1−/− mice
To rule out developmental abnormalities associated with FoxO1 ablation, we counted Pomc and Npy/Agrp neurons in the MBH. We did not detect differences in Pomc and Npy neuron number in 4–week–old mice (data not shown). In adult Pomc–Foxo1−/− mice, we found a ∼20% reduction of Pomc neurons (P = NS), and no difference in Npy/Agrp neurons (Supplementary Fig. 4a). Based on these data, we normalized measurements of Pomc and Agrp mRNA and peptide by average number of relevant neurons. (We show absolute values in Supplementary Figure 4b,c and Supplementary Table 1.)
FoxO1 deletion increases Pomc in young, but not in adult mice
To test whether hypophagia and leanness result from changes in hypothalamic neuropeptide mRNA, we measured MBH Pomc. We detected a significant increase of Pomc in 5–week–old Pomc–Foxo1−/− mice on refeeding, and no changes during ad libitum feeding (Supplementary Fig. 5a,b). Agrp expression was similar between the two groups under both conditions (data not shown). But in adult Pomc–Foxo1−/− mice, Pomc levels did not differ from controls under either condition (Supplementary Fig. 5c,d). This finding is consistent with prior data that FoxO1 gain–of–function fails to affect Pomc in the ARC of adult mice 8. In both ad libitum–fed and refed Pomc–Foxo1−/− mice, we detected a decrease of Agrp (Supplementary Fig. 5c,d) that significantly lowered the Agrp/Pomc ratio (Supplementary Fig. 5e,f), potentially resulting in an anorexigenic posture. But the data indicate that the observed changes in food intake cannot be accounted for by increased Pomc expression.
FoxO1 inactivation affects hypothalamic Pomc peptide processing
We next measured MBH content of Pomc peptide, its processing intermediates and cleavage products. Full–length Pomc and Pomc–derived neuropeptides in the MBH were comparable in ad libitum–fed Pomc–Foxo1−/− and WT mice (data not shown). Likewise, Pomc and its initial cleavage product adrenocorticotrophic hormone (Acth) were unaltered in Pomc–Foxo1−/− compared to WT after 6–h refeeding (Table 1). But the anorexigenic peptide α–Msh was significantly increased and total β–Ep inched upward after refeeding (Table 1). Agrp trended downward in ad libitum–fed (data not shown), and decreased significantly in refed Pomc–Foxo1−/− mice, resulting in reduced hypothalamic Agrp/α–Msh ratio (Table 1).
Table 1.
Neuropeptide levels in MBH lysates of refed WT and Pomc–Foxo1−/− males
| Peptide | WT | Pomc–Foxo1−/− | P |
|---|---|---|---|
| Acth | 0.102 ± 0.007 | 0.097 ± 0.016 | NS |
| Pomc | 0.118 ± 0.008 | 0.125 ± 0.008 | NS |
| β–Ep | 0.302 ± 0.022 | 0.354 ± 0.014 | NS |
| β–Ep1–31/β–Ep1–26, 1–27 | 1.052 ± 0.060 | 0.859 ± 0.058 | 0.01 (*) |
| α–Msh | 0.163 ± 0.012 | 0.200 ± 0.006 | 0.01 (*) |
| Agrp | 0.120 ± 0.005 | 0.088 ± 0.007 | 0.01 (*) |
| Agrp/α–Msh | 1.129 ± 0.036 | 0.987 ± 0.056 | 0.03 (*) |
Data are presented as mean fmol/cell ± SEM (n = 5–18).
(In Supplementary Results and Supplementary Fig. 6, we provide evidence of increased α–Msh activity and identify a mechanism to explain the dissociation of increased α–Msh from normal Acth.)
These data indicate that FoxO1 ablation increases Pomc processing into α–Msh, its anorexigenic product. Pro–hormone processing enzymes Pc2 and Cpe cleave Acth and β–lipotropin in the ARC (containing hypothalamic Pomc neurons) and PVN (containing projections and primary target neurons of ARC Pomc neurons) into α–Msh and β–Ep 13,14. The observed neuropeptide patterns are consistent with increased Cpe and/or Pc2 in the hypothalamus of Pomc–Foxo1−/− mice 34,35. But Pcsk2 was unaltered in whole MBH (Supplementary Fig. 7a), ARC, and PVN (data not shown), as was the active 64–66kDa Pc2 peptide in MBH of refed Pomc–Foxo1−/− mice (Supplementary Fig. 7b).
These observations led us to make the testable predictions that: (i) hypothalamic Cpe mRNA should be higher in Pomc–Foxo1−/− mice; (ii) hence, other products of Cpe–mediated cleavage (β–Ep1–26 and β–Ep1–27) should similarly be increased. Further, we reasoned that, if Cpe played a causal role in the observed phenotype, (iii) Cpe should be decreased by diet–induced obesity in WT, but not Pomc–Foxo1−/− mice–and the latter should be protected against weight gain; (iv) Cpe gain–of–function should phenocopy FoxO1 ablation in Pomc neurons; and (v) Pomc–Foxo1−/− mice should be inured to the homeostatic changes brought about by calorie restriction (CR). All predictions were borne out by the data.
Evidence of increased Cpe activity in Pomc–Foxo1−/− mice
Gene expression studies revealed that Cpe mRNA was increased in ARC, but not in PVN of Pomc–Foxo1−/− mice (Fig. 3a,b), consistent with a cell–autonomous effect of FoxO1 ablation.
Figure 3.
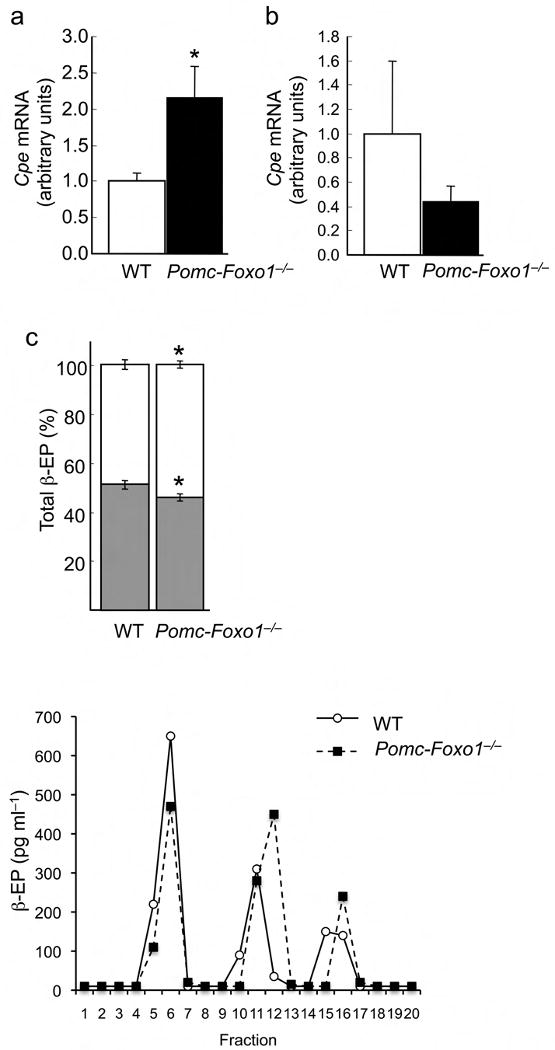
Evidence of altered Cpe activity in Pomc–Foxo1−/− mice. (a) Cpe levels in ARC (n = 12–13) and (b) PVN punches (n = 5–6) from refed male mice. (c) Relative abundance of β–Ep1–31 (shaded bars) and β–Ep1–27, 1–26 (open bars) (n = 5–6). (d) Reverse–phase HPLC elution profiles of β–Ep immunoreactivity in pooled MBH extracts from WT and Pomc–Foxo1−/−mice (n = 8). Arrows indicate the elution fraction of reference peptides encoding β–Ep1–31, β–Ep1–27 and β–Ep1–26.
Increased ARC Cpe activity is expected to increase generation of COOH–terminal cleavage products of β–Ep1–31: β–Ep1–26 and β–Ep1–27. Indeed, HPLC elution profiles of MBH protein extracts showed lower levels of β–Ep1–31, and higher levels of β–Ep1–26 and β–Ep1–27 in Pomc–Foxo1−/− mice (Fig. 3c,d; Table 1). (The increase of Cpe would also be expected to increase Pc2 enzymatic activity, by cleaving the COOH–terminal di–Lysine residue from the endogenous peptide inhibitor 7B2–CT1–18 36.)
FoxO1–dependent regulation of Cpe in diet–induced obesity
We predicted that, if changes in Cpe levels were causally linked to obesity, we should observe a decrease of Cpe in diet–induced obesity that should be relieved by somatic deletion of FoxO1 in Pomc neurons, protecting Pomc–Foxo1−/− mice from weight gain. Accordingly, we observed a 50% decrease of fasting Cpe mRNA in the ARC of mice rendered obese by high fat diet (HFD) (Fig. 4a). In contrast, adult Pomc–Foxo1−/− mice on a HFD were partly protected from weight gain (Fig. 4b,c), and showed a twofold rise in fasting Cpe levels in the ARC (Fig. 4d). These data are consistent with a contributory role of Cpe suppression in diet–induced obesity, and with a role of FoxO1 in regulating Cpe expression.
Figure 4.
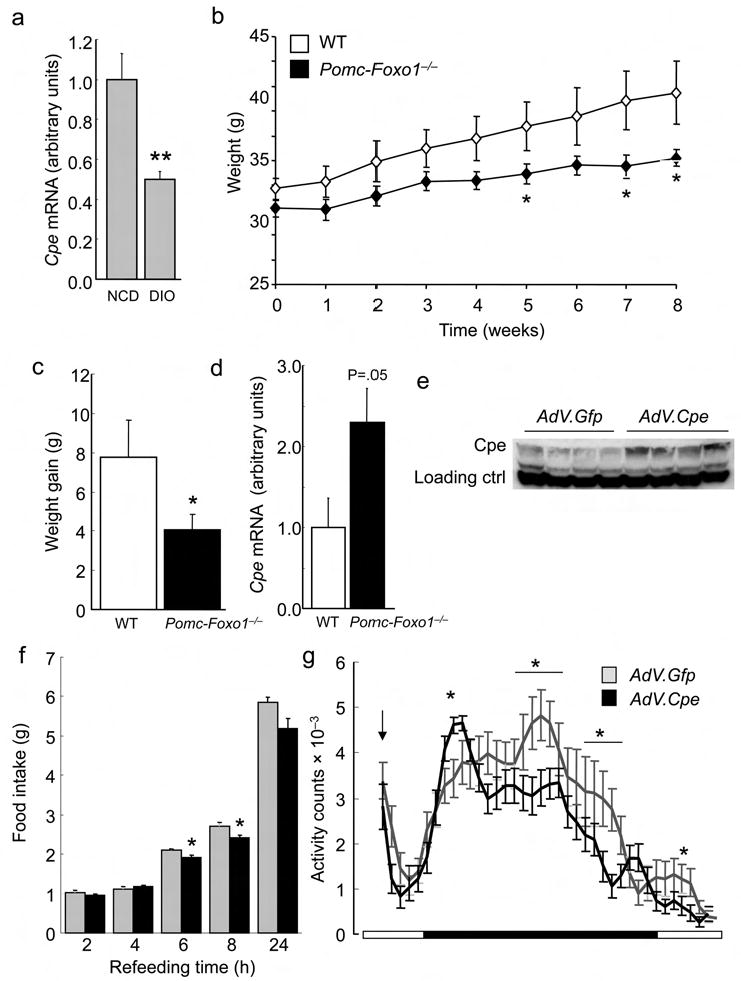
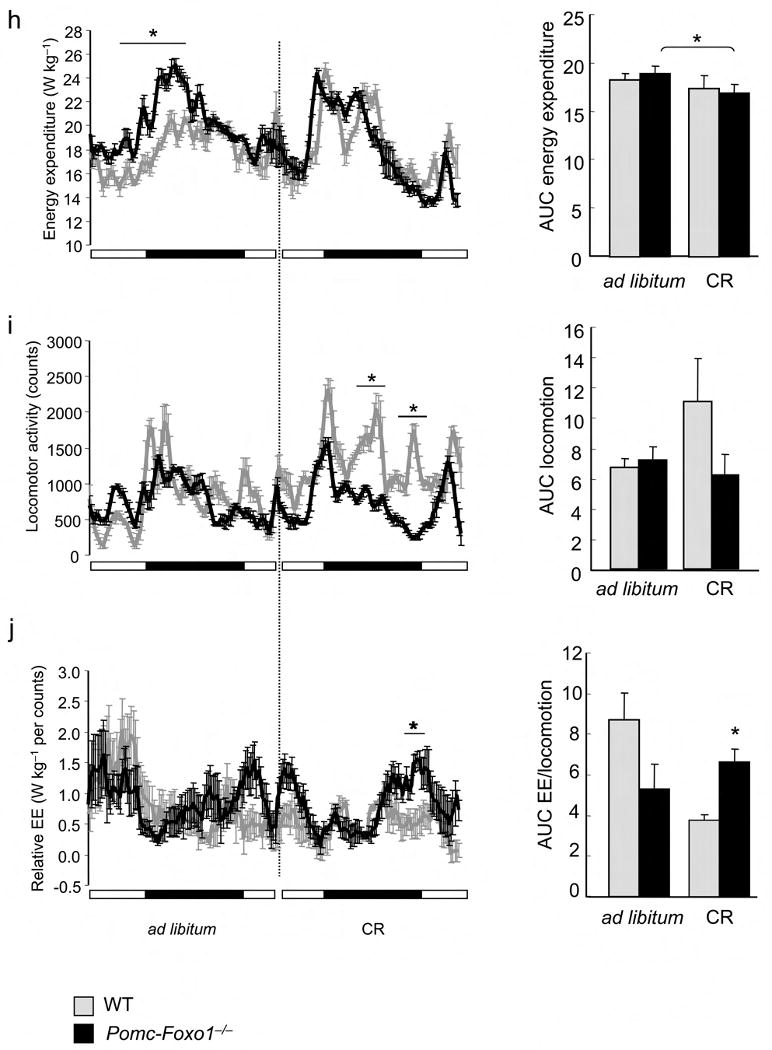
Regulation of Cpe by diet and CR. (a) ARC Cpe in 15–week–old fasted male mice on NCD or HFD (n = 5). (b) Body weight of 16–week–old male mice on HFD (n = 6–11). (c) Average weight gain in the two groups shown in (a) after 8–week HFD. (d) ARC Cpe after 4–week HFD (n = 4–5). (e) Cpe western blot in mouse MBH extracts one week after stereotactic adenovirus injection in the ARC. (f) Refeeding after food withdrawal in mice injected with AdV.Cpe or AdV.Gfp (n = 7–8). (g) Activity plots in the same mice shown in (d). The downward arrow indicates the time of food withdrawal. Data represent mean ± SEM. * = P <0.05; ** = P ≤0.01; *** = P ≤0.001 by unpaired t–test. (h) Energy expenditure plots and AUC, (i) activity plots and AUC, and (j) plots and AUC of energy expenditure divided by activity in 8–week–old male mice during ad libitum feeding and six–day CR (n = 3–4). Data represent mean ± SEM. * = P <0.05.
Targeted delivery of Cpe to the ARC phenocopies key features of Pomc–Foxo1−/− mice
To establish a causal link between Cpe and the phenotype of Pomc–Foxo1−/− mice, we delivered adenovirus encoding Cpe to mouse ARC and analyzed feeding behavior. One week after adenoviral delivery, MBH Cpe expression was ∼twofold higher in animals that had received the Cpe adenovirus compared to GFP controls (Fig. 4e). Post–surgical body weight and basal food intake did not differ between the two groups (data not shown). But mice overexpressing Cpe had lower refeeding response (Fig. 4f) and decreased nocturnal locomotor activity (Fig. 4g), consistent with the hypothesis that elevated Cpe levels have anorexient effects.
Reduced foraging in calorie–restricted Pomc–Foxo1−/− mice
CR promotes physical activity in the short term, likely as a result of foraging behavior 37. We reasoned that Pomc–Foxo1−/− mice would be protected against this adaptive change. After six–day CR (60–70% of ad libitum food intake), energy expenditure was similar in WT and Pomc–Foxo1−/− mice (Fig. 4h), but the latter failed to mount a foraging response (Fig. 4i). Consequently, energy expenditure relative to locomotor activity dropped markedly in WT mice, reflecting increased energy efficiency, but not in mutant mice (Fig. 4j). Notably, in the basal state Pomc–Foxo1−/− mice showed a trend toward increased serum ghrelin that also mimics CR (Supplementary Fig. 7c) 38. These data support the hypothesis that Pomc–Foxo1−/− mice are inured to behavioral changes associated with short–term CR.
FoxO1 corepresses Cpe transcription independent of DNA binding
We wanted to determine if FoxO1 regulated Cpe directly. ChIP assays showed occupancy of the Cpe promoter by FoxO1 in Neuro2A cells (Fig. 5a). Moreover, constitutively nuclear FoxO1 39 suppressed reporter gene activity driven by a Cpe promoter transfected in Neuro2A cells in a dose–dependent manner (Fig. 5b). The promoter fragment contains two putative FoxO1 consensus–binding sites, but mutations of neither site, alone or in combination, affected FoxO1's ability to repress transcription (data not shown). We then tested a DNA binding–deficient FoxO1 40, and found that it also repressed Cpe promoter activity (Fig. 5c), indicating that FoxO1 acts as a corepressor of Cpe transcription, independent of DNA binding.
Figure 5.
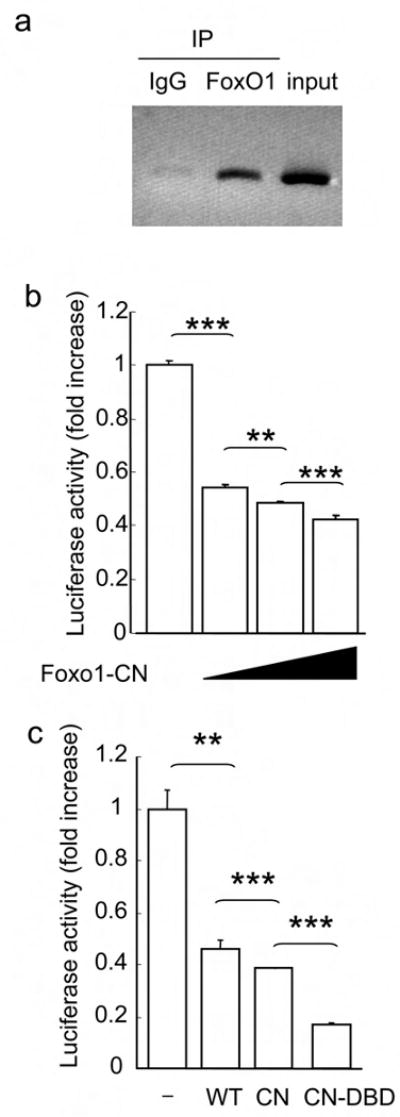
FoxO1 regulates Cpe expression. (a) ChIP assay of the Cpe promoter in Neuro2A cells transfected with FoxO1–CN. (b) Cpe–luciferase reporter gene activity in Neuro2A cells co–transfected with increasing amounts of pCMV.Foxo1–CN or control vector (n = 4). (c) Cpe–luciferase reporter gene activity in Neuro2A cells co–transfected with control vector (–), or vectors encoding WT, CN, or CN–DBD FoxO1 (n = 4). Luciferase activity is corrected by the values in cells expressing pCMV control vector alone.
Discussion
The key findings of this work are the demonstration that FoxO1 ablation in hypothalamic Pomc neurons reduces food intake without concurrently decreasing energy expenditure or leptin levels, and the identification of a mechanism linking this phenotype to the obesity susceptibility gene Cpe. The uncoupling of hypophagia and reduced body weight from energy expenditure/leptin levels is to our knowledge unprecedented. FoxO1's role in the modulation of neuropeptide function by regulation of their posttranslational processing is also new.
Multiple lines of evidence support the causal involvement of Cpe in the phenotype of Pomc–Foxo1−/− mice: (i) ARC–restricted increase of Cpe, (ii) protection from weight gain associated with reversal of diet–induced Cpe suppression, (iii) ability of Cpe gain–of–function in the ARC to phenocopy key features of the FoxO1 loss–of–function, (iv) lack of foraging behavior in calorie–restricted Pomc–Foxo1−/− mice, and (v) repression of Cpe promoter activity by FoxO1. Moreover, neuropeptide patterns in Pomc–Foxo1−/− mice (increased α–Msh, β–Ep1–26, and β–Ep1–27) are opposite to those seen in Cpe–deficient mice (decreased α–Msh, low β–Ep1–27, absent β–Ep1–26, and elevated β–Ep1–31) 34. β–Ep1–26 and β–Ep1–27 have markedly reduced opiatergic activity 41,42.
The dissociation of food intake from energy expenditure in Pomc–Foxo1−/− mice invites a comparison with the phenotype of transgenic mice overexpressing the FoxO1 deacetylase SirT1. In the latter, decreased food intake fails to affect body weight, owing to decreased locomotor activity 43. The two models are quite different in design, because the SirT1 gain–of–function is generalized, and thus the effects cannot be ascribed precisely to any one tissue or cell type. With this caveat, it's notable that changes in energy homeostasis are opposite in the two models, consistent with the notion that a gain of SirT1 function would promote FoxO1 activity via deacetylation 44, and thus with a specific role of SirT1/FoxO1 in linking food intake with energy expenditure.
The decrease of Agrp in Pomc–Foxo1−/− mice is likely to be secondary to increased leptin. The association of leanness, hyperleptinemia, and increased leptin sensitivity has been observed in mice with neuronal ablation of tyrosine phosphatase PTP1B, possibly as a result of increased leptin receptor signaling 45. But unlike neuron–specific Ptp1b−/−, Pomc–Foxo1−/− mice have normal energy balance and insulin sensitivity.
The relative increase in lean mass observed in Pomc–Foxo1−/− mice is reminiscent of the preservation of muscle mass observed in calorie–restricted mice carrying a Stat3 signaling–incompetent leptin receptor 32, and ostensibly indicates that hypothalamic FoxO1 inactivation is conducive to preservation of muscle mass during CR.
In addition to direct regulation of Cpe, the phenotype of Pomc–Foxo1−/− mice uncovered a new regulatory loop for neuropeptide processing. Pcsk1 is nutritionally regulated: leptin increases Pcsk1, while fasting decreases it 13,28,29. But the fasting signal required to suppress Pcsk1 was hitherto unknown. The reduction of hypothalamic Pc1 in Pomc–Foxo1−/− mice, despite their heightened leptin sensitivity, suggested to us a potential link between elevated α–Msh and low Pc1. In Supplementary Figure 6, we provide in vitro and in vivo evidence to support the conclusion that α–Msh is the fasting signal that inhibits Pcsk1. This interpretation can explain the paradoxical increase of Pc1 activity seen in the PVN of ob/ob mice with decreased hypothalamic α–Msh 46.
In conclusion, we demonstrate that the obesity susceptibility gene Cpe is regulated by FoxO1 in the hypothalamus to link food intake with energy expenditure. The separation of weight–reducing from energy expenditure effects in Pomc–Foxo1−/− mice is noteworthy, and suggests that Cpe stimulation can circumvent the primary adaptation associated with weight reduction, namely the increase in energy efficiency that predisposes to weight regain 47. Hence, Cpe may prove an attractive candidate for drug development in the treatment of obesity and insulin resistance.
Methods
Mice
C57BL/6 and Gt(ROSA)26Sortm2Sho mice were from Jackson Laboratories. The Columbia University Animal Care and Utilization Committee approved all procedures. Normal chow (NCD) had 62.1% calories from carbohydrates, 24.6% from protein, and 13.2% from fat (PicoLab rodent diet 20, #5053; Purina Mills); high–fat chow (HFD) had 20% calories from carbohydrates, 20% from protein, and 60% from fat (D12492; Research Diets). We measured weight, length, BMI, body composition by NMR (Bruker Optics). We generated Pomc–specific FoxO1 knockouts by mating Pomc(Cre) 30 with Foxo1lox/lox mice 48 and genotyped them as described 49. We excluded from analyses Pomc–Foxo1−/− mice that showed widespread recombination, owing to stochastic embryonic expression of Pomc(Cre) (∼5%).
Restraint stress
We handled mice as described to habituate them 49 and drew blood for basal corticosterone from the tail vein during the first 3 h of light. We restrained mice using a 3 cm–wide plastic cylinder.
Metabolic analyses
We measured food intake with feeding racks (Fa. Wenzel). For refeeding experiments, we habituated mice to feeding racks for three days, fasted them for 18 h, placed feeding racks 2 h after the start of the light phase, and measured food intake for 6 h thereafter. We used a TSE Labmaster Platform (TSE Systems) for indirect calorimetry and activity measurements 43. For CR experiments, we housed 8–week–old male mice on NCD in calorimetry chambers for one week for baseline recordings, after which we provided 60% of ad libitum food intake for three days (initiation period), followed by three days of 70% ad libitum intake (stabilization period) in two daily doses. We examined leptin sensitivity in 15–week–old, weight–matched male mice. After habituation (BID PBS injection), we injected leptin (2 mg/kg BID for three days) and measured body weight and food intake. We used ELISA for leptin (Millipore), corticosterone (Assay Designs), and ghrelin (RayBiotech) (measured 1 h prior to the dark phase).
Stereotactic injections
We performed stereotactic manipulations in 15–week–old male C57BL/6 mice 50. We injected NDP–α–Msh (1 nmol in 2 μl saline) (Phoenix Pharmaceuticals) or saline in the left brain ventricle (−0.2 mm anterior and 1.0 mm lateral to bregma and 2.3 mm below the skull surface). We sacrificed mice after 5 h, and harvested ARC and PVN using micro–punches. We verified correct positioning of ICV cannulae by injecting methylene blue (1%) after sacrifice. To deliver AdV–Cpe or AdV–GFP to the ARC, we injected 0.3μl of purified virus (1012 Pfu/ml) 51 −1.5 mm anterior and 0.5 mm lateral to bregma and 5.3 mm below the skull surface 8. After one week, we performed fasting and refeeding experiments.
Immunostaining and fluorescent in situ hybridization (FISH)
We perfused mice with saline, then formalin (immunostaining) or 4% paraformaldehyde (FISH). We froze brains in OCT (Sakura, CA, USA), and cut 30–μm–thick coronal sections for anti–GFP immunohistochemistry (Molecular Probes/Invitrogen), or serial 10–μm–thick coronal sections (bregma −0.8 mm to −2.4 mm) for FISH with Pomc and Npy riboprobes, using tyramide amplification reagents (PerkinElmer) and nuclear staining with Hoechst 33342 (Molecular Probes/Invitrogen). We acquired images to count neurons 49.
RNA procedures
We extracted RNA using Trizol® (Invitrogen) and performed quantitative PcR using LightCycler® 1.5 Carousell and LightCycler® FastStart DNA MasterPLUS SYBR Green I (Roche). Primer sequences are available on request.
Western blotting
We used T–PER Tissue Protein Extraction Reagent (Pierce) supplemented with protease and phosphatase inhibitors (Roche) to isolate proteins and blotted with the following antibodies: Pc1 (sc–33813; Santa Cruz Biotechnology), Pc2 (P9007–87; United States Biologicals), β–actin (CP01; Calbiochem), and Cpe (SC34321; Santa Cruz Biotechnology).
Chromatin Immunoprecipitation (ChIP) assays
We isolated intact chromatin and immunoprecipitated FoxO1 as described 8, and used primers 5′–TGGCTTCTAGCCAGACCTTG–3′ (−442 to −422) and 5′–GCCAGGGATGTTGGGTTTAT–3′ (−148 to −136) to amplify the Cpe promoter.
Hypothalamic neuropeptide assays and reverse–phase high–performance liquid chromatography
We extracted MBH in 0.1N HCl 52. We measured Pomc precursor by EIA 53, β–Ep and αMsh by RIA 52, Acth by RIA with antiserum against Acth7–18, (IgG Corp) 54 and Agrp by sandwich ELISA (R&D Systems). We performed HPLC as described 55.
Transfection and promoter studies
We performed luciferase assays in Neuro2A cells transfected with WT, constitutively nuclear (CN) or constitutively nuclear DNA binding–deficient (CN–DBD) FoxO1 40 as described 28, using α–Msh (Sigma–Aldrich) (10−6M). We harvested and cultured primary cells from pooled hypothalami of 7–day–old Pomc(Gfp) mice (n = 6), using Papain Dissociation System (Worthington Biochemical) as described 8.
Statistical Methods
We analyzed data using Student's t–test or repeated–measures ANOVA.
Supplementary Material
Acknowledgments
Supported by Deutsche Forschungsgemeinschaft PL542/1–1 (L.P.), US National Institutes of Health DK57539 and DK58282 (D.A.), DK80003 (S.W.), and DK63608 (Columbia Diabetes and Endocrinology Research Center). We thank R. Leibel for insightful discussions, L. Zeltser and S. Padilla for help with in situ hybridization, N. Seidah (Clinical Research Institute of Montreal), D. Good (University of Massachusetts), A. White (University of Manchester), and M. Low (Oregon Health Sciences University) for reagents, Y. Liu for technical assistance, and members of the Accili and Wardlaw laboratories for stimulating discussions. R.A.D. is an American Cancer Society Research Professor and an Ellison Medical Foundation Senior Scholar and is supported by the Robert A. and Renee E. Belfer Family Institute for Innovative Cancer Science.
Footnotes
Competing Financial Interest Statement: The Authors declare that they have no competing financial interest in the work described.
Author Contributions: L.P., H.V.L., R.D., J.T., K.S.A., M.M., A.J.K., and S.L.W. performed experiments and analyzed data. N.X.C. and J–H.P. generated reagents used for experiments. L.P., H.V.L., Y.P.L., R.A.D., S.L.W., and D.A. designed the studies, analyzed the data and wrote the manuscript.
References
- 1.Yach D, Stuckler D, Brownell KD. Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nat Med. 2006;12:62–66. doi: 10.1038/nm0106-62. [DOI] [PubMed] [Google Scholar]
- 2.Bray GA. Lifestyle and pharmacological approaches to weight loss: efficacy and safety. J Clin Endocrinol Metab. 2008;93:S81–88. doi: 10.1210/jc.2008-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plum L, Belgardt BF, Bruning JC. Central insulin action in energy and glucose homeostasis. J Clin Invest. 2006;116:1761–1766. doi: 10.1172/JCI29063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 5.McGowan MK, Andrews KM, Fenner D, Grossman SP. Chronic intrahypothalamic insulin infusion in the rat: behavioral specificity. Physiol Behav. 1993;54:1031–1034. doi: 10.1016/0031-9384(93)90320-f. [DOI] [PubMed] [Google Scholar]
- 6.Woods SC, Lotter EC, McKay LD, Porte D., Jr Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature. 1979;282:503–505. doi: 10.1038/282503a0. [DOI] [PubMed] [Google Scholar]
- 7.Benoit SC, et al. The catabolic action of insulin in the brain is mediated by melanocortins. J Neurosci. 2002;22:9048–9052. doi: 10.1523/JNEUROSCI.22-20-09048.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitamura T, et al. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med. 2006;12:534–540. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- 9.Accili D, Arden KC. FoxOs at the Crossroads of Cellular Metabolism, Differentiation, and Transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 10.Kim MS, et al. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci. 2006;9:901–906. doi: 10.1038/nn1731. [DOI] [PubMed] [Google Scholar]
- 11.Fukuda M, et al. Monitoring FoxO1 localization in chemically identified neurons. J Neurosci. 2008;28:13640–13648. doi: 10.1523/JNEUROSCI.4023-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Creemers JW, et al. Agouti-related protein is posttranslationally cleaved by proprotein convertase 1 to generate agouti-related protein (AGRP)83-132: interaction between AGRP83-132 and melanocortin receptors cannot be influenced by syndecan-3. Endocrinology. 2006;147:1621–1631. doi: 10.1210/en.2005-1373. [DOI] [PubMed] [Google Scholar]
- 13.Nillni EA. Regulation of prohormone convertases in hypothalamic neurons: implications for prothyrotropin-releasing hormone and proopiomelanocortin. Endocrinology. 2007;148:4191–4200. doi: 10.1210/en.2007-0173. [DOI] [PubMed] [Google Scholar]
- 14.Pritchard LE, White A. Neuropeptide processing and its impact on melanocortin pathways. Endocrinology. 2007;148:4201–4207. doi: 10.1210/en.2006-1686. [DOI] [PubMed] [Google Scholar]
- 15.Allen RG, et al. Altered processing of pro-orphanin FQ/nociceptin and pro-opiomelanocortin-derived peptides in the brains of mice expressing defective prohormone convertase 2. J Neurosci. 2001;21:5864–5870. doi: 10.1523/JNEUROSCI.21-16-05864.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furuta M, et al. Severe defect in proglucagon processing in islet A-cells of prohormone convertase 2 null mice. J Biol Chem. 2001;276:27197–27202. doi: 10.1074/jbc.M103362200. [DOI] [PubMed] [Google Scholar]
- 17.Zhu X, et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc Natl Acad Sci U S A. 2002;99:10293–10298. doi: 10.1073/pnas.162352599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lloyd DJ, Bohan S, Gekakis N. Obesity, hyperphagia and increased metabolic efficiency in Pc1 mutant mice. Hum Mol Genet. 2006;15:1884–1893. doi: 10.1093/hmg/ddl111. [DOI] [PubMed] [Google Scholar]
- 19.Jackson RS, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–306. doi: 10.1038/ng0797-303. see comments. [DOI] [PubMed] [Google Scholar]
- 20.Naggert JK, et al. Hyperproinsulinaemia in obese fat/fat mice associated with a carboxypeptidase E mutation which reduces enzyme activity. Nat Genet. 1995;10:135–142. doi: 10.1038/ng0695-135. [DOI] [PubMed] [Google Scholar]
- 21.Cawley NX, et al. The carboxypeptidase E knockout mouse exhibits endocrinological and behavioral deficits. Endocrinology. 2004;145:5807–5819. doi: 10.1210/en.2004-0847. [DOI] [PubMed] [Google Scholar]
- 22.Che FY, et al. Identification of peptides from brain and pituitary of Cpe(fat)/Cpe(fat) mice. Proc Natl Acad Sci U S A. 2001;98:9971–9976. doi: 10.1073/pnas.161542198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H, et al. Missense polymorphism in the human carboxypeptidase E gene alters enzymatic activity. Hum Mutat. 2001;18:120–131. doi: 10.1002/humu.1161. [DOI] [PubMed] [Google Scholar]
- 24.Benjannet S, Rondeau N, Day R, Chretien M, Seidah NG. PC1 and PC2 are proprotein convertases capable of cleaving proopiomelanocortin at distinct pairs of basic residues. Proc Natl Acad Sci U S A. 1991;88:3564–3568. doi: 10.1073/pnas.88.9.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perone MJ, Ahmed I, Linton EA, Castro MG. Procorticotrophin releasing hormone is endoproteolytically processed by the prohormone convertase PC2 but not by PC1 within stably transfected CHO-K1 cells. Biochem Soc Trans. 1996;24:497S. doi: 10.1042/bst024497s. [DOI] [PubMed] [Google Scholar]
- 26.Brakch N, et al. Role of prohormone convertases in pro-neuropeptide Y processing: coexpression and in vitro kinetic investigations. Biochemistry. 1997;36:16309–16320. doi: 10.1021/bi9714767. [DOI] [PubMed] [Google Scholar]
- 27.Viale A, et al. Cellular localization and role of prohormone convertases in the processing of pro-melanin concentrating hormone in mammals. J Biol Chem. 1999;274:6536–6545. doi: 10.1074/jbc.274.10.6536. [DOI] [PubMed] [Google Scholar]
- 28.Fox DL, Good DJ. Nescient helix-loop-helix 2 interacts with signal transducer and activator of transcription 3 to regulate transcription of prohormone convertase 1/3. Mol Endocrinol. 2008;22:1438–1448. doi: 10.1210/me.2008-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez VC, et al. Regulation of hypothalamic prohormone convertases 1 and 2 and effects on processing of prothyrotropin-releasing hormone. J Clin Invest. 2004;114:357–369. doi: 10.1172/JCI21620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balthasar N, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 31.Belgardt BF, et al. PDK1 Deficiency in POMC-Expressing Cells Reveals FOXO1-Dependent and -Independent Pathways in Control of Energy Homeostasis and Stress Response. Cell Metab. 2008;7:291–301. doi: 10.1016/j.cmet.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 32.Buettner C, et al. Leptin controls adipose tissue lipogenesis via central, STAT3-independent mechanisms. Nat Med. 2008;14:667–675. doi: 10.1038/nm1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med. 1995;332:621–628. doi: 10.1056/NEJM199503093321001. [DOI] [PubMed] [Google Scholar]
- 34.Berman Y, Mzhavia N, Polonskaia A, Devi LA. Impaired prohormone convertases in Cpe(fat)/Cpe(fat) mice. J Biol Chem. 2001;276:1466–1473. doi: 10.1074/jbc.M008499200. [DOI] [PubMed] [Google Scholar]
- 35.Miller R, et al. Obliteration of alpha-melanocyte-stimulating hormone derived from POMC in pituitary and brains of PC2-deficient mice. J Neurochem. 2003;86:556–563. doi: 10.1046/j.1471-4159.2003.01856.x. [DOI] [PubMed] [Google Scholar]
- 36.Zhu X, Rouille Y, Lamango NS, Steiner DF, Lindberg I. Internal cleavage of the inhibitory 7B2 carboxyl-terminal peptide by PC2: a potential mechanism for its inactivation. Proc Natl Acad Sci U S A. 1996;93:4919–4924. doi: 10.1073/pnas.93.10.4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Overton JM, Williams TD. Behavioral and physiologic responses to caloric restriction in mice. Physiol Behav. 2004;81:749–754. doi: 10.1016/j.physbeh.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 38.Wertz-Lutz AE, Daniel JA, Clapper JA, Trenkle A, Beitz DC. Prolonged, moderate nutrient restriction in beef cattle results in persistently elevated circulating ghrelin concentrations. J Anim Sci. 2008;86:564–575. doi: 10.2527/jas.2007-0556. [DOI] [PubMed] [Google Scholar]
- 39.Nakae J, et al. The forkhead transcription factor foxo1 regulates adipocyte differentiation. Dev Cell. 2003;4:119–129. doi: 10.1016/s1534-5807(02)00401-x. [DOI] [PubMed] [Google Scholar]
- 40.Kitamura T, et al. A Foxo/Notch pathway controls myogenic differentiation and fiber type specification. J Clin Invest. 2007;117:2477–2485. doi: 10.1172/JCI32054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yanagita K, Shiraishi J, Fujita M, Bungo T. Effects of N-terminal fragments of beta-endorphin on feeding in chicks. Neurosci Lett. 2008;442:140–142. doi: 10.1016/j.neulet.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 42.Nicolas P, Li CH. Beta-endorphin-(1-27) is a naturally occurring antagonist to etorphine-induced analgesia. Proc Natl Acad Sci U S A. 1985;82:3178–3181. doi: 10.1073/pnas.82.10.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banks AS, et al. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitamura YI, et al. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005;2:153–163. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Bence KK, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–924. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- 46.Nilaweera KN, Barrett P, Mercer JG, Morgan PJ. Precursor-protein convertase 1 gene expression in the mouse hypothalamus: differential regulation by ob gene mutation, energy deficit and administration of leptin, and coexpression with prepro-orexin. Neuroscience. 2003;119:713–720. doi: 10.1016/s0306-4522(02)00869-2. [DOI] [PubMed] [Google Scholar]
- 47.Rosenbaum M, et al. Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J Clin Invest. 2005;115:3579–3586. doi: 10.1172/JCI25977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paik JH, et al. FoxOs Are Lineage-Restricted Redundant Tumor Suppressors and Regulate Endothelial Cell Homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Plum L, et al. Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J Clin Invest. 2006;116:1886–1901. doi: 10.1172/JCI27123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Plum L, et al. Enhanced Leptin-Stimulated Pi3k Activation in the CNS Promotes White Adipose Tissue Transdifferentiation. Cell Metab. 2007;6:431–445. doi: 10.1016/j.cmet.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 51.Woronowicz A, et al. Absence of carboxypeptidase E leads to adult hippocampal neuronal degeneration and memory deficits. Hippocampus. 2008;18:1051–1063. doi: 10.1002/hipo.20462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wardlaw SL. Regulation of beta-endorphin, corticotropin-like intermediate lobe peptide, and alpha-melanotropin-stimulating hormone in the hypothalamus by testosterone. Endocrinology. 1986;119:19–24. doi: 10.1210/endo-119-1-19. [DOI] [PubMed] [Google Scholar]
- 53.Tsigos C, Crosby SR, Gibson S, Young RJ, White A. Proopiomelanocortin is the predominant adrenocorticotropin-related peptide in human cerebrospinal fluid. J Clin Endocrinol Metab. 1993;76:620–624. doi: 10.1210/jcem.76.3.8383142. [DOI] [PubMed] [Google Scholar]
- 54.Papadopoulos AD, Wardlaw SL. Endogenous alpha-MSH modulates the hypothalamic-pituitary-adrenal response to the cytokine interleukin-1beta. Journal of neuroendocrinology. 1999;11:315–319. doi: 10.1046/j.1365-2826.1999.00327.x. [DOI] [PubMed] [Google Scholar]
- 55.Jaffe SB, Sobieszczyk S, Wardlaw SL. Effect of opioid antagonism on beta-endorphin processing and proopiomelanocortin-peptide release in the hypothalamus. Brain Res. 1994;648:24–31. doi: 10.1016/0006-8993(94)91900-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.