

Abstract
Programmed death-1 (PD-1) is an inhibitory molecule expressed on activated T cells, however, the biological context in which PD-1 controls T cell tolerance remains unclear. Using two-photon laser-scanning microscopy, we showed that unlike naïve or activated islet antigen-specific T cells, tolerized islet antigen-specific T cells moved freely and did not swarm around antigen-bearing dendritic cells (DC) in pancreatic lymph nodes. Inhibition of T cell receptor (TCR)-driven stop signals depended on continued PD-1-PD-L1 interactions, as antibody blockade of PD-1 or PD-L1 decreased T cell motility, enhanced T cell-DC contacts, and caused autoimmune diabetes. CTLA-4 blockade did not alter T cell motility or abrogate tolerance. Thus, PD-1-PD-L1 interactions maintain peripheral tolerance by mechanisms fundamentally distinct from those of CTLA-4.
Introduction
Autoimmunity results from the breakdown of processes designed to maintain self-tolerance. The growing list of co-stimulatory and inhibitory receptors that influence immune cell activation has been a central concern in immunology for over two decades. CTLA-4 (also called CD152) (http://www.signaling-gateway.org/molecule/query?afcsid=A000706) is a well established negative regulator of T cell function 1. CTLA-4 is rapidly expressed on the surface of T cells following activation and is highly upregulated by engagement of the co-stimulatory molecule CD28 1. CTLA-4 ligation antagonizes early T cell activation leading to decreased interleukin 2 (IL-2) production, inhibition of cell cycle progression and modulation of TCR signaling 2,3. CTLA-4 and CD28 shares the ligands B7-1 (also called CD80) and B7-2 (also called CD86) 4. Mice deficient for CTLA-4 develop lymphoproliferative disease and die within 3-4 weeks of birth 5, adding further evidence for the critical role of CTLA-4 in controlling T cell responses and immune homeostasis.
Programmed death-1 (PD-1) is an inhibitory molecule found on the surface of activated B and T cells and has been implicated in immune tolerance 6. PD-1 (also called CD279) is a member of the CD28 and CTLA-4 immunoglobulin superfamily and interacts with two B7 family ligands, PD-L1 (also called CD274) and PD-L2 (also called CD273) 4. PD-L1 is widely distributed on leukocytes, non-hematopoietic cells, and in non-lymphoid tissues including pancreatic islets, while PD-L2 is expressed exclusively on dendritic cells (DC) and monocytes 7,8. The PD-1 signaling pathway limits viral clearance during chronic infections by creating an unresponsive state in virus-specific CD8+ T cells termed “exhaustion” 9. In an autoimmune setting, PD-1 is essential for maintaining T cell anergy and preventing autoimmunity 10,11. Genetic deletion of PD-1 results in profound and complex multi-organ autoimmune destruction 4. Similarly, blocking PD-1-PD-L1 interactions accelerates spontaneous autoimmune diabetes 12 and reverses immune tolerance 10,11. Together these data support a central role for PD-1 as a major inhibitory pathway controlling immunity and suggest that exhaustion and peripheral tolerance are linked mechanistically.
Several theories have emerged to explain the mechanism through which PD-1 suppresses T cell activation. PD-L1 may act passively by competing directly with CD28 for B7-1 binding 13. In addition, PD-1 may directly recruit phosphatases such as SHP-1, SHP-2 and PP2A, which interfere with TCR signaling 4,14-16. However, the effects of PD-1 engagement during T cell activation are unclear as in vitro models do not adequately demonstrate the inhibitory activity of PD-1 17. In vivo studies of PD-1–mediated inhibition have been limited to global effects on immunity with little direct evidence that PD-1 engagement directly controls T cell signaling.
Previously, we have used a well-defined, antigen-specific tolerance model to examine the cellular basis of immunological tolerance using antigen-pulsed and fixed antigen-presenting cells (APC) 10. We determined that the administration of islet antigen peptide mimic, p31, coupled to chemically fixed APCs reversed diabetes and induced a robust, long-term inactivation of islet-specific, BDC2.5 TCR transgenic T cells. Tolerized T cells did not proliferate or produce cytokines in response to TCR stimulation. Although both PD-1 and CTLA-4 interactions were critical for the induction of tolerance, the long term maintenance of the anergic state depended on PD-1-PD-L1 interactions, but not CTLA-4 interactions within the inflamed tissue 10. These results implied that a cell-intrinsic mechanism maintains tolerance in the setting of continual exposure to autoantigen.
Recent imaging advances have allowed the use of multi-photon laser-scanning microscopy (MPLSM) to visualize T cells in lymph nodes (LN) and non-lymphoid tissues in vivo on the single cell level 18. Several groups have described the highly dynamic movement of CD4+ and CD8+ T cells, as well as DCs during T cell priming and tolerance induction 19-23. Naïve circulating T cells enter the LN through high endothelial venules and move on a network of stromal cells called fibroblastic reticular cells (FRCs), which define the potential location and migratory range of T cells 24. The FRC also provide a substrate for resident DCs to sample and display antigen to passing T cells. As T cells migrate along the FRC highway they probe DCs for signals and antigen 24. Initial interactions may be transient, but antigen recognition results in T cell swarming, formation of stable T cell-DC conjugates, and T cell arrest 25,26. TCR ligation results in ‘stop signals’ that decrease T cell motility and are required for stable T cell-DC conjugate formation and development of the immunological synapse 21,26. Prolonged T cell-DC interactions are critical for full T cell activation, proliferation and cytokine production 27-29. However, T cell movement and velocity during the long term maintenance of tolerance has not been well described.
Here we examined the biological roles of the PD-1 and CTLA-4 inhibitory pathways during autoimmunity using multi-photon imaging techniques. PD-1 suppressed TCR-driven stop signals in the pancreatic islets and blockade of PD-1 or PD-L1 inhibited T cell migration, prolonged T cell-DC engagement, enhanced T cell cytokine production, boosted TCR signaling, and abrogated peripheral tolerance. CTLA-4 blockade left T cell mobility and tolerance unaltered. Thus, these findings suggest that PD-1 and CTLA-4 play fundamentally distinct roles in the maintenance of peripheral tolerance and autoimmunity.
Results
PD-1–dependent islet antigen-specific tolerance
Insulin-coupled, ethylene carbodiimide (ECDI) fixed APCs have a profound effect on the development of spontaneous type 1 diabetes (T1D) in the non-obese diabetic (NOD) mouse 10. A single injection of the antigen-pulsed fixed splenocytes prevents and even reverses T1D after disease onset. We previously identified a critical role for PD-1 but not CTLA-4 in the maintenance of tolerance induced by injection of insulin-coupled fixed splenocytes 10. Herein, we adapted this model using islet antigen-specific BDC2.5 TCR transgenic T cells to define the role of PD-1 and CTLA-4 in the maintenance of peripheral tolerance.
We tracked the movement of fluorochrome-labeled diabetogenic BDC2.5 CD4+ T cells in vivo by MPLSM. T1D was induced by the adoptive transfer of activated BDC2.5 T cells into naïve pre-diabetic NOD recipients. Control recipient animals injected with splenocytes coupled with an irrelevant antigen (SHAM-SP) developed severe T1D within 6 days of T cell transfer (Fig. 1a). Injection of splenocytes coupled to the p31 peptide (Ag-SP, p31-SP), an islet antigen mimetope recognized by the BDC2.5 TCR 30, prevented autoimmune diabetes and resulted in decreased T cell proliferation and cytokine production (Fig. 1a, data not shown). To test if T cell unresponsiveness was consistent with T cell anergy, we measured calcium flux 1 week following p31-SP tolerance induction. Previous work demonstrated that TCR-induced calcium signals are associated with T cell motility and stable interactions with APCs 25. BDC2.5 T cells isolated from p31-SP-tolerized, but not SHAM-SP-injected mice, did not flux calcium in response to strong secondary TCR stimulation with anti-CD3 (Fig. 1b). Furthermore, phosphorylation of the TCR ζ-chain and the MAP kinase Erk was decreased in T cells isolated from mice treated with p31-SP compared to SHAM-SP (data now shown); these data provide evidence that exposure to p31-SP in vivo results in a TCR proximal signaling defect.
Figure 1. Antigen-specific tolerance blocks diabetes, TCR signaling and Ca++ flux in a PD-L1-dependent manner.

(a) p31-SP tolerance prevents T1D induced by BDC2.5 T cell adoptive transfer. Activated BDC2.5 T cells were transferred to NOD mice followed with p31-SP (p31 Tolerized) or SHAM-SP (Activated control) treatment the same day. The percentages of diabetic mice receiving SHAM-SP (n=8) compared to p31-SP protected mice are shown (n=8). (b) p31-SP tolerized cells have decreased ability to flux calcium upon TCR ligation. BDC2.5 TCR CD4+ T cells treated in vivo with p31-SP (p31-Tolerized) or SHAM-SP (Control) were purified and loaded with Indo-1. Cells were activated with anti-CD3 (5μg/ml) and cross-linking antibody or ionomycin as indicated, and calcium flux was measured. (c) PD-L1 blockade breaks tolerance. 2×106 p31-SP tolerized BDC2.5 TCR transgenic T cells were transferred to naïve recipients followed by anti-PD-L1, anti-CTLA-4 or isotype control antibody treatments. Recipient mice were monitored for the development of T1D by blood glucose measurements. The percentage of diabetic mice receiving anti-PD-L1 (n=5), isotype control (n=5) and anti-CTLA-4 (n=5) are shown. Data are representative of three or more independent experiments except (c) which was from two independent experiments.
Next we investigated the role of PD-1 and CTLA-4 in the maintenance of this unresponsive state. p31-SP tolerized BDC2.5 TCR transgenic T cells were transferred into naïve recipients; these recipients were subsequently injected with anti-PD-L1, anti-CTLA-4 or isotype control antibody. Blocking PD-L1 led to the reversal of tolerance and rapid precipitation of clinical diabetes (Fig. 1c). In contrast, CTLA-4 blockade did not affect clinical disease (Fig. 1c). Anti– PD-L1, but not anti-CTLA-4, also led to an increase in antigen-specific BDC2.5 T cell accumulation in the pancreatic lymph node (PLN) (data not shown). Taken together, these results demonstrate that antigen-specific tolerance regulates autoimmune diabetes and that PD-L1, but not CTLA-4, plays a critical role for the maintenance of this anergic state.
PD-1 but not CTLA-4 prevents T cell stop signals
To ascertain the roles of PD-1 and CTLA-4 during the maintenance of tolerance, we developed a system to study the dynamic movement of tolerized T cells within intact mouse LNs by MPLSM. Since Ag-SP tolerance correlated with impaired calcium signaling and PD-1 but not CTLA-4 maintained this tolerant state, we tested the hypothesis that PD-L1 prevents the TCR mediated T cell stop signal associated with T cell activation and T1D. Antigen-specific T cells swarm and cluster when they enter antigen-containing PLNs 31. Thus, we compared the migration behavior of activated control BDC2.5 T cells with that of tolerized BDC2.5 T cells. BDC2.5 Thy1.1 CD4+ T cells from SHAM-SP or p31-SP-treated mice were harvested, CMTMR-labeled and transferred into NOD mice expressing a transgene encoding yellow fluorescent protein (YFP) driven by the CD11c promoter. This CD11c-YFP mouse allowed tracking of the in vivo movements of CD11c+ DC 32. The velocity and displacement of control and tolerized T cells in islet antigen-bearing PLN 18-24 hours following T cell transfer was compared. Control activated BDC2.5 T cells slowed down and swarmed in antigen-bearing PLN with a velocity of 2.2 ± 0.1 μm/min (Fig. 2a and Supplementary Movie 1), similar to what has been previously reported 31. In contrast, the tolerant T cells did not swarm in antigen-containing PLN, and exhibited a mean velocity of 5.2 ± 0.4 μm/min (Fig. 2a and Supplementary Movie 2), similar to what has been observed in antigen non-bearing LN 31.
Figure 2. PD-1-PD-L1 but not CTLA-4 prevents the T cell stop signal.

Dynamic migration of BDC2.5 diabetogenic CD4+ T cells in PLN. CMTMR-labeled control or tolerized BDC2.5 T cells were injected into NOD.CD11c-YFP recipient mice. (a-f) Multi-photon image analysis of T cells in islet antigen-containing PLN. (a) Mean velocity of adoptively transferred BDC2.5 T cells in mice subsequently injected with anti-PD-L1, anti-CTLA-4 or isotype control antibody. Horizontal lines illustrate the mean velocity for each group. (b) Mean displacement of BDC2.5 T cells plotted against the square root of time. Shown is the mean of multiple imaging data sets from PLN of mice receiving control T cells, tolerized T cells with isotype control antibody, tolerized T cells with anti-PD-L1, and tolerized T cells with anti-CTLA-4. Data show mean ± s.d.. A time-lapse recording corresponding to this region is shown for each group in Supplementary Movies 1-4. (c-f) Superimposed 10 min tracks of 40-60 randomly selected T cells from each treatment group in the xy plane, setting the starting coordinates from the origin 0,0. Units are in micrometers. Each line represents the path of one cell. Data are representative of three or more independent experiments.
Next, we designed experiments to test the role of PD-1 and CTLA-4 on T cell motility during the maintenance phase of tolerance. As PD-L1, but not PD-L2, is required for the maintenance of tolerance, we injected PD-L1-blocking antibodies 10. PD-L1 blockade led the tolerant T cells to slow and swarm with a velocity of 1.4 ± 0.1 μm/min, as compared to the 5.2 μm/min velocity observed after isotype control antibody treatment (P<0.0001) (Fig. 2a and Supplementary Movie 3). PD-1 blockade gave similar results with respect to effects upon T cell swarming and stop signals (data not shown). Together, these data support the notion that PD-1–PD-L1 interactions are required to mediate tolerance and prevent T cell stop signals.
CTLA-4 blockade, on the other hand, did not affect tolerized T cell movement, as tolerized T cells maintained a mean velocity of 5.4 ± 0.4 μm/min after CTLA-4 blockade (Fig. 2a and Supplementary Movie 4). We next measured T cell motility by measuring the displacement of T cells plotted against the square root of time [M=motility coefficient]. This measurement reflects not simply the total distance traveled, but rather displacement from the point of origin. Consistent with previous reports, antigen encounter resulted in low displacement of control T cells (Fig. 2b, M = 0.32 ± 0.16 μm2 min-1) 22,33,34. In contrast, the displacement of tolerized BDC2.5 T cells was significantly higher (M = 6.65 ± 0.21 μm2 min-1, Fig. 2b, P<0.0001), indicative of a process wherein activated antigen-specific T cells stop and are engaged in stable interactions with antigen-presenting cells while the tolerized T cells move freely in the PLN. As suggested by the change in velocity, blockade of PD-1-PD-L1 interactions resulted in decreased displacement and arrest of tolerant T cells (M = 0.19 ± 0.06 μm2 min-1; Fig. 2b). CTLA-4 blockade, on the other hand, did not decrease T cell displacement (Fig. 2b, M = 7.0 ± 4.6 μm2 min-1). Tracking individual cells and plotting the superimposed tracks from the origin for each group illustrates distance traveled and further demonstrated the limited movement of activated compared to tolerant T cells (Fig. 2c,d). PD-L1 but not CTLA-4 blockade induced T cell arrest and limited displacement (Fig. 2e,f), similar to that of activated T cells (Fig. 2c). These results demonstrate a fundamental distinction between the functions of PD-1 and CTLA-4, and suggest that PD-L1 blockade enhances T cell stop signals.
To investigate if PD-1-PD-L1 interactions function in an antigen-dependent or antigen-independent manner, we investigated the migration behavior of the BDC2.5 T cells in the antigen-deficient inguinal lymph nodes (ILN). The velocity and displacement of control and tolerized T cells 18-24 hours following T cell transfer were compared. In all cases, T cells migrated freely in the ILN with similar velocities, displacement and motility, and migration was independent of T cell activation or tolerance and the presence or absence of PD-L1 or CTLA-4 blockade (Fig. 3a-f and Supplementary Movies 5-8). Thus, the enhanced stop signal and confined displacement required TCR engagement with islet antigen(s) (PLN vs. ILN) as well as the disruption of PD-L1 ligation. It is important to note that anti-PD-L1 administration did not affect naïve, non-tolerant polyclonal T cell movement (Supplementary Movie 9). This is likely due to the fact that tolerized T cells express high amounts of PD-1 on their surface, while naïve T cells do not 10,35. These results reinforce the notion that antigen is required for the stop signal and that PD-1 normally functions to inhibit T cell activation by preventing T cell arrest.
Figure 3. PD-L1 inhibition requires antigen.

Dynamic migration of BDC2.5 diabetogenic CD4+ T cells in ILN. CMTMR-labeled control or tolerized BDC2.5 T cells were injected into NOD.CD11c-YFP recipient mice. (a-f) Multi-photon image analysis of T cells in non-islet antigen-containing ILN. (a) Mean velocity of adoptively transferred BDC2.5 T cells in mice subsequently injected with anti-PD-L1, andi-CTLA-4 or isotype control antibody. Horizontal lines illustrate the mean velocity for each group. (b) Mean displacement of BDC2.5 T cells plotted against the square root of time. Shown is the mean of multiple imaging data sets from ILN of mice receiving control T cells, tolerized T cells with isotype control antibody, tolerized T cells with anti-PD-L1, and tolerized T cells with anti-CTLA-4. Data show mean ± s.d.. A time-lapse recording corresponding to this region is shown for each group in Supplementary Movies 5-8c-f) Superimposed 10 min tracks of 40-50 randomly selected T cells from each treatment group in the xy plane, setting the starting coordinates from the origin 0,0. Units are in micrometers. Each line represents the path of one cell. Data are representative of three or more independent experiments.
PD-1 inhibits T cell movement within the islets
PD-1–mediated control of immune responses depends on interactions between PD-1 on T cells and PD-L1 ligand in tissues 10,11. Thus, we sought to determine if the effects of PD-1-PD-L1 blockade had similar effects in the autoimmune target tissue, the pancreatic islets, as was the case in antigen draining PLN. To accomplish this we developed a system to analyze T cell motility in vivo within pancreatic islets of Langerhans using MPLSM. Islets from NOD.MIP-GFP mice were transferred under the kidney capsule of recipient NOD.SCID mice. These transgenic mice express green fluorescent protein (GFP) under the control of mouse insulin 1 promoter. After 1 week these mice received CMTMR-labeled (red) tolerized BDC2.5.Thy1.1 T cells. Following T cell transfer, recipient mice received anti-PD-L1, anti-CTLA-4 or isotype control antibody and cell movement was tracked. As was the case in the antigen-containing PLN, tolerized BDC2.5 T cells moved rapidly within the islet transplants in isotype control antibody-treated mice (velocity = 6.2 ± 0.3 μm/min) (Fig. 4a, Supplementary Movie 10). PD-L1 blockade resulted in decreased velocity (3.3 ± 0.2 μm/min) (Fig. 4a, Supplementary Movie 11) whereas CTLA-4 blockade did not affect previously tolerized T cell migration despite the presence of antigen (velocity = 6.6 ± 0.3 μm/min) (Fig. 4a, Supplementary Movie 12). We also measured the T cell displacement of previously tolerized T cells within the islet transplants. We observed free and apparently unconstrained migration of tolerized T cells in recipient mice treated with isotype control antibody or anti-CTLA-4 (isotype control M= 15.8 ± 5.3 μm2 min-1, CTLA-4, M = 10.1 ± 2.3 μm2 min-1). The slope of these two curves is a straight line representing free and random walking within the target tissue and does not correspond to directed migration 33. PD-L1 blockade, however, attenuated T cell motility and decreased T cell displacement when compared to isotype control antibody-treated animals (Fig. 4b, M= 0.5 ± 0.3 μm2 min-1). Tracking individual cells confirmed these results (Fig. 4c-e). These data show that PD-L1-PD-1 interactions modulate TCR-induced swarming and arrest behaviors in autoimmune target tissues.
Figure 4. PD-L1 inhibits T cell movement within the islets.

Dynamics and motility of BDC2.5 diabetogenic CD4+ T cells in the pancreatic islets. NOD.SCID mice were transplanted with MIP-eGFP (green) islets under the kidney capsule, and were subsequently injected with CMTMR-labeled tolerized BDC2.5 T cells (red) and the indicated antibodies. Transplanted islets were imaged by multi-photon microscopy. (a) Average velocity of tolerized BDC2.5 T cells transferred into mice treated with isotype control antibody, anti-PD-L1, or anti-CTLA-4. Mean velocity is represented by the horizontal lines. A time-lapse recording corresponding to this region is shown for each treatment group in Supplementary Movies 10-12. (b) Mean displacement of BDC2.5 T cells plotted against the square root of time. Shown is the mean of multiple imaging data sets from mouse islets receiving tolerized T cells with anti-PD-L1, anti-CTLA-4 or isotype control antibody. Data show mean ± s.d. (c-e) Superimposed 10 min tracks of 43-60 randomly selected T cells from each treatment group in the xy plane, setting the starting coordinates from the origin 0,0. Units are in micrometers. Each line represents the path of one cell. Data are representative of at least three independent experiments.
PD-L1 blockade enhances T cell-DC stable interactions
The studies described above suggest that blocking PD-1-PD-L1 interactions leads to decreased T cell movement and thus may provide the opportunity for extended interactions between antigen-specific T cells and antigen-bearing APCs. Prolonged interactions between T cells and DCs is critical for full T cell activation 27,29. We hypothesized that PD-1 normally functions to prevent the stable T cell-DC contacts that are required for full T cell activation. To test this notion, we used the adoptive transfer and imaging system described above to measure the dwell time of antigen-specific T cells with DCs. BDC2.5 T cells were labeled red with CMTMR and transferred to NOD.CD11c-YFP recipient mice; T cell-DC interaction times were determined in antigen containing (PLN) and antigen-deficient (ILN) sites. Tolerized T cells did not slow down or form stable contacts with tissue DCs (Fig. 5a and Supplementary Movie 13 and 15). In sharp contrast, numerous stable conjugates between T cells and CD11c+ DCs were observed following PD-L1 blockade (Fig. 5b and Supplementary Movie 14 and 16). PD-L1 blockade was associated with increased duration of T cell–DC contacts (Fig. 5c). Notably, the majority of T cells from anti-PD-L1-treated mice interacted with tissue specific DCs for the entire duration of the imaging experiment (30 minutes) with a mean of 26.6 ± 6.8 minutes compared to a mean dwell time of 9.2 ± 8.5 minutes for tolerized T cells in isotype control-treated mice (Fig. 5c, P<0.0001). In addition, approximately 75% of labeled tolerized T cells that made contact with DCs maintained these contacts for the entire imaging sequence of 30 min in anti-PD-L1 treated mice, whereas only 8% of tolerized T cells maintained DC interactions in isotype control antibody treated mice (Fig. 5d). Stable conjugates in the ILN were not readily detected in either group, consistent with the idea that antigen is required for these interactions (data not shown). Together, these data suggest that PD-1-PD-L1 blockade fundamentally altered the way that diabetogenic T cells interacted with antigen-bearing CD11c+ DCs.
Figure 5. PD-L1 blockade promotes prolonged T cell-DC interactions and T cell activation.
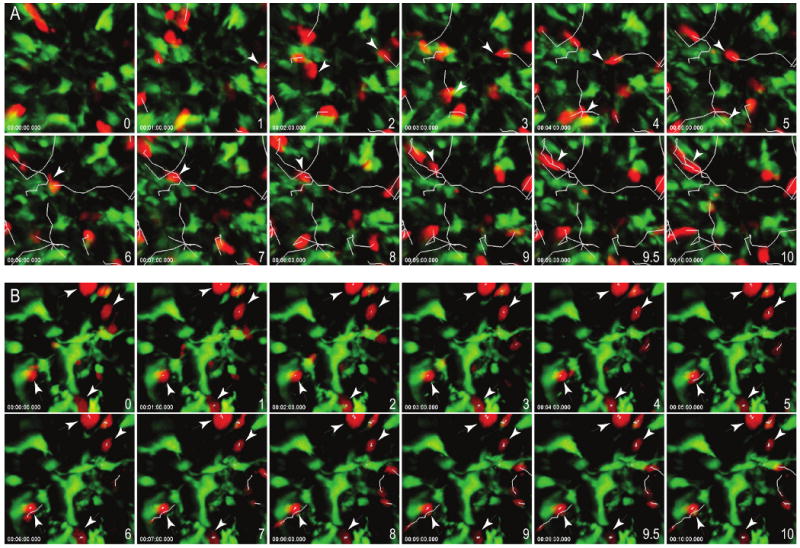
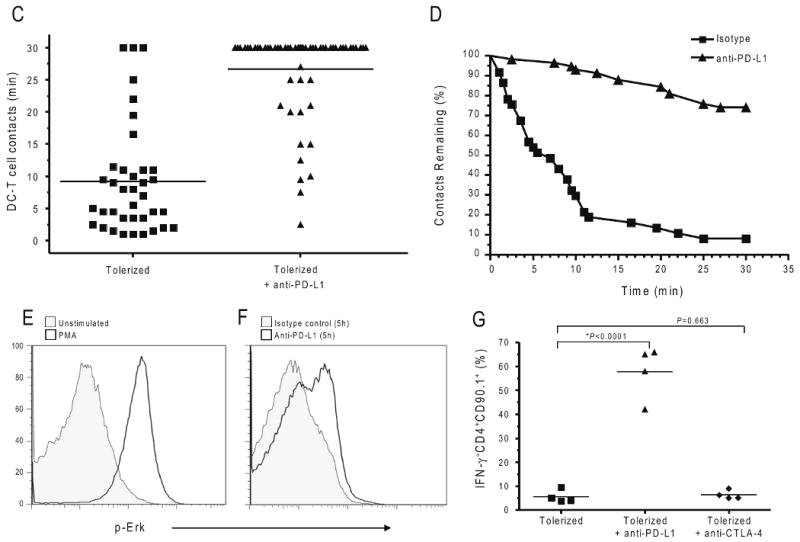
Time-lapse images of contacts between CD11c+ DC (green) and BDC2.5 T cells (red) from the PLN of (a) isotype control or (b) anti-PD-L1 treated recipients. Scale bars, 15 μm. Corresponding time-lapse recordings are shown in Supplementary Movies 13-16. (c) Contact times between antigen-specific tolerized T cells and antigen-bearing CD11c+ DCs following injection of isotype control antibody or anti-PD-L1. Each symbol represents an individual cell. The mean from each group is shown as a horizontal line. (d) Contact time decay curves illustrate the percentage of total T cell-DC contacts remaining after injection of isotype control or anti-PD-L1 antibody over time (min). (e) Intracellular phosphorylated Erk expression in tolerized BDC2.5 T cells that were isolated and left unstimulated or stimulated with PMA. (f) Intracellular phosphorylated Erk expression in tolerized BDC2.5 T cells directly ex vivo from mice treated with isotype control antibody or anti-PD-L1. (g) PD-L1 blockade restores effector cytokine production within the pancreatic islets. Interferon reporter BDC2.5.Thy1.1.Yeti mice were injected with p31-SP followed by anti-PD-L1, anti-CTLA-4, or isotype control antibody. Three days after antibody treatment, pancreas infiltrating CD4+IFN-γ+ (YFP+) cells were analyzed by flow cytometry. Shown is the percentage of CD4+IFN-γ+ BDC2.5 T cells. Data are representative from at least three independent experiments except (f) which was from two independent experiments.
PD-L1 blockade promotes T cell activation
Anergy induced in BDC2.5 T cells exposed to the p31-pulsed fixed APCs led to a defect in early TCR signaling events including Ca2+ flux (Fig. 1) and Erk phosphorylation 10,36. However, treatment of anergic T cells with PMA and ionophore can reverse tolerance and promote T cell activation (Fig. 1 and data not shown). Thus, any therapy that would break tolerance in this system would be expected to reverse this TCR signaling defect. To correlate our single cell imaging data with functional measures of T cell activation, we assayed Erk phosphorylation in tolerized BDC2.5 T cells using flow cytometric analysis. Antigen-specific tolerized BDC2.5 T cells were harvested 5 hours after in vivo anti-PD-L1 or isotype control antibody injection and stained for pErk activity both immediately and after PMA stimulation (Fig. 5e). Tolerized T cells did not exhibit Er phoksphorylation after in vivo APC engagement 36-38 (Fig. 5e). In contrast, treatment with anti-PD-L1 but not isotype control antibody during exposure of the tolerized T cells to antigen-bearing APCs resulted in substantial Erk phosphorylation in tolerized T cells (Fig. 5f). These results indicate that proximal TCR signaling components including Erk phosphorylation are restored following PD-1-PD-L1 blockade.
Effective TCR signaling culminates in the production of effector cytokines. Ag-SP tolerance resulted in a decrease in inflammatory cytokines including IL-2 and IFN-γ within the PLN 10. Here we bred antigen-specific NOD.BDC2.5 TCR transgenic mice to Yeti mice, which express a YFP reporter cassette under the control of the IFN-γ promoter 31. Tolerized BDC2.5.Thy1.1.Yeti T cells were transferred to recipient NOD mice and received isotype control antibody, anti-PD-L1, or anti-CTLA-4. PD-L1 blockade abrogated tolerance, and the majority of CD4+ T cells isolated directly from the islets of anti-PD-L1 injected mice produced IFN-γ (57.8 ± 5.5 %) (Fig. 5g). These results demonstrate a T cell-intrinsic abrogation of anergy, and this effect was not observed when anti-CTLA-4 or isotype antibodies were administered (6.3 ± 1.8%, 5.5 ± 1.3% P<0.0001) (Fig. 5g). Taken together, these findings indicate that PD-L1 blockade restored the ability of anergic T cells to engage in prolonged DC interactions, produce inflammatory cytokines and induce rapid development of T1D.
Discussion
In this study, we sought to determine the roles of the inhibitory receptors, PD-1 and CTLA-4, on T cell migration during the maintenance of tolerance. The results indicate that the disruption of PD-1-PD-L1 but not CTLA-4-B7 interactions enhances tolerized T cell interactions with antigen-bearing DCs, and facilitates the phosphorylation of key TCR signaling molecules. This T cell engagement with antigen-bearing DCs ultimately results in the production of effector cytokines and rapid progression of autoimmunity.
The duration of T cell-DCs contacts and its influence on T cell activation and the induction of tolerance has been a topic of great interest. Several other reports illustrated that during the induction of T cell tolerance both transient and stable DC interactions can occur 20,23. During the process of T cell activation the duration of T cell-DC contacts is highly variable in vivo, ranging from minutes to several hours 39. Early reports suggested that initial interactions during the first phase of T cell activation tend to be transient [5-10 minutes during the first 3-15 hours 20,21,40]. However, a more recent report found that long lived T cell-DC interactions could occur following the initial T cell-DC contact and that prolonged interactions were required for T cell activation 41. In this study, Erk phosphorylation occurred early after stable T cell-DC conjugates were formed, and IFN-γ production increased with longer T cell-DC interactions 41. These interactions required TCR engagement of peptide-MHCII complexes, as MHCII blockade in vivo terminated stable contacts, increased T cell motility, decreased T cell proliferation and prevented IFN-γ production. The second phase subsequently results in longer lived contacts where T cells form stable conjugates and begin to secrete cytokines which transitions into a third phase of high motility and rapid proliferation 21. Our studies demonstrated that PD-1 blockade restored stable T cell-DC contacts, Erk phosphorylation, IFN-γ production and, most importantly, T1D. These findings suggest that PD-1 normally functions to prevent the T cell stop signal and the formation of stable conjugates with antigen bearing DCs.
Anergic T cells have been shown in vitro to form unstable immunological synapses with allogeneic APCs and failed to recruit the signaling proteins necessary to initiate T-cell activation 42. We suggest that these transient interactions are required in our system (but impossible to discern in vivo), since the breakdown of tolerance following PD-1-PD-L1 blockade only occurs when antigen is present, as in the case of PLN and pancreas, and does not occur in the ILN. Thus, abrogation of tolerance allows the stabilization of these interactions allowing full T cell activation and clinical disease.
Recent reports suggested that B7-1 and PD-L1 interact directly with each other to negatively regulate T cells 13. Thus far, however, in vivo data to support the functionality of this interaction have not been reported. Our data do not suggest that this interaction was responsible for the tolerant phenotype observed in this study, as administration of anti-B7-1 had no effect on T cell velocity, track displacement, or movement trajectories when compared to isotype control antibody (data not shown). In addition, PD-1 blocking antibodies induced effects similar to those induced by PD-L1 blockade (data not shown). Finally, tolerized BDC2.5 T cells arrested and stopped when transferred to the PD-L1 deficient (PD-L1-/-) recipients similar to that observed in experiments presented here using PD-L1 antibody blockade, supporting the critical role for PD-1-PD-L1 interactions for tolerance (data not shown).
Blockade of CTLA-4–B7 interactions can prevent induction of tolerance by peptide-pulsed fixed APCs but could not reverse tolerance once established 10. It is important to note that the same dose of blocking CTLA-4–specific antibody was used in these MPLSM experiments as in a previous study that documented a critical role for CTLA-4 during the induction of tolerance 10. We detected no influence of CTLA-4 in the migratory behavior of tolerized T cells. Due to the complex nature of lattice formation with B7-1, we have also explored anti-CTLA-4 Fab fragments with similar results as shown using intact anti-CTLA-4. The results presented here suggest that CTLA-4 inhibition had different qualitative and quantitative biological consequences than PD-1-PD-L1 blockade.
Although CTLA-4 and PD-1 both limit T cell signaling, cytokine production, cell cycle progression, and may share potential targets, some key biochemical differences have been reported. Upon PD-L engagement, PD-1 can bind SH2-domain containing tyrosine phosphatase 1 (SHP-1) and SHP-2 4,43. The binding of SHP-1 and SHP-2 can terminate early TCR signals by dephosphorylating key signaling intermediates including the kinases Akt, PI3K, ZAP-70, and PKC-θ. Like PD-1, CTLA-4 can interact with SHP-1 and SHP-2 14. Unlike PD-1, CTLA-4 can also interact with the phosphatase PP2a 15. Another difference between these two inhibitory receptors is the structural motif used to bind phosphatases. CTLA-4 interacts through the immunoreceptor tyrosine-based inhibitory motifs (ITIM) while PD-1 contains an additional motif, the immunoreceptor tyrosine-based switch motif (ITSM) 43. Mutation of the ITIM motif had little effect on signaling, while ITSM mutations abrogated the ability of PD-1 to limit T cell population expansion 43. This suggests that PD-1 and CTLA-4 use different structural motifs to bind and recruit phosphatases for signal blockade. Future work is necessary to determine the precise biochemical relationship between these two potent negative regulatory molecules.
Genetic experiments may help to explain the different roles for PD-1 and CTLA-4 in immune homeostasis, breakdown of tolerance and establishment of autoimmunity. CTLA-4 deficiency results in rapid multi-organ tissue inflammation and death within 3-4 weeks of age, regardless of mouse genetic background 5, whereas autoimmunity in PD-1-deficient mice is slower and tissue specific in a manner dependent on genetic background 4. These reports suggest that deficiencies in PD-1/PD-L1 pathway may potentiate tissue specific autoimmune predispositions. CTLA-4 on the other hand, controls multi-organ infiltrate and autoimmunity irrespective of genetic background. CTLA-4 and PD-1 ligand expression and distribution may help explain these differences. The fundamental difference between the effects of CTLA-4 and PD-1 on T cell migration as described here may also help to explain these differences.
Two recent reports investigated the role of CTLA-4 on T cell stop signals 44,45. One study found that CTLA-4 positive T cells failed to stop or slow down in response to in vivo peptide challenge, and anti-CTLA-4 increased T cell motility 44. A follow-up study reported that CTLA-4-deficient T cells showed a marked resistance to a stop-signal induced by anti-CD3 45. It is difficult to explain the discrepancies between these previous studies, but it may be due to subtle differences between sorted T cell subpopulations, CTLA-4 surface stability, blocking antibodies, or use of knockout T cells. Here we found no influence of CTLA-4 blockade on TCR-driven stop signals. In our study, CTLA-4 blockade did not alter the DC-binding properties of the tolerant T cells, did not result in significant ERK phosphorylation, did not restore IFN-γ production, and did not result in the rapid development of autoimmune diabetes. One major difference is the previous reports tracked the migration of naïve T cells during primary stimulation 44,45 whereas the present studies focused on anergic T cells during the re-activation phase.
Finally, it is interesting to note that PD-1-PD-L1 blockade resulted in increased accumulation and/or enhanced proliferation of antigen-specific T cells within the target tissue 10. This finding supports a key role for T cell-DC interactions during tissue-specific reactivation. Future work in this area will determine if CD4+ T cells interact directly with islet target cells or through a tissue-specific MHC II+ DC. Further investigation of the signals that maintain tolerance in this and similar settings will aid in our understanding of how to exploit the PD-1-PD-L1 pathway in efforts to prevent and treat autoimmunity, promote transplant acceptance, and limit tumor growth.
Methods
Mice
All mice were housed and bred in specific pathogen–free conditions in Animal Barrier Facilities at the University of California, San Francisco or the University of Minnesota. C57BL/6.MIP.eGFP mice were obtained from M. Hara and G. Bell (University of Chicago, Chicago, IL) 46 and were backcrossed for more than 12 generations to NOD mice. C57BL/6 CD11c-YFP obtained from M. Nussenzweig (The Rockefeller University, New York, NY) 32 mice were backcrossed for more than 10 generations to NOD. C57BL/6.PD-L1 KO mice were obtained from A. Sharpe (Harvard, Boston, MA) 11 and were backcrossed for 10 generations to NOD. Female NOD mice were purchased from Taconic. NOD-BDC2.5 TCR Tg+ mice were provided by C. Benoist and D. Mathis (Harvard Medical School, Boston, MA) 47 and were crossed to NOD.Thy1.1. Interferon-γ reporter mice (Yeti) were provided by R. Locksley (UCSF) and were bred 10 generations to NOD and then crossed to NOD.BDC2.5.Thy1.1 TCR Tg+ mice to generate the NOD.BDC2.5.Thy1.1.Yeti mice 31. NOD.SCID mice were purchased from The Jackson Laboratory. Mice were 3–10 wk old at the initiation of the experiments. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco and the University of Minnesota.
Antibodies
FITC-conjugated-anti-CD4 (RM4-5), PerCP-conjugated-anti-CD8a (Ly-2), APC-conjugated-anti-CD90.1, PE-conjugated-anti-Vβ4 (KT4), APC-conjugated-anti-IFN-γ (XMG1.2), PE-conjugated-anti-Armenian hamster IgG1, PE-conjugated-anti-Armenian hamster control IgG2, PE-conjugated-anti-CD152 (UC10-4F10), PE-conjugated-anti-CD279 (J43), and isotype controls were purchased from BD Biosciences. Anti-Phospho-p44/42 MAPK (Thr202/Tyr204) (197G2) rabbit mAb was purchased from Cell Signaling Technology 48. Anti–PD-1 (RMP1-14), anti–PD-L1 (MIH5, MIH6), and anti–PD-L2 (TY25) were made as described previously 7,10. Anti-CTLA-4 (UC4F10), and anti-CTLA-4 Fab fragments were made as described previously 1. Mice were injected intraperitoneally with 250 μg anti–PD-L1, anti–PD-L2, anti-B7-1, anti-PD-1, or anti-CTLA-4 as indicated.
Antigens
1040-p31 peptide (YVRPLWVRME) was purchased from Genemed Synthesis Inc. The amino acid composition was verified by mass spectrometry, and purity (>98%) was assessed by HPLC.
Cell culture, transfer, and induction of tolerance
NOD.BDC2.5.Thy1.1 TCR Tg+ lymphocytes were harvested and pooled from brachial, axillary, peri-aortic, pancreatic and inguinal LN, and from the spleen. Cells were activated in vitro in the presence of 0.5 μM 1040-p31 peptide in complete DMEM containing 5 × 10−5 M 2-ME, 2 mM L-glutamine, 100 U/ml penicillin-streptomycin, 0.1 M nonessential amino acids (Invitrogen), and 10% FCS (Hy-clone). Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2. The cells were harvested after 96 h and washed, and 5 × 106 T cells were transferred i.v. to naive pre-diabetic NOD recipients. Tolerance was induced using i.v. injections of 50 × 106 chemically treated antigen-coupled syngeneic splenocytes (p31 or SHAM control), as described previously 10.
Assessment of diabetes
Blood glucose concentrations were measured from female NOD mice with OneTouch glucose meters (Lifescan, Inc.). Mice were considered diabetic with two consecutive readings of >250 mg/dL.
Flow cytometry and cell sorting
For assessment of surface molecule and cytokine expression, cells were labeled with predetermined optimal antibody concentrations according to the manufacturer's staining protocol and 0.5 × 106 events in the CD4+ gate were acquired, as described previously 10. Data acquisition was performed on an LSRII flow cytometer and analyzed using FACSDiva software (Becton Dickinson). NOD.BDC2.5.Thy1.1 LN cells and splenocytes were stained with FITC-conjugated-anti-CD4 and APC-conjugated-anti-CD90.1 and were sorted using a MoFlo cytometer high speed cell sorter (DakoCytomation). All sorted populations had ≥ 98% cell purity.
Pancreatic Islet Isolation and Transplantation
Islets were isolated as previously reported 49. Briefly, a 3-ml collagenase P (Roche) solution (0.75 mg/ml) was injected into the pancreatic duct of 4-wk-old NOD.MIP-eGFP mice. The distended pancreases were removed and incubated at 37°C for 17 min. The liberated free islets were purified by centrifugation on a Eurocollin-Ficoll gradient. 400-500 handpicked islets were transplanted beneath the left renal capsule of each recipient 50.
Multi-photon laser scanning microscopy acquisition and analysis
A custom built resonant-scanning instrument based on published designs containing four–photomultiplier tubes operating at video rate was used for multi-photon microscopy 31. For imaging of T cells, BDC2.5 CD4+ CD90.1 T cells were sorted and labeled with Cell Tracker™ Orange CMTMR (5-(and-6)-(((4-chloromethyl) benzoyl) amino) tetramethyl rhodamine) (Invitrogen) and transferred to recipient mice. Inguinal or pancreatic LNs were harvested from recipients and immobilized on coverslips with the hilum facing away from the objective. Lymph nodes were maintained at 36 °C in RPMI medium bubbled with 95% O2 and 5% CO2 and were imaged through the capsule distal to the efferent lymphatic. Transplanted islets were imaged under the exposed kidney capsule maintained at 36 °C in RPMI medium bubbled with 95% O2 and 5% CO2. Samples were excited with a 10-W MaiTai TiSaphire laser (Spectra-Physics) tuned to a wavelength of 800-810 nm, and emission wavelengths of 500–540 nm (for CFSE), 567–640 nm (for CMTMR) and 380–420 nm (for detection of second-harmonic emission) were collected. Each xy plane spanned 192 μm by 160 μm at a resolution of 0.4 μm per pixel and images of 44-46 xy planes with 2 μm z spacing were formed by averaging 10-12 video frames every 30 s for 10-30 min. Images acquired were between 70-250μm below the LN capsule identified by the second harmonic signal. Images were acquired by Video Savant software (IO Industries). The maximum intensity z-projection time-lapse image sequences were generated with MetaMorph software (Molecular Devices). 3-D rotations and time-lapse image sequences were generated in Imaris 5.7.2 ×64 (Bitplane). Semi-automated cell tracking in 3-D was verified manually. Tracking data were analyzed in Microsoft Excel with a custom macro written in Microsoft Visual Basic for Applications. The motility coefficient M=x2/6t was calculated from the slope (x/t1/2) obtained by regression analysis of the mean displacement (x) versus the square root of time (t1/2) as described previously 33.
Statistical analysis
Statistical analysis of the data was done with Graphpad Prizm software for comparisons of two groups, P values were calculated by the unpaired Student's T-test. For comparisons of multiple groups, P values were calculated by one-way ANOVA in GraphPad Prism. Values of P ≤0.05 were considered significant.
Supplementary Material
Activated antigen specific T cells stop in antigen-draining PLN. A time-lapse sequence of 40 μm z-projection images shows the dynamics of in vitro activated BDC2.5 CD4+ T cells (labeled red using CMTMR) in a PLN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient mouse. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
Dynamic movement of tolerized T cells in islet antigen-draining PLN. A time-lapse sequence of 40 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and isotype antibody injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 15 min in real-time, which is compressed to 3 sec in this video.
PD-L1 blockade causes tolerized T cells to stop in islet antigen-draining PLN. A time-lapse sequence of 36 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and anti-PD-L1 antibody injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 15 min in real-time, which is compressed to 3 sec in this video.
CTLA-4 blockade does not alter tolerized T cell movement in islet antigen-draining PLN. A time-lapse sequence of 44 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and anti-CTLA-4 antibody injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 15 min in real-time, which is compressed to 3 sec in this video.
Dynamic movement of activated antigen specific T cells in islet antigen-deficient ILN. A time-lapse sequence of 42 μm z-projection images shows the dynamics of in vitro activated BDC2.5 CD4+ T cells (labeled red using CMTMR) in a inguinal LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
Tolerized T cells move freely in islet antigen-deficient ILN. A time-lapse sequence of 44 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a inguinal LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and isotype control antibody injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
PD-L1 blockade does not alter tolerized T cell movement in islet antigen-deficient ILN. A time-lapse sequence of 46 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a inguinal LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and anti-PD-L1 injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
CTLA-4 blockade does not alter tolerized T cell movement in islet antigen-deficient ILN. A time-lapse sequence of 40 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a inguinal LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and anti-CTLA-4 injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
PD-L1 blockade causes tolerized T cells, but not naïve polyclonal T cells to stop in islet antigen-draining PLN. A time-lapse sequence of 28 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a pancreatic LN explant 18h after transfer to a NOD.CD4-eGFP (green naïve T cells) recipient and anti-PD-L1 antibody injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
Dynamic movement of tolerized T cells in pancreatic islet tissue. A time-lapse sequence of 46 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in pancreatic islet grafts of NOD.SCID transplant recipients receiving NOD.MIP-GFP islets (green β-cells) and isotype control antibody injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
PD-L1 blockade causes tolerized T cells to stop in pancreatic islet tissue. A time-lapse sequence of 42 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in pancreatic islet grafts of NOD.SCID transplant recipients receiving NOD.MIP-GFP islets (green β-cells) and anti-PD-L1 injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
CTLA-4 blockade does not alter tolerized T cell movement in pancreatic islet tissue. A time-lapse sequence of 44 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in pancreatic islet grafts of NOD.SCID transplant recipients receiving NOD.MIP-GFP islets (green β-cells) and anti-CTLA-4 injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
Investigation of T cell-DC conjugates in pancreatic LN. A time-lapse sequence of 34 μm 3D projection shows the dynamic movement of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and isotype control antibody injection. Elapsed time is shown as hh:mm:ss:sss. Randomly selected cells were tracked and the trailing line following the cells represents the cell path over time. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
PD-L1 blockade induces stable T cell-DC conjugates in pancreatic LN. A time-lapse sequence of 36 μm 3D projection shows the stable interactions of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) with DC (yellow and green) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP recipient and anti-PD-L1 injection. Elapsed time is shown as hh:mm:ss:sss. Randomly selected cells were tracked and the trailing line following the cells represents the cell path over time. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
Dynamic T cell-DC interactions following T cell tolerance. A magnified time-lapse sequence illustrating the dynamic and short interactions of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) with DC (yellow and green) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP recipient and isotype antibody injection. Elapsed time is shown as hh:mm:ss:sss. Brief conjugates can be seen with co localization of the cells in yellow. The duration of the imaging is 30 min in real-time, which is compressed to 6 sec in this video.
PD-L1 blockade induces stable T cell-DC conjugates and breaks tolerance. A magnified time-lapse sequence illustrating stable and long lasting interactions of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) with DC (yellow and green) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP recipient and anti-PD-L1 injection. Elapsed time is shown as hh:mm:ss:sss. Stable conjugates can be seen with co localization of the cells in yellow. The duration of the imaging is 30 min in real-time, which is compressed to 6 sec in this video.
Acknowledgments
The authors would like to thank N. Martenier for animal care, C. Allen, E. Finger, E. Peterson, R. Freidman, K. Hogquist, C. Penaranda and X. Zhou for scientific discussions, G. Szot and P. Koudria for islet transplantation, C. McArthur for cell sorting, and M. Jenkins for multi-photon imaging support. This research was supported by Lifescan, Inc., the National Institute of Health (NIH AI35297 to JAB), P30 DK63720 (for core support), the Juvenile Diabetes Research Foundation (10-2006-799 to BTF), the American Diabetes Association (7-09-JF-21 to BTF) and by start-up funds from the University of Minnesota Medical School (BTF).
Footnotes
Author contributions
B.T.F. and J.A.B designed and conceptualized the research project; B.T.F., K.E.P, T.O., and J.W. did the experiments; B.T.F., T.N.E., Q.T., and M.F.K. analyzed the data; B.T.F prepared the figures; B.T.F. and J.A.B. Interpreted the data and wrote the manuscript. M.A. provided anti-PD-L1 hybridoma.
References
- 1.Walunas TL, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–13. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 2.Luhder F, Chambers C, Allison JP, Benoist C, Mathis D. Pinpointing when T cell costimulatory receptor CTLA-4 must be engaged to dampen diabetogenic T cells. Proc Natl Acad Sci U S A. 2000;97:12204–9. doi: 10.1073/pnas.200348397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chikuma S, Imboden JB, Bluestone JA. Negative regulation of T cell receptor-lipid raft interaction by cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2003;197:129–35. doi: 10.1084/jem.20021646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and Its Ligands in Tolerance and Immunity. Annu Rev Immunol. 2008 doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tivol EA, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–7. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 6.Nishimura H, Minato N, Nakano T, Honjo T. Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int Immunol. 1998;10:1563–72. doi: 10.1093/intimm/10.10.1563. [DOI] [PubMed] [Google Scholar]
- 7.Yamazaki T, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. 2002;169:5538–45. doi: 10.4049/jimmunol.169.10.5538. [DOI] [PubMed] [Google Scholar]
- 8.Ishida M, et al. Differential expression of PD-L1 and PD-L2, ligands for an inhibitory receptor PD-1, in the cells of lymphohematopoietic tissues. Immunol Lett. 2002;84:57–62. doi: 10.1016/s0165-2478(02)00142-6. [DOI] [PubMed] [Google Scholar]
- 9.Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 10.Fife BT, et al. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J Exp Med. 2006;203:2737–47. doi: 10.1084/jem.20061577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keir ME, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–95. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ansari MJ, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med. 2003;198:63–9. doi: 10.1084/jem.20022125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–22. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chikuma S, Bluestone JA. CTLA-4 and tolerance: the biochemical point of view. Immunol Res. 2003;28:241–53. doi: 10.1385/IR:28:3:241. [DOI] [PubMed] [Google Scholar]
- 15.Parry RV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider H, Rudd CE. Tyrosine phosphatase SHP-2 binding to CTLA-4: absence of direct YVKM/YFIP motif recognition. Biochem Biophys Res Commun. 2000;269:279–83. doi: 10.1006/bbrc.2000.2234. [DOI] [PubMed] [Google Scholar]
- 17.Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–82. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- 18.Germain RN, Miller MJ, Dustin ML, Nussenzweig MC. Dynamic imaging of the immune system: progress, pitfalls and promise. Nat Rev Immunol. 2006;6:497–507. doi: 10.1038/nri1884. [DOI] [PubMed] [Google Scholar]
- 19.Bousso P, Robey EA. Dynamic behavior of T cells and thymocytes in lymphoid organs as revealed by two-photon microscopy. Immunity. 2004;21:349–55. doi: 10.1016/j.immuni.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 20.Hugues S, et al. Distinct T cell dynamics in lymph nodes during the induction of tolerance and immunity. Nat Immunol. 2004;5:1235–42. doi: 10.1038/ni1134. [DOI] [PubMed] [Google Scholar]
- 21.Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–9. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 22.Miller MJ, Wei SH, Parker I, Cahalan MD. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 2002;296:1869–73. doi: 10.1126/science.1070051. [DOI] [PubMed] [Google Scholar]
- 23.Shakhar G, et al. Stable T cell-dendritic cell interactions precede the development of both tolerance and immunity in vivo. Nat Immunol. 2005;6:707–14. doi: 10.1038/ni1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bajenoff M, et al. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 2006;25:989–1001. doi: 10.1016/j.immuni.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Negulescu PA, Krasieva TB, Khan A, Kerschbaum HH, Cahalan MD. Polarity of T cell shape, motility, and sensitivity to antigen. Immunity. 1996;4:421–30. doi: 10.1016/s1074-7613(00)80409-4. [DOI] [PubMed] [Google Scholar]
- 26.Dustin ML, Bromley SK, Kan Z, Peterson DA, Unanue ER. Antigen receptor engagement delivers a stop signal to migrating T lymphocytes. Proc Natl Acad Sci U S A. 1997;94:3909–13. doi: 10.1073/pnas.94.8.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hurez V, et al. Restricted clonal expression of IL-2 by naive T cells reflects differential dynamic interactions with dendritic cells. J Exp Med. 2003;198:123–32. doi: 10.1084/jem.20022230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benvenuti F, et al. Dendritic cell maturation controls adhesion, synapse formation, and the duration of the interactions with naive T lymphocytes. J Immunol. 2004;172:292–301. doi: 10.4049/jimmunol.172.1.292. [DOI] [PubMed] [Google Scholar]
- 29.Scholer A, Hugues S, Boissonnas A, Fetler L, Amigorena S. Intercellular adhesion molecule-1-dependent stable interactions between T cells and dendritic cells determine CD8+ T cell memory. Immunity. 2008;28:258–70. doi: 10.1016/j.immuni.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 30.Judkowski V, et al. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J Immunol. 2001;166:908–17. doi: 10.4049/jimmunol.166.2.908. [DOI] [PubMed] [Google Scholar]
- 31.Tang Q, et al. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006;7:83–92. doi: 10.1038/ni1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindquist RL, et al. Visualizing dendritic cell networks in vivo. Nat Immunol. 2004;5:1243–50. doi: 10.1038/ni1139. [DOI] [PubMed] [Google Scholar]
- 33.Sumen C, Mempel TR, Mazo IB, von Andrian UH. Intravital microscopy: visualizing immunity in context. Immunity. 2004;21:315–29. doi: 10.1016/j.immuni.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 34.Cahalan MD, Parker I, Wei SH, Miller MJ. Two-photon tissue imaging: seeing the immune system in a fresh light. Nat Rev Immunol. 2002;2:872–80. doi: 10.1038/nri935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishimura H, et al. Developmentally regulated expression of the PD-1 protein on the surface of double-negative (CD4-CD8-) thymocytes. Int Immunol. 1996;8:773–80. doi: 10.1093/intimm/8.5.773. [DOI] [PubMed] [Google Scholar]
- 36.Macian F, et al. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–31. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 37.Li W, Whaley CD, Mondino A, Mueller DL. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+ T cells. Science. 1996;271:1272–6. doi: 10.1126/science.271.5253.1272. [DOI] [PubMed] [Google Scholar]
- 38.Morton AM, McManus B, Garside P, Mowat AM, Harnett MM. Inverse Rap1 and phospho-ERK expression discriminate the maintenance phase of tolerance and priming of antigen-specific CD4+ T cells in vitro and in vivo. J Immunol. 2007;179:8026–34. doi: 10.4049/jimmunol.179.12.8026. [DOI] [PubMed] [Google Scholar]
- 39.Breart B, Bousso P. Cellular orchestration of T cell priming in lymph nodes. Curr Opin Immunol. 2006;18:483–90. doi: 10.1016/j.coi.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 40.Miller MJ, Safrina O, Parker I, Cahalan MD. Imaging the single cell dynamics of CD4+ T cell activation by dendritic cells in lymph nodes. J Exp Med. 2004;200:847–56. doi: 10.1084/jem.20041236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Celli S, Lemaitre F, Bousso P. Real-time manipulation of T cell-dendritic cell interactions in vivo reveals the importance of prolonged contacts for CD4+ T cell activation. Immunity. 2007;27:625–34. doi: 10.1016/j.immuni.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 42.Zambricki E, et al. In vivo anergized T cells form altered immunological synapses in vitro. Am J Transplant. 2006;6:2572–9. doi: 10.1111/j.1600-6143.2006.01517.x. [DOI] [PubMed] [Google Scholar]
- 43.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–54. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 44.Schneider H, et al. Reversal of the TCR stop signal by CTLA-4. Science. 2006;313:1972–5. doi: 10.1126/science.1131078. [DOI] [PubMed] [Google Scholar]
- 45.Downey J, Smith A, Schneider H, Hogg N, Rudd CE. TCR/CD3 mediated stop-signal is decoupled in T-cells from Ctla4 deficient mice. Immunol Lett. 2008;115:70–2. doi: 10.1016/j.imlet.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hara M, et al. Transgenic mice with green fluorescent protein-labeled pancreatic beta - cells. Am J Physiol Endocrinol Metab. 2003;284:E177–83. doi: 10.1152/ajpendo.00321.2002. [DOI] [PubMed] [Google Scholar]
- 47.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 48.Hale MB, Nolan GP. Phospho-specific flow cytometry: intersection of immunology and biochemistry at the single-cell level. Curr Opin Mol Ther. 2006;8:215–24. [PubMed] [Google Scholar]
- 49.Lenschow DJ, et al. Inhibition of transplant rejection following treatment with anti-B7-2 and anti-B7-1 antibodies. Transplantation. 1995;60:1171–8. doi: 10.1097/00007890-199511270-00019. [DOI] [PubMed] [Google Scholar]
- 50.Szot GL, Koudria P, Bluestone JA. Transplantation of pancreatic islets into the kidney capsule of diabetic mice. J Vis Exp. 2007;404 doi: 10.3791/404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Activated antigen specific T cells stop in antigen-draining PLN. A time-lapse sequence of 40 μm z-projection images shows the dynamics of in vitro activated BDC2.5 CD4+ T cells (labeled red using CMTMR) in a PLN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient mouse. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
Dynamic movement of tolerized T cells in islet antigen-draining PLN. A time-lapse sequence of 40 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and isotype antibody injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 15 min in real-time, which is compressed to 3 sec in this video.
PD-L1 blockade causes tolerized T cells to stop in islet antigen-draining PLN. A time-lapse sequence of 36 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and anti-PD-L1 antibody injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 15 min in real-time, which is compressed to 3 sec in this video.
CTLA-4 blockade does not alter tolerized T cell movement in islet antigen-draining PLN. A time-lapse sequence of 44 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and anti-CTLA-4 antibody injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 15 min in real-time, which is compressed to 3 sec in this video.
Dynamic movement of activated antigen specific T cells in islet antigen-deficient ILN. A time-lapse sequence of 42 μm z-projection images shows the dynamics of in vitro activated BDC2.5 CD4+ T cells (labeled red using CMTMR) in a inguinal LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
Tolerized T cells move freely in islet antigen-deficient ILN. A time-lapse sequence of 44 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a inguinal LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and isotype control antibody injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
PD-L1 blockade does not alter tolerized T cell movement in islet antigen-deficient ILN. A time-lapse sequence of 46 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a inguinal LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and anti-PD-L1 injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
CTLA-4 blockade does not alter tolerized T cell movement in islet antigen-deficient ILN. A time-lapse sequence of 40 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a inguinal LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and anti-CTLA-4 injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
PD-L1 blockade causes tolerized T cells, but not naïve polyclonal T cells to stop in islet antigen-draining PLN. A time-lapse sequence of 28 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a pancreatic LN explant 18h after transfer to a NOD.CD4-eGFP (green naïve T cells) recipient and anti-PD-L1 antibody injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
Dynamic movement of tolerized T cells in pancreatic islet tissue. A time-lapse sequence of 46 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in pancreatic islet grafts of NOD.SCID transplant recipients receiving NOD.MIP-GFP islets (green β-cells) and isotype control antibody injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
PD-L1 blockade causes tolerized T cells to stop in pancreatic islet tissue. A time-lapse sequence of 42 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in pancreatic islet grafts of NOD.SCID transplant recipients receiving NOD.MIP-GFP islets (green β-cells) and anti-PD-L1 injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
CTLA-4 blockade does not alter tolerized T cell movement in pancreatic islet tissue. A time-lapse sequence of 44 μm z-projection images shows the dynamics of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in pancreatic islet grafts of NOD.SCID transplant recipients receiving NOD.MIP-GFP islets (green β-cells) and anti-CTLA-4 injection. Elapsed time is shown as hh:mm:ss:sss. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
Investigation of T cell-DC conjugates in pancreatic LN. A time-lapse sequence of 34 μm 3D projection shows the dynamic movement of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP (yellow and green DC) recipient and isotype control antibody injection. Elapsed time is shown as hh:mm:ss:sss. Randomly selected cells were tracked and the trailing line following the cells represents the cell path over time. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
PD-L1 blockade induces stable T cell-DC conjugates in pancreatic LN. A time-lapse sequence of 36 μm 3D projection shows the stable interactions of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) with DC (yellow and green) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP recipient and anti-PD-L1 injection. Elapsed time is shown as hh:mm:ss:sss. Randomly selected cells were tracked and the trailing line following the cells represents the cell path over time. The duration of the imaging is 10 min in real-time, which is compressed to 2 sec in this video.
Dynamic T cell-DC interactions following T cell tolerance. A magnified time-lapse sequence illustrating the dynamic and short interactions of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) with DC (yellow and green) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP recipient and isotype antibody injection. Elapsed time is shown as hh:mm:ss:sss. Brief conjugates can be seen with co localization of the cells in yellow. The duration of the imaging is 30 min in real-time, which is compressed to 6 sec in this video.
PD-L1 blockade induces stable T cell-DC conjugates and breaks tolerance. A magnified time-lapse sequence illustrating stable and long lasting interactions of in vivo tolerized BDC2.5 CD4+ T cells (labeled red using CMTMR) with DC (yellow and green) in a pancreatic LN explant 18h after transfer to a NOD.CD11c-eYFP recipient and anti-PD-L1 injection. Elapsed time is shown as hh:mm:ss:sss. Stable conjugates can be seen with co localization of the cells in yellow. The duration of the imaging is 30 min in real-time, which is compressed to 6 sec in this video.