

Abstract
Motivation: Functional module detection within protein interaction networks is a challenging problem due to the sparsity of data and presence of errors. Computational techniques for this task range from purely graph theoretical approaches involving single networks to alignment of multiple networks from several species. Current network alignment methods all rely on protein sequence similarity to map proteins across species.
Results: Here we carry out network alignment using a protein functional similarity measure. We show that using functional similarity to map proteins across species improves network alignment in terms of functional coherence and overlap with experimentally verified protein complexes. Moreover, the results from functional similarity-based network alignment display little overlap (<15%) with sequence similarity-based alignment. Our combined approach integrating sequence and function-based network alignment alongside graph clustering properties offers a 200% increase in coverage of experimental datasets and comparable accuracy to current network alignment methods.
Availability: Program binaries and source code is freely available at http://www.stats.ox.ac.uk/research/bioinfo/resources
Contact: ali@stats.ox.ac.uk
Supplementary Information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The availability of large-scale protein interaction datasets has made it possible to carry out high level graph theoretic analysis of the interactomes of several species [see Bork et al. (2004) for a review]. Many studies have searched for higher level organization and modularity within the network by breaking it up into relatively independent modules, which may correspond to biologically relevant complexes (Han et al., 2004; Kashtan and Alon, 2005; Spirin et al., 2003). Several algorithms have been designed in recent years which use interaction information alone to identify functional modules and complexes. Most of these algorithms identify dense regions of high connectivity with relatively low connectivity to the rest of network (Bader and Hogue, 2003; Krogan et al., 2006). These dense sub-graphs are treated as potential functional modules. For instance, MCODE (Bader and Hogue, 2003) first identifies putative complexes using local network density and then filters away those which do not contain sub-graphs with minimal degree two. Another graph-based method, MCL (Dongen et al., 2000), simulates a large number of quenched random walks of varying length from each node of the graph using an expansion step and combines this with an inflation step to partition the graph into subsets that do not have paths between them.
Several methods have also been proposed for identifying functional modules by simultaneous analysis of the network and RNA expression data. Ideker et al. (2002) introduced a framework for identification of active sub-networks, that is, connected regions of the network that show significant changes in expression over a particular subset of the conditions. Segal et al. (2003) provided a probabilistic formulation, in which a module is a group of genes with high pair-wise similarities of expression profiles and with a significant fraction of possible interactions. Taking the combined approaches even further, Tanay et al. (2004) described an integrative framework allowing the integration of protein interaction data with gene expression, phenotypic sensitivity and transcription factor (TF) binding, using the SAMBA bi-clustering algorithm (Tanay et al., 2002).
As suggested by increasing evidence, protein interaction modules that are conserved across species may exist. Proteins in the same pathway have been found to be present or absent in a genome as a group (Kelly et al., 2003; Pellegrini et al., 1999), and many protein interactions in the yeast network have also been identified for the corresponding protein orthologs in worm (Matthews et al., 2001). These discoveries have led to research directed at identifying complexes and functional modules through network alignment, analogous to traditional sequence alignment (Dandekar et al., 1999; Kelly et al., 2004; Ogata et al., 2000). Given two or more networks, the aim of network alignment algorithms is to identify modules that are conserved across the networks. The premise is that patterns of interactions which are conserved across species have biological significance and hence are more likely to correspond to real protein complexes or functional modules. Most alignment algorithms first define an alignment graph where each node represents a set of orthologous proteins. The edges in the alignment graph represent conserved interactions. A search is then carried out over this alignment graph for high scoring sub-graphs.
One of the earliest network alignment algorithms, NetworkBlast (Sharan and Ideker, 2006) defines the network alignment graph by identifying sequence similar proteins from several species and carries out a search over this graph for dense clusters of interactions. NetworkBlast has been used to perform three-way comparisons of yeast, worm and fly which yielded conserved modules displaying good overlap with MIPS (Mewes et al., 2004) complexes. Graemlin (Flannick et al., 2006) use progressive pair-wise alignments to compare multiple networks. Graemlin's probabilistic formulation of the topology-matching problem eliminates restrictions on the possible architecture of conserved modules such as those imposed by NetworkBlast. However, it requires parameter learning through a training set of known alignments. The sensitivity of the method was assessed by counting the number of KEGG (Kanehisa et al., 2000) pathways in the alignments. The KEGG coverage of the alignment results was between 21 and 39%. In terms of speed, it far outperforms NetworkBlast with a running time approximately linear to the number of networks. Other alignment algorithms have tried to take into account the evolutionary forces shaping the interaction networks. For example, MaWISH (Koyuturk et al., 2006), which implements a duplication divergence model to carry out pair-wise network alignment. In one test, the yeast and human interaction networks were aligned using MaWish, identifying 151 modules. The identified modules were compared to MIPS complexes of size 3–25, and the reported MIPS coverage was 20%. More recently an evolutionary-based multiple network alignment algorithm CAPPI (Dutkowski and Tiuryn, 2007) was developed which tries to reconstruct the ancestral network for the input species and maps it back onto the extant networks to identify common modules. In a comparison with NetworkBlast, CAPPI identified a lower number of conserved modules when aligning the yeast, worm and fly networks but the results were more functionally pure. Graemlin 2.0 (Flannick et al., 2008) is also a multiple network aligner, with a scoring function that can use evolutionary events. Some other network alignment methods proposed recently include IsoRank (Singh et al., 2008) and IsoRankN (Liao et al., 2009), GNA and PATH (Zaslavskiy et al., 2009) and DOMAIN (Guo and Hartemink, 2009). DOMAIN is the first algorithm to introduce protein domains into the network alignment problem and uses a novel direct-edge-alignment paradigm to directly detect equivalent interaction pairs across species. IsoRankN is a global multiple network alignment based on spectral clustering on the induced graph of pairwise alignment scores. GNA formulates alignment as two different graph matching problems depending on whether strict constraints on protein matches based on sequence similarity are given, or whether an optimal compromise between sequence similarity and interaction conservation in the alignment is desired. It should be noted that global network alignment methods such as IsoRank and GNA do not directly address the conserved module detection problem.
A disadvantage of network alignment is that despite its success in identification of conserved modules in multiple species, it offers limited coverage compared to graph clustering methods. It is also highly dependent on the graph topology for correct results, thus error rates pose a special challenge. This is a critical issue due to the unusually high percentage of false positive and false negative interactions in current networks. Recent estimates have put these numbers as high as 70 and 90%, respectively (Hart et al., 2006; Saeed and Deane, 2008). A common theme in all previous studies of protein interaction network alignment has been the use of protein sequence similarity to map orthologous proteins across different species. However, this does not necessarily provide a complete picture of orthologous relationships in the context of interaction networks. When aligning networks from species that are very distant in evolutionary terms, the proteins may not display enough sequence similarity to achieve a reasonable degree of mapping. This would result in a severely restricted alignment graph that may miss biologically conserved regions in the networks. Here, we explore the possibility of using a different measure of protein similarity. Since the goal of alignment is to extract modules that correspond to specific biological processes, we examined the use of functional similarity of proteins across networks to aid alignment. We present a novel functional similarity-based measure to carry out network alignment that increases the number of conserved interactions found by more than 30%. The modules found using our measure display 15% higher functional coherence on average compared to sequence-based alignment. Module detection was carried out purely through alignment of functionally similar proteins across species. Specifically, functional similarity of proteins within a species was not used to guide module detection. We also go on to investigate the benefits of combining network alignment with clustering techniques to identify larger modules. The combined method improves the coverage of experimentally verified complex datasets by nearly 200% compared to either sequence or function-based network alignment alone.
Finally, we present a novel representation that attempts to perform simultaneous clustering of multiple networks constrained by the similarity links between them. This method accounts for the errors in interaction data by completely relaxing the restrictions on the module topology and can identify conserved and non-conserved modules at the same time.
2 METHODS
2.1 Functional similarity score
To be useful for network alignment, a subjective concept like functional similarity must be expressed in a quantitative form that reflects the closeness in the biological functions of the proteins being compared. Functional annotation of proteins is an ongoing scientific activity and one of the most widely used resources is Gene Ontology (GO; Ashburner et al., 2000). GO offers substantial coverage of major protein databases and provides a species-specific, structured set of terms describing gene products. We devised a simple measure of functional similarity which is based on the most specific and hence most informative GO annotation of each protein. For simplicity we focus here only on the Biological Process category of GO, the method being identical for the other top categories of Molecular Function and Cellular Component.
Let there be a total of N proteins in the dataset under consideration and the GO functional annotation of each protein be defined as a set of terms SA. We define a multi-set of size n as a pair (S, σ) where σ : S→ℕ, with the conditions:
Here, σ is a function that maps a GO term to the number of times it occurs in the dataset. Terms having fewer proteins annotated to them occur less frequently in the dataset and are thus classified as more specific. For any two proteins A and B with annotation sets SA and SB, the functional similarity score (funsim) is then calculated as follows:
| (1) |
The above scoring scheme assigns higher functional similarity to protein pairs that share more specific GO annotations. It should be noted that other, more sophisticated scoring schemes for functional similarity based on GO are possible. Several measures of functional similarity have been proposed in recent years making use of the information content of GO terms as well as the semantics ( ‘is a’, ‘part of’) of the GO relationships (Pandey et al., 2008; Resnik et al., 1995; Schlicker et al., 2006). We compared our score to the one proposed by Pandey et al. and found the lists of functionally similar proteins generated in both cases to be in good agreement. We carried out pair-wise alignments of the human and yeast as well as human and fly interaction networks using our score. The results were compared to sequence-based alignment using several existing methods.
2.2 Combining function and sequence
Analysis of the results from function and sequence-based alignment led us to the development of an extended algorithm. This method combines high quality sequence and function-based alignment results with common clustering measures to identify larger modules in a network. A seed set of edges is first identified from the weighted interaction network, where the weights are based on the degree of conservation and other complementary measures. Modules are then expanded from this set in a greedy fashion as described later.
2.2.1 Alignment-based edge score
Intuitively, an interaction that is conserved across many species should have a high score. We align the query network with each of the other input networks separately using the Match-and-Split (MAS) algorithm (Narayanan et al., 2007). For each edge in the query network a track is kept of the number of alignments, x, in which it was found to be conserved. The alignment-based score is found through a logistic scoring function,
| (2) |
This choice is motivated by the initial exponential growth of the function followed by saturation. This models the requirement that the score increases rapidly initially as an edge is found to be conserved in multiple species and then should slowly approach a limiting value with increasing evidence of conservation. Here, the parameters k, c and t can be adjusted to set an upper limit on the score as well as the saturation point. In our implementation these have been set to 1, 20 and 2, respectively. This choice limits the alignment score to a maximum of 1, like the graph and co-expression-based scores, and also ensures that the score saturates when an edge is found to be conserved in two species (as we carried out our tests on pair-wise alignments).
We align the networks using both sequence and our functional similarity measures. Each edge will therefore have two alignment scores assigned to it, ASseq (for sequence-based alignment) and ASfunc (for function-based alignment).
2.2.2 Graph-based edge score
The graph-based score is constructed from two commonly used graph statistics. The clustering coefficient is a local network measure of how close a vertex and its neighbors are to being a clique. Consider a selected node i in a network, having ki neighbors. The value of the clustering coefficient of the node i is given by the ratio between the number of edges Ei that actually exist between these ki nodes and the total number ki(ki−1)/2 of such edges that could exist in the neighborhood of i
| (3) |
As the clustering coefficient is a node-based score and our algorithm is based on edge scoring, we assigned to each edge the average clustering coefficient of its endpoints, so that an edge which links two nodes with high clustering coefficients is more likely to be within a dense cluster.
The betweenness (B) of an edge is defined as the number of shortest paths between pairs of nodes that run along it (Girvan and Newman, 2002). If a network contains groups or communities that are only loosely connected by a few inter-group edges then all the shortest paths between different communities must go along one of these edges. Thus, the edges connecting communities will have high betweenness while edges inside the clusters would tend to have lower betweenness.
To favor edges that have both a high clustering coefficient and low betweenness, we use the product of normalized edge betweenness values (NB) and edge clustering coefficients to calculate the graph-based edge score:
| (4) |
2.2.3 Co-expression-based edge score
Co-expression by itself is not necessarily an effective measure of co-membership in a module, though it is still a useful indicator of biological coordination between proteins. Combined with the other measures presented above, it may contribute to better module detection. Co-expression data for human proteins was obtained from Obayashi et al. (2008). We use the Pearson correlation coefficient values (ranging from −1 to 1) as the Co-expression Score (CS). Where no co-expression data is available, a CS of 0 is assigned.
2.2.4 Combined edge score and module expansion
The four different scores for each edge in the network are finally integrated through a weighted linear combination:
| (5) |
The weights (α, β, γ, δ) can be adjusted to assign relative importance to the different techniques, depending upon the confidence level attributed to them and the type of results sought. In our implementation we assigned the set of weights (α = 9, β = 7, γ = 2, δ = 1) based on regression tests. The weights were learned through edges extracted from a set of 100 randomly selected experimentally verified complexes (details in Supplementary Material). This set of complexes was subsequently removed from the validation set. To test the robustness of the combined score, weights were inferred from the human-yeast analysis and the same set of values was then used for all subsequent analyses, including human–fly, fly–yeast and human–worm comparisons (In the Supplementary Material, we also determined optimized weights for yeast–fly analysis to test how the weights differ for different comparisons). We found that using weights optimized for one species can be used for the analysis of other species with little effect on the results. This can of course be better tested as data for more species becomes available. After edge-weighting, modules are extracted from the network using the highest-scoring edges (Algorithm 1). First, a seed set is selected consisting of edges with scores ≥3μ , μ being the average edge score of the network. These seeds are then expanded stepwise into modules. At each step, the highest scoring neighbor of a seed is added to the module if it does not decrease the average score, S, of the module (averaged over all edge scores in the module) by more than a user defined threshold, t. We carried out all our tests with t set to 0.75. The algorithm terminates when no more edges can be added. At this point, the input network is divided into a set of modules which potentially correspond to real biological complexes.
2.3 Simultaneous clustering
An inherent issue with alignment-based methods is low coverage due to emphasis on only conserved modules along with sensitivity to errors in network topologies. To tackle these problems we introduce the concept of simultaneous clustering. A basic implementation of this concept takes as input multiple interaction networks along with the similarity relationships between proteins from different species. A ‘global’ graph is then built with all the nodes present in the input networks and two types of links: inter- and intra-species edges. We stress here that the global graph is different from the alignment graph used in network alignment algorithms. In particular, the alignment graph is a product of the input species networks where each node is a merged representation of orthology relationships. In contrast, the global graph does not involve merging of nodes and edges and all orthologs are represented individually. The alignment task can then be reduced to a clustering of this global graph based on edge density. Proteins in a species that are highly connected to each other as well as to a highly connected group of proteins in another species through similarity links would be clustered together and identified as meta-clusters (Fig. 1). These meta-clusters represent putative conserved modules.
Fig. 1.
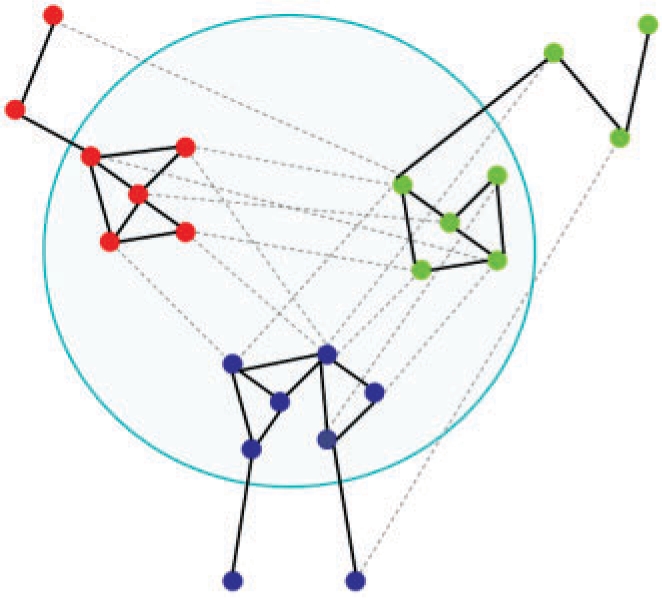
A meta-cluster within the global graph composed of networks of multiple species. The meta-clusters are characterized by relatively higher number of intra-species links (protein interactions, bold lines) as well as high number of inter-species links (orthology relationships, dotted lines).

The crucial difference to network alignment is that to be part of the same meta-cluster, clusters from different species need not be very similar in edge topology: they only need to be well-connected within as well as with each other. Moreover, unlike network alignment, module detection using this technique is not limited to only conserved regions. Dense regions in the interaction network of one species that do not have sufficient similarity links to any other species will still be clustered into modules. In this case the meta-cluster would only contain proteins from that particular species. For the clustering process, any of the myriad of already available algorithms can be used, provided they can deal with the presence of two different types of links in the global graph. We used an implementation of the popular clustering algorithm, MCL to carry out our tests, where the inter- and intra-species links are differentiated by their weights.
2.4 Data sources
The species selected for analysis were Homo Sapiens (human), Saccharomyces Cerevisiae (yeast), Drosophila Melanogaster (fly) and Caenorhabdidits Elegans (worm). Interaction data for yeast (4941 proteins, 17 387 interactions), fly (6701 proteins, 20 092 interactions) and worm (2328 proteins, 3495 interactions) was downloaded from the Database of Interacting Proteins (DIP; Xenarios et al., 2002), while data for human (9305 proteins, 35 458 interactions) was taken from the Human Protein Reference Database (HPRD; Prasad et al., 2008).
2.5 Alignment algorithm
We tested our new similarity measure using the MAS network alignment algorithm. Instead of creating an alignment graph, MAS uses a recursive process that alternately identifies locally matching nodes across two networks and then splits the matching sub-graphs into connected components. Nodes are deemed locally matching if they share sequence similarity as well as network neighborhood. In the case of multiple orthologs for a node, the orthologs are aligned independently of each other. They can therefore be part of different sub-graphs in the alignment. MAS is relatively fast and uses a flexible node similarity component that uses BLAST (Altschul et al., 1990) E-values in the original implementation. We modified it to use our functional similarity scores. A cutoff score of 0.9 was used to select highly similar proteins. We found that this particular choice of cutoff identifies functional matches for a significant proportion of proteins across two species whilst keeping multiple hits within reasonable limits.
2.6 Testing criteria
The modules recovered were analyzed in terms of their functional coherence and compared to experimentally determined complexes to assess their quality. We define the functional coherence of a module M, as the average functional similarity of all possible protein pairs (i, j) in the module. Pairs for which functional similarity could not be calculated due to lack of GO annotation for one or both proteins were not included.
| (6) |
To check whether the extracted modules corresponded to real complexes we compared them to high quality datasets containing experimentally identified complexes. Experimentally determined complexes for yeast and human were downloaded from MIPS CYGD (Guldener et al., 2005) and MIPS CORUM (Ruepp et al., 2008) databases, respectively. For each MIPS complex (A), we identify its best matching complex in the solutions (M) as the one having the greatest value of the following comparison score:
| (7) |
We compared the performance of function and sequence-based network alignment by aligning the human network with yeast and fly networks. Function-based alignment was carried out using MAS only, while sequence-based alignment was done using MAS, MaWISH and NetworkBlast. For sequence-based alignment, the orthology file was generated by selecting pairs of proteins with BLAST E-values ≤1e−7 following the cut-off used by Sharan et al. (2005). The combined method was tested by using alignment results from MAS (both sequence and function based) as input. To observe the effect of multiple networks combined method was executed using sets of two (human–yeast, human–fly, fly–yeast and human–worm) as well as three (human–yeast–fly) networks.
In addition to comparisons with alignment-based methods, we also compared our methods to the more commonly used clustering of single networks. This was done by weighting the edges using GO functional similarity of interacting proteins, co-expression and graph properties and then using our combined method for module detection. This method (GO clustering) is in contrast to our alignment-based combined method, which makes use of interaction conservation.
3 RESULTS
Here we discuss results for the human network aligned with yeast. Detailed results including the human–fly and other comparisons can be found in the Supplementary Material (Results in the main text and Supplementary Material follow the same pattern. Any significant differences are pointed out in the detailed results below).
Function-based alignment using our similarity score was successful in uncovering a larger number of proteins participating in conserved interactions than sequence-based alignment. As shown in Table 1, the number of conserved modules discovered in the human network increased from 74 (spanning 457 unique proteins) to 94 (spanning 727 unique proteins). Moreover, the two sets share only 58 proteins (<15%), indicating that the modules targeted by the two methods are nearly disjoint. We successfully exploit this observation in our combined method.
Table 1.
Summary of results from all methods used (human network)
| Sequence-based alignment | Function-based alignment | Combined method | MaWISH | Network blast | Combined method 3-way | Simultaneous clustering | GO clustering | |
|---|---|---|---|---|---|---|---|---|
| Number of clusters | 74 | 94 | 303 | 242 | 2353 | 430 | 1197 | 1093 |
| Number of proteins | 457 | 727 | 1479 | 543 | 894 | 1603 | 7371 | 6663 |
| MIPS coverage | 96 | 126 | 283 | 83 | 153 | 327 | 424 | 346 |
| MIPS accuracy | 0.18 | 0.24 | 0.21 | 0.1 | 0.08 | 0.17 | 0.05 | 0.03 |
| Functional coherence | 0.36 | 0.51 | 0.43 | 0.32 | 0.3 | 0.40 | 0.23 | 0.29 |
| Run time (s) | 3061 | 5432 | 21 | 663 | 68 977 | 29 | 369 | 112 |
In addition to greater coverage, modules identified using function-based alignment displayed higher biological coherence than sequence-based alignment using any of the other methods (Fig. 2a). Almost 50% of the modules identified by our technique scored 0.5 or more compared to only 30% when using sequence-based MAS; MaWISH and NetworkBLAST both performed far worse. As illustrated in Figure 2b, function-based alignment also identifies modules that correlate better with experimentally verified complexes. Around one–third of the modules correspond well (overlap >50%) with a MIPS complex as opposed to only one–tenth of the modules from MaWISH and NetworkBlast.
Fig. 2.

Comparison of methods on extracted from the Human network: cumulative frequency distribution of (a) functional homogeneity and (b) MIPS comparison scores. (Results for the simultaneous clustering method not included.) Plots shifted towards right (higher values of functional homogeneity and MIPS score) indicate better results.
Using the combined method, in the human–yeast case a total of 303 modules (1479 proteins) were identified, far higher than either sequence or function-based alignment alone. This increased overage does not affect the module quality as both the functional coherence and overlap with MIPS complexes is still better than sequence-based alignment methods. An even greater number of modules are found when the fly network is also added for a three-way analysis (Table 1, combined method three-way), accompanied by an increase in MIPS coverage (Fig. 3).Figure 3 plots the coverage and accuracy of the various methods in terms of the total number of MIPS interactions captured by the identified modules. The simultaneous clustering approach exhibits the highest coverage although its accuracy is relatively low. This is probably because this method clusters the entire network, instead of identifying only the conserved regions. This would extract many modules that are not yet present in MIPS and thus drive down the accuracy of the method. As illustrated in Figure 3, our combined method offers a superior coverage-accuracy trade-off amongst all techniques. The plots for yeast (against human) show higher values (Supplementary Fig. 2), while those for human (against fly) show lower values (Supplementary Fig. 3), though the combined method performs best in all cases. The coverage of MIPS interactions using this approach is better than any of the other alignment-based methods, accompanied by a higher accuracy than MaWISH and NetworkBlast. Finally, GO clustering of single networks performs worse than all methods based on multiple networks. Specifically, this method suffers a 5-fold drop in accuracy compared to our combined method (with three networks) for a marginal increase in coverage. Upon detailed inspection of the results we found that while some of the modules extracted using this technique are highly functionally homogeneous (by construction), this comes at the cost of a substantial number of spurious clusters. These results also support the view that module detection based on evidence from multiple species leads to more reliable, though fewer results.
Fig. 3.

Coverage of interactions in MIPS plotted against accuracy for each method (Human network). The combined method clearly offers the best coverage-accuracy tradeoff.
Table 1 also shows the execution time for each of the methods on a machine with a 1.66 GHz processor with 1 GB RAM. Further analysis of other species (Supplementary Material) indicates that the run times of the alignment algorithms are extremely sensitive to the number of orthologs between two species. The simultaneous clustering approach is an exception to this, as it does not experience an explosion in the number of possible alignments due to multiple possible orthologs, experienced by MAS, NetworkBlast and MaWISH.
3.1 An example: the human DAB complex
The human DAB (Moldanodo et al., 1990) is a multi-protein complex involved in transcription initiation and consists of the TFIID complex, TFIIA complex and TFIIB. It is present in the MIPS database as complex ID 493 which lists 16 proteins as its subunits, 15 of which are transcription initiation factors and 1 is a TATA-box binding protein. It is a typical target of module detection methods both in terms of the number of proteins involved as well as the tight functional relationships between them. Figure 4 shows the performance of our combined approach along with NetworkBlast and MaWish in terms of their ability to correctly identify this complex in the human network.
Fig. 4.

Identification of the human DAB complex (MIPS ID 493) using MaWISH, NetworkBlast and the Combined method. Circular nodes in the figure represent components of the DAB complex while other nodes represent proteins that were mistakenly classified as part of the same complex by any of the methods.
It must be noted that the human network from HPRD on which all module detection methods were tested was missing four out of the 16 protein subunits of the DAB complex. We have found this to be a widespread problem with over 30% of MIPS proteins missing from the HPRD network. Circular nodes in the figure represent components of the DAB complex while other nodes represent proteins that were mistakenly classified as part of the same complex by any of the methods. MaWish and NetworkBlast which use only sequence similarity to carry out network alignment manage to capture <50% of the complex. Both these methods capture almost the same set of proteins while missing the rest. This probably indicates an inherent weakness of using just sequence information, rather than the alignment algorithms themselves. Note that the combined method which uses sequence and functional similarity with additional module expansion correctly identifies almost the entire complex except two proteins, one of which is not identified as part of the complex by any of the methods.
The above example is just one of several cases in which the combined method improves the coverage of real complexes demonstrating the ability of functional mapping to capture conserved interactions independently of sequence similarity. In addition to a comparison of alignment-based methods, this example also demonstrates the benefits of cross-species analysis. When we carried out GO clustering of the single human network, the DAB complex was detected as part of a much larger cluster consisting of 23 proteins, of which only nine are present in the real complex (results not shown in Fig. 4). Similarly, analysis of the human Rap1 complex (MIPS complex ID 1204) using the combined method detects all six component proteins, while GO clustering misclassifies three of the components as part of a different cluster. The use of interaction evidence from multiple species can therefore not only identify modules at a higher resolution, but can also detect components missed by single-network clustering.
4 DISCUSSION
At present, all protein network alignment studies use sequence similar proteins across species to aid the network comparison process. Here, we have for the first time used a quantitative measure of functional similarity to align protein interaction networks. Our results indicate that modules found by alignment using functional similarity exhibit higher functional coherence compared to sequence similarity-based alignment. This is encouraging because functionally coherent modules are more likely to be biologically relevant. These observations were further confirmed by the comparison of identified modules to experimentally determined complexes in the MIPS database. Again, the modules found using our functional similarity score displayed higher levels of overlap with real complexes. Given that <15% of interactions were common in the modules from the functional similarity and sequence similarity-based alignments, a question that arises naturally is whether using both techniques simultaneously can increase the power of computational complex detection.
Our combined method that uses network alignment based on both function and sequence similarity led to several improvements in the module detection results. First, the combined approach produced better results in terms of agreement with experimental datasets. The coverage of MIPS was more than twice that of using sequence-based alignment alone. In terms of the functional coherence of the detected modules, the combined method performs far better than sequence-based alignment. Adding simple clustering measures from graph theoretic methods and gene co-expression information improves the results further by increasing the size of the solution set. While these two measures alone are not powerful enough to produce high quality results, they can be used to expand the solution sets of alignment-based methods and thus increase their coverage. Finally, the weighted combination of different techniques in our method provides a natural way of optimizing the results for a particular measure of goodness. Modules with high functional coherence can be produced by assigning a relatively high weight to the functional similarity-based alignment component (see Supplementary Fig. 5), while higher weights for the graph-based component will identify larger modules.
Results for our simultaneous clustering-based alignment method are less conclusive. The coverage of MIPS is naturally much higher in this case, though the modules are not of comparable quality to the other approaches. This could be a consequence of the larger sample size, more likely to contain highly connected sub-graphs with no biological relevance. Furthermore, not all real modules are expected to be completely functionally homogeneous (Spirin et al., 2003). Still, several improvements in our method are possible with the potential to improve the results. We differentiated between inter- and intra-species links by assigning them different weights whereas they might need to be treated entirely differently, for instance as a bi-partite graph. Also, currently all networks in the global graph are treated at the same level. One way forward could be to take a more evolutionary realistic approach and assign relative ordering to the protein orthology links based on how evolutionary distant the respective species are.
In conclusion, we have demonstrated that using function as a metric for protein network alignment offers improved performance over traditional sequence-based network comparisons. The two measures manage to identify an almost disjoint set of conserved interactions which indicates that network alignment methods may benefit by exploiting still other ways of mapping similar proteins across species. We have also simultaneously clustered entire networks from several species using both protein similarity and interaction links as constraints. This method offers far greater coverage than any network alignment approach and fewer restrictions on module topology make it more suitable for error-prone data.
Funding: Teaching Assistant Bursary from the Department of Statistics, University of Oxford (to W.A.); University of Oxford Systems Biology Doctoral Training Centre (to C.D.).
Conflict of interest: none declared.
Supplementary Material
REFERENCES
- Altschul S, et al. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Ashburner M, et al. Gene Ontology tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader GD, Hogue WV. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P, et al. Protein interaction networks from yeast to human. Curr. Opin. Struct. Biol. 2004;14:292–299. doi: 10.1016/j.sbi.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Dandekar T, et al. Pathway alignment: application to the comparative analysis of glycolytic enzymes. Biochem. J. 1999;343:115–124. [PMC free article] [PubMed] [Google Scholar]
- Dongen SV. A cluster algorithm for graphs. Technical Report INS-R0010. 2000 [Google Scholar]
- Dutkowski J, Tiuryn J. Identification of functional modules from conserved ancestral protein–protein interactions. Bioinformatics. 2007;23:149–158. doi: 10.1093/bioinformatics/btm194. [DOI] [PubMed] [Google Scholar]
- Flannick J, et al. Graemlin General and robust alignment of multiple large interaction networks. Genome Res. 2006;16:1169–1181. doi: 10.1101/gr.5235706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannick J, et al. Automatic parameter learning for multiple network alignment. RECOMB. 2008;4955:214–231. doi: 10.1089/cmb.2009.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girvan M, Newman MEJ. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA. 2002;99:7821–7826. doi: 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güldener U, et al. CYGD the comprehensive yeast genome eatabase. Nucleic Acids Res. 2005;33:33. doi: 10.1093/nar/gki053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Hartemink JA. Domain-oriented edge-based alignment of protein interaction networks. Bioinformatics. 2009;25:i240–i246. doi: 10.1093/bioinformatics/btp202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JD, et al. Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature. 2004;430:88–93. doi: 10.1038/nature02555. [DOI] [PubMed] [Google Scholar]
- Hart GT, et al. How complete are current yeast and human protein-interaction networks? Genome Biol. 2006;7:120. doi: 10.1186/gb-2006-7-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, et al. Discovering regulatory and signaling circuits in molecular interaction networks. Bioinformatics. 2002;18:S233–S240. doi: 10.1093/bioinformatics/18.suppl_1.s233. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashtan N, Alon U. Spontaneous evolution of modularity and network motifs. Proc. Natl Acad. Sci. USA. 2005;102:13773–13778. doi: 10.1073/pnas.0503610102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley BP, et al. Conserved pathways within bacteria and yeast as revealed by global protein network alignment. Proc. Natl. Acad. Sci. USA. 2003;100:11394–1399. doi: 10.1073/pnas.1534710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley BP, et al. PathBLAST: a tool for alignment of protein interaction networks. Nucleic Acids Res. 2004;32(Suppl. 2):W83–W88. doi: 10.1093/nar/gkh411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyutürk M, et al. Pairwise alignment of protein interaction networks. J. Comput. Biol. 2006;13:182–199. doi: 10.1089/cmb.2006.13.182. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- Liao CS, et al. IsoRankN: spectral methods for global alignment of multiple protein networks. Bioinformatics. 2009;25:i253–i258. doi: 10.1093/bioinformatics/btp203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado E, et al. Factors involved in specific transcription by mammalian RNA polymerase II role of transcription factors IIA, IID, and IIB during formation of a transcription-competent complex. Mol. Cell Biol. 1990;12:6335–6347. doi: 10.1128/mcb.10.12.6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews LR, et al. Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or ‘interologs’. Genome Res. 2001;11:2120–2126. doi: 10.1101/gr.205301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewes HW, et al. MIPS: analysis and annotation of proteins from whole genomes. Nucleic Acids Res. 2004;32:D41–D44. doi: 10.1093/nar/gkh092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan M, et al. Comparing protein interaction networks via a graph match and split algorithm. J. Comput. Biol. 2007;14:892–907. doi: 10.1089/cmb.2007.0025. [DOI] [PubMed] [Google Scholar]
- Obayashi T, et al. COXPRESdb a database of coexpressed gene networks in mammals. Nucleic Acids Res. 2008;36:D77–D82. doi: 10.1093/nar/gkm840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, et al. A heuristic graph comparison algorithm and its application to detect functionally related enzyme clusters. Nucleic Acids Res. 2000;28:4021–4028. doi: 10.1093/nar/28.20.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini M, et al. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl. Acad. Sci. USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad SK, et al. Human protein reference database—2009 update. Nucleic Acids Res. 2008;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnik P, et al. Using information content to evaluate semantic similarity in a taxonomy. IJCAI. 1995;95:448–453. [Google Scholar]
- Ruepp A, et al. CORUM the comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 2008;36:D646–D650. doi: 10.1093/nar/gkm936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed R, Deane CM. An assessment of the uses of homologous interactions. Bioinformatics. 2008;24:689–695. doi: 10.1093/bioinformatics/btm576. [DOI] [PubMed] [Google Scholar]
- Schlicker A, et al. A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinformatics. 2006;7:302. doi: 10.1186/1471-2105-7-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, et al. Discovering molecular pathways from protein interaction and gene expression data. Bioinformatics. 2003;19:i264–i271. doi: 10.1093/bioinformatics/btg1037. [DOI] [PubMed] [Google Scholar]
- Sharan R, Ideker T. Modeling cellular machinery through biological network comparison. Nat. Biotechnol. 2006;24:427–433. doi: 10.1038/nbt1196. [DOI] [PubMed] [Google Scholar]
- Sharan R, et al. Conserved patterns of protein interaction in multiple species. Proc. Natl Acad. Sci. USA. 2005;102:1974–1979. doi: 10.1073/pnas.0409522102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, et al. Global alignment of multiple protein interaction networks with application to functional orthology detection. Proc. Natl Acad. Sci. USA. 2008;105:12763–12768. doi: 10.1073/pnas.0806627105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spirin V, Mirny LA. Protein complexes and functional modules in molecular networks. Proc. Natl Acad. Sci. USA. 2003;100:2123–12128. doi: 10.1073/pnas.2032324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, et al. Discovering statistically significant biclusters in gene expression data. Bioinformatics. 2002;18:S136–S144. doi: 10.1093/bioinformatics/18.suppl_1.s136. [DOI] [PubMed] [Google Scholar]
- Tanay A, et al. Revealing modularity and organization in the yeast molecular network by integrated analysis of highly heterogeneous genomewide data. Proc. Natl Acad. Sci. USA. 2004;101:2981–2986. doi: 10.1073/pnas.0308661100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xenarios I, et al. DIP the database of interacting proteins a research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002;30:303–305. doi: 10.1093/nar/30.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaslavskiy M, et al. Global alignment of protein–protein interaction networks by graph matching methods. Bioinformatics. 2009;25:i259–i1267. doi: 10.1093/bioinformatics/btp196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.