

Abstract
The explosion of new discoveries in the field of immunology has provided new insights into mechanisms that promote an immune response directed against a transplanted organ. Central to the allograft response are T lymphocytes. This review summarizes the current literature on allorecognition, costimulation, memory T cells, T cell migration, and their role in both acute and chronic graft destruction. An in depth understanding of the cellular mechanisms that result in both acute and chronic allograft rejection will provide new strategies and targeted therapeutics capable of inducing long-lasting, allograft-specific tolerance.
Keywords: Allograft, Children, Rejection, T lymphocytes, Transplantation
Learning objectives:
To review recent advances in understanding the mechanisms of allograft rejection
To outline the current data on allorecognition and its role in allograft rejection
To discuss current therapeutics targeting costimulatory pathways
To briefly discuss recent data on the role T regulatory and memory T cells play in alloimmune responses
Introduction
Transplantation of solid organs has emerged as a viable therapeutic modality for the treatment of a variety of ailments, such as end stage renal disease. Acute allograft rejection is understood as an impediment to long-term allograft survival, increasing the risk of developing chronic rejection and decreasing allograft half-life by 34% [1]. With the widespread use of potent immunosuppressive drugs, early graft loss due to acute rejection has decreased dramatically; however, current immunosuppressive protocols have not reduced the rates of graft loss due to chronic rejection and have increased the risk of serious complications, such as life-threatening infections and cancers [2].
Rejection of solid organ allografts is the result of a complex series of interactions involving coordination between both the innate and adaptive immune system with T cells central to this process. The ability of recipient T cells to recognize donor-derived antigens, called allorecognition, initiates allograft rejection. Once recipient T cells become activated, they undergo clonal expansion, differentiate into effector cells, and migrate into the graft where they promote tissue destruction. In addition, CD4 T cells help B cells produce alloantibodies. Here, we will review the components of an anti-allograft adaptive immune response.
Allorecognition
Antigens that activate the immune system against the allograft, i.e. alloantigens, are both major and minor histocompatibility antigens. The major histocompatibility complex (MHC), located on chromosome 6 in humans, encodes the human leukocyte antigens (HLA), which are polymorphic molecules responsible for eliciting the strongest of responses to allogeneic tissues. The genes in this region encode for class I (HLA-A, -B, -C) and class II (HLA-DR, -DP, -DQ) molecules. The function of MHC molecules is to present foreign antigens to T cells. It has been known for more than 30 years that the T cell receptor (TCR) present on the surface of the T cell interacts with a peptide bound in the groove of the MHC molecule present on the surface of the antigen presenting cell. CD8 T cells recognize peptide/MHC class I complexes. MHC class I molecules are constitutively expressed on the surface of virtually all nucleated cells. CD4 T cells recognize peptide/MHC class II complexes. MHC class II molecules are constitutively expressed on the surface of professional antigen presenting cells, but expression can be induced on many cell types with activation.
Minor histocompatibility antigens are proteins that are expressed in some individuals in the population but not others, thereby creating potential antigenic differences between donors and recipients. This occurs, for example, when proteins encoded on the Y chromosome (H-Y) from male grafts induce an anti-Y response in females [3]. In theory, a polymorphism of any protein between donor and recipient, as is the case for certain enzymes and surface receptors that can be processed and presented on self-MHC, can potentially elicit an anti-graft response. Any non-MHC gene that encodes epitopes capable of binding to both MHC class I and class II molecules and inducing both CD4 and CD8 T cell responses can be considered a minor histocompatibility gene. CD8 T cells [4, 5] and, more recently, CD4 T cells [6] specific for minor antigens have been isolated from humans and rodents and have been shown to play an important role in the rejection of solid organs and corneal transplants as well as causing graft-versus-host disease after bone marrow transplantation [3, 7].
Unique to transplant immunobiology is the idea that alloantigen recognition can occur via two distinct pathways, both of which focus on the source of the antigen presenting cells (donor versus recipient). The direct pathway of allorecognition describes the ability of T cells to “directly” recognize intact non-self MHC molecules present on the surface of donor cells (Fig. 1a). The indirect pathway of allorecognition describes the ability of T cells to recognize donor MHC molecules that are processed and presented as peptides by self-MHC molecules (Fig. 1b). The recognition of intact donor MHC molecule(s) elicits a potent anti-graft immune response while processed MHC peptides and minor histocompatibility antigens elicit a slower tempo, less intense immune response.
Fig. 1.
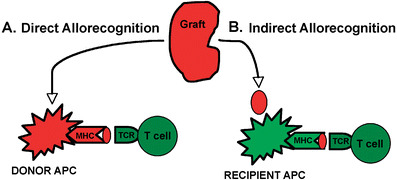
Two distinct pathways of allorecognition. a Direct pathway of allorecognition. Dendritic cells migrate from the graft to secondary lymphoid tissues to activate T cells. b Indirect pathway of allorecognition. Graft proteins are processed by recipient dendritic cells and presented to T cells. APC Antigen-presenting cell, TCR T cell receptor, MHC major histocompatibility complex
Direct pathway of allorecognition
T cells will respond vigorously when mixed in culture with MHC-disparate stimulator cells, i.e. the mixed lymphocyte reaction [8]. This in vitro response is thought to reflect the propensity for acute rejection [9], and the ability to detect an alloresponse with the mixed lymphocyte culture is generally believed to be due to the high precursor frequency of alloreactive T cells within the periphery. Mature, naïve T cells in circulation survive a selection process in the thymus that ensures that their TCR has a low but significant affinity for a self-peptide/MHC molecule but a high affinity for foreign peptides associated with self-MHC molecules [10, 11]. Recent studies have shown that the weak interaction between self-MHC and the TCR is required for the survival of naive T cells in the periphery [12]. The inherent affinity of the TCR on mature T cells for self peptide-MHC complexes probably explains the high frequency of T cells within any individual that cross react with high affinity to a closely related allo-MHC molecule [13, 14].
Direct recognition does not conform to the classic rules of self-MHC restriction. Mounting evidence suggests that the structural similarity between certain MHC molecules is ‘close enough’ to allow T cell receptor ligation and to trigger TCR signaling and subsequent activation [13, 15]. Alloreactive T cells are thought to recognize polymorphic residues on allogeneic MHC regardless of the peptide bound to it [16–18]. However, evidence also exists to support the notion that peptide binding facilitates a diverse T cell response [19, 20]. It is possible that, in the setting where the donor MHC is structurally very different from the recipient MHC, recognition may occur regardless of the peptide bound. Alternatively, if the donor MHC is structurally similar to the recipient MHC, recognition may occur through the peptide/MHC complex [21–23].
In order for recipient T cells to directly respond to intact allo-MHC molecules, cells from within the graft must migrate out of the graft [24] to make direct contact with recipient T cells within secondary lymphoid tissue. The first evidence that graft-derived cells participate in the alloimmune response was reported by Lafferty and colleagues [25] and was termed the passenger leukocyte theory. In their studies, the culture of thyroid cells prior to transplant prolonged graft survival, an effect that was thought to be due to the removal of the donor-derived passenger leukocytes from the graft [25–28]. Follow-up studies in rat renal allografts confirmed prolonged survival upon removal of passenger leukocytes but provided evidence that acute rejection could be induced with the injection of donor dendritic cells (DC) [29, 30].
Dendritic cells are professional antigen-presenting cells (APC) [31] that have been implicated as the passenger leukocyte responsible for inducing an acute anti-allograft response [32]. In their immature state, DC are abundant within peripheral tissues and organs where they are ideally positioned to capture antigens. Upon receiving inflammatory signals, such as interleukin (IL)-1β [33], tumor necrosis factor (TNF)-α [34], and CD40 [35–37], these cells undergo a maturation process and migrate via afferent lymphatics to the paracortex of lymph nodes [38, 39] where naïve and central memory T cells primarily reside [40, 41]. Dendritic cells, unlike macrophages and B cells, are potent stimulators of naïve T cells due in part to their high levels of class I and class II MHC and costimulatory molecules. Once activated, graft-specific T cells infiltrate the graft where they are capable of recognizing the alloantigens directly on the graft parenchyma. Over time, however, donor APC are depleted from the graft, and the response is predominated by recipient DC that migrate into the graft and continuously pick up antigens from the graft and present processed peptides to T cells through the indirect pathway [42, 43].
Indirect pathway of allorecognition
In contrast to the direct pathway, the indirect pathway of allorecognition describes recipient APC presenting foreign MHC molecules in the form of peptides associated with self-MHC molecules [29]. This is the means by which most exogenous antigens enter the immune system and are recognized by T cells. Therefore, all proteins in donor grafts that differ from the recipient are potential antigens capable of inducing an anti-graft response. Three mechanisms of antigen delivery can be postulated to occur via this allorecognition pathway. First, antigens from the graft can be shed into the circulation and engulfed by recipient DC that reside within secondary lymphoid tissue. Second, donor cells can migrate to secondary lymphoid tissue where they are engulfed by recipient DC. Third, recipient APC can migrate into the graft, pick up antigens, and then migrate to secondary lymphoid tissue.
Evidence to support the indirect pathway as a viable means by which rejection is initiated comes from studies in which peptides derived from donor MHC molecules have been eluted from the binding grooves of recipient MHC molecules [44]. Furthermore, in vitro detection of an indirect response has been found to correlate with clinical rejection episodes in solid organ recipients [45, 46]. In addition, Auchincloss and colleagues [42] showed that in a situation where APC from a skin graft were incapable of activating CD4 T cells, rejection involving recipient CD4 T cells still occurred. In these studies, it was presumed that recipient MHC class II+ cells activated the recipient CD4 T cells. Immature DC have the unique ability to produce not only peptide/MHC class II complexes from exogenous antigens, but also peptide/MHC class I complexes [44, 47–49]. It is therefore conceivable that both kinds of peptide/MHC complexes derived from donor antigens could be presented to both CD4 and CD8 recipient T cells via this pathway.
There are two distinct differences between the direct and indirect pathway that merit clarification: first, the precursor frequency for T cells activated through the indirect pathway is significantly lower; second, the effector arm of the immune response within the graft differs from the direct pathway. When the donor and recipient differ, for example, at the MHC class I level, cytotoxic CD8 T cells specific for donor peptides bound to recipient MHC class I molecules (indirect pathway) would be unable to kill parenchymal cells of the graft because the graft cells express donor and not recipient MHC class I molecules. Therefore, recipient APC would have to migrate into the graft and take up residence, or the graft would have to share MHC identity with the recipient [50]. T cells specific for donor peptides bound to recipient MHC molecules could damage the graft indirectly by producing cytokines that through a bystander effect would damage graft cells [51].
In the setting of MHC-identical transplantation, the expression of the same MHC molecules by the donor and recipient blurs the distinction between direct and indirect donor antigen presentation. The stimulus for rejection in this situation is donor minor antigen peptide MHC class I and class II complexes. These complexes can theoretically be produced by donor cells themselves or by recipient phagocytes after engulfing donor cells or debris. The magnitude of the T cell response would be small at first because the frequency of T cell clones reactive to processed peptides from donor MHC proteins presented by recipient DC is orders of magnitude lower than the frequency of T cells specific for allogeneic MHC molecules [52]. However, a recent study demonstrated that the frequency of graft-specific T cells activated via the indirect pathway influences the ability of the costimulatory blockade to be effective in promoting graft survival [53].
Innate alloimmunity
Every renal allograft undergoes a degree of ischemic reperfusion injury during transplantation and, as a result of this injury, the innate immune system is activated. Activation of the innate immune response can initiate acute rejection and contribute to the development of chronic allograft nephropathy. The mechanism by which ischemia reperfusion injury promotes rejection is likely to be multifactorial. Studies have shown that reperfusion injury activates both a cellular response and humoral factors of the innate immune system.
Central to the ischemia injury are reactive oxygen species (ROS) [54]. Reactive oxygen species are directly toxic to cells inducing apoptosis and/or necrosis. The greater the ischemic insult, the more ROS generated and, consequently, the greater the toxic effect to the graft. The ROS trigger activation of caspases, such as caspase 3, resulting in apoptosis [55]. In addition, ROS induce activation of chaperoning proteins, which are ligands to toll-like receptors (TLRs). These proteins can be secreted from stressed or damaged cells (i.e. heat shock protein 72 and high-mobility group box 1), or they can be altered matrix proteins (i.e. hyaluronan fragments) [56, 57]. By binding to TLR4 or TLR2, these ligands activate immature TLR-expressing DCs and/or vascular endothelium [58–60]. Toll-like receptor-mediated DC activation induces DC to migrate from grafts to secondary lymphoid tissues to initiate an adaptive alloimmune response [61].
Oxidative injury also facilitates signaling through adaptor molecules. Adaptive molecules, such as MyD88 and TRIF, have been shown to play a role in the development of acute rejection [62, 63]. Signaling through these adaptor molecules has been reported to promote chemokine expression within grafts, such as IP-10. IP-10 is a central chemokine that promotes T cell recruitment into allografts [64]. Studies using MyD88- and/or TRIF-deficient allografts demonstrate impaired donor-derived DC migration and less graft cell damage [65]. Redundancy within the innate immune response exists. Dendritic cells can also be activated upon reperfusion by activated natural killer (NK) cells, NK T cells, and Tγδ cells. Blocking any of these signaling pathways during reperfusion of a transplanted allograft could blunt activation of the adaptive immune response and prevent graft rejection.
Costimulation
T cell activation is central to graft rejection. Tissue destruction occurs due to direct T cell-mediated lysis of graft cells, T cell activation of accessory cells, alloantibody production, and/or complement activation. Some studies have implicated CD4 T cells as sufficient on their own to result in complete graft destruction [66], while other studies have suggested that CD8 T cell activation alone results in acute rejection [67]. It is now understood that T cells require at least two signals to become optimally activated and develop effector function [68–70]. Alloantigen-specific signals are delivered through the T cell receptor (Signal 1), and antigen-nonspecific signals are delivered through accessory or costimulatory molecules (Signal 2). Although not graft specific, these costimulatory signals are essential for the development of potent anti-graft responses. Blocking costimulatory pathways at the time of T cell activation with the intention of prolonging graft survival and inducing tolerance has been an area of intense research over the past two decades. Lack of costimulation at the time of antigen presentation has been shown to induce T cell deletion, unresponsiveness (anergy), suppression, regulation, and/or immune deviation.
One of the most intensely studied costimulatory pathways involved in allo-T cell activation is the CD28/B7 pathway. CD28 is expressed on resting T cells, and its ligands B7.1 (CD80) and B7.2 (CD86) are expressed on APC. Signaling through CD28 lowers the threshold of TCR signaling to promote T cell proliferation, cytokine production, and differentiation. Several groups have shown in animal models that blocking CD28 signaling on T cells prevents both acute [71–73] and chronic [74] allograft rejection and can induce anergy [75, 76]. Cytotoxic T lymphocyte-associated antigen 4 (CTLA4), a homolog to CD28, is up-regulated on activated T cells and binds to CD80 and CD86 with greater affinity than CD28 [77]. This molecule is antagonistic to CD28 and transmits an inhibitory signal turning off T cell activation [78, 79]. However, blocking the CD28 pathway alone has been less effective in promoting tolerance in certain situations [80, 81]. This may be explained by recent data suggesting that Signal 1 and Signal 2 can be sufficient to stimulate CD8 T cell proliferation and clonal expansion, but that a third signal delivered early in the response is essential for naïve CD8 T cells to develop optimal effector function [82, 83]—especially if the T cells are activated through the indirect pathway [84].
For the rejection of grafts mismatched for minor histocompatibility antigens, cooperation between CD4 and CD8 T cells is thought to be required for maximal graft rejection. CD4 T cells have been shown to facilitate CD8 T cell differentiation by direct cell-to-cell contact or by producing effector cytokines, such as IL-2 and IFN-γ, that directly support CD8 T cell differentiation and killing [85]. Alternatively, it is possible that CD4 T cells act indirectly through a dendritic cell to be a more potent stimulator [35–37] or suppressor of CD8 T cell responses [86]. This indirect effect could be mediated through CD154 expression on CD4 T cells and CD40 expression on dendritic cells [36, 84].
The CD154/CD40 costimulatory pathway has been widely studied in animal models of transplantation. CD154 is expressed on activated T cells, while CD40 is constitutively expressed on APC. CD154/CD40 interaction was initially shown to be important for humoral immune responses, but it has also been shown to enhance T cell responses [87]. Blocking CD154 alone has been shown to inhibit both acute and chronic rejection in animal models [88–94], but when used in combination with CD28/B7 blockade, the effect on prolonging graft survival can be synergistic [90, 95].
Costimulatory blockade
Building upon the knowledge learned from animal studies, researchers have been able to develop novel therapeutics currently in clinical trials for transplantation. Costimulatory blockade offers selective but long-lasting, graft-specific immunosuppression without nephrotoxicity and the possibility of inducing tolerance. The first pathway targeted was the CD28 pathway. In an attempt to block CD28 signaling, a soluble fusion protein was developed that consists of the extracellular binding domain of CTLA4 fused with the Fc domain of human immunoglobulin (Ig)G1, creating abatacept (CTLA4Ig) [96]. Abatacept binds to both CD80 and CD86, blocking CD28 engagement and T cell activation [73]. However, although transplantation studies in rodents demonstrated efficacy, studies in nonhuman primates did not live up to the expectation of inducing tolerance [97]. The failure of abatacept was thought to be secondary to a fast off-rate from CD86; consequently, a second generation agent, LEA29Y or belatacept, was created by codon-based mutagenesis, and it did demonstrate superior binding to CD80 and CD86 than abatacept [98]. In nonhuman primate renal transplant studies, belatacept was better at preventing acute rejection episodes than abatacept [99]. Belatacept is currently in phase III human clinical trials to determine if blocking this pathway in humans can promote graft survival and allow reduced exposure to calcinurin inhibitors [100]. Preliminary data suggests it may also prevent the development of chronic rejection [101]. Other agents, such as agonists to CD28, TGN1412, have been developed to target this pathway with the intention of expanding a regulatory T cell population. These studies have been abandoned at present due to the resultant cytokine storm and shock-like symptoms [102].
Targeting other costimulatory pathways, such as the CD154/CD40 pathway, are very appealing because of the potent ability to block T cell activation as well as antibody production that has been demonstrated in small animal models. Initial studies in nonhuman primates demonstrated long-term kidney allograft survival using anti-CD154 [93]. However, anti-CD154 (hu5C8) treatment in humans and nonhuman primates resulted in thromboembolic complications not observed rodent studies. This has been attributed to the expression of CD154 on human but not mouse platelets [103]. Current areas of intense investigation are focused on alternative costimulatory and inhibitory molecules that would target T cell adhesion and T cell memory [104, 105].
Regulatory T cells
Regulatory T cells are considered to be essential mediators of peripheral tolerance by maintaining immune homeostasis, preventing autoimmunity, and regulating inflammation. Studies have shown a positive correlation between regulatory T cell function and allograft survival [106–108]. T regulatory cells suppress immune responses by a number of mechanisms: production of suppressor cytokines, direct suppression of effector cells, and modulation of DC maturation and function. Harnessing the power of T regulatory cells is appealing as a potential tolerizing strategy in transplant recipients; however, markers that consistently identify and isolate regulatory T cells in vivo have been elusive.
Although both CD4 and CD8 T cells have been shown to demonstrate suppressive function, much attention has focused on a subpopulation of CD4 T cells that express high levels of CD25, the a subunit of the IL-2 receptor. CD4+CD25+ T regulatory cells have been identified in peripheral blood samples of tolerant liver allograft recipients [109] and within tolerated allografts [110]. Adoptive transfer of CD4+CD25+ T regulatory cells has been shown to prevent graft rejection and graft-versus-host-disease (GVHD) in animal models [111]. There are difficulties, however, in using CD25 as a marker for regulation. For example, because CD25 is up-regulated on activated T cells, its sustained expression on regulatory T cells could be confused with recently activated T cells. A transcription factor known as forkhead box P3 (FoxP3) was recently identified in regulatory T cells. This transcription factor is required for the development, maintenance, and function of T regulatory cells [112, 113]. It has proven to be a consistent marker to identify T regulatory cells in murine models but has not been as consistent in humans. In humans, transient expression of FoxP3 has been observed during T cell activation [114], and FoxP3 has recently been identified in inflamed and rejecting allografts [115, 116]. The expression of FoxP3 on graft infiltrating cells has also been associated with donor-specific hyporesponsiveness and less chronic changes on biopsy [117]. This conundrum emphasizes the continued need for further characterization of T regulatory cells to identify an exclusive marker of regulatory cells in humans. Despite this difficulty, CD4+CD25+FoxP3+ T cells with suppressive function can be generated de novo with costimulatory blockade, such as CTLA4Ig, anti-CD154, and non-depleting anti-CD4 [118–120], and in vitro with rapamycin [121, 122]. It has been recently shown that donor-derived T regulatory cells can inhibit CD4 T cells responses as well as recipient-derived T regulatory cells [123]. These data entertain the possibility of cell therapy using regulatory T cells generated in vitro.
Memory T cells
Memory T cells can be divided into central memory and effector memory subsets based on their circulation pattern and functional responsiveness. Memory T cells have been shown to be more sensitive to antigen, function more rapidly, produce effector cytokines, survive longer, and show less dependence on CD28 costimulation than their naïve counterparts [124–129].
Memory T cells specific for alloantigens can be generated after exposure to blood transfusions, pregnancy, rejection of a previous transplant, homeostatic proliferation, and heterologous immunity. Homeostatic proliferation refers to the division of peripheral T cells in a lymphopenic environment in the absence of antigenic stimulus. This occurs after a situation where T cells are depleted, i.e. after viral infection or immunotherapies, and has been shown to be dependent upon recognition of self antigens [130] and the presence of factors such as IL-7 [131]. After undergoing homeostatic proliferation, naïve T cells will change their phenotype to that of a memory cell and display some of the functional properties of memory cells [132–134]. Heterologous immunity refers to the generation of memory cells to infectious antigens that cross react with alloantigens [135, 136]. This would result in allo-specific memory in the absence of specific exposure. In transplant studies, it is clearly understood that memory T cells, however they are generated, pose a significant barrier to inducing tolerance to allografts [129, 137–140]. As humans age, the proportion of memory phenotype T cells increases. Thus, a better understanding of how to target this cell population and the designing novel of therapies that inhibit these cells would be beneficial.
T cell migration
Naïve T cells and central memory cells circulate between blood and secondary lymphoid tissue and are excluded from non-lymphoid tissues, such as the skin, gut, and lung. This migration pattern is guided mainly by the cell surface expression of specific homing molecules, such as selectins, integrins, and chemokine receptors. Activation of naïve lymphocytes occurs within secondary lymphoid tissue [40, 87, 141, 142]. Upon activation and differentiation, marked changes in the homing behavior of lymphocytes are observed as a direct result of changes in the cell surface expression of homing molecules. The interactions between these molecules and their ligands or receptors triggers a sequential and coordinated series of events; leukocyte rolling, stopping, and transmigration enable T cells to move from the blood across endothelial cells into peripheral tissues.
The new combination of cell surface molecules expressed on differentiated T cells enables access to tissues that were previously ‘off limits’. For example, activated T cells lose the expression of CD62L and CCR7, which prevents cells from re-entering peripheral lymph nodes. At the same time, they express increased levels of VLA-4 and LFA-1, which facilitates binding to endothelial cells at sites of inflammation. Different sites of inflammation express different adhesion molecules to select for different cell populations. This is evident from studies in which blocking CD62E and CD62P inhibited T cell infiltration into the skin [143–149]. In addition, much work has been done to characterize the chemokines expressed in the rejection of heart allografts [150]. They have been divided into early events, related to the ischemia and reperfusion injury of grafts, and late events, which are related to the immune response [151]. The specific chemokines found to be important for lymphocyte trafficking in rejecting heart grafts are CXCL9 (MIG), CXCL10 (IP-10), and CXCL11 (I-TAC) [150]. Neutralizing chemokines or blocking their receptors has been shown to prolong graft survival and prevent graft infiltration in animal models [152, 153].
The change in homing phenotypes appears to be determined during the transition from a naïve to memory T cell [154]. In fact, recent data suggest that it occurs during the initial activation and differentiation in secondary lymphoid tissue and that the unique microenvironment of the secondary lymphoid tissue draining various tissue sites directs the homing phenotype imprinted on T cells activated at that site [154–156]. Further studies have linked this education to the resident DC within lymphoid tissue [156]. Thus, the difference in T cell responses between vascularized and nonvascularized grafts could be explained, in part, by differences in the migratory capacity and thus the ability of cells to infiltrate grafts.
Chronic rejection
Chronic rejection is now the leading cause of graft failure in pediatric renal transplant recipients. Organs undergoing chronic rejection display many of the features of healing wounds, including fibroblast, endothelial cell, or epithelial cell proliferation and collagen deposition within the graft parenchyma and blood vessels; all of these processes result in interstitial fibrosis, ischemia, and the loss of graft function [157, 158]. Although risk factors can be identified, the pathophysiology of chronic rejection remains poorly understood. Both immunologic and nonimmunologic injuries have been shown to play a role in the development of chronic rejection.
Major histocompatibility complex-mismatched grafts, which undergo acute rejection in the absence of immunosuppression, undergo chronic rejection in rodents even if acute rejection is prevented [159, 160]. Allografts that are depleted of passenger leukocytes survive acute rejection only to succumb to chronic rejection [42]. It is postulated that chronic rejection occurs after donor DC are replaced by recipient DC within the allograft. Thus, chronic rejection of vascularized organs is thought to occur via the indirect pathway [45, 46]. As mentioned above, this type of T cell stimulation cannot damage parenchymal cells of graft origin directly because these cells express donor MHC molecules. Recipient T cells responding to allopeptide/self-MHC complexes on recipient APC that enter the graft can only cause bystander damage by producing cytokines or other soluble mediators. Several studies implicate the Th2 cytokines (IL-4, IL-5, IL-6, IL-10, and IL-13) as having a role in chronic rejection or fibrosis [161, 162]. Injection of Th2 cells induces chronic allograft rejection in immunodeficient recipients [163]. Dermal fibrosis in skin grafts undergoing chronic rejection has been shown to be blocked by treatment with anti-IL-4 antibodies [164]. Similarly, fibrosis induced by chemical injury is dependent on IL-4 [165]. These effects could be explained by the findings that IL-4 stimulates the production of extracellular matrix proteins by fibroblasts and that IL-4 and IL-10 inhibit macrophage production of metalloproteinases that digest extracellular matrix proteins [166, 167]. Type 2 cytokine-producing T cells would also be expected to promote antibody production by B cells [168]. Results from several studies indicate that chronic rejection-related fibrosis is dependent on anti-graft antibodies, which may cross-link surface antigens on endothelial cells and thereby cause the production of growth factors and complement activation [169, 170]. Although these studies suggest that Th2 cytokines enhance fibrosis, another study found that chronic heart graft rejection was inhibited in Stat4-deficient mice that are impaired with respect to the generation of Th1 cells [171]. Given the therapeutic implications, it is essential to resolve the nature of the relationships between graft antigen-reactive T cells, their products, and fibroblast proliferation and collagen production. One aspect of chronic rejection that is currently under intense investigation is the role of alloantibodies in the development and progression of chronic rejection.
B cells and alloantibodies
B cells and anti-HLA antibodies have recently been shown to play an important role in both acute and chronic allograft rejection. The presence of CD20+ B cells and plasma cells infiltrating allografts has been found to correlate with irreversible acute rejection episodes [172, 173]. The ability to detect circulating anti-HLA antibodies in patients and the association of tissue deposition of C4d, a complement split product, has revitalized the study of alloantibodies. Acute antibody-mediated rejection was added to the Banff criteria in 2003 [174] and has been recently updated [175]. The diagnosis of acute (or chronic) antibody-mediated rejection depends upon the presence of three criteria: (1) serologic evidence for circulating anti-donor antibodies, (2) C4d+ staining in peritubular capillaries, and (3) morphological evidence of acute (or chronic) tissue injury. Intense (>1+), widespread (>50%) C4d staining of peritubular capillaries is both a sensitive and specific marker of acute antibody-mediated rejection. In rodent models, C4d deposition within kidney allografts is alloantibody dependent and begins as a focal process progressing to diffuse peritubular capillary staining. In nonhuman primate studies, circulating donor-specific alloantibodies and deposition of C4d in peritubular capillaries lead, in most cases, to chronic transplant glomerulopathy and arteriopathy [176].
Rituximab, a chimeric anti-CD20 monoclonal antibody therapy, has been shown to be effective in some but not all cases of acute humoral rejection [177, 178]. This may be due in part to the elimination of CD20-expressing B cells but not plasma cells, which do not express CD20 [179]. In chronic inflammatory situations, ectopic lymphoid structures can form within grafts and are not responsive to rituximab therapy even though circulating alloantibodies are reduced [180].
B cells not only damage grafts by producing anti-graft antibodies, but they have recently been shown to infiltrate grafts and present graft-derived antigens to alloreactive T cells via the indirect pathway of allorecognition [181]. Stimulation of B cells by antigen in the presence of T cells helps drive naïve B cell proliferation and differentiation into memory B cells and plasma cells [182]. Memory B cells survive in lymphoid tissue in the absence of antigen and, upon challenge with antigen, they respond rapidly with robust proliferation and antibody production. Plasma cells, in contrast, home to the spleen and bone marrow, are terminally differentiated, and are thought to be responsible for the circulating levels of antibodies. It is debated whether human plasma cells last a lifetime or, alternatively, need to be replenished from the B cell pool [183]. In contrast to plasma cells, memory B cells require reactivation to produce antibodies. Thus, the lack of circulating alloantibodies may not reflect lack of sensitization but rather may reflect the lack of active antibody-producing plasma cells. New therapies aimed at targeting alloantigen-specific memory B cell activation and alloantibody production would be advantageous.
Immunological monitoring
Analysis of gene expression using genomic microarrays can complement clinical research and may provide insights into disease pathogenesis. A microarray is a high-throughput technology that consists of a series of small nucleic acid sequences or oligonucleotides (called probes) that are attached to a solid surface (i.e. glass slide or a silicon chip). Millions of probes can be contained in one array. DNA and RNA of experimental and/or control samples can be extracted, labeled, and hybridized to the probes under high-stringency conditions (Fig. 2). Probe–target hybridization is usually detected and quantified by fluorescence-based detection methods to determine relative abundance of DNA sequences in the target (ratio of test to reference sample). DNA microarrays can be used to measure changes in the levels of gene expression or to detect single nucleotide polymorphisms. The technology is reproducible across multiple samples and when large numbers of genes are analyzed, it can be used to identify patterns of gene expression within disease states that are believed to correlate with functional changes at the protein level. This technology can produce an overwhelming amount of data that must be analyzed using sophisticated data analysis software. The current cost and complexity of this technology precludes its use as a screening tool in the clinical setting.
Fig. 2.
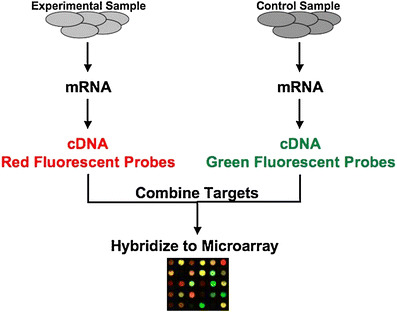
DNA microarray analysis DNA from experimental samples is purified and fluorescently labeled and then hybridized to immobilized probes on the array to determine alterations in gene expression
DNA microarrays have been applied to peripheral blood and renal allograft biopsy samples of pediatric patients with and without graft dysfunction in the posttransplant period [184]. Several gene expression patterns are altered during acute rejection episodes, chronic allograft nephropathy, and infection. Three molecular categories have been identified during acute rejection episodes that appear to correlate with graft function and survival. For example, biopsy samples that demonstrate enhanced B cell-related genes appear to have the worst outcomes. Since this initial study, many studies have subsequently been performed in both humans and mice investigating acute rejection, chronic allograft nephropathy, operational tolerance, minimum immunosuppression, infections, and drug toxicity [185]. In some cases, these have resulted in the identification of molecular subtypes that can predict outcome and response to treatment; in other cases, potential novel therapeutic targets have been identified. The expectation from these studies is to move from a ‘one-size fits all’ to a more personalized approach to posttransplant immunosuppressive regimens.
Summary
The benefits learned at the bench and, in particular, in small animal models are beginning to translate to the bedside. Novel therapeutics currently in clinical trials in humans have originated from basic studies investigating the requirements of T cell activation. It is clear that costimulatory blockade alone, while highly effective at blocking activation of naïve T cells, may be effective in blocking memory T cell responses, and current T cell depletion-based therapies may, in fact, promote memory T cell development. Recent studies have suggested that memory T cells pose the next barrier to overcome in the quest to induce allograft-specific tolerance. Once again, we turn to the bench.
Questions
(Answers appear following question list)
- Which of the following statements is true?
- A six-antigen matched kidney is at risk for chronic rejection.
- Naïve T cells are activated directly in the allografts where they mediate acute rejection.
- Memory T cells have increased susceptibility to costimulatory blockade.
- Plasma cells are depleted with rituximab therapy.
- Which statement is true of the indirect pathway of allorecognition?
- Activation of T cells occurs by recognition of intact donor MHC molecules.
- The pathway thought to be involved in chronic rejection.
- Passenger leukocytes migrate from the graft to activate T cells in lymphoid tissue.
- The frequency of T cells specific for a given alloantigen activated via this pathway is high.
- Costimulation
- is the synergy between CD4 and CD8 T cells that results in acute rejection.
- is the signaling that results in optimal T cell activation.
- can easily be blocked in humans to result in tolerance.
- refers to induction therapy.
- Memory T cells directed against an allograft can be formed after
- pregnancy
- a viral infection
- depletional anti-T cell antibodies
- all of the above
- none of the above
Answers
a
b
b
d
References
- 1.Matas AJ, Gillingham KJ, Payne WD, Najarian JS. The impact of an acute rejection episode on long-term renal allograft survival (t1/2) Transplantation. 1994;57:857–859. doi: 10.1097/00007890-199403270-00015. [DOI] [PubMed] [Google Scholar]
- 2.Meier-Kriesche HU, Schold JD, Kaplan B. Long-term renal allograft survival: have we made significant progress or is it time to rethink our analytic and therapeutic strategies? Am J Transplant. 2004;4:1289–1295. doi: 10.1111/j.1600-6143.2004.00515.x. [DOI] [PubMed] [Google Scholar]
- 3.Roopenian D, Choi EY, Brown A. The immunogenomics of minor histocompatibility antigens. Immunol Rev. 2002;190:86–94. doi: 10.1034/j.1600-065x.2002.19007.x. [DOI] [PubMed] [Google Scholar]
- 4.Akatsuka Y, Nishida T, Kondo E, Miyazaki M, Taji H, Iida H, Tsujimura K, Yazaki M, Naoe T, Morishima Y, Kodera Y, Kuzushima K, Takahashi T. Identification of a polymorphic gene, BCL2A1, encoding two novel hematopoietic lineage-specific minor histocompatibility antigens. J Exp Med. 2003;197:1489–1500. doi: 10.1084/jem.20021925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simpson E, Scott D, Chandler P. The male-specific histocompatibility antigen, H-Y: a history of transplantation, immune response genes, sex determination and expression cloning. Annu Rev Immunol. 1997;15:39–61. doi: 10.1146/annurev.immunol.15.1.39. [DOI] [PubMed] [Google Scholar]
- 6.Spierings E, Vermeulen CJ, Vogt MH, Doerner LE, Falkenburg JH, Mutis T, Goulmy E. Identification of HLA class II-restricted H-Y-specific T-helper epitope evoking CD4+ T-helper cells in H-Y-mismatched transplantation. Lancet. 2003;362:610–615. doi: 10.1016/S0140-6736(03)14191-8. [DOI] [PubMed] [Google Scholar]
- 7.Haskova Z, Sproule TJ, Roopenian DC, Ksander AB. An immunodominant minor histocompatibility alloantigen that initiates corneal allograft rejection. Transplantation. 2003;75:1368–1374. doi: 10.1097/01.TP.0000063708.26443.3B. [DOI] [PubMed] [Google Scholar]
- 8.Bach FH, Hirschhorn K. Lymphocyte interaction: A potential histocompatibility test in vitro. Science. 1964;143:813–814. doi: 10.1126/science.143.3608.813. [DOI] [PubMed] [Google Scholar]
- 9.Braun MY, McCormack A, Webb G, Batchelor JR. Mediation of acute but not chronic rejection of MHC-incompatible rat kidney grafts by alloreactive CD4 T cells activated by the direct pathway of sensitization. Transplantation. 1993;55:177–182. doi: 10.1097/00007890-199301000-00033. [DOI] [PubMed] [Google Scholar]
- 10.Jameson SC, Hogquist KA, Bevan MJ. Positive selection of thymocytes. Annu Rev Immunol. 1995;13:93–126. doi: 10.1146/annurev.iy.13.040195.000521. [DOI] [PubMed] [Google Scholar]
- 11.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol. 2003;21:139–176. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 12.Grandjean I, Duban L, Bonney EA, Corcuff E, Di Santo JP, Matzinger P, Lantz O. Are major histocompatibility complex molecules involved in the survival of naive CD4+ T cells? J Exp Med. 2003;198:1089–1102. doi: 10.1084/jem.20030963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaye J, Hedrick SM. Analysis of specificity for antigen, Mls, and allogenic MHC by transfer of T-cell receptor alpha- and beta-chain genes. Nature. 1988;336:580–583. doi: 10.1038/336580a0. [DOI] [PubMed] [Google Scholar]
- 14.Matzinger P, Bevan MJ. Hypothesis: why do so many lymphocytes respond to major histocompatibility antigens? Cell Immunol. 1977;29:1–5. doi: 10.1016/0008-8749(77)90269-6. [DOI] [PubMed] [Google Scholar]
- 15.Daniel C, Horvath S, Allen PM. A basis for alloreactivity: MHC helical residues broaden peptide recognition by the TCR. Immunity. 1998;8:543–552. doi: 10.1016/s1074-7613(00)80559-2. [DOI] [PubMed] [Google Scholar]
- 16.Lombardi G, Barber L, Sidhu S, Batchelor JR, Lechler RI. The specificity of alloreactive T cells is determined by MHC polymorphisms which contact the T cell receptor and which influence peptide binding. Int Immunol. 1991;3:769–775. doi: 10.1093/intimm/3.8.769. [DOI] [PubMed] [Google Scholar]
- 17.Schneck J, Munitz T, Coligan JE, Maloy WL, Margulies DH, Singer A. Inhibition of allorecognition by an H-2Kb-derived peptide is evidence for a T-cell binding region on a major histocompatibility complex molecule. Proc Natl Acad Sci USA. 1989;86:8516–8520. doi: 10.1073/pnas.86.21.8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villadangos JA, Galocha B, Lopez de Castro JA. Unusual topology of an HLA-B27 allospecific T cell epitope lacking peptide specificity. J Immunol. 1994;152:2317–2323. [PubMed] [Google Scholar]
- 19.Eckels DD, Gorski J, Rothbard J, Lamb JR. Peptide-mediated modulation of T-cell allorecognition. Proc Natl Acad Sci USA. 1988;85:8191–8195. doi: 10.1073/pnas.85.21.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weber DA, Terrel NK, Zhang Y, Strindberg G, Martin J, Rudensky A, Braunstein NS. Requirement for peptide in alloreactive CD4+ T cell recognition of class II MHC molecules. J Immunol. 1995;154:5153–5164. [PubMed] [Google Scholar]
- 21.Berkowitz N, Braunstein NS. T cell responses specific for subregions of allogeneic MHC molecules. J Immunol. 1992;148:309–317. [PubMed] [Google Scholar]
- 22.Bluestone JA, Jameson S, Miller S, Dick R., 2nd Peptide-induced conformational changes in class I heavy chains alter major histocompatibility complex recognition. J Exp Med. 1992;176:1757–1761. doi: 10.1084/jem.176.6.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Catipovic B, Dal Porto J, Mage M, Johansen TE, Schneck JP. Major histocompatibility complex conformational epitopes are peptide specific. J Exp Med. 1992;176:1611–1618. doi: 10.1084/jem.176.6.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larsen CP, Austyn JM, Morris PJ. The role of graft-derived dendritic leukocytes in the rejection of vascularized organ allografts. Recent findings on the migration and function of dendritic leukocytes after transplantation. Ann Surg. 1990;212:308–315. doi: 10.1097/00000658-199009000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Talmage DW, Dart G, Radovich J, Lafferty KJ. Activation of transplant immunity: effect of donor leukocytes on thyroid allograft rejection. Science. 1976;191:385–388. doi: 10.1126/science.1082167. [DOI] [PubMed] [Google Scholar]
- 26.Lafferty KJ, Bootes A, Dart G, Talmage DW. Effect of organ culture on the survival of thyroid allografts in mice. Transplantation. 1976;22:138–149. doi: 10.1097/00007890-197608000-00009. [DOI] [PubMed] [Google Scholar]
- 27.Lafferty KJ, Bootes A, Killby VA, Burch W. Mechanism of thyroid allograft rejection. Aust J Exp Biol Med Sci. 1976;54:573–586. doi: 10.1038/icb.1976.58. [DOI] [PubMed] [Google Scholar]
- 28.Lafferty KJ, Talmage DW. Theory of allogeneic reactivity and its relevance to transplantation biology. Transplant Proc. 1976;8:349–353. [PubMed] [Google Scholar]
- 29.Lechler RI, Batchelor JR. Restoration of immunogenicity to passenger cell-depleted kidney allografts by the addition of donor strain dendritic cells. J Exp Med. 1982;155:31–41. doi: 10.1084/jem.155.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lechler RI, Batchelor JR. Immunogenicity of retransplanted rat kidney allografts. Effect of inducing chimerism in the first recipient and quantitative studies on immunosuppression of the second recipient. J Exp Med. 1982;156:1835–1841. doi: 10.1084/jem.156.6.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137:1142–1162. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schuler G, Steinman RM. Murine epidermal Langerhans cells mature into potent immunostimulatory dendritic cells in vitro. J Exp Med. 1985;161:526–546. doi: 10.1084/jem.161.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Enk AH, Angeloni VL, Udey MC, Katz SI. An essential role for Langerhans cell-derived IL-1 beta in the initiation of primary immune responses in skin. J Immunol. 1993;150:3698–3704. [PubMed] [Google Scholar]
- 34.Kimber I, Cumberbatch M. Stimulation of Langerhans cell migration by tumor necrosis factor alpha (TNF-alpha) J Invest Dermatol. 1992;99:48S–50S. doi: 10.1111/1523-1747.ep12668986. [DOI] [PubMed] [Google Scholar]
- 35.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 36.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 37.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. (T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 38.Barker CF, Billingham RE. The role of afferent lymphatics in the rejection of skin homografts. J Exp Med. 1968;128:197–221. doi: 10.1084/jem.128.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larsen CP, Steinman RM, Whitmer-Pack M, Hankins DF, Morris PJ, Austyn JM. Migration and maturation of Langerhans cells in skin transplants and explants. J Exp Med. 1990;172:1483–1493. doi: 10.1084/jem.172.5.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ingulli E, Mondino A, Khoruts A, Jenkins MK. In vivo detection of dendritic cell antigen presentation to CD4(+) T cells. J Exp Med. 1997;185:2133–2141. doi: 10.1084/jem.185.12.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinman RM, Pack M, Inaba K. Dendritic cells in the T-cell areas of lymphoid organs. Immunol Rev. 1997;156:25–37. doi: 10.1111/j.1600-065x.1997.tb00956.x. [DOI] [PubMed] [Google Scholar]
- 42.Auchincloss H, Jr, Lee R, Shea S, Markowitz JS, Grusby MJ, Glimcher LH. The role of “indirect” recognition in initiating rejection of skin grafts from major histocompatibility complex class II-deficient mice. Proc Natl Acad Sci USA. 1993;90:3373–3377. doi: 10.1073/pnas.90.8.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inaba K, Turley S, Yamaide F, Iyoda T, Mahnke K, Inaba M, Pack M, Subklewe M, Sauter B, Sheff D, Albert M, Bhardwaj N, Mellman I, Steinman RM. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J Exp Med. 1998;188:2163–2173. doi: 10.1084/jem.188.11.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benichou G, Takizawa PA, Olson CA, McMillan M, Sercarz EE. Donor major histocompatibility complex (MHC) peptides are presented by recipient MHC molecules during graft rejection. J Exp Med. 1992;175:305–308. doi: 10.1084/jem.175.1.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ciubotariu R, Liu Z, Colovai AI, Ho E, Itescu S, Ravalli S, Hardy MA, Cortesini R, Rose EA, Suciu-Foca N. Persistent allopeptide reactivity and epitope spreading in chronic rejection of organ allografts. J Clin Invest. 1998;101:398–405. doi: 10.1172/JCI1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vella JP, Spadafora-Ferreira M, Murphy B, Alexander SI, Harmon W, Carpenter CB, Sayegh MH. Indirect allorecognition of major histocompatibility complex allopeptides in human renal transplant recipients with chronic graft dysfunction. Transplantation. 1997;64:795–800. doi: 10.1097/00007890-199709270-00001. [DOI] [PubMed] [Google Scholar]
- 47.Yewdell JW, Norbury CC, Bennink JR. Mechanisms of exogenous antigen presentation by MHC class I molecules in vitro and in vivo: implications for generating CD8+ T cell responses to infectious agents, tumors, transplants, and vaccines. Adv Immunol. 1999;73:1–77. doi: 10.1016/s0065-2776(08)60785-3. [DOI] [PubMed] [Google Scholar]
- 48.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I- restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 49.Brossart P, Bevan MJ. Presentation of exogenous protein antigens on major histocompatibility complex class I molecules by dendritic cells: pathway of presentation and regulation by cytokines. Blood. 1997;90:1594–1599. [PMC free article] [PubMed] [Google Scholar]
- 50.Grimm PC, Nickerson P, Jeffery J, Savani RC, Gough J, McKenna RM, Stern E, Rush DN. Neointimal and tubulointerstitial infiltration by recipient mesenchymal cells in chronic renal-allograft rejection. N Engl J Med. 2001;12:93–97. doi: 10.1056/NEJM200107123450203. [DOI] [PubMed] [Google Scholar]
- 51.Arakelov A, Lakkis FG. The alloimmune response and effector mechanisms of allograft rejection. Semin Nephrol. 2000;20:95–102. [PubMed] [Google Scholar]
- 52.Liu Z, Sun YK, Xi YP, Harris P, Suciu-Foca N. T cell recognition of self-human histocompatibility leukocyte antigens (HLA)-DR peptides in context of syngeneic HLA-DR molecules. J Exp Med. 1992;175:1663–1668. doi: 10.1084/jem.175.6.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ford ML, Koehn BH, Wagener ME, Jiang W, Gangappa S, Pearson TC, Larsen CP. Antigen-specific precursor frequency impacts T cell proliferation, differentiation, and requirement for costimulation. J Exp Med. 2007;204:299–309. doi: 10.1084/jem.20062319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Land W. Postischemic reperfusion injury to allografts—a case for ‘innate immunity’? Eur Surg Res. 2002;34:160–169. doi: 10.1159/000048904. [DOI] [PubMed] [Google Scholar]
- 55.Lopez-Neblina F, Toledo AH, Toledo-Pereyra LH. Molecular biology of apoptosis in ischemia and reperfusion. J Invest Surg. 2005;18:335–350. doi: 10.1080/08941930500328862. [DOI] [PubMed] [Google Scholar]
- 56.Mueller AR, Platz KP, Heckert C, Hausler M, Guckelberger O, Schuppan D, Lobeck H, Neuhaus P. The extracellular matrix: an early target of preservation/reperfusion injury and acute rejection after small bowel transplantation. Transplantation. 1998;65:770–776. doi: 10.1097/00007890-199803270-00002. [DOI] [PubMed] [Google Scholar]
- 57.Warger T, Hilf N, Rechtsteiner G, Haselmayer P, Carrick DM, Jonuleit H, von Landenberg P, Rammensee HG, Nicchitta CV, Radsak MP, Schild H. Interaction of TLR2 and TLR4 ligands with the N-terminal domain of Gp96 amplifies innate and adaptive immune responses. J Biol Chem. 2006;281:22545–22553. doi: 10.1074/jbc.M502900200. [DOI] [PubMed] [Google Scholar]
- 58.Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tesar BM, Jiang D, Liang J, Palmer SM, Noble PW, Goldstein DR. The role of hyaluronan degradation products as innate alloimmune agonists. Am J Transplant. 2006;6:2622–2635. doi: 10.1111/j.1600-6143.2006.01537.x. [DOI] [PubMed] [Google Scholar]
- 60.Yang D, Chen Q, Yang H, Tracey KJ, Bustin M, Oppenheim JJ. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J Leukoc Biol. 2007;81:59–66. doi: 10.1189/jlb.0306180. [DOI] [PubMed] [Google Scholar]
- 61.Oh KH, Kim JY, Kim D, Lee EM, Oh HY, Seo JS, Han JS, Kim S, Lee JS, Ahn C. Targeted gene disruption of the heat shock protein 72 gene (hsp70.1) in the donor tissue is associated with a prolonged rejection-free survival in the murine skin allograft model. Transplant Immunol. 2004;13:273–281. doi: 10.1016/j.trim.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 62.McKay D, Shigeoka A, Rubinstein M, Surh C, Sprent J. Simultaneous deletion of MyD88 and Trif delays major histocompatibility and minor antigen mismatch allograft rejection. Eur J Immunol. 2006;36:1994–2002. doi: 10.1002/eji.200636249. [DOI] [PubMed] [Google Scholar]
- 63.Goldstein DR, Tesar BM, Akira S, Lakkis FG. Critical role of the Toll-like receptor signal adaptor protein MyD88 in acute allograft rejection. J Clin Invest. 2003;111:1571–1578. doi: 10.1172/JCI17573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhai Y, Shen XD, O’Connell R, Gao F, Lassman C, Busuttil RW, Cheng G, Kupiec-Weglinski JW. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173:7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 65.Shigeoka AA, Holscher TD, King AJ, Hall FW, Kiosses WB, Tobias PS, Mackman N, McKay DB. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J Immunol. 2007;178:6252–6258. doi: 10.4049/jimmunol.178.10.6252. [DOI] [PubMed] [Google Scholar]
- 66.Krieger NR, Yin DP, Fathman CG. CD4+ but not CD8+ cells are essential for allorejection. J Exp Med. 1996;184:2013–2018. doi: 10.1084/jem.184.5.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hall BM. Cells mediating allograft rejection. Transplantation. 1991;51:1141–1151. doi: 10.1097/00007890-199106000-00001. [DOI] [PubMed] [Google Scholar]
- 68.Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med. 1987;165:302–319. doi: 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jenkins MK, Taylor PS, Norton SD, Urdahl KB. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol. 1991;147:2461–2466. [PubMed] [Google Scholar]
- 70.Kearney ER, Walunas TL, Karr RW, Morton PA, Loh DY, Bluestone JA, Jenkins MK. Antigen-dependent clonal expansion of a trace population of antigen-specific CD4+ T cells in vivo is dependent on CD28 costimulation and inhibited by CTLA-4. J Immunol. 1995;155:1032–1036. [PubMed] [Google Scholar]
- 71.Baliga P, Chavin KD, Qin L, Woodward J, Lin J, Linsley PS, Bromberg JS. CTLA4Ig prolongs allograft survival while suppressing cell-mediated immunity. Transplantation. 1994;58:1082–1090. [PubMed] [Google Scholar]
- 72.Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL, Mittler RS. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity. 1996;4:535–543. doi: 10.1016/s1074-7613(00)80480-x. [DOI] [PubMed] [Google Scholar]
- 73.Lenschow DJ, Zeng Y, Thistlethwaite JR, Montag A, Brady W, Gibson MG, Linsley PS, Bluestone JA. Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4lg. Science. 1992;257:789–792. doi: 10.1126/science.1323143. [DOI] [PubMed] [Google Scholar]
- 74.Glysing-Jensen T, Raisanen-Sokolowski A, Sayegh MH, Russell ME. Chronic blockade of CD28-B7-mediated T-cell costimulation by CTLA4Ig reduces intimal thickening in MHC class I and II incompatible mouse heart allografts. Transplantation. 1997;64:1641–1645. doi: 10.1097/00007890-199712270-00002. [DOI] [PubMed] [Google Scholar]
- 75.Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH. CTLA-4 regulates induction of anergy in vivo. Immunity. 2001;14:145–155. doi: 10.1016/s1074-7613(01)00097-8. [DOI] [PubMed] [Google Scholar]
- 76.Wells AD, Walsh MC, Bluestone JA, Turka LA. Signaling through CD28 and CTLA-4 controls two distinct forms of T cell anergy. J Clin Invest. 2001;108:895–903. doi: 10.1172/JCI13220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174:561–569. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med. 1996;183:2541–2550. doi: 10.1084/jem.183.6.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 80.Trambley J, Bingaman AW, Lin A, Elwood ET, Waitze SY, Ha J, Durham MM, Corbascio M, Cowan SR, Pearson TC, Larsen CP. Asialo GM1(+) CD8(+) T cells play a critical role in costimulation blockade-resistant allograft rejection. J Clin Invest. 1999;104:1715–1722. doi: 10.1172/JCI8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Newell KA, He G, Guo Z, Kim O, Szot GL, Rulifson I, Zhou P, Hart J, Thistlethwaite JR, Bluestone JA. Cutting edge: blockade of the CD28/B7 costimulatory pathway inhibits intestinal allograft rejection mediated by CD4+ but not CD8+ T cells. J Immunol. 1999;163:2358–2362. [PubMed] [Google Scholar]
- 82.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141–1151. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, Mescher MF. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol. 1999;162:3256–3262. [PubMed] [Google Scholar]
- 84.Filatenkov AA, Jacovetty EL, Fischer UB, Curtsinger JM, Mescher MF, Ingulli E. CD4 T cell-dependent conditioning of dendritic cells to produce IL-12 results in CD8-mediated graft rejection and avoidance of tolerance. J Immunol. 2005;174:6909–6917. doi: 10.4049/jimmunol.174.11.6909. [DOI] [PubMed] [Google Scholar]
- 85.Shrikant P, Khoruts A, Mescher MF. CTLA-4 blockade reverses CD8+ T cell tolerance to tumor by a CD4+ T cell- and IL-2-dependent mechanism. Immunity. 1999;11:483–493. doi: 10.1016/s1074-7613(00)80123-5. [DOI] [PubMed] [Google Scholar]
- 86.Taylor PA, Friedman TM, Korngold R, Noelle RJ, Blazar BR. Tolerance induction of alloreactive T cells via ex vivo blockade of the CD40:CD40L costimulatory pathway results in the generation of a potent immune regulatory cell. Blood. 2002;99:4601–4609. doi: 10.1182/blood.v99.12.4601. [DOI] [PubMed] [Google Scholar]
- 87.Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK. Visualization of specific B and T lymphocyte interactions in the lymph node. Science. 1998;281:96–99. doi: 10.1126/science.281.5373.96. [DOI] [PubMed] [Google Scholar]
- 88.Larsen CP, Alexander DZ, Hollenbaugh D, Elwood ET, Ritchie SC, Aruffo A, Hendrix R, Pearson TC. CD40-gp39 interactions play a critical role during allograft rejection. Suppression of allograft rejection by blockade of the CD40-gp39 pathway. Transplantation. 1996;61:4–9. doi: 10.1097/00007890-199601150-00002. [DOI] [PubMed] [Google Scholar]
- 89.Parker DC, Greiner DL, Phillips NE, Appel MC, Steele AW, Durie FH, Noelle RJ, Mordes JP, Rossini AA. Survival of mouse pancreatic islet allografts in recipients treated with allogeneic small lymphocytes and antibody to CD40 ligand. Proc Natl Acad Sci USA. 1995;92:9560–9564. doi: 10.1073/pnas.92.21.9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hancock WW, Sayegh MH, Zheng XG, Peach R, Linsley PS, Turka LA. Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc Natl Acad Sci USA. 1996;93:13967–13972. doi: 10.1073/pnas.93.24.13967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Blazar BR, Taylor PA, Panoskaltsis-Mortari A, Buhlman J, Xu J, Flavell RA, Korngold R, Noelle R, Vallera DA. Blockade of CD40 ligand-CD40 interaction impairs CD4+ T cell-mediated alloreactivity by inhibiting mature donor T cell expansion and function after bone marrow transplantation. J Immunol. 1997;158:29–39. [PubMed] [Google Scholar]
- 92.Markees TG, Phillips NE, Noelle RJ, Shultz LD, Mordes JP, Greiner DL, Rossini AA. Prolonged survival of mouse skin allografts in recipients treated with donor splenocytes and antibody to CD40 ligand. Transplantation. 1997;64:329–335. doi: 10.1097/00007890-199707270-00026. [DOI] [PubMed] [Google Scholar]
- 93.Kirk AD, Burkly LC, Batty DS, Baumgartner RE, Berning JD, Buchanan K, Fechner JH, Jr, Germond RL, Kampen RL, Patterson NB, Swanson SJ, Tadaki DK, TenHoor CN, White L, Knechtle SJ, Harlan DM. Treatment with humanized monoclonal antibody against CD154 prevents acute renal allograft rejection in nonhuman primates. Nat Med. 1999;5:686–693. doi: 10.1038/9536. [DOI] [PubMed] [Google Scholar]
- 94.Huddleston SJ, Hays WS, Filatenkov A, Ingulli E, Jenkins MK. CD154+ graft antigen-specific CD4+ T cells are sufficient for chronic rejection of minor antigen incompatible heart grafts. Am J Transplant. 2006;6:1312–1319. doi: 10.1111/j.1600-6143.2006.01309.x. [DOI] [PubMed] [Google Scholar]
- 95.Larsen CP, Elwood ET, Alexander DZ, Ritchie SC, Hendrix R, Tucker-Burden C, Cho HR, Aruffo A, Hollenbaugh D, Linsley PS, Winn KJ, Pearson TC. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381:434–438. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 96.Linsley PS, Wallace PM, Johnson J, Gibson MG, Greene JL, Ledbetter JA, Singh C, Tepper MA. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science. 1992;257:792–795. doi: 10.1126/science.1496399. [DOI] [PubMed] [Google Scholar]
- 97.Kirk AD, Harlan DM, Armstrong NN, Davis TA, Dong Y, Gray GS, Hong X, Thomas D, Fechner JH, Jr, Knechtle SJ. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci USA. 1997;94:8789–8794. doi: 10.1073/pnas.94.16.8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vincenti F. Costimulation blockade in autoimmunity and transplantation. J Allergy Clin Immunol. 2008;121:299–306. doi: 10.1016/j.jaci.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 99.Larsen CP, Pearson TC, Adams AB, Tso P, Shirasugi N, Strobertm E, Anderson D, Cowan S, Price K, Naemura J, Emswiler J, Greene J, Turk LA, Bajorath J, Townsend R, Hagerty D, Linsley PS, Peach RJ. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant. 2005;5:443–453. doi: 10.1111/j.1600-6143.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- 100.Vincenti F. New monoclonal antibodies in renal transplantation. Minerva Urol Nefrol. 2003;55:57–66. [PubMed] [Google Scholar]
- 101.Vincenti F, Larsen C, Muhlbacher F, Charpentier B (2007) Interium report of phase 2 long-term safety of belatacept (abstr 0253). European Society of Organ Transplatation, Prague
- 102.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 103.Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med. 2000;6:114. doi: 10.1038/72162. [DOI] [PubMed] [Google Scholar]
- 104.Vincenti F, Mendez R, Pescovitz M, Rajagopalan PR, Wilkinson AH, Butt K, Laskow D, Slakey DP, Lorber MI, Garg JP, Garovoy M. A phase I/II randomized open-label multicenter trial of efalizumab, a humanized anti-CD11a, anti-LFA-1 in renal transplantation. Am J Transplant. 2007;7:1770–1777. doi: 10.1111/j.1600-6143.2007.01845.x. [DOI] [PubMed] [Google Scholar]
- 105.St Clair EW, Turka LA, Saxon A, Matthews JB, Sayegh MH, Eisenbarth GS, Bluestone J. New reagents on the horizon for immune tolerance. Annu Rev Med. 2007;58:329–346. doi: 10.1146/annurev.med.58.061705.145449. [DOI] [PubMed] [Google Scholar]
- 106.Demirkiran A, Kok A, Kwekkeboom J, Kusters JG, Metselaar HJ, Tilanus HW, van der Laan LJ. Low circulating regulatory T-cell levels after acute rejection in liver transplantation. Liver Transpl. 2006;12:277–284. doi: 10.1002/lt.20612. [DOI] [PubMed] [Google Scholar]
- 107.Meloni F, Vitulo P, Bianco AM, Paschetto E, Morosini M, Cascina A, Mazzucchelli I, Ciardelli L, Oggionni T, Fietta AM, Pozzi E, Vigano M. Regulatory CD4+CD25+ T cells in the peripheral blood of lung transplant recipients: correlation with transplant outcome. Transplantation. 2004;77:762–766. doi: 10.1097/01.tp.0000116565.86752.6b. [DOI] [PubMed] [Google Scholar]
- 108.Salama AD, Najafian N, Clarkson MR, Harmon WE, Sayegh MH. Regulatory CD25+ T cells in human kidney transplant recipients. J Am Soc Nephrol. 2003;14:1643–1651. doi: 10.1097/01.asn.0000057540.98231.c1. [DOI] [PubMed] [Google Scholar]
- 109.Yoshizawa A, Ito A, Li Y, Koshiba T, Sakaguchi S, Wood KJ, Tanaka K. The roles of CD25+CD4+ regulatory T cells in operational tolerance after living donor liver transplantation. Transplant Proc. 2005;37:37–39. doi: 10.1016/j.transproceed.2004.12.259. [DOI] [PubMed] [Google Scholar]
- 110.Graca L, Cobbold SP, Waldmann H. Identification of regulatory T cells in tolerated allografts. J Exp Med. 2002;195:1641–1646. doi: 10.1084/jem.20012097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Taylor PA, Noelle RJ, Blazar BR. CD4(+)CD25(+) immune regulatory cells are required for induction of tolerance to alloantigen via costimulatory blockade. J Exp Med. 2001;193:1311–1318. doi: 10.1084/jem.193.11.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 113.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 114.Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, Roncarolo MG, Levings MK. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. 2007;19:345–354. doi: 10.1093/intimm/dxm014. [DOI] [PubMed] [Google Scholar]
- 115.Bunnag S, Allanach K, Jhangri GS, Sis B, Einecke G, Mengel M, Mueller TF, Halloran PF. FOXP3 expression in human kidney transplant biopsies is associated with rejection and time post transplant but not with favorable outcomes. Am J Transplant. 2008;8:1423–1433. doi: 10.1111/j.1600-6143.2008.02268.x. [DOI] [PubMed] [Google Scholar]
- 116.Dijke IE, Caliskan K, Korevaar SS, Maat AP, Zondervan PE, Balk AH, Weimar W, Baan CC. FOXP3 mRNA expression analysis in the peripheral blood and allograft of heart transplant patients. Transplant Immunol. 2008;18:250–254. doi: 10.1016/j.trim.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 117.Bestard O, Cruzado JM, Mestre M, Caldes A, Bas J, Carrera M, Torras J, Rama I, Moreso F, Seron D, Grinyo JM. Achieving donor-specific hyporesponsiveness is associated with FOXP3+ regulatory T cell recruitment in human renal allograft infiltrates. J Immunol. 2007;179:4901–4909. doi: 10.4049/jimmunol.179.7.4901. [DOI] [PubMed] [Google Scholar]
- 118.Honey K, Cobbold SP, Waldmann H. CD40 ligand blockade induces CD4+ T cell tolerance and linked suppression. J Immunol. 1999;163:4805–4810. [PubMed] [Google Scholar]
- 119.Yi H, Zhen Y, Zeng C, Zhang L, Zhao Y. Depleting anti-CD4 monoclonal antibody (GK1.5) treatment: influence on regulatory CD4+CD25+Foxp3+ T cells in mice. Transplantation. 2008;85:1167–1174. doi: 10.1097/TP.0b013e31816a1242. [DOI] [PubMed] [Google Scholar]
- 120.Cobbold SP, Castejon R, Adams E, Zelenika D, Graca L, Humm S, Waldmann H. Induction of foxP3+ regulatory T cells in the periphery of T cell receptor transgenic mice tolerized to transplants. J Immunol. 2004;172:6003–6010. doi: 10.4049/jimmunol.172.10.6003. [DOI] [PubMed] [Google Scholar]
- 121.Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 2006;177:8338–8347. doi: 10.4049/jimmunol.177.12.8338. [DOI] [PubMed] [Google Scholar]
- 122.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105:4743–4748. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- 123.Velasquez-Lopera MM, Eaton VL, Lerret NM, Correa LA, Decresce RP, Garcia LF, Jaramillo A. Induction of transplantation tolerance by allogeneic donor-derived CD4(+)CD25(+)Foxp3(+) regulatory T cells. Transpl Immunol. 2008;19:127–135. doi: 10.1016/j.trim.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 124.Ku CC, Murakami M, Sakamoto A, Kappler J, Marrack P. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science. 2000;288:675–678. doi: 10.1126/science.288.5466.675. [DOI] [PubMed] [Google Scholar]
- 125.Garcia S, DiSanto J, Stockinger B. Following the development of a CD4 T cell response in vivo: from activation to memory formation. Immunity. 1999;11:163–171. doi: 10.1016/s1074-7613(00)80091-6. [DOI] [PubMed] [Google Scholar]
- 126.Rogers PR, Dubey C, Swain SL. Qualitative changes accompany memory T cell generation: faster, more effective responses at lower doses of antigen. J Immunol. 2000;164:2338–2346. doi: 10.4049/jimmunol.164.5.2338. [DOI] [PubMed] [Google Scholar]
- 127.Veiga-Fernandes H, Walter U, Bourgeois C, McLean A, Rocha B. Response of naive and memory CD8+ T cells to antigen stimulation in vivo. Nat Immunol. 2000;1:47–53. doi: 10.1038/76907. [DOI] [PubMed] [Google Scholar]
- 128.Tough DF, Sprent J. Turnover of naive- and memory-phenotype T cells. J Exp Med. 1994;179:1127–1135. doi: 10.1084/jem.179.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Valujskikh A, Pantenburg B, Heeger PS. Primed allospecific T cells prevent the effects of costimulatory blockade on prolonged cardiac allograft survival in mice. Am J Transplant. 2002;2:501–509. doi: 10.1034/j.1600-6143.2002.20603.x. [DOI] [PubMed] [Google Scholar]
- 130.Kieper WC, Jameson SC. Homeostatic expansion and phenotypic conversion of naive T cells in response to self peptide/MHC ligands. Proc Natl Acad Sci USA. 1999;96:13306–13311. doi: 10.1073/pnas.96.23.13306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schluns KS, Kieper WC, Jameson SC, Lefrancois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 132.Cho BK, Rao VP, Ge Q, Eisen HN, Chen J. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J Exp Med. 2000;192:549–556. doi: 10.1084/jem.192.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Murali-Krishna K, Ahmed R. Cutting edge: naive T cells masquerading as memory cells. J Immunol. 2000;165:1733–1737. doi: 10.4049/jimmunol.165.4.1733. [DOI] [PubMed] [Google Scholar]
- 134.Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med. 2000;192:557–564. doi: 10.1084/jem.192.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Welsh RM, Selin LK. No one is naive: the significance of heterologous T-cell immunity. Nat Rev Immunol. 2002;2:417–426. doi: 10.1038/nri820. [DOI] [PubMed] [Google Scholar]
- 136.Misko IS, Cross SM, Khanna R, Elliott SL, Schmidt C, Pye SJ, Silins SL. Crossreactive recognition of viral, self, and bacterial peptide ligands by human class I-restricted cytotoxic T lymphocyte clonotypes: implications for molecular mimicry in autoimmune disease. Proc Natl Acad Sci USA. 1999;96:2279–2284. doi: 10.1073/pnas.96.5.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chalasani G, Dai Z, Konieczny BT, Baddoura FK, Lakkis FG. Recall and propagation of allospecific memory T cells independent of secondary lymphoid organs. Proc Natl Acad Sci USA. 2002;99:6175–6180. doi: 10.1073/pnas.092596999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhai Y, Meng L, Gao F, Busuttil RW, Kupiec-Weglinski JW. Allograft rejection by primed/memory CD8+ T cells is CD154 blockade resistant: therapeutic implications for sensitized transplant recipients. J Immunol. 2002;169:4667–4673. doi: 10.4049/jimmunol.169.8.4667. [DOI] [PubMed] [Google Scholar]
- 139.Adams AB, Williams MA, Jones TR, Shirasugi N, Durham MM, Kaech SM, Wherry EJ, Onami T, Lanier JG, Kokko KE, Pearson TC, Ahmed R, Larsen CP. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. 2003;111:1887–1895. doi: 10.1172/JCI17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Williams MA, Onami TM, Adams AB, Durham MM, Pearson TC, Ahmed R, Larsen CP. Cutting edge: persistent viral infection prevents tolerance induction and escapes immune control following CD28/CD40 blockade-based regimen. J Immunol. 2002;169:5387–5391. doi: 10.4049/jimmunol.169.10.5387. [DOI] [PubMed] [Google Scholar]
- 141.Lakkis FG, Arakelov A, Konieczny BT, Inoue Y. Immunologic ‘ignorance’ of vascularized organ transplants in the absence of secondary lymphoid tissue. Nat Med. 2000;6:686–688. doi: 10.1038/76267. [DOI] [PubMed] [Google Scholar]
- 142.Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualization of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity. 1994;1:327–339. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 143.Austrup F, Vestweber D, Borges E, Lohning M, Brauer R, Herz U, Renz H, Hallmann R, Scheffold A, Radbruch A, Hamann A. P- and E-selectin mediate recruitment of T-helper-1 but not T-helper-2 cells into inflamed tissues. Nature. 1997;385:81–83. doi: 10.1038/385081a0. [DOI] [PubMed] [Google Scholar]
- 144.Binns RM, Whyte A, Licence ST, Harrison AA, Tsang YT, Haskard DO, Robinson MK. The role of E-selectin in lymphocyte and polymorphonuclear cell recruitment into cutaneous delayed hypersensitivity reactions in sensitized pigs. J Immunol. 1996;157:4094–4099. [PubMed] [Google Scholar]
- 145.Erdmann I, Scheidegger EP, Koch FK, Heinzerling L, Odermatt B, Burg G, Lowe JB, Kundig TM. Fucosyltransferase VII-deficient mice with defective E-, P-, and L-selectin ligands show impaired CD4+ and CD8+ T cell migration into the skin, but normal extravasation into visceral organs. J Immunol. 2002;168:2139–2146. doi: 10.4049/jimmunol.168.5.2139. [DOI] [PubMed] [Google Scholar]
- 146.Hirata T, Merrill-Skoloff G, Aab M, Yang J, Furie BC, Furie B. P-Selectin glycoprotein ligand 1 (PSGL-1) is a physiological ligand for E-selectin in mediating T helper 1 lymphocyte migration. J Exp Med. 2000;192:1669–1676. doi: 10.1084/jem.192.11.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Silber A, Newman W, Sasseville VG, Pauley D, Beall D, Walsh DG, Ringler DJ. Recruitment of lymphocytes during cutaneous delayed hypersensitivity in nonhuman primates is dependent on E-selectin and vascular cell adhesion molecule 1. J Clin Invest. 1994;93:1554–1563. doi: 10.1172/JCI117134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Staite ND, Justen JM, Sly LM, Beaudet AL, Bullard DC. Inhibition of delayed-type contact hypersensitivity in mice deficient in both E-selectin and P-selectin. Blood. 1996;88:2973–2979. [PubMed] [Google Scholar]
- 149.Subramaniam M, Saffaripour S, Watson SR, Mayadas TN, Hynes RO, Wagner DD. Reduced recruitment of inflammatory cells in a contact hypersensitivity response in P-selectin-deficient mice. J Exp Med. 1995;181:2277–2282. doi: 10.1084/jem.181.6.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.el-Sawy T, Fahmy NM, Fairchild RL. Chemokines: directing leukocyte infiltration into allografts. Curr Opin Immunol. 2002;14:562–568. doi: 10.1016/s0952-7915(02)00382-5. [DOI] [PubMed] [Google Scholar]
- 151.Morita K, Miura M, Paolone DR, Engeman TM, Kapoor A, Remick DG, Fairchild RL. Early chemokine cascades in murine cardiac grafts regulate T cell recruitment and progression of acute allograft rejection. J Immunol. 2001;167:2979–2984. doi: 10.4049/jimmunol.167.5.2979. [DOI] [PubMed] [Google Scholar]
- 152.Hancock WW, Lu B, Gao W, Csizmadia V, Faia K, King JA, Smiley ST, Ling M, Gerard NP, Gerard C. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med. 2000;192:1515–1520. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Miura M, Morita K, Kobayashi H, Hamilton TA, Burdick MD, Strieter RM, Fairchild RL. Monokine induced by IFN-gamma is a dominant factor directing T cells into murine cardiac allografts during acute rejection. J Immunol. 2001;167:3494–3504. doi: 10.4049/jimmunol.167.6.3494. [DOI] [PubMed] [Google Scholar]
- 154.Picker LJ, Treer JR, Ferguson-Darnell B, Collins PA, Bergstresser PR, Terstappen LW. Control of lymphocyte recirculation in man. II. Differential regulation of the cutaneous lymphocyte-associated antigen, a tissue-selective homing receptor for skin-homing T cells. J Immunol. 1993;150:1122–1136. [PubMed] [Google Scholar]
- 155.Campbell DJ, Butcher EC. Rapid acquisition of tissue-specific homing phenotypes by CD4(+) T cells activated in cutaneous or mucosal lymphoid tissues. J Exp Med. 2002;195:135–141. doi: 10.1084/jem.20011502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, Von Andrian UH. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 157.Racusen LC, Solez K, Colvin R. Fibrosis and atrophy in the renal allograft: interim report and new directions. Am J Transplant. 2002;2:203–206. doi: 10.1034/j.1600-6143.2002.20303.x. [DOI] [PubMed] [Google Scholar]
- 158.Solez K, Colvin RB, Racusen LC, Sis B, Halloran PF, Birk PE, Campbell PM, Cascalho M, Collins AB, Demetris AJ, Drachenberg CB, Gibson IW, Grimm PC, Haas M, Lerut E, Liapis H, Mannon RB, Marcus PB, Mengel M, Mihatsch MJ, Nankivell BJ, Nickeleit V, Papadimitriou JC, Platt JL, Randhawa P, Roberts I, Salinas-Madriga L, Salomon DR, Seron D, Sheaff M, Weening JJ. Banff ‘05 Meeting Report: differential diagnosis of chronic allograft injury and elimination of chronic allograft nephropathy (‘CAN’) Am J Transplant. 2007;7:518–526. doi: 10.1111/j.1600-6143.2006.01688.x. [DOI] [PubMed] [Google Scholar]
- 159.Hancock WH, Whitley WD, Tullius SG, Heemann UW, Wasowska B, Baldwin WM, Tilney NL. Cytokines, adhesion molecules, and the pathogenesis of chronic rejection of rat renal allografts. Transplantation. 1993;56:643–650. doi: 10.1097/00007890-199309000-00028. [DOI] [PubMed] [Google Scholar]
- 160.Orloff SL, Yin Q, Corless CL, Loomis CB, Rabkin JM, Wagner CR. A rat small bowel transplant model of chronic rejection: histopathologic characteristics. Transplantation. 1999;68:766–779. doi: 10.1097/00007890-199909270-00008. [DOI] [PubMed] [Google Scholar]
- 161.Chan SY, DeBruyne LA, Goodman RE, Eichwald EJ, Bishop DK. In vivo depletion of CD8+ T cells results in Th2 cytokine production and alternate mechanisms of allograft rejection. Transplantation. 1995;59:1155–1161. [PubMed] [Google Scholar]
- 162.Shirwan H, Barwari L, Khan NS. Predominant expression of T helper 2 cytokines and altered expression of T helper 1 cytokines in long-term allograft survival induced by intrathymic immune modulation with donor class I major histocompatibility complex peptides. Transplantation. 1998;66:1802–1809. doi: 10.1097/00007890-199812270-00039. [DOI] [PubMed] [Google Scholar]
- 163.VanBuskirk AM, Wakely ME, Orosz CG. Transfusion of polarized TH2-like cell populations into SCID mouse cardiac allograft recipients results in acute allograft rejection. Transplantation. 1996;62:229–238. doi: 10.1097/00007890-199607270-00014. [DOI] [PubMed] [Google Scholar]
- 164.Le Moine A, Flamand V, Demoor FX, Noel JC, Surquin M, Kiss R, Nahori MA, Pretolani M, Goldman M, Abramowicz D. Critical roles for IL-4, IL-5, and eosinophils in chronic skin allograft rejection. J Clin Invest. 1999;103:1659–1667. doi: 10.1172/JCI5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shi Z, Wakil AE, Rockey DC. Strain-specific differences in mouse hepatic wound healing are mediated by divergent T helper cytokine responses. Proc Natl Acad Sci USA. 1997;94:10663–10668. doi: 10.1073/pnas.94.20.10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Postlethwaite AE, Holness MA, Katai H, Raghow R. Human fibroblasts synthesize elevated levels of extracellular matrix proteins in response to interleukin 4. J Clin Invest. 1992;90:1479–1485. doi: 10.1172/JCI116015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lacraz S, Nicod LP, Chicheportiche R, Welgus HG, Dayer JM. IL-10 inhibits metalloproteinase and stimulates TIMP-1 production in human mononuclear phagocytes. J Clin Invest. 1995;96:2304–2310. doi: 10.1172/JCI118286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 169.Shi C, Lee WS, He Q, Zhang D, Fletcher DL, Jr, Newell JB, Haber E. Immunologic basis of transplant-associated arteriosclerosis. Proc Natl Acad Sci USA. 1996;93:4051–4056. doi: 10.1073/pnas.93.9.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Harris PE, Bian H, Reed EF. Induction of high affinity fibroblast growth factor receptor expression and proliferation in human endothelial cells by anti-HLA antibodies: a possible mechanism for transplant atherosclerosis. J Immunol. 1997;159:5697–5704. [PubMed] [Google Scholar]
- 171.Koglin J, Glysing-Jensen T, Gadiraju S, Russell ME. Attenuated cardiac allograft vasculopathy in mice with targeted deletion of the transcription factor STAT4. Circulation. 2000;101:1034–1039. doi: 10.1161/01.cir.101.9.1034. [DOI] [PubMed] [Google Scholar]
- 172.Hippen BE, DeMattos A, Cook WJ, Kew CE, 2nd, Gaston RS. Association of CD20+ infiltrates with poorer clinical outcomes in acute cellular rejection of renal allografts. Am J Transplant. 2005;5:2248–2252. doi: 10.1111/j.1600-6143.2005.01009.x. [DOI] [PubMed] [Google Scholar]
- 173.Lehnhardt A, Mengel M, Pape L, Ehrich JH, Offner G, Strehlau J. Nodular B-cell aggregates associated with treatment refractory renal transplant rejection resolved by rituximab. Am J Transplant. 2006;6:847–851. doi: 10.1111/j.1600-6143.2006.01246.x. [DOI] [PubMed] [Google Scholar]
- 174.Racusen LC, Colvin RB, Solez K, Mihatsch MJ, Halloran PF, Campbell PM, Cecka MJ, Cosyns JP, Demetris AJ, Fishbein MC, Fogo A, Furness P, Gibson IW, Glotz D, Hayry P, Hunsickern L, Kashgarian M, Kerman R, Magil AJ, Montgomery R, Morozumi K, Nickeleit V, Randhawa P, Regele H, Seron D, Seshan S, Sund S, Trpkov K. Antibody-mediated rejection criteria - an addition to the Banff 97 classification of renal allograft rejection. Am J Transplant. 2003;3:708–714. doi: 10.1034/j.1600-6143.2003.00072.x. [DOI] [PubMed] [Google Scholar]
- 175.Solez K, Colvin RB, Racusen LC, Haas M, Sis B, Mengel M, Halloran PF, Baldwin W, Banfi G, Collins AB, Cosio F, David DS, Drachenberg C, Einecke G, Fogo AB, Gibson IW, Glotz D, Iskandar SS, Kraus E, Lerut E, Mannon RB, Mihatsch M, Nankivell BJ, Nickeleit V, Papadimitriou JC, Randhawa P, Regele H, Renaudin K, Roberts I, Seron D, Smith RN, Valente M. Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant. 2008;8:753–760. doi: 10.1111/j.1600-6143.2008.02159.x. [DOI] [PubMed] [Google Scholar]
- 176.Truong LD, Barrios R, Adrogue HE, Gaber LW. Acute antibody-mediated rejection of renal transplant: pathogenetic and diagnostic considerations. Arch Pathol Lab Med. 2007;131:1200–1208. doi: 10.5858/2007-131-1200-AARORT. [DOI] [PubMed] [Google Scholar]
- 177.Becker YT, Becker BN, Pirsch JD, Sollinger HW. Rituximab as treatment for refractory kidney transplant rejection. Am J Transplant. 2004;4:996–1001. doi: 10.1111/j.1600-6143.2004.00454.x. [DOI] [PubMed] [Google Scholar]
- 178.Faguer S, Kamar N, Guilbeaud-Frugier C, Fort M, Modesto A, Mari A, Ribes D, Cointault O, Lavayssiere L, Guitard J, Durand D, Rostaing L. Rituximab therapy for acute humoral rejection after kidney transplantation. Transplantation. 2007;83:1277–1280. doi: 10.1097/01.tp.0000261113.30757.d1. [DOI] [PubMed] [Google Scholar]
- 179.Pescovitz MD. Rituximab, an anti-cd20 monoclonal antibody: history and mechanism of action. Am J Transplant. 2006;6:859–866. doi: 10.1111/j.1600-6143.2006.01288.x. [DOI] [PubMed] [Google Scholar]
- 180.Thaunat O, Patey N, Gautreau C, Lechaton S, Fremeaux-Bacchi V, Dieu-Nosjean MC, Cassuto-Viguier E, Legendre C, Delahousse M, Lang P, Michel JB, Nicoletti A. B cell survival in intragraft tertiary lymphoid organs after rituximab therapy. Transplantation. 2008;85:1648–1653. doi: 10.1097/TP.0b013e3181735723. [DOI] [PubMed] [Google Scholar]
- 181.Alegre ML, Florquin S, Goldman M. Cellular mechanisms underlying acute graft rejection: time for reassessment. Curr Opin Immunol. 2007;19:563–568. doi: 10.1016/j.coi.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 182.Lanzavecchia A, Bernasconi N, Traggiai E, Ruprecht CR, Corti D, Sallusto F. Understanding and making use of human memory B cells. Immunol Rev. 2006;211:303–309. doi: 10.1111/j.0105-2896.2006.00403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science. 2002;298:2199–2202. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- 184.Sarwal M, Chua MS, Kambham N, Hsieh SC, Satterwhite T, Masek M, Salvatierra O., Jr Molecular heterogeneity in acute renal allograft rejection identified by DNA microarray profiling. N Engl J Med. 2003;349:125–138. doi: 10.1056/NEJMoa035588. [DOI] [PubMed] [Google Scholar]
- 185.Ying L, Sarwal M (2008) In praise of arrays. Pediatr Nephrol doi: 10.1007/s00467-008-0808-z [DOI] [PMC free article] [PubMed]