

Summary
Collapsin response mediator proteins (CRMPs) mediate signal transduction of neurite outgrowth and axonal guidance during neuronal development. Voltage-gated Ca2+ channels and interacting proteins are essential in neuronal signaling and synaptic transmission during this period. We recently identified the presynaptic N-type voltage-gated Ca2+ channel (Cav2.2) as a CRMP-2-interacting partner. Here, we investigated the effects of a functional association of CRMP-2 with Cav2.2 in sensory neurons. Cav2.2 colocalized with CRMP-2 at immature synapses and growth cones, in mature synapses and in cell bodies of dorsal root ganglion (DRG) neurons. Co-immunoprecipitation experiments showed that CRMP-2 associates with Cav2.2 from DRG lysates. Overexpression of CRMP-2 fused to enhanced green fluorescent protein (EGFP) in DRG neurons, via nucleofection, resulted in a significant increase in Cav2.2 current density compared with cells expressing EGFP. CRMP-2 manipulation changed the surface levels of Cav2.2. Because CRMP-2 is localized to synaptophysin-positive puncta in dense DRG cultures, we tested whether this CRMP-2-mediated alteration of Ca2+ currents culminated in changes in synaptic transmission. Following a brief high-K+-induced stimulation, these puncta became loaded with FM4-64 dye. In EGFP and neurons expressing CRMP-2–EGFP, similar densities of FM-loaded puncta were observed. Finally, CRMP-2 overexpression in DRG increased release of the immunoreactive neurotransmitter calcitonin gene-related peptide (iCGRP) by ∼70%, whereas siRNA targeting CRMP-2 significantly reduced release of iCGRP by ∼54% compared with control cultures. These findings support a novel role for CRMP-2 in the regulation of N-type Ca2+ channels and in transmitter release.
Keywords: Presynaptic Ca2+ channels, α1B/Cav2.2, CRMP-2, DRG, CGRP, Transmitter release
Introduction
At the presynaptic terminal, influx of Ca2+ and release of neurotransmitter are tightly coupled. This is achieved by the close spatial and temporal proximity of voltage-gated Ca2+ channels and the vesicular release machinery (Catterall and Few, 2008; Stanley, 1997). A variety of interactions between Ca2+ channels and presynaptic proteins contribute to the molecular and mechanistic complexities of the vesicle release cycle and synaptic transmission (Sudhof, 1995). In addition to a key role in triggering transmitter release, Ca2+ signals contribute to the dynamics of neuronal growth cones (Spitzer, 1995). That Ca2+ channels are present on growth cones (Soeda et al., 1997; Soeda et al., 1998) and support transmitter release (Hirning et al., 1988; Igarashi et al., 2000; Westenbroek et al., 1992) suggests that this `pre' synapse-like structure is endowed with all the proteins necessary for vesicular release. However, it remains unclear as to how these Ca2+ channels are targeted to release sites in growth cones. Protein-protein interactions with modular adaptor proteins (Khanna et al., 2006a; Leenders et al., 2008; Maximov et al., 1999; Szabo et al., 2006) and post-translational modifications (Zhang et al., 2008) facilitate trafficking and targeting of the voltage-gated N-type Ca2+ channel (Cav2.2) in neurons. We recently identified a novel protein, the axonal regulator collapsin response mediator protein-2 (CRMP-2) in a screen for proteins that interact with the Cav2.2 from fractions obtained from either growth cones (Brittain et al., 2009) or synaptosomes isolated from rat brains (Khanna et al., 2007c).
CRMPs were identified as a cytosolic component of semaphorin-3A (Sema 3A)-induced growth-cone collapse in dorsal root ganglia (DRG) neurons (Goshima et al., 1995). Whereas it is well established that CRMPs 1-5 promote axonal growth, neurite branching and growth-cone formation (Fukata et al., 2002; Hotta et al., 2005; Inagaki et al., 2001; Schmidt and Strittmatter, 2007), the role of CRMPs in sensory signaling is largely unexplored. Studies indicate that Sema 3A might produce its effects by causing opening of Ca2+ channels (Behar et al., 1999). In DRG neurons, most of the total Ca2+ current responsible for synaptic transmission is conducted via Cav2.2 (Fox et al., 1987; Gross and Macdonald, 1987; Scroggs and Fox, 1992a; Scroggs and Fox, 1992b). Cav2.2 is crucial for pain transduction because blocking Cav2.2 elicits an analgesic effect (Bowersox et al., 1996) and Cav2.2 knockout mice have a higher pain threshold (Hatakeyama et al., 2001; Kim et al., 2001; Saegusa et al., 2002). Regulation of presynaptic Ca2+ channels and interacting proteins has a significant impact on synaptic strength. Improper function and regulation of these Ca2+ channels contributes to epilepsy, migraine, ataxia and other neurological diseases. In this study, we have used immunocytochemistry, immunoprecipitation, biotinylation, electrophysiology and a transmitter-release radioimmunoassay to demonstrate the effect(s) of CRMP-2 on Ca2+ channel function and transmitter release in sensory neurons.
Results
CRMP-2 associates with Cav2.2 in DRG neurons
CRMP-2 is expressed in growth cones, axonal shafts and cell bodies of neurons (Inagaki et al., 2000; Yoshimura et al., 2005), where it is believed to play an important role in axonogenesis. We first confirmed that CRMP-2 was expressed in growth cones of neurons isolated from the DRG. Neurons isolated from rat DRG were fixed at 2 days in vitro (DIV) and then immunostained with anti-CRMP-2 and F-actin, the latter protein being enriched in the lamellipodia and filopodia of growth cones (Fan et al., 1993). Robust expression of CRMP-2 and F-actin was detected in both collapsed or spread growth cones (Fig. 1A).
Fig. 1.

CRMP-2 colocalizes with Cav2.2 in DRG growth cones. (A) Immunocytochemistry of DRG growth cones double-labeled with the growth-cone-enriched protein F-actin (red) and CRMP-2 (green). Note the extensive colocalization of both proteins in the lamellipodia of the collapsed (asterisk) and spread (closed arrowhead) growth cones. Staining of both proteins at the edges of the filopodia is also evident (open arrowheads). (B) Pseudocolor images of a DRG growth cone labeled with CRMP-2 (Bi) and Cav2.2 (Bii), with the merged image shown in (Biii). Inset: enlarged region of the peripheral zone of the growth cone showing extensive colocalization of CRMP-2 and Cav2.2. (C,D) The region demarcated by the white broken line in Bi generated scatter plots with strong right skews for CRMP-2 (C) and Cav2.2 (D). The ICQ value for the region shown was 0.11 (P=0<0.05).
Because growth cones are capable of Ca2+-dependent neurotransmitter release (Young and Poo, 1983) and Cav2.2 has been localized to growth cones (Bahls et al., 1998; Lipscombe et al., 1988; Vigers and Pfenninger, 1991), we next tested whether CRMP-2 and Cav2.2 proteins were present in growth cones of isolated neurons from rat DRG. Double-labeling with antibodies against Cav2.2 and CRMP-2 revealed the presence of both proteins in the peripheral, central and transitional regions of the growth cones (Fig. 1B). Cav2.2 was enriched at the distal ends of the filopodia in conjunction with CRMP-2 (Fig. 1Biii, inset). The intensity correlation analysis (ICA) was used to quantitatively assess the colocalization of Cav2.2 and CRMP-2 (Li et al., 2004). This method determines whether staining for two proteins co-vary, as would be predicted if the proteins were part of a complex. This test generates an intensity correlation quotient (ICQ), which ranges from –0.5 to +0.5 and can be used for statistical comparison. ICA plots of CRMP-2 and Cav2.2 staining of the growth cone peripheral, central and transitional zones (Fig. 1Bi, region demarcated by the broken line) exhibited positive skews (see Fig. 1C,D), suggesting a moderate level of covariance. The mean ICQ values were 0.124±0.017 (n=8, P=0<0.001), which is moderately positive within the theoretical –0.5 to 0.5 range. Thus, the staining intensities for CRMP-2 and Cav2.2 vary in synchrony, consistent with two proteins existing in a complex.
CRMP-2 and Cav2.2 were colocalized in the soma and neurites of DRG neurons grown in culture for 5-7 DIV (Fig. 2A-C), at which time a majority of the neurons exhibited extensive neurite formation. ICA plots of CRMP-2 and Cav2.2 in the membrane proximal region of the soma (Fig. 2C inset) exhibited very strong skews, consistent with two proteins that are components of a common complex. The mean CRMP-2:Cav2.2 ICQ values were 0.349±0.019 (n=8, P=0<0.001) (Fig. 2F), indicating that CRMP-2 and Cav2.2 staining are strongly correlated at the soma surface.
Fig. 2.

CRMP-2 colocalizes with Cav2.2 in DRG neurons. (A,B) Immunocytochemistry with anti-CRMP-2 (A) and anti-Cav2.2 (B) antibodies in adult rat DRG neurons cultured for 7 DIV. (C) Merged pseudocolor image (CRMP-2, red; Cav2.2, green). (D,E) ICA of the soma surface (inset in C) generated scatter plots with strong right skews for both CRMP-2 (D) and Cav2.2 (E). The intensity correlation quotient (ICQ) values for the regions shown in the inset in C was 0.295 (P=0<0.05; juxtamembrane region of DRG soma). (F) Boxplot showing the ICQ values for CRMP-2:Cav2.2 staining from cell-surface soma (n=8). (G) V5-epitope-tagged CRMP-1 and CRMP-2 proteins were expressed in heterologous cells, immunoprecipitated with V5-tag antibody and immunoblotted with CRMP-2 (top blot) or V5 antibody (lower blot). The CRMP-2 antibody recognized a single band for CRMP-2 in immunoprecipitations from cells expressing CRMP-2 but not CRMP-1. (H,I) DRG lysates were immunoprecipitated (IP) with Cav2.2 and CRMP-2 antibodies and immunoblotted with Cav2.2 (H) and CRMP-2 antibodies (I). Input lane represents 5% of the starting material used for IP. Only the light chains (LC) of the IgG were observed with an anti-rabbit IgG (light-chain-specific) secondary antibody. A representative blot from four experiments is shown. Molecular weight markers are indicated in kDa.
Co-immunoprecipitation experiments were performed to test for a biochemical complex between CRMP-2 and Cav2.2. Prior to the immunoprecipitation experiments, we established that the antibody used for the CRMP-2 immunoprecipitations was selective for CRMP-2 only because it did not cross-react with the highly homologous CRMP-1 in a heterologous expression system (Fig. 2G). Precipitation of CRMP-2 or Cav2.2 from whole rat DRG (n>60) recovered the other protein (Fig. 2H,I). No proteins were observed in control immunoprecipitations using rabbit or mouse isotype-specific IgG antibodies (Fig. 2H,I). Thus, immunocytochemical and biochemical evidence showed that CRMP-2 and Cav2.2 co-exist in a complex in vivo.
CRMP-2 increases calcium currents in DRG neurons
Using whole-cell patch-clamp recordings, the potential effects of CRMP-2 on the capacity of Cav2.2 to conduct current was examined. Current-voltage relationships in enhanced green fluorescent protein (EGFP) and neurons transfected with CRMP-2–EGFP (Zeitelhofer et al., 2007) were examined by application of 50-ms voltage steps to various test potentials from a potential of –80 mV. Peak inward Ca2+ currents were measured and expressed as peak current density (pA/pF) to account for variations in cell size. Representative current traces and examples of transfected DRG are shown in Fig. 3A and 3B, respectively. EGFP-expressing neurons showed a peak density of –124.4±25.2 pA/pF (n=6), whereas neurons expressing CRMP-2–EGFP had a maximum density of –334.3±51.2 pA/pF (n=11), a significant increase of ∼2.7-fold (Fig. 3D, P<0.05, Student's t-test). A significant amount of the Ca2+ current in these neurons was conducted via Cav2.2, as assessed from the block observed on perfusion of the Cav2.2-selective blocker ω-conotoxin GVIA (ω-CTX; Fig. 3E). After 5 minutes of application of 1 μM ω-CTX, the Ca2+ current was inhibited by 51.1±8.9% (n=4) versus 66.3±7.1% (n=9) in neurons expressing EGFP and CRMP-2–EGFP, respectively (P>0.2, Student's t-test). Treatment with short interfering RNA (siRNA) against endogenous CRMP-2 reduced CRMP-2 protein by >90% (see later) and Ca2+ currents by ∼70% (peak density of –38.1±7.3 pA/pF, n=6; Fig. 3D) compared with EGFP (peak density of –124.4±25.2 pA/pF, n=6) or ∼57% compared with scramble controls (peak density of –88.6±12.5 pA/pF, n=6). To rule out off-target effects of CRMP-2 siRNA, we measured voltage-gated outward K+ currents, which were not changed between the two groups: 768.1±197.5 (n=6; scramble siRNA) and 709.3±94.8 (n=6; CRMP-2 siRNA) pA/pF at +40 mV (P>0.5, Student's t-test; Fig. 3F,G).
Fig. 3.

CRMP-2 increases Ca2+ current density in DRG neurons. (A) Exemplar current traces obtained from DRG neurons transfected with EGFP, CRMP-2–EGFP or CRMP-2 siRNA. Currents were evoked by 50-millisecond steps in 10 mV increments applied from a holding potential of –80 mV (see voltage protocol above the traces). Current traces are shown for every 10-mV step between –80 and +30 mV. Bath solutions contained 1 μM TTX, 10 mM TEA and 1 μM nifedipine to block Na+, K+ and L-type voltage-gated Ca2+ channels, respectively. Lines labeled 0 indicate the zero-current level. (B) Representative fluorescence and differential interference contrast (DIC) images of CRMP-2–EGFP- and EGFP-transfected DRG neurons. Scale bar: 20 μm. (C) Current-voltage relationships for the currents shown in A as well as for currents from cells transfected with scramble siRNA. Peak currents were normalized to cell capacitance. The Ca2+ current density was significantly greater in CRMP-2-EGFP neurons than that in EGFP between 0 to +50 mV (P<0.05, Student's t-test). Values are mean ± s.e.m. and some error bars are smaller than the symbols. (D) Peak current density (pA/pF) measured at +10 mV for EGFP (n=6) is significantly smaller than that for neurons transfected with CRMP-2–EGFP (n=11) (**P<0.01, Student's t-test). Peak current density for CRMP-2 siRNA (n=6) is significantly smaller than for scramble siRNA (*P<0.05, Student's t-test). Peak current density values are not different between EGFP and scramble siRNA (P>0.2, Student's t-test). (E) Percent inhibition of Ca2+ current via Cav2.2, at 5 minutes post-perfusion with 1 μM ω-CTX, in DRGs expressing EGFP (n=4) and CRMP-2–EGFP (n=9). (F) Representative K+ currents from a DRG neuron transfected with CRMP-2 siRNA (left) and scramble siRNA (right) in response to 10-mV voltage steps from –80 to +60 mV from a holding potential of –60 mV. (G) Current-voltage relationship for DRG neurons treated with scramble and CRMP-2 siRNA (n=6 each). Peak K+ currents were not different between the two conditions for any of the voltages tested.
Surprisingly, overexpression of CRMP-2 produced a significant shift in the reversal potential for the Ca2+ current (see Fig. 3C). In recordings from those neurons overexpressing CRMP-2, the reversal potential was 52±2 mV (n=11) whereas the reversal potential was 34±4 mV (n=6), 34±3 mV (n=6), and 35±4 mV (n=11) for neurons expressing EGFP, CRMP-2 siRNA, and scramble siRNA, respectively (P<0.05, ANOVA, Tukey post-hoc). The origins of this shift are presently unclear (see Discussion).
Because changes in current amplitude could result from changes in channel gating, the voltage-dependent properties of Ca2+ currents were assessed in these neurons. The steady-state activation of Ca2+ channels from neurons overexpressing EGFP and CRMP-2–EGFP was well described by the Boltzmann relation: G/Gmax=1/{1+exp[(V-V50%)/kn]}, where G is peak conductance, Gmax is fitted maximal G, V50% is half-activation voltage, and kn is the slope factor. As shown in Fig. 4A, for EGFP-expressing neurons, V50% was –7.8±1.1 mV (n=6), which was not significantly different from that of CRMP-2–EGFP-transfected neurons (–6.8±1.1 mV, n=9; P>0.05, Student's t-test). The slope factors for the Boltzmann fits (mV/e-fold change in conductance) were also similar (not shown). Activation time constants, measured from single exponential fits to the activation phase of the inward current, were significantly different at 0, 10 and 20 mV between the two groups of neurons (Fig. 4B; P<0.05, Student's t-test). At 10 mV, the mean activation time constant for EGFP-expressing neurons was 13.7±5.9 milliseconds (n=5), whereas that of CRMP-2–EGFP neurons was significantly faster, measuring 2.2±0.2 milliseconds (n=9).
Fig. 4.
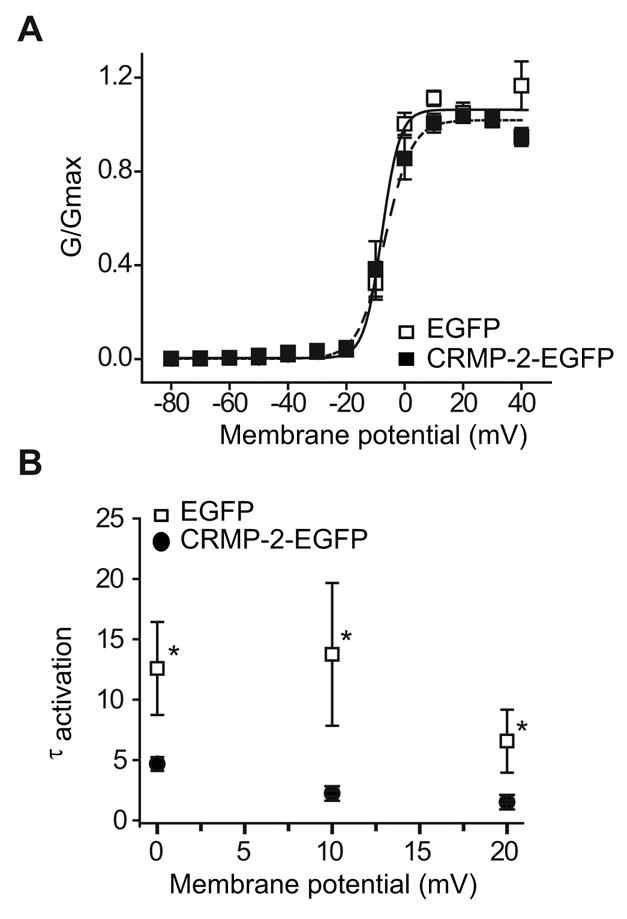
Effect of CRMP-2 on DRG Cav2.2 activation. (A) Normalized conductance (G) versus voltage for DRG neurons transfected with EGFP or CRMP-2–EGFP. Values are mean ± s.e.m. and most error bars are smaller than the symbols. (B) Plots of activation time constants (τactivation) were calculated from single exponential fits to the falling phase of the voltage-activated inward current. The mean activation time constants for CRMP-2–EGFP currents were significantly faster than those for EGFP (P<0.05, Student's t-test).
The voltage-dependent properties of inactivation were determined by applying 5-second conditioning pre-pulses that ranged successively from –80 mV to +40 mV in 10-mV voltage steps, followed by a 50-millisecond second step depolarization to 10 mV (Fig. 5A). The relationship of normalized test pulse voltage to peak current amplitude was plotted against its corresponding holding potential and fitted with the Boltzmann equation. The voltage dependence of inactivation was similar for currents recorded from neurons expressing EGFP and CRMP-2–EGFP, as shown by the conductance versus voltage relationship (Fig. 5B). For EGFP-expressing neurons, V50% was –19.1±3.2 mV (n=5), which was not significantly different from that of CRMP-2–EGFP-transfected neurons (–16.1±2.2 mV, n=9; P>0.05; Fig. 5C). The slope factors were also similar: 5.8±1.1 mV (n=6) for EGFP-transfected neurons and 8.3±0.9 (n=9; P>0.05) for CRMP-2–EGFP-transfected neurons. These results indicate that CRMP-2 augmented the density for currents carried by ω-CTX-sensitive channels but had no effect on either the activation or inactivation properties.
Fig. 5.

Effect of CRMP-2 on DRG Cav2.2 inactivation. (A) Top panel: voltage protocol. Currents were evoked by voltage steps from –80 to +40 mV in 10-mV increments prior to delivering a test pulse to +10 mV. Bottom two panels: exemplar current traces from EGFP and CRMP-2–EGFP transfected neurons. (B) Normalized test pulse peak current amplitude plotted against its preceding holding potential and fitted with the Boltzmann relation. (C) Plot of mean V50% for EGFP and CRMP-2–EGFP showed no difference between the groups.
CRMP-2 localizes to functional synapses
To evaluate the effect of CRMP-2 on synaptic transmission, we first examined the distribution of endogenous CRMP-2 in cultured DRG neurons. Sensory neurons grown for 5-7 DIV exhibited extensive neurites that were labeled with an antibody against βIII tubulin, a neuron-specific protein (Fig. 6A). Co-labeling with an antibody against CRMP-2 revealed punctate distribution of CRMP-2 along axonal neurites (Fig. 6A, green). Double-labeling of sensory neurons with antibodies against CRMP-2 and the synaptic vesicle marker synaptophysin (Ichikawa et al., 1991) revealed extensive colocalization of CRMP-2 in synaptophysin-positive puncta (Fig. 6B), wherein 85±12% (n=468) of synapses were labeled with both proteins (Fig. 6D). To test whether endogenous CRMP-2 was targeted to functionally active synapses and/or release sites, neurons were loaded with the membrane-bound dye FM4-64 by stimulation with 90 mM KCl. Neurons loaded with FM dye were then fixed and labeled with an antibody against CRMP-2 (Fig. 6Ci). Of the functionally active FM-dye-loaded release sites, 78±17% (n=347) were positive for CRMP-2, thus indicating that CRMP-2 is targeted to functional synapses (Fig. 6Ciii,D).
Fig. 6.

Endogenous CRMP-2 colocalizes to synaptic boutons. (A) Representative confocal immunofluorescence image of DRGs stained with a monoclonal antibody against βIII tubulin (red) and polyclonal (p) CRMP-2 (green). CRMP-2 stains distinct punctate structures (arrowheads) along DRG neurites. (B) Confocal black and white image of DRG neurites double-labeled with a monoclonal CRMP-2 antibody (green) and a polyclonal synaptophysin antibody (red). The merged image shows that CRMP-2 protein localizes to synapses identified by the synaptophysin antibody. (C) DRGs grown for 7 DIV were loaded with FM4-64 (red) and then fixed and stained with a monoclonal (m) antibody against CRMP-2 and identified with an FITC-conjugated goat anti-mouse secondary antibody (green). Note the colocalization of CRMP-2 with FM4-64 in active synapses. (D) Quantification of the percentage of CRMP-2-positive puncta that were also labeled with synaptophysin (synp.) or FM4-64. A total of 468 (n=7 neurons) and 347 puncta (n=8 neurons) were counted for synaptophysin and FM4-64, respectively.
We next determined the density of release sites in DRG neurons overexpressing EGFP or CRMP-2–EGFP, because a change in the density could potentially impact synaptic transmission. Functionally active release sites were loaded with FM4-64 by a 1-minute stimulation with 90 mM KCl. Cells were then fixed and imaged for EGFP and FM dye fluorescence (Fig. 7). No FM dye was taken up by synapses prior to stimulation (Fig. 7B). Following stimulation, FM4-64 was observed in synapses of neurons expressing CRMP-2–EGFP (Fig. 7D-F) or EGFP (Fig. 7G-I). The number of FM4-64-loaded release sites and the EGFP fluorescence along neurites were counted in seven neurons expressing EGFP and eight neurons expressing CRMP-2–EGFP. The density of FM-dye-loaded synapses was expressed as the number of release sites per micrometer of axonal process. Neurons expressing CRMP-2–EGFP exhibited 0.19±0.045 sites/μm, which was not significantly different from those expressing EGFP alone (0.13±0.029 sites/μm; P>0.05, Wilcoxon-Mann-Whitney test). These results indicate that CRMP-2 is targeted to synapses but CRMP-2 overexpression does not affect the density of functional synapses.
Fig. 7.

CRMP-2 does not affect synapse density in DRG neurons. (A-I) DRGs grown in culture for 5 DIV were transfected with CRMP-2–EGFP (A–F) or EGFP (G–I) and then incubated for 1 minute without (A–C) or with 90 mM KCl (D–I) for 1 minute to label release sites. Representative confocal images of neurites are shown. Note the lack of FM4-64 staining in the absence of stimulation. In high-K+-stimulated neurons, EGFP fluorescence of cells transfected with EGFP and CRMP-2–EGFP colocalizes with FM4-64 (I and F). (J) Quantification of the number of FM dye-loaded synapses in neurons transfected with EGFP or in cells transfected with CRMP-2–EGFP (CRMP-2). A total of 175 (n=9 EGFP neurons) and 184 puncta (n=13 CRMP-2–EGFP neurons) were counted.
CRMP-2 modulates evoked transmitter release from DRG neurons
Because sensory neurons overexpressing CRMP-2 had increased Ca2+ current density, we tested whether this increased intracellular Ca2+ affected transmitter release. It has been previously shown that Ca2+ influx via Cav2.2 underlies release of the neuropeptide transmitter calcitonin gene-related peptide (CGRP) (Evans et al., 1996). Therefore, to determine whether CRMP-2 can modulate transmitter release, the KCl-stimulated release of CGRP was measured from sensory neurons transfected with CRMP-2–EGFP or EGFP. Additionally, to test the role of CRMP-2, sensory neurons were grown in the absence or presence of siRNA targeted to CRMP-2 or control scramble siRNA. The levels of basal or resting release of immunoreactive CGRP (iCGRP) were not significantly different between the groups: 1.03±0.10% total peptide content/10 minutes (n=12 wells) in untreated neurons, 0.84±0.07% total peptide content/10 minutes (n=16 wells) in EGFP-transfected neurons, 0.80±0.07 total peptide content/10 minutes (n=16 wells) in CRMP-2–EGFP-transfected neurons, 0.95±0.07% total peptide content/10 minutes (n=24 wells) in scramble-treated neurons, and 0.74±0.04% total peptide content/10 minutes (n=24 wells) in CRMP-2 siRNA-treated DRG. A 10-minute stimulation with 50 mM KCl evoked a robust increase in iCGRP release in both EGFP and CRMP-2–EGFP-transfected neurons: 12.33±0.71% total peptide content/10 minutes (n=16) and 19.74±1.00% total peptide content/10 minutes (n=16), respectively. Thus, CRMP-2 overexpression caused a significant ∼1.7-fold increase in stimulated release compared with EGFP-transfected cells (Fig. 8A) and is consistent with the effects of CRMP-2 on the Ca2+ currents. The increase in KCl-stimulated iCGRP release observed in CRMP-2–EGFP-transfected neurons was not caused by an increase in the total cellular content of iCGRP as there was no significant difference in neuropeptide content between the two conditions (Fig. 8B): 1436.6±51.1 fmol/well for EGFP-transfected DRG and 1428.2±76.2 fmol/well for CRMP-2–EGFP-transfected DRG (P>0.9, Student's t-test; n=16 wells each).
Fig. 8.

CRMP-2 affects K+-stimulated transmitter release in DRG neurons. Adult mouse DRG neurons were maintained in culture for 5-7 days prior to the release experiments. (A) Bar graph of iCGRP release expressed as mean percent total iCGRP content of cells in each well ± s.e.m. (n=12-24 wells per condition). Neuropeptide release was measured from cells treated with normal HEPES buffer containing 3.5 mM KCl (basal, B), HEPES buffer containing 50 mM KCl (S), and again with HEPES buffer containing 3.5 mM KCl. The Cav2.2 inhibitor, ω-CTX, at 5.0 μM or 500 nM, was included in the 10 minutes prior to and throughout the high K+ exposures. Total inhibitor exposure time was 20 minutes. Asterisks (*) indicate statistically significant differences in iCGRP release between treatment groups and the control (no treatment) using an ANOVA with Dunnett's post-hoc test (P<0.05). The hash mark (#) indicates a statistically significant difference between the scramble- and CRMP-2-siRNA-treated cells (P<0.05; Student's t-test). In all cases, release stimulated by high extracellular K+ was significantly higher than basal release. (B) Total content of iCGRP measured at the end of the release experiment. There were no significant differences in iCGRP content between the conditions tested. (C) Western blot analysis with the indicated antibodies showing successful knockdown of CRMP-2 protein in neurons treated with 200 nM siRNAs for 4 days. Under the same conditions, expression of the neuronal protein β-tubulin remained unchanged. Lysates from untreated and scramble-siRNA-treated DRGs showed no differences in protein expression for either CRMP-2 or β-tubulin. Knockdown was assessed by quantifying densities of CRMP-2 for each treatment and then normalizing them to their respective β-tubulin densities (treatment CRMP-2 density/β-tubulin). This value was then divided by the normalized density obtained for the parallel untreated control (control CRMP-2 density/β-tubulin). For the blot shown, treatment with CRMP-2siRNA reduced CRMP-2 by ∼90% compared to treatment with scramble (Sc) siRNA. A representative blot from two experiments is shown.
A 10-minute stimulation with 50 mM KCl evoked a robust increase (∼12-fold over basal) in iCGRP release in both untreated and scramble siRNA-treated neurons: 12.52±2.72% total peptide content/10 minutes (n=12) and 12.24±0.54% total peptide content/10 minutes (n=24), respectively, demonstrating that treatment with siRNA does not impact the ability of these neurons to release neurotransmitter. By contrast, KCl-stimulated iCGRP release from DRG neurons maintained in culture for 7 days and treated with CRMP-2 siRNA for 48 hours preceding the radioimmunoassay (see Materials and Methods), reduced the release to 54% less than that observed in DRG treated with scramble siRNA or in untreated DRG (5.96±2.13% total peptide content/10 minutes, n=24). This indicates that a major portion of the K+-stimulated neuropeptide transmitter occurs via a CRMP-2-associated mechanism. The decrease in KCl-stimulated iCGRP release observed in neurons treated with CRMP-2 siRNA was not caused by a decrease in the total cellular content of iCGRP because there was no significant difference in neuropeptide content in any of the three conditions (Fig. 8B): 1367.53±99.78 fmol/well for untreated DRG, 1241.53±49.44 fmol/well for scramble siRNA-treated DRG, and 1319.72±43.75 fmol/well for CRMP-2-siRNA-treated DRG, n=12–24 wells. The KCl-stimulated increase in iCGRP occurred via N-type Ca2+ channels, and ω-CTX inhibited iCGRP release by ∼63% at 500 nM and ∼90% at 5 μM compared with untreated or scramble siRNA-treated DRG (Fig. 8A). These results indicate that CRMP-2 expression levels have an effect on the release of the neuropeptide transmitter iCGRP in sensory neurons.
CRMP-2 increases surface expressed Cav2.2
To test whether there is a change in cell surface Cav2.2 following manipulation of CRMP-2 levels, biotinylation experiments were performed as previously described (Chan et al., 2007). Immunoblotting with Cav2.2 for streptavidin-enriched complexes from biotinylated DRG neurons grown in culture for 7 DIV showed an ∼60% increase in cell-surface expression of Cav2.2 in neurons expressing CRMP-2-EGFP compared with EGFP neurons (Fig. 9A, top panel, and Fig. 9B). Knockdown of CRMP-2 using siRNA targeted to CRMP-2 reduced surface Cav2.2 levels by ∼84% compared with scramble siRNA and by ∼90% compared with overexpressed CRMP-2 (Fig. 9B). To confirm that CRMP-2-mediated increase in channel expression was specific to Cav2.2, we measured the cell-surface expression of the voltage-gated K+ channel Kv1.1, a channel that is expressed at robust levels in DRG neurons (Chi and Nicol, 2007). The cell-surface expression of Kv1.1 was not affected by CRMP-2 manipulation (Fig. 9B). Taken together, these results suggest that CRMP-2 affects cell-surface trafficking of Cav2.2.
Fig. 9.
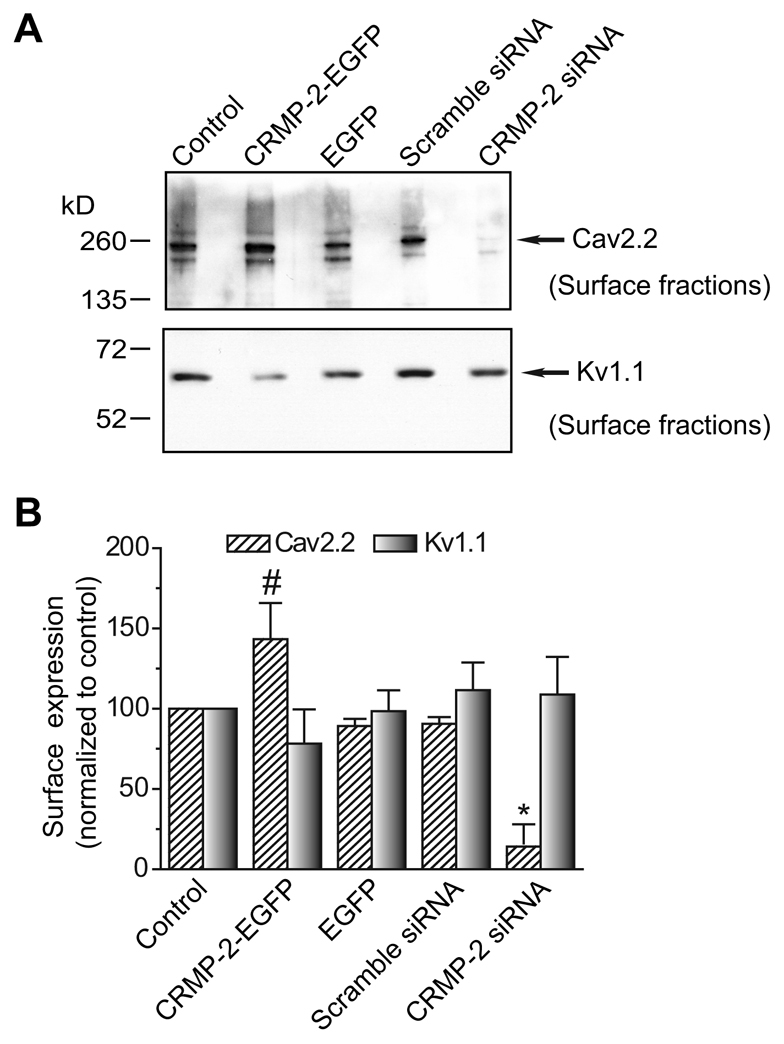
CRMP-2 affects Cav2.2 surface expression. (A) DRG neurons grown in culture were transfected as indicated and then biotinylated with EZ-link Sulfo NHS-SS-biotin 2 days after transfection. Equal amounts of biotinylated proteins precipitated with streptavidin agarose beads were resolved by electrophoresis and subjected to immunoblot analysis with Cav2.2 (top blot) and Kv1.1 (bottom blot). (B) Amount of biotinylated (surface) Cav2.2 or Kv1.1 protein, calculated by densitometry from three experiments. Values were normalized to the untransfected (control) cells for each condition. The surface levels of Cav2.2 were significantly higher in CRMP-2-EGFP neurons than that in EGFP neurons (#P<0.05, Student's t-test). The surface Cav2.2 levels were significantly lower in neurons treated with CRMP-2 siRNA than in neurons treated with scramble siRNA (*P<0.05, Student's t-test). Levels of Kv1.1 were not significantly different between any of the conditions tested.
Discussion
In this study, we identify a novel role for CRMP-2 in the modulation of the properties of Ca2+ channels, leading to a change in the release of a neuropeptide transmitter. The axonal regulator CRMP-2 colocalized and associated with Cav2.2, increased Ca2+ current density and accelerated activation kinetics of this current. CRMP-2 manipulation affected Cav2.2 expression at the plasma membrane. CRMP-2, both endogenous and overexpressed, was targeted to synapses. Notably, CRMP-2 overexpression and siRNA-targeted knockdown, resulted in a significant enhancement and reduction, respectively, in Cav2.2-mediated transmitter release of CGRP. Thus, by functionally coupling to Ca2+ channels in synapses, CRMP-2 might serve as a mechanistic link between the synaptic vesicle machinery and presynaptic Ca2+ channels that has significant modulatory effects on transmitter release.
A novel mechanism for presynaptic targeting of Ca2+ channels by CRMP-2
It has been established that Ca2+ influx via presynaptic Ca2+ channels is crucial for the release of neurotransmitters at the nerve terminal (Augustine, 2001; Catterall and Few, 2008; Stanley, 1997). Functional Ca2+ channels contributing to transmitter release have been observed very early during synaptogenesis (Hall and Sanes, 1993; Igarashi et al., 2000; Poo et al., 1985; Vigers and Pfenninger, 1991), suggesting that precursors to synaptic terminals (i.e. growth cones) contain the synaptic vesicle machinery obligatory for synaptic transmission. Our finding that CRMP-2 colocalized with Cav2.2 at the lamellipodia and filopodia in growth cones suggests that this interaction might serve to target Ca2+ channels to presynaptic release sites. This interaction is further supported by our finding that CRMP-2 is present in synapses through its colocalization with synaptophysin (Fig. 6) as well as in functional synapses, as identified by loading with the synaptic vesicle recycling dye FM4-64 (Fig. 7).
Whether the channels are trafficked to synapses by CRMP-2 is unclear and warrants further investigation. From our studies and those of others, it is clear, however, that this targeting is achieved through interaction of the presynaptic Ca2+ channel with a growing number of proteins known to be involved in synaptic vesicle exocytosis and endocytosis pathways (Catterall and Few, 2008; Chan et al., 2007; el Far et al., 1995; Evans and Zamponi, 2006; Khanna et al., 2007c; Khanna et al., 2007a; Khanna et al., 2007b; Kisilevsky and Zamponi, 2008; Stanley, 1997; Szabo et al., 2006). For example, syntaxin 1, the cardinal Cav2.2 partner protein (Sheng et al., 1994), is important for the incorporation of Ca2+ channels into the release site apparatus. A region in the cytoplasmic II-III loop of Cav2.2 is involved in presynaptic targeting of Cav2.2 into active zones in hippocampal neurons (Szabo et al., 2006). However, this region is neither necessary nor sufficient for axonal targeting of Cav2.2, implying that additional mechanisms, such as protein-protein interactions of Cav2.2 with other synaptic proteins, are needed for targeting of these channels to axons. Cav2.2 is found in a complex with proteins of the vesicle fusion module, a multimolecular complex that forms the core of the transmitter release site (Khanna et al., 2007a) as well as proteins associated with endocytosis (Chen et al., 2003; Khanna et al., 2007b). CRMP-4b, a CRMP-2-variant, is localized to synaptic vesicles in growth cones and interacts with intersectin (Quinn et al., 2003); the latter protein also interacts with Cav2.2 (Khanna et al., 2007b). Notably, intersectin interacts with other Cav2.2-interacting proteins (SNAP-25 and dynamin) that are involved in endocytosis (Khanna et al., 2007b; Okamoto et al., 1999; Yamabhai et al., 1998). Recently, light chain 2 of microtubule-associated protein (MAP1A) was shown to anchor Cav2.2 to the actin cytoskeleton in synapses made by hippocampal neurons (Leenders et al., 2008). Cav2.2 also associates with actin and β-tubulin (Khanna et al., 2007c; Phillips et al., 2001). The finding that CRMP-2 associates with β-tubulin (Fukata et al., 2002; Nishimura et al., 2003) and causes actin reorganization as well as axonal protein trafficking via interaction with the light chain of the motor protein kinesin-1 (Kimura et al., 2005) suggests that cytoskeletal interactions might contribute to presynaptic trafficking of channels as well. Thus, by binding to Ca2+ channels, other synaptic and/or cytoskeletal proteins and motor proteins, CRMP-2 might serve to bridge exocytosis and endocytosis machineries, thereby facilitating synaptic transmission.
Presynaptic Ca2+ channel activity can be modulated by CRMP-2
CRMP-2 overexpression caused an increase in current density whereas knockdown resulted in a decrease, suggesting a direct functional link between the two proteins. The biophysical parameters to be significantly affected by CRMP-2 expression were the shift in the reversal potential to more depolarized voltages (Fig. 3C) and the kinetics of activation, wherein Ca2+ currents activated faster in CRMP-2-overexpressing neurons (Fig. 4B). It is not clear why overexpression of CRMP-2 shifts the reversal potential ∼15 mV to the right. In support of this observation, overexpression of the neuronal calcium sensor-1 protein in growth cones produced a large leftward shift in the reversal potential of the Ca2+ current in Lymnaea neurons [see figure 8C2 in Hui and Feng (Hui and Feng, 2008)]. The authors did not discuss the mechanism of this shift. One can speculate that CRMP-2 overexpression might alter the intracellular Ca2+ concentration immediately around the channel. The Nernst equation was used to perform some elementary calculations (using 2 mM extracellular [Ca2+]), which would indicate that overexpression of CRMP-2 could reduce intracellular [Ca2+] by about fourfold and might lower the effective [Ca2+] around the channel. In addition, it is possible that association of CRMP-2 with Cav2.2 somehow alters the screening of membrane surface charge around the channel, potentially altering its permeability. Elevations in extracellular [Ca2+] or a lowering of intracellular [Ca2+] can produce a small rightward shift in the reversal potential for the sodium current, which was attributed to alterations in screening of surface charge. However, this shift was also accompanied by a significant decrease in the amplitude of the sodium current (Kostyuk and Krishtal, 1977; Ohmori and Yoshii, 1977). It is possible that overexpression of CRMP-2 might have a similar action in the DRG neurons and remains an area of future investigation.
Whereas changes in current density cannot be fully accounted for by changes in activation kinetics, they might be related to an increased insertion of Cav2.2 channels in the membrane and/or result from changes in the open probability of the channel. In support of the first possibility, we observed a CRMP-2-dependent regulation of Cav2.2 trafficking to the cell surface (Fig. 9). Although an increase in CaV2.2 incorporation at the surface was observed in CRMP-2-overexpressing cells, this increase was not of the same magnitude as the robust decrease of CaV2.2 incorporation observed when CRMP-2 was knocked down. The difference in the magnitude of these changes can be attributed to the moderate level of transfection of CRMP-2–GFP achieved in our cultures, as opposed to the efficiency of the siRNA construct. Surprisingly, despite an increase in Ca2+ currents in CRMP-2 overexpressing neurons, we did not observe a corresponding increase in ω-CTX sensitivity (Fig. 3E). This could be due, in part, to CRMP-2 regulation of non N-type Ca2+ channels in DRG neurons, such as the L-, P/Q- and T-type channels (Fox et al., 1987; Scroggs and Fox, 1992a). L-type channels were blocked with nifedipine under our recording conditions, whereas T-type channels, which are conotoxin- and nifedipine-resistant (Fox et al., 1987), are also biophysically distinct (Fox et al., 1987); thus ruling out these channel types. Although we did not explore the possibility that CRMP-2 regulates P/Q-type Ca2+ channels in this study, P/Q channels might be involved because block of N-type channels with 500 nM ω-CTX reduced evoked iCGRP release by ∼63% compared with untreated neurons. This observation leaves about a third of the evoked release unaccounted for (Fig. 8A). Alternative splicing of the Cav2.2 has been reported and might also account for the lack of sensitivity (Bell et al., 2004; Lin et al., 2004; Pan and Lipscombe, 2000). However, further studies are needed to address whether the mechanism of this CRMP-2-mediated Ca2+ increase can be explained by increased stability, post-translational modifications and/or other factors affecting the two proteins.
CRMP-2 is a multi-phosphorylated protein in neurons that can be regulated by several kinases, such as glycogen synthase kinase-3β (GSK-3β), cyclin-dependent kinase 5 (cdk5), and Rho kinase (Schmidt and Strittmatter, 2007). Phosphorylation by GSK-3β and/or Rho kinase reduces the affinity of CRMP-2 for tubulin, leading to arrest of axonal growth and collapse of growth cones (Schmidt and Strittmatter, 2007). Like tubulin (Fukata et al., 2002), Cav2.2 seems to bind preferentially to the presumably non-phosphorylated, but active, form of CRMP-2. Binding of Cav2.2 and tubulin to the active form of CRMP-2 in the distal parts of the growth cone are entirely consistent with the roles served by these proteins at these locations – Ca2+ influx and synaptic transmission, and axon growth, respectively. Finally, whereas the modulator adaptor protein Ca2+/calmodulin-dependent serine protein kinase (CASK) binds to Cav2.2 (Khanna et al., 2006b; Maximov et al., 1999), it was recently shown that cdk5 phosphorylation of CASK frees it to interact with proteins of the presynaptic machinery, including Cav2.2 (Samuels et al., 2007). Because cdk5-mediated phosphorylation of CASK increased Ca2+ currents via Cav2.2 (Samuels et al., 2007), it is possible that cdk5 affects Cav2.2 indirectly by modulating CRMP-2 phosphorylation and thus Cav2.2 activity.
CRMP-2 modulates transmitter release
Our findings that CRMP-2 localizes to functional synapses (Figs 6, 7) suggest a potential role for CRMP-2 in transmitter release. Because the power-law relationship between Ca2+ and transmitter release varies from an exponent of 1 to 5, depending on the synapse, even minute changes in Ca2+ concentration can have a dramatic impact on transmitter release (Dodge, Jr and Rahamimoff, 1967; Llinas, 1982). Several neurotransmitters are known to be present in and responsible for the actions of sensory neurons, including serotonin, glutamate, substance P (SP) and CGRP (Hokfelt et al., 1988; Schoenen et al., 1989; Wiesenfeld-Hallin et al., 1984). However, glutamate and the peptide transmitters, including SP and CGRP, are the major transmitters used by the small- and medium-diameter sensory neurons (Garry et al., 1989; Kangrga and Randic, 1991; Wiesenfeld-Hallin et al., 1984). Therefore, we tested the release of the 37-amino acid neuropeptide, CGRP. CGRP is released from the peripheral and central branches of primary sensory nerves (Skofitsch and Jacobowitz, 1985a; Skofitsch and Jacobowitz, 1985b; Skofitsch and Jacobowitz, 1985c). Considerable evidence suggests that CGRP plays a key role in neurogenic inflammation, as well as in modulation of pain transmission (Miletic and Tan, 1988; Ryu et al., 1988a; Ryu et al., 1988b; Schon et al., 1985). High concentrations of K+ can be used as a non-selective stimulus to depolarize primary sensory afferents of the DRG and cause robust release of CGRP, which can be reliably measured using radioimmunoassay (Hingtgen and Vasko, 1994b). Because CRMP-2 overexpression produced an increase in Ca2+ current density and accelerated the activation kinetics of Cav2.2 channels, we sought to determine whether this increased Ca2+ translated into a change in transmitter release. Indeed, we observed a 1.7-fold increase in KCl-stimulated iCGRP from sensory neurons overexpressing CRMP-2 compared with control EGFP overexpressing DRG (Fig. 8A). Probably, this is an underestimate of the ability of CRMP-2 to augment evoked iCGRP release because consistently high levels of transfected cells were not attained using various transfection techniques (Metafectene, Lipofectamine, Ca2+ phosphate, nucleofection electroporation), whereas the size of the CRMP-2 protein precluded use of lentivirus overexpression.
In addition to the overexpression studies, an approach using siRNA reduced CRMP-2 levels to ∼5% of normal. This reduction produced a corresponding significant reduction in both the Ca2+ current density and reduction in iCGRP release in DRG neurons (Fig. 3D and Fig. 8A). The KCl-evoked iCGRP release occurred primarily via Cav2.2 because ω-CTX blocked the release (Fig. 8A), consistent with an important role of Cav2.2 in mediating transmitter release. Interestingly, only evoked release was affected; basal release or outflow of iCGRP was not changed. Thus, it is probable that CRMP-2 affects activity-dependent (depolarization-induced) transmitter release, as would be expected from its effect on the activation kinetics of the Ca2+ currents. Upon depolarization, CRMP-2 might preferentially interact with the synaptic vesicle machinery to increase transmitter release. In support of this idea, in preliminary immunoprecipitation experiments (J.M.B. and R.K., unpublished data), depolarization with 50 mM KCl (5 minutes) of synaptosomes from neonatal rat brains caused a dramatic increase in the amount of CRMP-2 binding to the presynaptic cytomatrix active zone proteins bassoon and piccolo (Dresbach et al., 2001).
It is important to note that the release of neuropeptide transmitters, such as CGRP, from large dense-core vesicles (LDCVs), does not always occur at classical presynaptic active zone structures. Whereas SVs might be located at a distance of ∼20 nm from the calcium channel, LDCVs might reside as far as 300 nm away from the channels (Park and Kim, 2009), a spatial separation suggestive of differences in the calcium-dependence of release. However, FM4-64 has been used to examine cycling of LDCVs in trigeminal ganglia, in which CGRP is a major peptide transmitter and where the physiology is similar to the DRG (Matsuka et al., 2007). In addition, the stimulus-evoked release of CGRP from isolated sensory neurons occurs in a calcium-dependent manner (Hingtgen and Vasko, 1994a). For these reasons, we suggest that the stimulus-evoked release of CGRP occurs in a calcium-dependant manner from areas of LDCV exocytosis in a regulated fashion. This is consistent with electron microscopy studies (Alvarez et al., 1993; Hayes and Carlton, 1992) and functional investigations in the literature (Hingtgen and Vasko, 1994a; Kitamura et al., 2009; Matsuka et al., 2007). Because synaptic vesicles and LDCVs share similarities in the core aspects of the machinery involved in transmitter release (Edwards, 1998), our results showing the synaptic localization of CRMP-2 suggests that a CRMP-2-mediated increase in neurosecretion might occur via both classical and non-classical mechanisms. However, it remains possible that CRMP-2 regulation of calcium-channel function can impact these release processes in different ways.
Potential link of CRMP-2 with `calciumopathies' and pain
CRMP proteins have been implicated in both developmental and adult neurological diseases (Schmidt and Strittmatter, 2007). A common element of these diseases is alteration of Ca2+ homeostasis, which appears to play a key role in the mechanisms of the neuronal or axonal injury underlying these diseases. These `calciumopathies' include Alzheimer's disease (Yoshida et al., 1998), paraneoplastic neurological syndromes (Honnorat et al., 1999), cerebral ischemia and stroke (Chen et al., 2007), neuroinflammation (Chen et al., 2007) and Down's syndrome (Weitzdoerfer et al., 2001). CRMP-2 levels are altered in people with epilepsy and in animal models of epilepsy (Ryu et al., 2008). CRMP-2 levels were found to correlate with increased depression in an animal model of chronic stress (Bisgaard et al., 2007). Calcium-dependent CRMP-2 proteolysis has been observed in traumatic brain injury and cerebral ischemia (Hou et al., 2006) and might be a limiting factor for post-injury axonal regeneration. Excitotoxicity and oxidative stress induce Ca2+ influx via voltage-gated Ca2+ channels (Kowara et al., 2005), which elicits CRMP cleavage, thus providing an additional functional link between CRMPs and Ca2+ channels. Cav2.2 is crucial for pain transduction; blocking Cav2.2 relieves hyperalgesia (Bowersox et al., 1996) and Cav2.2 knockout mice have a higher pain threshold (Hatakeyama et al., 2001). Injection of CGRP into thecal space also produces hyperalgesia (Oku et al., 1987). Furthermore, inflammatory hyperalgesia can be abolished by a CGRP-blocking antibody (Ambalavanar et al., 2006). Experimental and clinical studies have shown that CGRP release contributes to the generation of pain during migraine, suggesting that blocking CGRP release has promising therapeutic potential (Durham, 2006; Durham, 2008). Proteins interacting with Ca2+ channels, such as CRMP-2, thus represent a novel target for manipulation of Ca2+ homeostasis in neurological diseases. In addition, the role of CRMP-2 in primary sensory afferent signaling suggests that the CRMP-2–Cav2.2 complex represents a novel target for pain regulation and future therapeutics.
Materials and Methods
Materials
C57BL/6 mice (3-6 months old) and Sprague-Dawley rats (100-150 g) were purchased from Harlan Laboratories (Indianapolis, IN). Media, serum and antibiotics were purchased from Invitrogen, whereas nerve growth factor (NGF) was from Harlan Bioproducts. Antibody to CGRP was provided by Michael Vasko (Indiana University School of Medicine, Indianapolis, IN and originally produced by Michael J. Iadarola, NIH, Bethesda, MD). Other antibodies were from commercial suppliers: synaptophysin and polyclonal CRMP-2 (Cell Signaling Technology, MA), monoclonal CRMP-2 (Abcam, Cambridge, MA), α1B/Cav2.2 (Calbiochem, San Diego, CA), Kv1.1 (Alomone Labs, Israel), V5-epitope (GKPIPNPLLGLDST) tag (Invitrogen,), rabbit isotype-specific control IgG (Sigma). Horseradish peroxidase (HRP) and Alexa-Fluor-conjugated secondary antibodies were from Sigma Aldrich and Invitrogen, respectively. Scramble siRNA, with the same GC% but no sequence homology, designed as a scramble for apurinic/apyrimidinic endonuclease 1, was from Dharmacon (Lafayette, CO). Validated siRNAs against the rat or mouse CRMP-2 [5′-ACTCCTTCCTCGTGTACAT-3′] sequence (Nishimura et al., 2003) were from Invitrogen. All other reagents were from Sigma.
Co-immunoprecipitation and immunoblotting
Lysate from ∼60 ganglia were lysed in RIPA buffer (50 mM Tris-HCl pH 8, 1% nonidet P-40, 150 mM NaCl, 0.5% sodium deoxycholate, and 1 mM EDTA) and supplemented with protease inhibitors: 1 μg/ml leupeptin, 2 μg/ml aprotinin and 1 mM PMSF (Sigma). Lysates were pre-cleared by a 1-hour incubation with 30 μl of a 50% slurry of protein A or protein G beads (Pierce, Rockford, IL) and then subjected to immunoprecipitation (2 hours) with various antibodies. The antibody-captured complexes were recovered with protein A agarose beads by incubation with lysate-antibody mixture at 4°C for 1-2 hours and washed three times with lysis buffer. Immunoblotting was performed as described (Khanna et al., 2007a) except that a mouse anti-rabbit light-chain specific secondary antibody (Jackson ImmunoResearch, West Grove, PA) was used for detecting proteins following immunoprecipitations.
Immunocytochemistry, confocal microscopy and iterative deconvolution deblurring
Immunocytochemistry was performed as described (Chan et al., 2007; Khanna et al., 2006b; Khanna et al., 2007a; Khanna et al., 2007b). Cultured neurons from DRG were imaged on a Nikon Ti swept-field confocal microscope. Z-stack image pairs were captured every 200 nm through the sample. Images were deblurred off-line by an iterative deconvolution protocol (Nikon Elements v3.0).
Intensity correlation analysis (ICA), intensity correlation quotient (ICQ) and image presentation
Developed by Elise Stanley (University of Toronto, Ontario, Canada), ICA was performed exactly as described (Khanna et al., 2006b; Li et al., 2004). Images shown in Figs 1, 2, 6 and 7 were enhanced using the smart sharpen mask filter and brightness-contrast adjustment functions in Adobe Photoshop.
FM4-64 imaging
FM4-64 imaging was performed as described previously (Brittain et al., 2009; Pan et al., 2005) on DRG cultures grown for 5-7 DIV. Axonal terminals were loaded with the fluorescent styryl dye FM4-64 (15 μM) by incubating the cells for 1 minute in high-KCl solution containing 58 mM NaCl, 90 mM KCl, 10 mM HEPES, 3 mM CaCl2·2H2O, 8 mM glucose, and 2 mM MgCl2·6H2O (pH 7.3) and then washed in Ca2+-free solution for 10 minutes to reduce nonspecific staining. The cells were then fixed and processed for immunocytochemistry as above (Khanna et al., 2007a).
Transfection of rat DRG
Isolated rat DRG neurons (∼1.5×106) were transfected with 3 μg of EGFP or CRMP-2–EGFP cDNA using the rat neuron Nucleofector solution (Amaxa Biosystems) (Leclere et al., 2005).
Sensory neuron isolation and maintenance
Sensory neurons were isolated from adult rodents (Sprague-Dawley rats or adult C57BL/6 mice) using procedures developed previously (Lindsay, 1988; Winter et al., 1988) with modifications (Chi and Nicol, 2007). Isolated rat cells (∼1.5×105 cells/ml) were plated on coverslips coated with poly-d-lysine and laminin and maintained at 37°C and 3% CO2 in F-12 media supplemented with NGF (30 ng/ml). Isolated mouse neurons were plated in coated wells of 24-well dishes at a density of 3-5×104 cells per well and maintained at 37°C in a 5% CO2 atmosphere in supplemented F12 media.
Electrophysiology
Whole-cell voltage clamp recordings were obtained from sensory neurons 48 hours after transfection or siRNA treatment (Chi et al., 2007). To isolate N-type Ca2+ currents, the Na+, K+ currents and L-type Ca2+ currents were blocked with 1 μM tetrodotoxin (TTX; Alomone Laboratories), 30 mM tetraethylammonium-Cl (TEA-Cl; Sigma) and 1 μM nifedipine (Calbiochem), respectively. Neurons were bathed in normal Ringers solution and Ca2+ currents were recorded in 110 mM N-methyl glucamine (NMG), 2 mM CaCl2, 30 mM TEA-Cl, 10 mM HEPES, 10 mM glucose, 0.001 mM nifedipine or 0.001 mM TTX. The recording pipettes, 2-5 MΩ resistances, contained 150 mM CsCl2, 10 mM HEPES, 5 mM Mg-ATP, 5 mM BAPTA, pH at 7.2 with KOH. Whole-cell recordings were obtained with an Axopatch 200B amplifier; data were acquired and analyzed using pCLAMP 8.2 (Molecular Devices). Capacitive artifacts were fully compensated and the series resistance was compensated by ∼80%. All experiments were performed at ∼23°C. The currents were filtered at 5 kHz and sampled at 2 kHz using Clampex 8.2 (Molecular Devices) with a standard P/4 leak subtraction protocol.
Voltage-gated K+ currents were recorded exactly as described (Chi and Nicol, 2007).
CRMP-2 siRNA transfection of mouse DRG
Mouse DRG neurons were transfected with 100 or 200 nM CRMP-2 siRNA using the reagent Metafectene (Biontex, Planegg, Germany), and release experiments were performed 72 hours later.
Stimulated-release of iCGRP
A radioimmunoassay was used to measure stimulus-evoked release and content of immunoreactive CGRP (iCGRP) from isolated sensory neurons (Hingtgen et al., 2006). Briefly, at 5-7 DIV, the basal (resting) release of iCGRP was measured from cells incubated for 10 minutes in HEPES buffer. The cells were incubated in HEPES buffer containing stimulus (50 mM KCl) for 10 minutes, and then incubated again with HEPES buffer containing 3.5 mM KCl to re-establish resting release levels. The remaining peptide content in each well was determined by exposing the cells to 2 N acetic acid for 10 minutes. The release of iCGRP during the 10-minute incubation period is expressed as percent of the total content.
Biotinylation of DRG neurons in culture
Biotinylation of live DRG neurons grown in culture was performed as described (Chan et al., 2007; Joiner et al., 2001). DRG neurons grown for 5 DIV were transfected with EGFP or CRMP-2–EGFP or treated with scramble or CRMP-2 siRNA for 2 days and then processed for biotinylation experiments.
Statistical analyses
Imaging values were either tested for two means between conditions (P) or for one mean for a difference from 0 (P=0). Each DRG ICQ value reflects the mean of two determinations from image planes at least 400 nm apart (two z-axis increments) (Li et al., 2004). The normal approximation to the sign test was used for statistical tests of ICQ values for single experiments. Differences in iCGRP release and total content were compared with analyses of variance (ANOVAs) and Dunnett's post-hoc analysis or Student's t-tests, as indicated. A P value of <0.05 was used to indicate statistical significance between treatment and non-treatment groups.
We thank Alyson Fournier (McGill University, Montreal, Canada) for V5-tagged CRMP constructs. Supported by grants from Indiana State Department of Health – Spinal Cord and Brain Injury Fund 4786219 (R.K.), the NIH NINDS R01 NS051668 (C.M.H.) and NS046084 (G.D.N.), and the Children's Tumor Foundation Young Investigator's Award (B.S.S.). Deposited in PMC for release after 12 months.
References
- Alvarez, F. J., Kavookjian, A. M. and Light, A. R. (1993). Ultrastructural morphology, synaptic relationships, and CGRP immunoreactivity of physiologically identified C-fiber terminals in the monkey spinal cord. J. Comp Neurol. 329, 472-490. [DOI] [PubMed] [Google Scholar]
- Ambalavanar, R., Moritani, M., Moutanni, A., Gangula, P., Yallampalli, C. and Dessem, D. (2006). Deep tissue inflammation upregulates neuropeptides and evokes nociceptive behaviors which are modulated by a neuropeptide antagonist. Pain 120, 53-68. [DOI] [PubMed] [Google Scholar]
- Augustine, G. J. (2001). How does calcium trigger neurotransmitter release? Curr. Opin. Neurobiol. 11, 320-326. [DOI] [PubMed] [Google Scholar]
- Bahls, F. H., Lartius, R., Trudeau, L. E., Doyle, R. T., Fang, Y., Witcher, D., Campbell, K. and Haydon, P. G. (1998). Contact-dependent regulation of N-type calcium channel subunits during synaptogenesis. J. Neurobiol. 35, 198-208. [DOI] [PubMed] [Google Scholar]
- Behar, O., Mizuno, K., Badminton, M. and Woolf, C. J. (1999). Semaphorin 3A growth cone collapse requires a sequence homologous to tarantula hanatoxin. Proc. Natl. Acad. Sci. USA 96, 13501-13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, T. J., Thaler, C., Castiglioni, A. J., Helton, T. D. and Lipscombe, D. (2004). Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron 41, 127-138. [DOI] [PubMed] [Google Scholar]
- Bisgaard, C. F., Jayatissa, M. N., Enghild, J. J., Sanchez, C., Artemychyn, R. and Wiborg, O. (2007). Proteomic investigation of the ventral rat hippocampus links DRP-2 to escitalopram treatment resistance and SNAP to stress resilience in the chronic mild stress model of depression. J. Mol. Neurosci. 32, 132-144. [DOI] [PubMed] [Google Scholar]
- Bowersox, S. S., Gadbois, T., Singh, T., Pettus, M., Wang, Y. X. and Luther, R. R. (1996). Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent and neuropathic pain. J. Pharmacol. Exp. Ther. 279, 1243-1249. [PubMed] [Google Scholar]
- Brittain, J. M., Piekarz, A. D., Wang, Y., Kondo, T., Cummins, T. R. and Khanna, R. (2009). An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J. Biol. Chem. 284, 31375-31390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall, W. A. and Few, A. P. (2008). Calcium channel regulation and presynaptic plasticity. Neuron 59, 882-901. [DOI] [PubMed] [Google Scholar]
- Chan, A. W., Khanna, R., Li, Q. and Stanley, E. F. (2007). Munc18: a presynaptic transmitter release site N type (CaV2.2) calcium channel interacting protein. Channels (Austin.) 1, 11-20. [PubMed] [Google Scholar]
- Chen, A., Liao, W. P., Lu, Q., Wong, W. S. and Wong, P. T. (2007). Upregulation of dihydropyrimidinase-related protein 2, spectrin alpha II chain, heat shock cognate protein 70 pseudogene 1 and tropomodulin 2 after focal cerebral ischemia in rats-a proteomics approach. Neurochem. Int. 50, 1078-1086. [DOI] [PubMed] [Google Scholar]
- Chen, Y., Deng, L., Maeno-Hikichi, Y., Lai, M., Chang, S., Chen, G. and Zhang, J. F. (2003). Formation of an endophilin-Ca2+ channel complex is critical for clathrin-mediated synaptic vesicle endocytosis. Cell 115, 37-48. [DOI] [PubMed] [Google Scholar]
- Chi, X. X. and Nicol, G. D. (2007). Manipulation of the potassium channel Kv1.1 and its effect on neuronal excitability in rat sensory neurons. J. Neurophysiol. 98, 2683-2692. [DOI] [PubMed] [Google Scholar]
- Chi, X. X., Jiang, X. and Nicol, G. D. (2007). ATP-sensitive potassium currents reduce the PGE2-mediated enhancement of excitability in adult rat sensory neurons. Brain Res. 1145, 28-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge, F. A., Jr and Rahamimoff, R. (1967). On the relationship between calcium concentration and the amplitude of the end-plate potential. J. Physiol. 189, 90P-92P. [PubMed] [Google Scholar]
- Dresbach, T., Qualmann, B., Kessels, M. M., Garner, C. C. and Gundelfinger, E. D. (2001). The presynaptic cytomatrix of brain synapses. Cell Mol. Life Sci. 58, 94-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham, P. L. (2006). Calcitonin gene-related peptide (CGRP) and migraine. Headache. 46 Suppl. 1, S3-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham, P. L. (2008). Inhibition of calcitonin gene-related peptide function: a promising strategy for treating migraine. Headache. 48, 1269-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, R. H. (1998). Neurotransmitter release: variations on a theme. Curr. Biol. 8, R883-R885. [DOI] [PubMed] [Google Scholar]
- el Far, O., Charvin, N., Leveque, C., Martin-Moutot, N., Takahashi, M. and Seagar, M. J. (1995). Interaction of a synaptobrevin (VAMP)-syntaxin complex with presynaptic calcium channels. FEBS Lett. 361, 101-105. [DOI] [PubMed] [Google Scholar]
- Evans, A. R., Nicol, G. D. and Vasko, M. R. (1996). Differential regulation of evoked peptide release by voltage-sensitive calcium channels in rat sensory neurons. Brain Res. 712, 265-273. [DOI] [PubMed] [Google Scholar]
- Evans, R. M. and Zamponi, G. W. (2006). Presynaptic Ca2+ channels-integration centers for neuronal signaling pathways. Trends Neurosci. 29, 617-624. [DOI] [PubMed] [Google Scholar]
- Fan, J., Mansfield, S. G., Redmond, T., Gordon-Weeks, P. R. and Raper, J. A. (1993). The organization of F-actin and microtubules in growth cones exposed to a brain-derived collapsing factor. J. Cell Biol. 121, 867-878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox, A. P., Nowycky, M. C. and Tsien, R. W. (1987). Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. J. Physiol. 394, 149-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata, Y., Itoh, T. J., Kimura, T., Menager, C., Nishimura, T., Shiromizu, T., Watanabe, H., Inagaki, N., Iwamatsu, A., Hotani, H. et al. (2002). CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 4, 583-591. [DOI] [PubMed] [Google Scholar]
- Garry, M. G., Miller, K. E. and Seybold, V. S. (1989). Lumbar dorsal root ganglia of the cat: a quantitative study of peptide immunoreactivity and cell size. J. Comp Neurol. 284, 36-47. [DOI] [PubMed] [Google Scholar]
- Goshima, Y., Nakamura, F., Strittmatter, P. and Strittmatter, S. M. (1995). Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature. 376, 509-514. [DOI] [PubMed] [Google Scholar]
- Gross, R. A. and Macdonald, R. L. (1987). Dynorphin A selectively reduces a large transient (N-type) calcium current of mouse dorsal root ganglion neurons in cell culture. Proc. Natl. Acad. Sci. USA 84, 5469-5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, Z. W. and Sanes, J. R. (1993). Synaptic structure and development: the neuromuscular junction. Cell 72 Suppl., 99-121. [DOI] [PubMed] [Google Scholar]
- Hatakeyama, S., Wakamori, M., Ino, M., Miyamoto, N., Takahashi, E., Yoshinaga, T., Sawada, K., Imoto, K., Tanaka, I., Yoshizawa, T. et al. (2001). Differential nociceptive responses in mice lacking the alpha(1B) subunit of N-type Ca(2+) channels. Neuroreport 12, 2423-2427. [DOI] [PubMed] [Google Scholar]
- Hayes, E. S. and Carlton, S. M. (1992). Primary afferent interactions: analysis of calcitonin gene-related peptide-immunoreactive terminals in contact with unlabeled and GABA-immunoreactive profiles in the monkey dorsal horn. Neuroscience 47, 873-896. [DOI] [PubMed] [Google Scholar]
- Hingtgen, C. M. and Vasko, M. R. (1994a). Prostacyclin enhances the evoked-release of substance P and calcitonin gene-related peptide from rat sensory neurons. Brain Res. 655, 51-60. [DOI] [PubMed] [Google Scholar]
- Hingtgen, C. M. and Vasko, M. R. (1994b). The phosphatase inhibitor, okadaic acid, increases peptide release from rat sensory neurons in culture. Neurosci. Lett. 178, 135-138. [DOI] [PubMed] [Google Scholar]
- Hingtgen, C. M., Roy, S. L. and Clapp, D. W. (2006). Stimulus-evoked release of neuropeptides is enhanced in sensory neurons from mice with a heterozygous mutation of the Nf1 gene. Neuroscience 137, 637-645. [DOI] [PubMed] [Google Scholar]
- Hirning, L. D., Fox, A. P., McCleskey, E. W., Olivera, B. M., Thayer, S. A., Miller, R. J. and Tsien, R. W. (1988). Dominant role of N-type Ca2+ channels in evoked release of norepinephrine from sympathetic neurons. Science 239, 57-61. [DOI] [PubMed] [Google Scholar]
- Hokfelt, T., Herrera-Marschitz, M., Seroogy, K., Ju, G., Staines, W. A., Holets, V., Schalling, M., Ungerstedt, U., Post, C., Rehfeld, J. F. et al. (1988). Immunohistochemical studies on cholecystokinin (CCK)-immunoreactive neurons in the rat using sequence specific antisera and with special reference to the caudate nucleus and primary sensory neurons. J. Chem. Neuroanat. 1, 11-51. [PubMed] [Google Scholar]
- Honnorat, J., Byk, T., Kusters, I., Aguera, M., Ricard, D., Rogemond, V., Quach, T., Aunis, D., Sobel, A., Mattei, M. G. et al. (1999). Ulip/CRMP proteins are recognized by autoantibodies in paraneoplastic neurological syndromes. Eur. J. Neurosci. 11, 4226-4232. [DOI] [PubMed] [Google Scholar]
- Hotta, A., Inatome, R., Yuasa-Kawada, J., Qin, Q., Yamamura, H. and Yanagi, S. (2005). Critical role of collapsin response mediator protein-associated molecule CRAM for filopodia and growth cone development in neurons. Mol. Biol. Cell 16, 32-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, S. T., Jiang, S. X., Desbois, A., Huang, D., Kelly, J., Tessier, L., Karchewski, L. and Kappler, J. (2006). Calpain-cleaved collapsin response mediator protein-3 induces neuronal death after glutamate toxicity and cerebral ischemia. J. Neurosci. 26, 2241-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui, K. and Feng, Z. P. (2008). NCS-1 differentially regulates growth cone and somata calcium channels in Lymnaea neurons. Eur. J. Neurosci. 27, 631-643. [DOI] [PubMed] [Google Scholar]
- Ichikawa, M., Kimura-Kuroda, J., Yasui, K. and Kuroda, Y. (1991). Expression of synaptophysin during synapse formation between dissociated cortical neurons. Neurosci. Res. 12, 452-458. [DOI] [PubMed] [Google Scholar]
- Igarashi, M., Ohyama, A., Ohbayashi, K., Kozaki, S. and Komiya, Y. (2000). The mechanism of the neurotransmitter release in growth cones. J. Neurosci. Res. 60, 743-753. [DOI] [PubMed] [Google Scholar]
- Inagaki, H., Kato, Y., Hamajima, N., Nonaka, M., Sasaki, M. and Eimoto, T. (2000). Differential expression of dihydropyrimidinase-related protein genes in developing and adult enteric nervous system. Histochem. Cell Biol. 113, 37-41. [DOI] [PubMed] [Google Scholar]
- Inagaki, N., Chihara, K., Arimura, N., Menager, C., Kawano, Y., Matsuo, N., Nishimura, T., Amano, M. and Kaibuchi, K. (2001). CRMP-2 induces axons in cultured hippocampal neurons. Nat. Neurosci. 4, 781-782. [DOI] [PubMed] [Google Scholar]
- Joiner, W. J., Khanna, R., Schlichter, L. C. and Kaczmarek, L. K. (2001). Calmodulin regulates assembly and trafficking of SK4/IK1 Ca2+-activated K+ channels. J. Biol. Chem. 276, 37980-37985. [DOI] [PubMed] [Google Scholar]
- Kangrga, I. and Randic, M. (1991). Outflow of endogenous aspartate and glutamate from the rat spinal dorsal horn in vitro by activation of low- and high-threshold primary afferent fibers. Modulation by mu-opioids. Brain Res. 553, 347-352. [DOI] [PubMed] [Google Scholar]
- Khanna, R., Li, Q., Sun, L., Collins, T. J. and Stanley, E. F. (2006a). N type Ca2+ channels and RIM scaffold protein covary at the presynaptic transmitter release face but are components of independent protein complexes. Neuroscience 140, 1201-1208. [DOI] [PubMed] [Google Scholar]
- Khanna, R., Sun, L., Li, Q., Guo, L. and Stanley, E. F. (2006b). Long splice variant N type calcium channels are clustered at presynaptic transmitter release sites without modular adaptor proteins. Neuroscience 138, 1115-1125. [DOI] [PubMed] [Google Scholar]
- Khanna, R., Li, Q., Bewersdorf, J. and Stanley, E. F. (2007a). The presynaptic CaV2.2 channel-transmitter release site core complex. Eur. J. Neurosci. 26, 547-559. [DOI] [PubMed] [Google Scholar]
- Khanna, R., Li, Q., Schlichter, L. C. and Stanley, E. F. (2007b). The transmitter release-site CaV2.2 channel cluster is linked to an endocytosis coat protein complex. Eur. J. Neurosci. 26, 560-574. [DOI] [PubMed] [Google Scholar]
- Khanna, R., Zougman, A. and Stanley, E. F. (2007c). A proteomic screen for presynaptic terminal N-type calcium channel (CaV2.2) binding partners. J. Biochem. Mol. Biol. 40, 302-314. [DOI] [PubMed] [Google Scholar]
- Kim, C., Jun, K., Lee, T., Kim, S. S., McEnery, M. W., Chin, H., Kim, H. L., Park, J. M., Kim, D. K., Jung, S. J. et al. (2001). Altered nociceptive response in mice deficient in the alpha(1B) subunit of the voltage-dependent calcium channel. Mol. Cell Neurosci. 18, 235-245. [DOI] [PubMed] [Google Scholar]
- Kimura, T., Watanabe, H., Iwamatsu, A. and Kaibuchi, K. (2005). Tubulin and CRMP-2 complex is transported via Kinesin-1. J. Neurochem. 93, 1371-1382. [DOI] [PubMed] [Google Scholar]
- Kisilevsky, A. E. and Zamponi, G. W. (2008). Presynaptic calcium channels: structure, regulators, and blockers. Handb. Exp. Pharmacol. 184, 45-75. [DOI] [PubMed] [Google Scholar]
- Kitamura, Y., Matsuka, Y., Spigelman, I., Ishihara, Y., Yamamoto, Y., Sonoyama, W., Kuboki, T. and Oguma, K. (2009). Botulinum toxin type a (150 kDa) decreases exaggerated neurotransmitter release from trigeminal ganglion neurons and relieves neuropathy behaviors induced by infraorbital nerve constriction. Neuroscience 159, 1422-1429. [DOI] [PubMed] [Google Scholar]
- Kostyuk, P. G. and Krishtal, O. A. (1977). Effects of calcium and calcium-chelating agents on the inward and outward current in the membrane of mollusc neurones. J. Physiol. 270, 569-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowara, R., Chen, Q., Milliken, M. and Chakravarthy, B. (2005). Calpain-mediated truncation of dihydropyrimidinase-like 3 protein (DPYSL3) in response to NMDA and H2O2 toxicity. J. Neurochem. 95, 466-474. [DOI] [PubMed] [Google Scholar]
- Leclere, P. G., Panjwani, A., Docherty, R., Berry, M., Pizzey, J. and Tonge, D. A. (2005). Effective gene delivery to adult neurons by a modified form of electroporation. J. Neurosci. Methods. 142, 137-143. [DOI] [PubMed] [Google Scholar]
- Leenders, A. G., Lin, L., Huang, L. D., Gerwin, C., Lu, P. H. and Sheng, Z. H. (2008). The role of MAP1A light chain 2 in synaptic surface retention of Cav2.2 channels in hippocampal neurons. J. Neurosci. 28, 11333-11346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q., Lau, A., Morris, T. J., Guo, L., Fordyce, C. B. and Stanley, E. F. (2004). A syntaxin 1, Galpha(o), and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J. Neurosci. 24, 4070-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y., McDonough, S. I. and Lipscombe, D. (2004). Alternative splicing in the voltage-sensing region of N-Type CaV2.2 channels modulates channel kinetics. J. Neurophysiol. 92, 2820-2830. [DOI] [PubMed] [Google Scholar]
- Lindsay, R. M. (1988). Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J. Neurosci. 8, 2394-2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe, D., Madison, D. V., Poenie, M., Reuter, H., Tsien, R. Y. and Tsien, R. W. (1988). Spatial distribution of calcium channels and cytosolic calcium transients in growth cones and cell bodies of sympathetic neurons. Proc. Natl. Acad. Sci. USA 85, 2398-2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas, R. R. (1982). Calcium in synaptic transmission. Sci. Am. 247, 56-65. [DOI] [PubMed] [Google Scholar]
- Matsuka, Y., Edmonds, B., Mitrirattanakul, S., Schweizer, F. E. and Spigelman, I. (2007). Two types of neurotransmitter release patterns in isolectin B4-positive and negative trigeminal ganglion neurons. Neuroscience 144, 665-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov, A., Sudhof, T. C. and Bezprozvanny, I. (1999). Association of neuronal calcium channels with modular adaptor proteins. J. Biol. Chem. 274, 24453-24456. [DOI] [PubMed] [Google Scholar]
- Miletic, V. and Tan, H. (1988). Iontophoretic application of calcitonin gene-related peptide produces a slow and prolonged excitation of neurons in the cat lumbar dorsal horn. Brain Res. 446, 169-172. [DOI] [PubMed] [Google Scholar]
- Nishimura, T., Fukata, Y., Kato, K., Yamaguchi, T., Matsuura, Y., Kamiguchi, H. and Kaibuchi, K. (2003). CRMP-2 regulates polarized Numb-mediated endocytosis for axon growth. Nat. Cell Biol. 5, 819-826. [DOI] [PubMed] [Google Scholar]
- Ohmori, H. and Yoshii, M. (1977). Surface potential reflected in both gating and permeation mechanisms of sodium and calcium channels of the tunicate egg cell membrane. J. Physiol. 267, 429-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto, M., Schoch, S. and Sudhof, T. C. (1999). EHSH1/intersectin, a protein that contains EH and SH3 domains and binds to dynamin and SNAP-25. A protein connection between exocytosis and endocytosis? J. Biol. Chem. 274, 18446-18454. [DOI] [PubMed] [Google Scholar]
- Oku, R., Satoh, M., Fujii, N., Otaka, A., Yajima, H. and Takagi, H. (1987). Calcitonin gene-related peptide promotes mechanical nociception by potentiating release of substance P from the spinal dorsal horn in rats. Brain Res. 403, 350-354. [DOI] [PubMed] [Google Scholar]
- Pan, J. Q. and Lipscombe, D. (2000). Alternative splicing in the cytoplasmic II-III loop of the N-type Ca channel alpha 1B subunit: functional differences are beta subunit-specific. J. Neurosci. 20, 4769-4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, P. Y., Cai, Q., Lin, L., Lu, P. H., Duan, S. and Sheng, Z. H. (2005). SNAP-29-mediated modulation of synaptic transmission in cultured hippocampal neurons. J. Biol. Chem. 280, 25769-25779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, Y. and Kim, K. T. (2009). Short-term plasticity of small synaptic vesicle (SSV) and large dense-core vesicle (LDCV) exocytosis. Cell Signal 21, 1465-1470. [DOI] [PubMed] [Google Scholar]
- Phillips, G. R., Huang, J. K., Wang, Y., Tanaka, H., Shapiro, L., Zhang, W., Shan, W. S., Arndt, K., Frank, M., Gordon, R. E. et al. (2001). The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron 32, 63-77. [DOI] [PubMed] [Google Scholar]
- Poo, M. M., Sun, Y. A. and Young, S. H. (1985). Three types of transmitter release from embryonic neurons. J. Physiol. (Paris) 80, 283-289. [PubMed] [Google Scholar]
- Quinn, C. C., Chen, E., Kinjo, T. G., Kelly, G., Bell, A. W., Elliott, R. C., McPherson, P. S. and Hockfield, S. (2003). TUC-4b, a novel TUC family variant, regulates neurite outgrowth and associates with vesicles in the growth cone. J. Neurosci. 23, 2815-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu, M. J., Lee, C., Kim, J., Shin, H. S. and Yu, M. H. (2008). Proteomic analysis of stargazer mutant mouse neuronal proteins involved in absence seizure. J. Neurochem. 104, 1260-1270. [DOI] [PubMed] [Google Scholar]
- Ryu, P. D., Gerber, G., Murase, K. and Randic, M. (1988a). Actions of calcitonin gene-related peptide on rat spinal dorsal horn neurons. Brain Res. 441, 357-361. [DOI] [PubMed] [Google Scholar]
- Ryu, P. D., Gerber, G., Murase, K. and Randic, M. (1988b). Calcitonin gene-related peptide enhances calcium current of rat dorsal root ganglion neurons and spinal excitatory synaptic transmission. Neurosci. Lett. 89, 305-312. [DOI] [PubMed] [Google Scholar]
- Saegusa, H., Matsuda, Y. and Tanabe, T. (2002). Effects of ablation of N- and R-type Ca(2+) channels on pain transmission. Neurosci. Res. 43, 1-7. [DOI] [PubMed] [Google Scholar]
- Samuels, B. A., Hsueh, Y. P., Shu, T., Liang, H., Tseng, H. C., Hong, C. J., Su, S. C., Volker, J., Neve, R. L., Yue, D. T. et al. (2007). Cdk5 promotes synaptogenesis by regulating the subcellular distribution of the MAGUK family member CASK. Neuron. 56, 823-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, E. F. and Strittmatter, S. M. (2007). The CRMP family of proteins and their role in Sema3A signaling. Adv. Exp. Med. Biol. 600, 1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenen, J., Delree, P., Leprince, P. and Moonen, G. (1989). Neurotransmitter phenotype plasticity in cultured dissociated adult rat dorsal root ganglia: an immunocytochemical study. J. Neurosci. Res. 22, 473-487. [DOI] [PubMed] [Google Scholar]
- Schon, F., Ghatei, M., Allen, J. M., Mulderry, P. K., Kelly, J. S. and Bloom, S. R. (1985). The effect of sympathectomy on calcitonin gene-related peptide levels in the rat trigeminovascular system. Brain Res. 348, 197-200. [DOI] [PubMed] [Google Scholar]
- Scroggs, R. S. and Fox, A. P. (1992a). Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. J. Physiol. 445, 639-658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scroggs, R. S. and Fox, A. P. (1992b). Multiple Ca2+ currents elicited by action potential waveforms in acutely isolated adult rat dorsal root ganglion neurons. J. Neurosci. 12, 1789-1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, Z. H., Rettig, J., Takahashi, M. and Catterall, W. A. (1994). Identification of a syntaxin-binding site on N-type calcium channels. Neuron 13, 1303-1313. [DOI] [PubMed] [Google Scholar]
- Skofitsch, G. and Jacobowitz, D. M. (1985a). Calcitonin gene-related peptide coexists with substance P in capsaicin sensitive neurons and sensory ganglia of the rat. Peptides 6, 747-754. [DOI] [PubMed] [Google Scholar]
- Skofitsch, G. and Jacobowitz, D. M. (1985b). Calcitonin gene-related peptide: detailed immunohistochemical distribution in the central nervous system. Peptides 6, 721-745. [DOI] [PubMed] [Google Scholar]
- Skofitsch, G. and Jacobowitz, D. M. (1985c). Quantitative distribution of calcitonin gene-related peptide in the rat central nervous system. Peptides 6, 1069-1073. [DOI] [PubMed] [Google Scholar]
- Soeda, H., Tatsumi, H. and Katayama, Y. (1997). Neurotransmitter release from growth cones of rat dorsal root ganglion neurons in culture. Neuroscience 77, 1187-1199. [DOI] [PubMed] [Google Scholar]
- Soeda, H., Tatsumi, H., Kozawa, Y., Mishima, H. and Katayama, Y. (1998). Visualization of calcium channels involved in transmitter release from neuronal growth cones. Neurosci. Lett. 251, 93-96. [DOI] [PubMed] [Google Scholar]
- Spitzer, N. C. (1995). Spontaneous activity: functions of calcium transients in neuronal differentiation. Perspect. Dev. Neurobiol. 2, 379-386. [PubMed] [Google Scholar]
- Stanley, E. F. (1997). The calcium channel and the organization of the presynaptic transmitter release face. Trends Neurosci. 20, 404-409. [DOI] [PubMed] [Google Scholar]
- Sudhof, T. C. (1995). The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature 375, 645-653. [DOI] [PubMed] [Google Scholar]
- Szabo, Z., Obermair, G. J., Cooper, C. B., Zamponi, G. W. and Flucher, B. E. (2006). Role of the synprint site in presynaptic targeting of the calcium channel CaV2.2 in hippocampal neurons. Eur. J. Neurosci. 24, 709-718. [DOI] [PubMed] [Google Scholar]
- Vigers, A. J. and Pfenninger, K. H. (1991). N-type and L-type calcium channels are present in nerve growth cones. Numbers increase on synaptogenesis. Brain Res. Dev. Brain Res. 60, 197-203. [DOI] [PubMed] [Google Scholar]
- Weitzdoerfer, R., Fountoulakis, M. and Lubec, G. (2001). Aberrant expression of dihydropyrimidinase related proteins-2,-3 and -4 in fetal Down syndrome brain. J. Neural Transm. Suppl., 95-107. [DOI] [PubMed]
- Westenbroek, R. E., Hell, J. W., Warner, C., Dubel, S. J., Snutch, T. P. and Catterall, W. A. (1992). Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron 9, 1099-1115. [DOI] [PubMed] [Google Scholar]
- Wiesenfeld-Hallin, Z., Hokfelt, T., Lundberg, J. M., Forssmann, W. G., Reinecke, M., Tschopp, F. A. and Fischer, J. A. (1984). Immunoreactive calcitonin gene-related peptide and substance P coexist in sensory neurons to the spinal cord and interact in spinal behavioral responses of the rat. Neurosci. Lett. 52, 199-204. [DOI] [PubMed] [Google Scholar]
- Winter, J., Forbes, C. A., Sternberg, J. and Lindsay, R. M. (1988). Nerve growth factor (NGF) regulates adult rat cultured dorsal root ganglion neuron responses to the excitotoxin capsaicin. Neuron 1, 973-981. [DOI] [PubMed] [Google Scholar]
- Yamabhai, M., Hoffman, N. G., Hardison, N. L., McPherson, P. S., Castagnoli, L., Cesareni, G. and Kay, B. K. (1998). Intersectin, a novel adaptor protein with two Eps15 homology and five Src homology 3 domains. J. Biol. Chem. 273, 31401-31407. [DOI] [PubMed] [Google Scholar]
- Yoshida, H., Watanabe, A. and Ihara, Y. (1998). Collapsin response mediator protein-2 is associated with neurofibrillary tangles in Alzheimer's disease. J. Biol. Chem. 273, 9761-9768. [DOI] [PubMed] [Google Scholar]
- Yoshimura, T., Kawano, Y., Arimura, N., Kawabata, S., Kikuchi, A. and Kaibuchi, K. (2005). GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 120, 137-149. [DOI] [PubMed] [Google Scholar]
- Young, S. H. and Poo, M. M. (1983). Spontaneous release of transmitter from growth cones of embryonic neurones. Nature 305, 634-637. [DOI] [PubMed] [Google Scholar]
- Zeitelhofer, M., Vessey, J. P., Xie, Y., Tubing, F., Thomas, S., Kiebler, M. and Dahm, R. (2007). High-efficiency transfection of mammalian neurons via nucleofection. Nat. Protoc. 2, 1692-1704. [DOI] [PubMed] [Google Scholar]
- Zhang, Y., Helm, J. S., Senatore, A., Spafford, J. D., Kaczmarek, L. K. and Jonas, E. A. (2008). PKC-induced intracellular trafficking of Ca(V)2 precedes its rapid recruitment to the plasma membrane. J. Neurosci. 28, 2601-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]