

Abstract
The proposed sst1 pharmacophore1 derived from the NMR structures of a family of mono- and dicyclic undecamers was used to design octa-, hepta- and hexamers with high affinity and selectivity for the somatostatin sst1 receptor. These compounds were tested for their in vitro binding properties to all five somatostatin (SRIF) receptors using receptor autoradiography; those with high SRIF receptor subtype 1 (sst1) affinity and selectivity were shown to be agonists when tested functionally in a luciferase reporter gene assay. Des-AA1,4-6,10,12,13-[DTyr2,DAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-Thr-NH2 (25) was radio-iodinated (125I-25) and specifically labeled sst1-expressing cells and tissues. 3D NMR structures were calculated for des-AA1,4-6,10,12,13-[DPhe2,DTrp8,IAmp9]-SRIF-Thr-NH2 (16), des-AA1,2,4-6,10,12,13-[DAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-Thr-NH2 (23) and des-AA1,2,4-6,10,12,13-[DAgl(NMe,2naphthoyl)8,IAmp9,Tyr11]-SRIF-NH2 (27) in DMSO. Though the analogues have the sst1 pharmacophore residues at the previously determined distances from each other, the positioning of the aromatic residues in 16, 23 and 27 is different from that described earlier suggesting an induced fit mechanism for sst1 binding of these novel, less constrained sst1-selective family members.
Keywords: somatostatin analogues, radio-iodinatable SRIF analogues, SAR, NMR structures, somatostatin receptor 1 selectivity
Introduction
Somatostatin (SRIF) is a cyclic tetradecapeptide widely distributed throughout the body with important regulatory effects (mostly inhibitory) on a variety of endocrine and exocrine functions. It was originally isolated from the hypothalamus and characterized in 1973.2, 3 Somatostatin is also found in the gut, pancreas, in the nervous system, and in various exocrine and endocrine glands. Somatostatin inhibits the release of growth hormone from the anterior pituitary, insulin and glucagon from the pancreas, and gastrin from the gastrointestinal tract. It also has anti-proliferative activity, and acts as a neurotransmitter, or neuromodulator in the brain.4-7
SRIF interacts with five specific receptor subtypes (sst1 to sst5) that have been cloned and characterized, all belonging to the G-protein-coupled receptor family.8-10 Cell lines recombinantly expressing these cloned receptors are available to test SRIF analogues for binding affinity, selectivity and functional effects.7, 11-13 So far, only sst2 and sst5 receptors have been clearly linked to specific physiological functions.14, 15 For sst1 receptors, however, the cellular localization as well as the distinct functions are still not fully understood, similarly to our current lack of knowledge of sst3 or sst4 receptor function and physiology.
Sst1 receptors have been found in human cerebral cortex,16 human retina,17 neuroendocrine cells,14 endothelial cells of human blood vessels (arteries and veins)18 and human tumors.19-24 According to Lanneau25 and Olias,14 sst1 receptors are involved in the intrahypothalamic regulation of growth hormone (GH) secretion. The studies of Kreienkamp et al. suggested an important role for sst1 in the control of basal GH release in somatotrophs.26 Zatelli et al. have demonstrated that sst1-selective activation inhibits hormone secretion and cell viability in GH- and prolactin-secreting adenomas in vitro and suggested that somatostatin analogues with affinity for sst1 receptors may be useful to control hormone hyper secretion and reduce neoplastic growth of pituitary adenomas.27 Sst1 receptor activation also modulates somatostatin release in basal ganglia.28 In hypothalamic, basal ganglia, and retinal functions, the sst1 receptor appears to act as an inhibitory auto-receptor located on somatostatin neurons, whereas in the hippocampus, such a role is still based on circumstantial evidence.
Sst1-selective analogues could possibly play a role in various diseases. For instance, retinal disease therapeutics has been a suggested indication for sst1.17, 28-30 Recently, an sst1 antagonist was shown to promote social interactions, reduce aggressive behavior and stimulate learning.31, 32 Matrone et al. demonstrated the role of sst1-selective analogues in mediating the inhibitory effect of SRIF on growth hormone secreting pituitary tumors. They suggested that the sst1-selective analogues might represent a further useful approach for the treatment of acromegaly in patients resistant or partially responsive to octreotide-LAR or lanreotide treatment in vivo.33 It has also been reported that in medullary thyroid carcinoma, calcitonin secretion and gene expression can be reduced by treatment with sst1-selective agonists by the reduction of cAMP response element binding phosphorylation, suggesting that potent sst1-selective agonists could have a therapeutic role in medullary thyroid carcinoma.34, 35 Furthermore, since sst1-selective agonists were able to inhibit endothelial activities, a potential therapeutic utility for administration of sst1-selective agonists in the proliferative diseases involving angiogenesis has been suggested.18
Although numerous reports on the localization, physiological and therapeutic functions of sst1 receptors have been published, it is still not clear which is the main sst1 receptor function and the main sst1 related pathology. The design of more potent and more (>100-fold) sst1-selective agonists and antagonists with radio-labelable properties for in vitro binding assays and in vivo scintigraphy studies as well as greater metabolic stability in biological fluids than the native hormone could help to further understand sst1-related biology and pathobiology. Originally, we had identified the first generation of a peptidic scaffold with selected amino acids (AA) deletions (des-AA1,2,5-SRIF) that in combination with DTrp at position 8 and 4-(N-isopropyl)-aminomethylphenylalanine (IAmp) at position 9, yielded des-AA1,2,5-[DTrp8,IAmp9]-SRIF (CH-275)36 (2), (Table 1) a SRIF agonist that was 30-fold more selective for sst1 versus sst2/4/5 and 10-fold versus sst3, respectively.36 Our standard drug design approaches led to additional very potent sst1-selective mono- and dicyclic undecapeptides des-AA1,2,5-[DTrp8,IAmp9, Tyr11]-SRIF (3)37, des-AA1,2,5-[DTrp8,IAmp9, ITyr11]-SRIF (4)37, des-AA1,2,5-[DTrp8,IAmp9, ITyr11]-Cbm-SRIF (5)1, des-AA1,2,5-cyclo(7-12)[Glu7,DTrp8,IAmp9, ITyr11, hhLys12]-SRIF38(6), and des-AA1,2,5-[DAgl(NMe,2naphthoyl)8, IAmp9, Tyr11]-SRIF39 (7) (Table 1).1, 37-40
Table 1.
Physico-chemical properties and sst1-5 binding affinities (IC50s, nM) of sst1-selective analogues and control peptides
| Compound | Purity (%) | MSc | IC50 nMd | Luciferase Assay |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HPLCa | CZE | M (mono) calc. |
MH+ (mono) obs. |
sst1 | sst2 | sst3 | sst4 | sst5 | |||
| 1 | SRIF-28 | 98 | 93 | 1636.72 | 1637.70 | 2.9 ± 0.2 | 2.6 ± 0.11 | 4.3 ± 0.39 | 2.9 ± 0.2 | 2.9 ± 0.24 | agonist |
| 2 | des-AA1,2,5-[DTrp8,IAmp9]-SRIF36 | 94 | 97 | 1484.66 | 1485.5 | 31 ± 13 | >1K | 540; 150 | >1000 | >1K | |
| 3 | des-AA1,2,5-[DTrp8,IAmp9, Tyr11]-SRIF37 | 98 | 97 | 1500.66 | 1501.5 | 17 ± 6.0 | >1K | >1K | >1K | >1K | |
| 4 | des-AA1,2,5-[DTrp8,IAmp9, ITyr11]-SRIF37 | 94 | 96 | 1626.55 | 1627.5 | 3.6 ± 0.69 | >1K | >1K | >1K | >1K | |
| 5 | des-AA1,2,5-[DTrp8,IAmp9, ITyr11]-Cbm-SRIF1 | 91 | 97 | 1669.56 | 1670.56 | 2.5 ± 0.2 | >1K | 618 ± 125 | >1K | >1K | |
| 6 | des-AA1,2,5-cyclo(7-12)[Glu7,DTrp8,IAmp9, ITyr11, hhLys12]-SRIF38 | 98 | 98 | 1645.57 | 1646.50 | 6.1 ± 0.6 | >1K | >1K | >1K | >1K | |
| 7 | des-AA1,2,5-[DAgl(NMe,2naphthoyl)8,IAmp9, Tyr11]-SRIF39 | 89 | 93 | 1554.67 | 1555.70 | 3.3 ± 1.0 | >1K | >1K | 562 ± 135 | >1K | |
| 8 | des-AA1,2,4,5,12,13-[DTrp8]-SRIF50 | 95 | 98 | 1078.45 | 1078.90 | 27 ± 3.4 | 41 ± 8.7 | 13 ± 3.2 | 1.8 ± 0.7 | 46 ± 27 | |
| 9 | des-AA1,2,4,5,12,13-[DTrp8, IAmp9]-SRIF | 98 | 98 | 1168.58 | 1169.5 | >1K | >1K | >1K | >1K | >1K | |
| 10 | des-AA1,2,5,12,13-[DTrp8,IAmp9]-SRIF | 99 | 99 | 1296.58 | 1297.1 | 189 ± 31 | >1K | 789 ± 231 | 932 ± 125 | >1K | |
| 11 | des-AA1,4-6,11-13-[DPhe2, DTrp8]-SRIF-Thr-NH2 | 95 | 99 | 1031.4 | 1032.0 | >1K | 1.9 ± 0.33 | 39 ± 14 | >1K | 5.1 ± 1.1 | |
| 12 | des-AA1,4-6,11-13-[Cpa2,DCys3,Tyr7,DTrp8]-SRIF-2Nal-NH260 | 99 | 99 | 1177.43 | 1178.43 | >1K | 5.7 ± 1.5 | 112 ± 32 | 296 ± 19 | 218 ± 63 | |
| 13 | des-AA1,4-6,11-13-[Cpa2,DCys3,Tyr7,DTrp8,IAmp9]-SRIF-2Nal-NH2 | 98 | 98 | 1267.48 | 1268.45 | >1K | 240; 665 | 300; 234 | >1K | >1K | |
| 14 | des-AA1,4-6,12,13-[Tyr2,DTrp8,IAmp9]-SRIF-Thr-NH2 | 98 | 98 | 1284.55 | 1285.6 | 150; 186 | >1K | 60; 284 | >1K | >1K | |
| 15 | des-AA1,2,4-6,10,12,13-[DTrp8,IAmp9]-SRIF-Thr-NH2 | 99 | 99 | 1020.45 | 1021.51 | 108; 120 | >1K | >1K | 585; 250 | >1K | |
| 16 | des-AA1,4-6,10,12,13-[DPhe2,DTrp8,IAmp9]-SRIF-Thr-NH2 | 99 | 99 | 1167.50 | 1168.38 | 41; 14 | >1K | 448; 72 | 1349; 252 | >1K | |
| 17 | des-AA1,2,4-6,10,12,13-[DTrp8,IAmp9,ITyr11]-SRIF-Thr-NH2 | 99 | 99 | 1162.33 | 1163.24 | 591; 889 | >1K | 701; 989 | 468; 1273 | >1K | |
| 18 | des-AA1,4-6,10,12,13-[DPhe2,DTrp8,IAmp9, ITyr11]-SRIF-Thr-NH2 | 99 | 98 | 1309.40 | 1310.43 | 449; 565 | >1K | 514; 864 | >1K | >1K | |
| 19 | des-AA1,2,4-6,10,12,13-[DCys3,DTrp8,IAmp9, ITyr11]-SRIF-2Nal-NH2 | 97 | 98 | 1258.36 | 1259.33 | 356; 689 | >1K | 162; 438 | 329; 489 | >1K | |
| 20 | des-AA1,4-6,10,12,13-[Cpa2,DCys3,DTrp8,IAmp9, ITyr11]-SRIF-2Nal-NH2 | 95 | 95 | 1439.39 | 1440.21 | >1K | >1K | >1K | >1K | >1K | |
| 21 | des-AA1,4-6,10,12,13-[Cpa2,DCys3,DAgl(NMe,2naphthoyl)8]-SRIF-2Nal-NH2 | 98 | 96 | 1262.46 | 1262.3 | 20; 29 | 100; 881 | 17; 23 | 71; 72 | 9.9; 29 | |
| 22 | des-AA1,4-6,10,12,13-[Cpa2,DCys3,LAgl(NMe,2naphthoyl)8]-SRIF-2Nal-NH2 | 91 | 93 | 1262.46 | 1262.3 | >1K | >1K | 312 | >1K | >1K | |
| 23 | des-AA1,2,4-6,10,12,13-[DAAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-Thr-NH2 | 94 | 95 | 1074.45 | 1075.60 | 1.0 ± 0.25 | >1K | 681 ± 52 | 95 ± 18 | >1K | agonist |
| 24 | des-AA1,2,4-6,10,12,13-[LAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-Thr-NH2 | 97 | 98 | 1074.45 | 1075.59 | 37; 42 | >1K | 848; 253 | 833; 525 | >1K | |
| 25 | des-AA1,4-6,10,12,13-[DTyr2,DAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-Thr-NH2 | 99 | 99 | 1237.52 | 1238.52 | 0.19 ± 0.04 | >1K | 158 ± 14 | 27 ± 7.5 | >1K | agonist |
| 26 | des-AA1,4-6,10,12,13-[DTyr2,LAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-Thr-NH2 | 99 | 99 | 1237.52 | 1238.25 | 124; 97 | >1K | >1K | >1K | >1K | |
| 27 | des-AA1,2,4-6,10,12,13-[DAgl(NMe,2naphthoyl)8,IAmp9,Tyr11]-SRIF-NH2 | 80 | 87 | 989.40 | 990.18 | 4.7; 7.3 | >1K | 252; 381 | 695; 266 | >1K | agonist |
| 28 | des-AA1,2,4-6,10,12,13-[LAgl(NMe,2naphthoyl)8,IAmp9,Tyr11]-SRIF-NH2 | 81 | 94 | 989.40 | 990.18 | 530; 631 | >1K | >1K | >1K | >1K | |
| 29 | des-AA1,2,4-6,10,12,13-[DCys3,DAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-2Nal-NH2 | 91 | 93 | 1170.48 | 1171.70 | 1.2 ± 0.54 | >1K | 112 ± 23 | 63 ± 32 | 181 ± 5 | agonist |
| 30 | des-AA1,2,4-6,10,12,13-[DCys3,LAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-2Nal-NH2 | 98 | 85 | 1170.48 | 1171.50 | 267; 316 | >1K | 329; 729 | 535; 214 | 279; 635 | |
| 31 | des-AA1,4-6,10,12,13-[Tyr2,DCys3,DAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-2Nal-NH2 | 81 | 87 | 1333.55 | 1334.81 | 8.2 ± 4.9 | >1K | 410 ± 114 | 270 ± 122 | >1K | agonist |
| 32 | des-AA1,4-6,10,12,13-[Tyr2,DCys3,LAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-2Nal-NH2 | 73 | 86 | 1333.55 | 1334.61 | 189; 365 | >1K | 463; 1169 | >1K | >1K | |
| 33 | des-AA1,5-[Tyr2,DTrp8,IAmp9]-SRIF37 | 97 | 97 | 1647.73 | 1648.8 | 14 ± 3 | >1K | >1K | >1K | >1K | agonist |
| 34 | des-AA1,4-6,10,12,13-[DnatITyr2,DAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-Thr-NH2 | 88 | 99 | 1363.41 | 1364.29 | 0.8 ± 0.14 | >1K | 344 ± 123 | 45 ± 6.7 | >1K | |
Legend Table 1: Structure of SRIF: (Cyclo 3-14)H-Ala1-Gly2-Cys3-Lys4-Asn5-Phe6-Phe7-Trp8-Lys9-Thr10-Phe11-Thr12-Ser13-Cys14-OH
Percent purity determined by HPLC using buffer system: A = TEAP (pH 2.5) and B = 60% CH3CN/40% A with a gradient slope of 1% B/min, at flow rate of 0.2 mL/min on a Vydac C18 column (0.21 × 15 cm, 5-μm particle size, 300 Å pore size). Detection at 214 nm.
Capillary zone electrophoresis (CZE) was done using a Beckman P/ACE System 2050 controlled by an IBM Personal System/2 Model 50Z and using a ChromJet integrator. Field strength of 15 kV at 30 °C, mobile phase: 100 mM sodium phosphate (85:15, H2O:CH3CN) pH 2.50, on a Supelco P175 capillary (363 μm OD × 75μm ID × 50 cm length). Detection at 214 nm.
The calculated m/z of the monoisotope compared with the observed [M + H]+ monoisotopic mass.
The IC50 values (in nM) were derived from competitive radioligand displacement assays reflecting the affinities of the analogues for the five cloned somatostatin receptors using the non-selective 125I-[Leu8,DTrp22,Tyr25]SRIF-28 as radioligand. Mean value ± SEM when n ≥ 3. When n<3, individual values of two assays are listed. 1K corresponds to 1000.
In several drug design studies of different hypothalamic releasing hormones (gonadotropin-releasing hormone (GnRH),41 corticotropin-releasing factor (CRF),42 SRIF,37, 43, 44), we had demonstrated that substitutions of natural amino acids with betidamino acids were compatible with biological activity. Betidamino acids are monoacylated derivatives of α-aminoglycine. The synthesis of α-Fmoc,β-Boc-aminoglycine was originally described by Qasmi et al.,45 and the synthesis of the methylated derivatives was published by our group.46 As we demonstrated earlier, the substitution of DTrp8 by DAgl(NMe,2naphthoyl)8 in the undecamer scaffold37 (3 versus 7, Table 1) had resulted in a 5-fold increase in binding affinity at sst1.39

Here, we describe the rational design and optimization of a novel class of peptidic somatostatin sst1-selective analogues derived from the use of our published sst1-pharmacophore model.1
Results and Discussion
Peptide synthesis
All analogues shown in Table 1 were synthesized either manually or automatically on a 4-methylbenzhydrylamine (MBHA) or chloromethylated (CM) resin using the Boc-strategy and N,N’-diisopropylcarbodiimide (DIC)/1-hydroxybenzotriazole (HOBt) for amide bond formation.
We used an unresolved aminoglycine (Agl) derivative Fmoc-D/LAgl(NMe,Boc)-OH46, 47 as template for the introduction of a betidamino acid in the scaffolds (des-AA1,4-6,10,12,13-SRIF-Thr/Nal-NH2, des-AA1,2,4-6,10,12,13-SRIF-Thr/Nal-NH2 and des-AA1,2,4-6,10,12,13-SRIF-NH2), which resulted in diastereomeric mixtures that were separated by RP-HPLC48-50 and were fully characterized. The absolute configuration of the Agl residue in the diastereomers was deduced from enzymatic hydrolysis studies with aminopeptidase M and proteinase K, respectively. Comparison of the enzymatic cleavage patterns of the diastereomers showed that the LAgl-containing analogue was hydrolyzed while the DAgl-containing analogue was not.50
The peptide resins were treated with anhydrous hydrogen fluoride in the presence of scavengers (anisole and dimethylsulfide) to liberate the fully deblocked crude peptides. Cyclization of the cysteines was mediated by iodine in an acidic milieu. Purification was achieved using multiple RP-HPLC steps.49 Analytical RP-HPLC,49 capillary zone electrophoresis (CZE)51 and mass spectrometry were used to determine the purity and identity of the analogues.
NMR studies
The NMR samples of 16, 23 and 27 were prepared by dissolving 2.5 mg of the peptide in 500 μl of DMSO-d6. Assignment of the various proton resonances was carried out using the standard procedure, using DQF-COSY, TOCSY and NOESY experiments. Though 16 showed a single set of resonances for all the protons, two sets of resonances were observed for most of the protons for 23 and 27 in the ratio 60:40, except for the HN and αH resonances of IAmp9. This is due to the cis/trans isomerization of the amide bond present in the side chain of the betidamino acid DAgl at position 8 of 23 and 27, as reported earlier.52 Almost complete assignment of the chemical shifts of the various proton resonances is carried out for 16, 23 and 27 and is shown in Table 2A-2C. Amide resonances of Cys3 were not observed for 23 and 27 in the NMR spectra due to fast exchange with the solvent.
Table 2A. Chemical shifts (in ppm) of 16 in DMSO-d6.
| Residue | NH | αH | βH | Others |
|---|---|---|---|---|
| DPhe2 | 8.00 | 4.19 | 3.26, 2.97 | QD: 7.40, QE: 7.36 |
| Cys3 | 9.28 | 5.39 | 2.88, 2.76 | |
| Phe7 | 8.55 | 4.71 | 2.87, 2.76 | QD: 6.99, QE: 7.03 |
| DTrp8 | 8.66 | 4.23 | 2.66, 2.50 | HD1: 6.86, HE3: 7.49, HE1: 10.73 HZ3: 7.04, HZ2: 7.36, HH2: 7.11 |
| IAmp9 | 8.88 | 4.24 | 3.09, 2.56 | QD: 7.29, QE: 7.36, QT: 4.06, HH: 8.59, QK1: 1.15, QK2: 1.15, HI: 3.15 |
| Phe11 | 7.72 | 4.90 | 3.19, 3.00 | QD: 7.30, QE: 7.20 |
| Cys14 | 8.65 | 5.20 | 2.84, 2.84 | |
| Thr15 | 8.11 | 4.25 | 4.05 | γCH3: 1.07, OH: 5.21 |
| NH2 | 7.58,7.39 |
Table 2C. Chemical shifts (in ppm) of 27 in DMSO-d6.
| Residue | Conf | NH | αH | βH | Others |
|---|---|---|---|---|---|
| Cys3 | Major | - | 3.95 | 3.03,2.82 | |
| Minor | - | ||||
| Phe7 | Major | 8.75 | 5.02 | 3.01,2.91 | QD: 7.28, QE: 7.41 |
| Minor | - | 4.75 | 3.00,2.87 | ||
| DAgl8 | Major | 8.86 | 6.53 | QG: 2.34, H1: 7.81, H3: 7.62, H4: 8.00 H5: 7.89, H6: 7.64, H7: 8.00, H8: 7.28 |
|
| Minor | 9.23 | 5.71 | QG: 2.62, H5: 8.01 | ||
| IAmp9 | Major | 8.35 | 4.24 | 2.83,2.83 | QD: 7.10, QE: 7.21, QT: 3.81, HH: 7.34, QK1: 1.23, QK2: 1.11, HI: 3.07 |
| Minor | - | - | HH: 7.21, QT: 4.07, HI: 3.23 | ||
| Tyr11 | Major | 8.30 | 4.41 | 3.08,2.83 | QD: 7.07, QE: 6.69 |
| Minor | 8.02 | 4.35 | QD: 7.27 | ||
| Cys14 | Major | 7.62 | 4.41 | 3.02,2.80 | |
| Minor | |||||
| NH2 | Major | 7.27,7.08 |
A reasonably large number of experimental NOEs is observed for the three analogues in the NOESY spectrum measured with a mixing time of 100 msec, leading to over 70 meaningful distance restraints per analogue (Table 3, Figure 1). For all analogues, structural restraints from NOEs were used as input for the structure calculation with the program CYANA53 followed by restrained energy minimization using the program DISCOVER.54 The resulting bundle of 20 conformers per analogue represents the 3D structure of each analogue. For each analogue, the small residual violations in the distance constraints only for the 20 refined conformers (Table 3) and the coincidence of experimental NOEs and short interatomic distances (data not shown) indicate that the input data represent a self-consistent set, and that the restraints are well satisfied in the calculated conformers (Table 3). The deviations from ideal geometry are minimal, and similar energy values were obtained for all 20 conformers of each analogue. The quality of the structures determined is reflected by the small backbone RMSD values relative to the mean coordinates of ~0.5 Å (see Table 3 and Figure 2).
Table 3. Characterization of the NMR structures of the analogues studied by NMRa.
| Parameters | 16 | 23 | 27 |
|---|---|---|---|
| Restraints | |||
| NOE Distances | 93 | 72 | 58 |
| Angles | 23 | 16 | 7 |
| CYANA Target function | 0.29 | 0.25 | 0.27 |
| RMSD (in Å) | |||
| Backbone | 0.35 ± 0.15 | 0.75 ± 0.18 | 0.12 ± 0.06 |
| Overall | 1.12 ± 0.20 | 1.47 ± 0.31 | 1.92 ± 0.37 |
| Residual Violations on | |||
| Distances | |||
| No ≥ 0.1 Å | 0.50 ± 0.20 | 0.20 ± 0.06 | 0.40 ± 0.08 |
| Max (Å) | 0.80 ± 0.15 | 0.06 ± 0.02 | 0.08 ± 0.01 |
| Dihedral Angles | |||
| No ≥ 1.5° | 0.0 ± 0.01 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Max (°) | 0.1 ± 0.10 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| CFF91 energies (kcal/mol) | |||
| Total energy | 218 ± 9 | 233 ± 8 | 202 ± 4 |
| Van der Waals | 168 ± 8 | 181 ± 5 | 174 ± 6 |
| Electro-static | 50 ± 7 | 51 ± 4 | 28 ± 2 |
Figure 1.

Survey of characteristic NOEs describing the secondary structure of analogues (A) 16, (B) 23 and (C) 27. Thin, medium and thick bars represent weak (4.5-6 Å), medium (3-4.5 Å) and strong (< 3 Å) NOEs observed in the NOESY spectrum. One letter code represents the amino acids in the sequence of the peptide, where f, g, w and X represents DPhe, DAgl(NMe, 2naphthoyl), DTrp and IAmp.
Figure 2.

3D NMR structures of analogues (A) H-DPhe-c[Cys-Phe-DTrp-IAmp-Phe-Cys]-Thr-NH2 (16), (B) H-c[Cys-Phe-DAgl(NMe,2naphthoyl)-IAmp-Phe-Cys]-Thr-NH2 (23) and (C) H-c[Cys-Phe-DAgl(NMe,2naphthoyl)-IAmp-Tyr-Cys]-NH2 (27). For each analogue, 20 energy-minimized conformers with the lowest target function are used to represent the 3D NMR structure. The bundle is obtained by overlapping the Cα atoms of all the residues. The backbone and the side chains are displayed including the disulphide bridge. The amino acid side chains that are proposed to be involved in sst1 binding are highlighted: light green, DTrp/DAgl at position 8; blue, IAmp at position 9; yellow, Phe at positions 7 and 11. In (D) the side chains of sst1 binding analogue 5 (in dark green) are superimposed on the structure of 16 (in grey), 23 (in cyan) and 27 (in magenta). It should be noted that the aromatic side chains at position 7 and 11 of analogue 5 are in the other side of the peptide backbone compared to 16, 23 and 27.
Three-dimensional structure of H-DPhe2-c[Cys3-Phe7-DTrp8-IAmp9-Phe11-Cys14]-Thr15-NH2 (16)
Analogue 16 binds selectively to sst1 with moderately high affinity (IC50 ~ 30 nM). It differs from octreotide by IAmp9, Phe11 and Thr15-NH2 and the 3D NMR structure shows that the backbone has a β-turn of type III’ around DTrp8 and IAmp9 (Figure 2A, Table 4). In all of the calculated 20 conformers, there is a hydrogen bond between the amide protons of Phe11 to the carbonyls of Phe7 that stabilizes the β-turn. The side chains of DPhe2, Phe7 and DTrp8 are in the gauche+ configuration, the side chains of IAmp9 and Phe11 are in the gauche- configuration (Table 4).
Table 4. Torsion angles ϕ, ψ and χ1 (in degrees) of the bundle of 20 energy minimized conformers.
| Analogue | Angle | DPhe2 | Cys3 | Phe7 | DTrp8 | IAmp9 | Phe11 | Cys14 | Thr15 |
|---|---|---|---|---|---|---|---|---|---|
| 16 | ϕ | - | -174 ± 5 | -108 ± 12 | 60 ± 7 | 63 ± 1 | -68 ± 11 | 52 ± 34 | -54 ±75 |
| ψ | -147 ± 1 | 46 ± 2 | 145 ± 5 | 31 ± 4 | 14 ± 2 | -68 ± 24 | 100 ± 18 | 134 ± 47 | |
| χ1 | 9 ± 30 | -125 ± 11 | 1 ± 32 | 27 ± 31 | -109 ± 1 | -168 ± 19 | -100 ± 37 | 102 ± 76 | |
| 23 | DAgl8 | ||||||||
| ϕ | - | -84 ± 62 | -13 ± 62 | -117 ± 33 | 48 ± 7 | -96 ± 26 | 146 ± 64 | ||
| ψ | 179 ± 60 | 170 ± 10 | -74 ± 13 | -158 ± 11 | 26 ± 2 | -172 ± 2 | -98 ± 81 | ||
| χ1 | 99 ± 61 | -41 ± 50 | -59 ± 93 | -150 ± 6 | -155 ± 4 | -102 ± 12 | -137 ± 40 | ||
| 27 | DAgl8 | Tyr11 | |||||||
| ϕ | - | -156 ± 8 | 95 ± 4 | 166 ± 3 | -110 ± 8 | -57 ± 6 | |||
| ψ | 132 ± 9 | 157 ± 1 | -54 ± 1 | 42 ± 1 | -35 ± 3 | -85 ± 0 | |||
| χ1 | 34 ± 13 | 27 ± 1 | -112 ± 102 | -18 ± 80 | 141 ± 9 | -109 ± 0 | |||
Three-dimensional structure of H-c[Cys3-Phe7-DAgl(NMe,2naphthoyl)8-IAmp9-Phe11-Cys14]-Thr15-NH2 (23)
Analogue 23 differs from 16 by the substitution of DTrp8 by DAgl(NMe,2naphthoyl)8 and the deletion of DPhe2 at the N-teminus (Table 1). With those modifications, 23 binds selectively to sst1 with high affinity (IC50 = 1 nM). The 3D NMR structure of 23 shows that the angles around DAgl(NMe,2naphthoyl)8 and IAmp9 do not represent any standard β-turns (Figure 2B, Table 4). The side chains of Phe7, DAgl(NMe,2naphthoyl)8, IAmp9 and Phe11 are in the gauche- configuration (Table 4). In addition, the side chains of Phe7 and DAgl(NMe,2naphthoyl)8 span a large conformational space (Figure 2B, Table 4). In the major conformer, the side chain methyl protons of DAgl(NMe,2naphthoyl)8 give rise to NOE/ROEs to both the H1 and H8 protons of the naphthoyl group suggesting that DAgl(NMe,2naphthoyl)8 is in trans isomer in the major conformer. Based on the large number of NOEs observed, the 3D NMR structure of the major conformer was calculated and this is assumed to be the bioactive conformation.
Three-dimensional structure of H-c[Cys3-Phe7-DAgl(NMe,2naphthoyl)8-IAmp9-Tyr11-Cys14]-NH2 (27)
The composition of 27 is different from that of 23 with Tyr11 replacing Phe11 and the deletion of Thr15 at the C-terminus (Table 1), yet it binds selectively to sst1 with high affinity (IC50 = 6 nM). The 3D NMR structure shows that the angles around DAgl(NMe,2naphthoyl)8 and IAmp9 do not represent any standard β-turns (Figure 2C, Table 4). The side chains of Phe7 and Tyr11 are in the gauche+ configuration, the side chains of DAgl(NMe,2naphthoyl)8 and IAmp9 are in the gauche- configuration (Table 4). The side chains of DAgl(NMe,2naphthoyl)8 and IAmp9 span a large conformational space (Figure 2C, Table 4). In the major conformer, the side chain methyl protons of DAgl(NMe,2naphthoyl)8 give rise to NOE/ROEs to both the H1 and H8 protons of the naphthoyl group suggesting that DAgl(NMe,2naphthoyl)8 is in trans isomer in the major conformer. Based on the large number of NOEs observed, the 3D NMR structure of the major conformer was calculated and this is assumed to be the bioactive conformation. Moreover, the minor conformer was poorly defined due to the few number of NOEs that could be identified for this conformation.
Biological testing
To determine their SRIF receptor-binding properties, the compounds were tested for their ability to bind to cryostat sections of a membrane pellet of cells expressing the five human SRIF receptor subtypes (Table 1). For each of the tested compounds, complete competition experiments were carried out with the universal SRIF radioligand [Leu8,DTrp22,125ITyr25]SRIF-28 (125I-[LTT]-SRIF-28).55 The results are shown in Table 1. The most potent and selective analogues were functionally evaluated for their agonist/antagonist properties using a reporter gene assay that determines the biological activity of the human sst1 receptor in CCL39-sst1-Luci cells constitutively expressing the human sst1 receptor as well as the luciferase gene under the control of the serum response element (SRE). The SRE is regulated by transcription factors and is activated by many extracellular signals including ligands acting at G-protein-coupled receptors.56, 57 It has been shown that upon agonist binding, SRIF receptors mediate an increase of luciferase expression via SRE in this reporter gene assay.32, 58 Figure 3 shows that stimulating CCL39-sst1-Luci cells with somatostatin analogues activates the luciferase gene in a concentration-dependant manner. SRIF-14, 23 and 25 exhibit EC50 values of 0.93 nM, 3.9 nM and 0.37 nM, respectively while the sst1- agonist des-AA1,5-[Tyr2,DTrp8,IAmp9]-SRIF (CH-288)37 (33) exhibits an EC50 value of 33 nM. Des-AA1,2,4,5,12,13[DCys3,Tyr7,DAgl8(NMe,2naphthoyl)]-Cbm-SRIF (Sst3-ODN-8)43 (35), a short-sized somatostatin analog with no affinity to sst1 receptor used as negative control was unable to stimulate luciferase gene expression in CCL39-sst1-Luci cells. The results of the reporter gene assay are summarized in Table 1.
Figure 3.
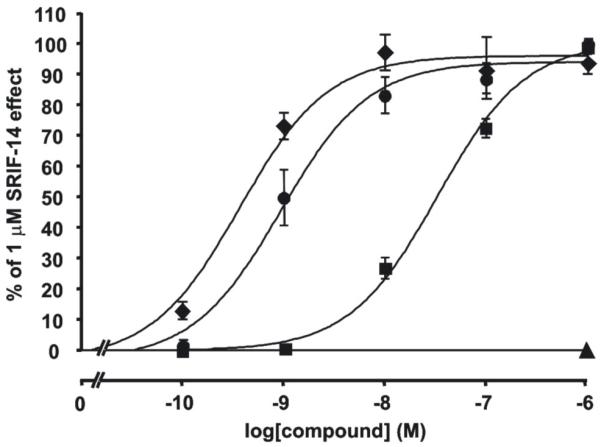
The SRIF analogue 25 is a potent sst1 agonist when tested in the luciferase reporter gene assay. The assay was performed as described in Materials and Methods. CCL39-sst1-Luci cells were treated with increasing concentrations (0.1 nmol/L, 1 nmol/L, 10 nmol/L, 100 nmol/L, and 1 μmol/mL) of SRIF (●), 33 (■), or 25 (◆). The stimulation of the luciferase reporter gene activity by the compounds is expressed as % stimulation of the 1 μmol/mL SRIF effect. Shown are the dose-response curves of the compounds. The sst3-antagonist 35 (▲), used as negative control at 1 μmol/mL, shows no effect in the luciferase reporter gene assay.
Structure activity relationship studies
The proposed sst1 receptor pharmacophore1 derived from the NMR structures of the family of the undecamers (mono and dicyclic)37, 38 compared with the previously proposed sst2, sst2,5 and sst4 pharmacophores are shown in Figure 4.
Figure 4.

Schematic drawings of agonist pharmacophores for receptor-selective analogues binding to the somatostatin receptors: (A) sst1, (B) sst2, (C) sst2/5 and (D) sst4. The amino acid side chains, which are part of the pharmacophores and the range of distances between the corresponding Cγ atoms of the side chains are shown.
With a well-defined pharmacophore, we were now in a position to design improved analogues following a rational approach. In this regard, we designed putative sst1-selective octa-, hepta-, and hexamers derived from the known undecamers.1 Figure 4A shows that the sst1-selective agonist-pharmacophore has two aromatic side chains, at position 6 or 7 and 11, in addition to the DTrp8 and IAmp9 pair. The sst1 selectivity is achieved mainly through the amino acid IAmp at position 9, which replaces Lys, present in most of the somatostatin analogues. The side chain of IAmp is longer than the side chain of Lys, it has an aromatic group (aminomethylphenylalanine) extended by a relatively bulky aliphatic group (isopropyl). Hence, we conclude that IAmp cannot be accommodated in a smaller binding pocket that will fit a smaller side chain as that of Lys9 critical for sst2-5 binding. We propose that only in the sst1 receptor structure is the binding pocket large enough to accommodate the side chain of IAmp (comparing the residues in the models of the trans-membrane regions of the somatostatin receptors based on the crystal structure of Rhodopsin) and therefore analogues with IAmp at position 9 show selectivity for sst1.
We wanted to identify smaller peptidic molecules than the undecamers, which would retain selectivity as well as good affinity at sst1 receptors and possibly would act as antagonists having greater metabolic stability than the previously published analogues.36-39, 59 As we published earlier, the hypothesis that IAmp9 by itself might establish sst1 selectivity in the otherwise potent pan-somatostatin octapeptide des-AA1,2,4,5,12,13-[DTrp8]-SRIF (ODT-8)50 (8) failed (Table 1). Substitution of Lys9 in 8 by IAmp9 resulted in des-AA1,2,4,5,12,13-[DTrp8, IAmp9]-SRIF (9), which showed no binding affinity to any SRIF receptor,37 most probably due to steric effects perturbing the alignment of the DTrp8-IAmp9 side chains with respect to the other aromatic side chains.
Since the backbone conformations of the sst1-selective analogues have a hairpin-like structure similar to that of the sst2/3/5-selective octreotide-based analogues, we decided to introduce IAmp at position 9 in the octreotide scaffold with the additional substitutions (an aromatic side chain at positions 6 or 7 and at position 11) indispensable to fulfill the sst1 pharmacophore’s requirements (see below).
With the aim of searching for sst1-selective antagonists, we first substituted Lys9 with IAmp9 in the published sst2-antagonist des-AA1,4-6,11-13-[Cpa2,DCys3,Tyr7,DTrp8 ]-SRIF-2Nal-NH2 (12),60, 61 which resulted in (des-AA1,4-6,11-13-[Cpa2,DCys3,Tyr7,DTrp8,IAmp9]-SRIF-2Nal-NH2 (13) with no binding affinity to sst1/4/5 and low binding affinity to sst2/3 (ca. 350 nM). Based on the sst1 pharmacophore, it was expected that the introduction of IAmp alone would not be sufficient for sst1 binding (Table 1) since 13 does not have two of the aromatic residues at positions 6 or 7 and 11 necessary for sst1 binding. Though there is an aromatic residue at position 7 in octreotide-based analogues, they do not have Phe at position 11. Hence, we replaced Thr at position 10 by Phe at 11 (as in SRIF), leaving 4 residues and the two Cys residues in the cycle. This analogue (16) exhibited similar affinity to sst1 (IC50 = ~30 nM) as 8 but it is more selective since it shows low affinity to the other four receptors (see Table 1). Therefore analogue 16 was selected as lead for further derivatization.
Our SAR studies focused on positions 2, 3, 8, 9, 11, and 15 of the analogue 16 (SRIF numbering). Affinities to the five SRIF receptors were determined in receptor binding assay and are summarized in Table 1.
SAR at position 2 where DPhe, Cpa, Tyr and DTyr were substituted show that these residues can be eliminated with only small differences in binding affinity to sst1 (des-AA1,2,4-6,10,12,13-[DTrp8,IAmp9]-SRIF-Thr-NH2 (15) versus 16, des-AA1,2,4-6,10,12,13-[DTrp8,IAmp9,ITyr11]-SRIF-Thr-NH2 (17) versus des-AA1,4-6,10,12,13-[DPhe2,DTrp8,IAmp9, ITyr11]-SRIF-Thr-NH2 (18), des-AA1,2,4-6,10,12,13-[DCys3,DTrp8,IAmp9, ITyr11]-SRIF-2Nal-NH2 (19) versus des-AA1,4-6,10,12,13-[Cpa2,DCys3,DTrp8,IAmp9, ITyr11]-SRIF-2Nal-NH2 (20), des-AA1,2,4-6,10,12,13-[LAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-Thr-NH2 (24) versus des-AA1,4-6,10,12,13-[DTyr2,LAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-Thr-NH2 (26) and des-AA1,2,4-6,10,12,13-[DCys3,DAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-2Nal-NH2 (29) versus des-AA1,4-6,10,12,13-[Tyr2,DCys3,DAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-2Nal-NH2 (31)). We suggest that the individual nature of these amino acids (despite the fact that their side chains are not involved in binding) is responsible for the differences in affinity. SAR at position 3 suggests that the substitution of Cys with DCys has little effect on sst1 binding affinity and function. SAR at position 8 shows the unique nature of Agl8-containing analogues that are all highly selective for sst1 over the other four receptor subtypes (23, 25, 27, 29). Interestingly, for this scaffold, a D-configuration is significantly more favorable than the L-conformation. SAR at position 9 shows that the introduction of IAmp results in more active and selective analogues (des-AA1,4-6,10,12,13-[Cpa2,DCys3,DAgl(NMe,2naphthoyl)8]-SRIF-2Nal-NH2 (21) versus 31). SAR at position 11 shows that the substitution of Phe as in 15 or 16 with I-Tyr results in some loss in sst1 binding affinity (17 and 18). SAR at position 15 shows that this residue can also be eliminated from some structures without loss of binding affinity and selectivity as in 27. This deletion along with other most favorable substitutions yielded the hexapeptide H-c[Cys3-Phe7-DAgl(NMe,2naphthoyl)8-IAmp9-Tyr11-Cys14]-NH2 (27). This peptide selectively binds to sst1 with an average IC50 = 6.0 nM, (IC50 >1000 nM at sst2, 317 nM at sst3, 481 nM at sst4, and >1000 nM at sst5, respectively). Compared to our previously published analogues, which contained 11 residues, 27, containing only 6 residues, is a much smaller and appealing molecule.
Though des-AA1,4-6,12,13-[Tyr2,DTrp8,IAmp9]-SRIF-Thr-NH2 (14), 15 and 17-20 (Table 1) showed IC50 for sst1 in the 500 nM range, their binding affinities are significantly lower compared to that of SRIF-28 (IC50 for sst1 = 2.9 nM), 8 (IC50 for sst1 = 27 nM) and 5 (IC50 for sst1 = 2.5 nM).1 The lower affinities of these analogues could be explained based on the knowledge of the sst1 pharmacophore and be due to one or more of the following reasons: (1) the distances between the side chains of the residues involved in binding may be slightly different from the distances required by the sst1 pharmacophore (the distances reported for efficient receptor-ligand interactions are as follows: between DTrp8 and IAmp9 is 7-8 Å, between DTrp8 and Phe6/7 is 6-7.5 Å, between DTrp8 and Phe11 is 9.5-12 Å, between IAmp9 and Phe6/7 is 9-11 Å, between IAmp9 and Phe11 is 8-10 Å and between Phe6/7 and Phe11 is 6-7.5 Å, (Figure 4A)1) that the side chains possibly occupy the binding pocket partially; (2) the conformational rigidity in the side chain of DTrp at position 8 is in such a way that it cannot occupy completely the available binding pocket; (3) though the analogues have two aromatic residues, the aromatic side chains could be on the front side of the peptide backbone (when displayed from N to the C-terminus) resulting in μM binding affinity.
In the octreotide pharmacophore, the side chains of DTrp8 and Lys9 are in close proximity (4-6 Å),62, 63 (Figure 4C) whereas in the sst1-pharmacophore the side chains of DTrp8 and IAmp9 are farther apart (7-8 Å) (Figure 4A). Since the side chain of aminoglycine (Agl) has been shown to span a larger volume than the DTrp side chain, we introduced acylated Agl at position 8.52 This substitution in the shortened analogues (21, 23, 25, 27, 29 and 31) improved the binding affinity and selectivity for sst1 as well. Some of these analogues also exhibit binding to sst4. The introduction of Phe11 (21, 23, 25, 29 and 31) results in an aromatic group close to Lys9 /IAmp9 as well as DTrp/DAgl8, which satisfies the sst4 pharmacophore partially and allows binding (Figure 4D, Table 5).
Table 5. Distances between Cγ atoms (in Å) of selected residues for the analogues studied by NMR and the sst1/sst4- pharmacophores.
| F7 - X8 | F7 - IAmp9 | F7 - F11 | X8 - IAmp9 | X8 - F11 | IAmp9-F11 | |
|---|---|---|---|---|---|---|
| Sst1 pharmacophore | 6.0-7.5 | 9.0-11.0 | 6.0-7.5 | 7.0-8.0 | 9.5-12.0 | 8.0-10.0 |
| Sst4 pharmacophore | - | - | - | 4.5-6.5 | 5.5-9.5 | 4.5-6.5 |
| 16 | 5.9-7.0 | 9.4-9.8 | 5.1-6.8 | 5.3-6.7 | 8.2-8.8 | 8.0-8.6 |
| 23 | 5.9-7.8 | 7.3-10.6 | 6.6-11.0 | 6.9-8.1 | 9.6-11.2 | 5.8-6.6 |
| 27 | 5.2-6.8 | 8.5-10.0 | 6.4-7.7 | 4.5-7.1 | 9.0-9.8 | 6.4-8.3 |
X refers to either DTrp or DAgl(NMe, 2naphthoyl) residues.
Following up on the observation by Bass et al.64, 65 and Hocart et al.61 that one could turn an octreotide-based sst2 agonist scaffold characterized by a DAaa2-LCys3 to an antagonist scaffold characterized by a LAaa2-DCys3, we tested 31 for agonism/antagonism. In our functional luciferase reporter gene assay, 31 along with 23, 25, 27 and 29, were agonists at human sst1 (Table 1). The best analogue in this series is 25 with an EC50 value of 0.37 nM in the luciferase reporter gene assay, with a binding IC50 of 0.19 nM, and 5,000-fold selectivity versus sst2/5, 500-fold selectivity versus sst3, and 100-fold selectivity versus sst4, respectively. SRIF and the sst1-agonist 33 exhibit EC50 values of 0.93 nM and 33 nM, respectively. Figure 3 shows that SRIF, 25 and 33 are agonists since they effectively stimulate luciferase expression in the reporter gene assay. It is noteworthy to mention that 25 is more potent than SRIF (EC50: 0.37 nM versus EC50: 0.93 nM), whereas the standard sst1 agonist 33 is approximately 100-fold less potent than either of them. We have used 354,3 a short-sized somatostatin analog with no affinity to sst1 receptors, as a negative control, in this test; it was found to be inactive at a concentration of 1 μM.
Analogue 25 was radio-iodinated with 125I and found to bind to sections of membrane pellet of sst1-expressing transfected cells with high specificity, as shown in Figure 5. Moreover, we evaluated the ability of the 125I-25 to bind to sst1-expressing human cancers. Table 6 shows that this radioligand was able to label virtually all tested sst1-expressing tumors, including a selection of well characterized sst1-expressing prostate cancers, mesenchymal cancers, bronchial carcinoids and gastroenteropancreatic tumors. Figure 6 and Table 6 show that compared to the strong 125I-[LTT]-SRIF-28 labeling, found to be fully displaceable by 33 in these tissues and therefore indicative of sst1 expression, the 125I-25 labeling was noticeably weaker when performed on adjacent sections of sst1 expressing human prostate cancer. The relatively weak 125I-25-labeling is difficult to explain, since the binding affinity of the cold iodinated compound des-AA1,4-6,10,12,13-[DnatITyr2,DAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-Thr-NH2 (34) is in the low nanomolar range (IC50 ≅ 1.0 nM for 34). Sst2-expressing tumors, used as negative controls, were completely negative for 125I-25 binding (Table 6), despite an extremely high density of sst2 receptors in these tumors. This further indicates the high sst1 specificity of the tracer.
Figure 5.
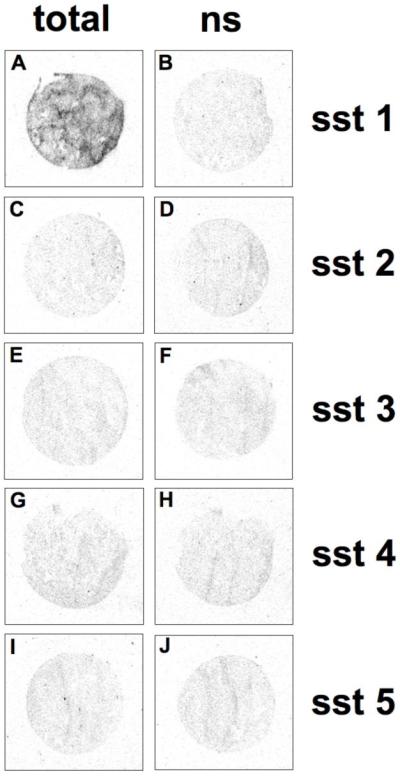
125I-25 specifically labels the sst1 expressing cell line. Autoradiograms showing total binding of 125I-25 in sst1 (A), sst2 (C), sst3 (E), sst4 (G), and sst5 (I) receptor expressing cells. Only sst1 cells are labeled. Autoradiograms showing non-specific binding of 125I-25 in sst1 (B), sst2 (D), sst3 (F), sst4 (H), and sst5 (J) receptor expressing cells (in the presence of an excess of unlabeled 25).
Table 6.
125I-25 specifically labels sst1-expressing human cancers
| # of | 125I-[LTT]-SRIF-28 binding displaceable by 33 | 125I-25 binding displaceable by 33 | |||
|---|---|---|---|---|---|
| Tumor type | cases | Incidence | Density | Incidence | Density |
| A: Sst1-expressing tumors | |||||
| - Prostate Cancer | 8 | 8/8 | +++ | 7/8 | + |
| -Mesenchymal Tumors | 7 | 7/7 | ++ / +++ | 6/7 | ++ |
| -Bronchial Carcinoids | 6 | 6/6 | ++ | 6/6 | + /++ |
| -Gastroenteropancreatic Tumors | 4 | 4/4 | ++ | 4/4 | + / ++ |
| B: Sst2-expressing tumors | 8 | 0/8* | - | 0/8 | - |
+++, ++, + means high, moderate or low receptor density, respectively.
125I-[LTT]-SRIF-28-binding in these tumors is fully displaceable by octreotide.
Figure 6.
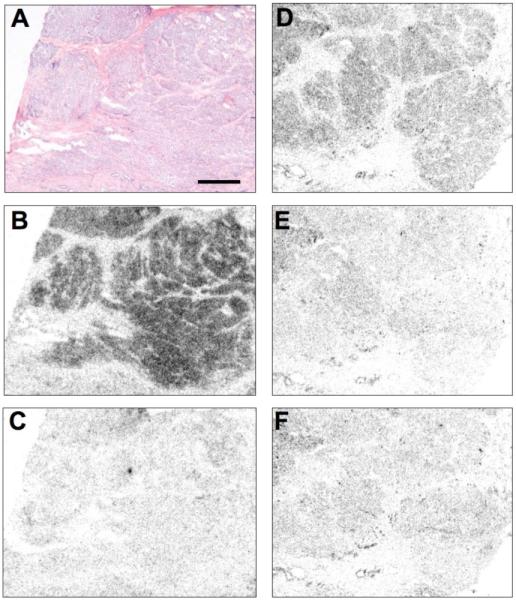
125I-25 labels an sst1 receptor-expressing human prostate cancer. (A) Hematoxylin eosin stained tumor section (bar: 1 mm). (B, C) Autoradiograms showing total binding of 125I-[LTT]-SRIF-28 with strong tumor labeling (B) that is displaceable by the sst1-selective 33 (C) indicating sst1 expression. (D,E,F) Autoradiograms showing total binding of 125I-25 with clear tumor labeling (D) that is displaceable by an excess of unlabeled 25 (E) or 33 (F).
The sst1 pharmacophore reported previously by our group showed that the side chains of four different residues are important for the binding of these analogues.1 They are the indole ring of DTrp at position 8, IAmp side chain at position 9, and two aromatic side chains at positions 6 or 7 and 11. The distances between the side chains of these residues should be close to the values given in Table 5.1 In addition, the sst1 pharmacophore was different from the other SRIF receptor pharmacophores63, 66, 67 in the positioning of the aromatic side chains. In all of the other SRIF receptor pharmacophores, the aromatic side chains are present at the front side of the peptide backbone, when the structure of the peptide is oriented from N- to C-terminus. In contrast, in the sst1 pharmacophore, both of the aromatic side chains are present at the back side of the peptide backbone.1 Therefore, any peptide that has the sst1 pharmacophore, that involves the four residues at the proposed distance, should bind to sst1.
To verify the identity of the sst1 pharmacophore, the 3D structures of three analogues, 16, 23 and 27, that bind selectively to sst1 with nM affinity were studied. All these analogues have IAmp at position 9, which is crucial for selectivity to sst1. As shown in Figure 2, the other residues important for sst1 binding, namely DTrp/DAgl8, Phe7 and Phe/Tyr11 are present in all of the three analogues. The distances between the corresponding Cγ atoms of the side chains of the four residues are given in Table 5. Comparing the distances with the sst1 pharmacophore suggests that all of the three analogues have the sst1 pharmacophore, except for a small discrepancy. But a detailed comparison of the structures show that the positioning of the side chains of the aromatic groups at position 7 and 11 is not exactly identical to that described for the sst1 pharmacophore.1 In the structures of the three analogues studied here, the aromatic side chains are present in the front side of the peptide backbone (Figure 2). Since these peptides have high binding affinity for sst1, the binding of these peptides to sst1 could be due to the inherent flexibility observed in these peptides. The presence of DAgl at position 8 introduces a large flexibility to the peptide backbone and in turn to the side chains of 23 and 27, compared to 16 that has DTrp at position 8. This is also reflected in the number of NOEs observed for 23 and 27, which is less compared to the number of NOEs observed for 16 (Table 3). In addition, the chemical shift values observed for the aromatic protons of Phe7 and Phe11 are very similar for 23 and 27 (Tables 2B, 2C) indicating a random coil or less structured elements compared to 16 (Table 2A). Comparing the chemical shift dispersion of the aromatic region of similar sst2-selective octreotide-type SRIF analogues,63 with these sst1-selective analogues suggests that the latter are less structured in DMSO (Figure 7). Based on the conformational flexibility of 23 and 27 (Tables 2B, 2C), it is suggested that the aromatic side chains could move to the backside of the peptide backbone for sst1 binding. The higher binding affinity of 23 and 27 compared to that of 16, could be due to the larger flexibility of the peptide backbone of the former, resulting in a better fit of 23 and 27 into the sst1 binding pocket compared to 16, that shows a single conformation with much less flexibility for its peptide backbone (Figure 2A-D, Table 5).
Table 2B. Chemical shifts (in ppm) of 23 in DMSO-d6.
| Residue | Conf | NH | αH | βH | Others |
|---|---|---|---|---|---|
| Cys3 | Major | - | 4.01 | 3.07, 2.89 | |
| Minor | - | ||||
| Phe7 | Major | 8.87 | 5.03 | 3.01, 2.91 | QD: 7.28, QE: 7.17 |
| Minor | - | 4.75 | |||
| DAgl8 | Major | 8.80 | 6.53 | QG: 2.27, H1: 7.78, H3: 7.65, H4: 8.02 H5: 8.01, H6: 7.65, H7: 8.02, H8: 7.25 |
|
| Minor | 9.21 | 5.74 | QG: 2.55, H1: 7.86, H3: 7.69, H4: 7.93 H5: 7.92, H6: 7.69, H7: 7.93, H8: 7.36 |
||
| IAmp9 | Major | 8.24 | 4.27 | 2.92, 2.80 | QD: 7.12, QE: 7.21, QT: 3.78, HH: 7.37, QK1: 1.23, QK2: 1.10, HI: 3.04 |
| Minor | - | - | QE: 7.36, QT: 4.06, HI: 3.24 | ||
| Phe11 | Major | 8.39 | 4.57 | 3.21, 2.93 | QD: 7.29, QE: 7.21 |
| Minor | 8.12 | 4.48 | |||
| Cys14 | Major | 7.81 | 4.63 | 3.07, 2.83 | |
| Minor | 8.12 | 4.55 | |||
| Thr15 | Major | 7.67 | 4.10 | 4.04 | γCH3: 1.03, OH: 4.90 |
| Minor | 7.60 | ||||
| NH2 | 7.17,7.29 |
Figure 7.

Amide and aromatic region of the 1D proton NMR spectra of analogues des-AA1,4-6,11-13-[DPhe2, DTrp8]-SRIF-Thr-NH2 (octreotide amide) (11), 16, 23 and 27. The chemical shift dispersion of the aromatic region of analogs 11 and 16 are comparable, while that of 23 and 27 suggests that these peptides are less structured in DMSO compared to analogues 11 and 16.
Conclusions
To conclude, using a pharmacophore derived from the structures of undecamers,1 we were able to design somatostatin heptamers and hexamers with improved or similar affinity and sst1 selectivity compared to the previously published undecamers. Though the positioning of the aromatic side chains in these shortened analogues is not exactly identical to that found for the sst1 pharmacophore proposed earlier, backbone flexibility is suggested as a mechanism for induced fit necessary for sst1 binding.
These results demonstrate that rational and efficient SAR in peptides is possible. In this case, it was critical that the structure of the original pharmacophore be well defined and accurate. This was the case since it was based on the structures of several high affinity ligands of differing primary sequences.1 The most potent of these sst1-selective analogues (25) in respect of affinity, selectivity and functionality could be radio-iodinated. While this tracer is highly specific for labeling sst1-expressing tumors with very high binding affinity, the sensitivity of the assay using this radioligand remains below expectations. This is somewhat disappointing, but it should be kept in mind that agonist radioligands are not devoid of caveats. We have shown previously that although 125I-[LTT]-SRIF-28, [125I]Tyr10-CST14, or [125I]Tyr3-octreotide are excellent radioligands showing high affinity and high specific binding in studies using membranes of recombinantly expressed SRIF receptors in CCL-39 or other cells,68, 69 it is clear that [125I]Tyr10-CST14 is by far inferior as a radioligand compared to 125I-[LTT]-SRIF-28 or [125I]Tyr3-octreotide in receptor autoradiography when using slices of brain or peripheral tissues known to express SRIF receptors,70 illustrating the fact that all agonists are not equal.
EXPERIMENTAL PROCEDURES
Chemistry: Starting Materials
The Boc-Cys(Mob)-CM resin with a capacity of 0.3-0.5 mequiv/g was obtained according to published procedures.71 All Nα-tert-butoxycarbonyl (BOC) protected amino acids with side chain protection were purchased from Bachem Inc. (Torrance, CA), Chem-Impex Intl. (Wood Dale, IL), Novabiochem (San Diego, CA) or Reanal (Budapest, Hungary). The side chain protecting groups were as follows: Cys(Mob), Lys[Z(2Cl)] Ser(Bzl), Thr(Bzl), Tyr[Z(2Br)], and m-I-Tyr[Bzl(3Br)]. Boc-IAmp(Z),72 Fmoc-d/lAgl(NMe,Boc)46 and Fmoc-dAgl(Boc)73 were synthesized in our laboratory. All reagents and solvents were reagent grade or better and used without further purification.
Peptide Synthesis
Peptides were synthesized by the solid-phase approach with Boc chemistry either manually or on a CS-Bio Peptide Synthesizer Model CS536. Boc-Cys(Mob)-CM resin with a capacity of 0.3-0.5 mequiv/g or 4-Methylbenzhydrylamine (MBHA) resin with a capacity of 0.4 mequiv/g was used, respectively. Couplings of the protected amino acids were mediated by diisopropylcarbodiimide (DIC) and (HOBt) in DMF for 1 h and monitored by the qualitative ninhydrin test.74 A 3-equivalent excess of the protected amino acids based on the original substitution of the resin was used in most cases. Boc removal was achieved with trifluoroacetic acid (60% in CH2Cl2, 1-2% ethanedithiol or m-cresol) for 20 min. An isopropyl alcohol (1% m-cresol) wash followed TFA treatment and then successive washes with triethylamine solution (10% in CH2Cl2), methanol, triethylamine solution, methanol and CH2Cl2 completed the neutralization sequence. D/L-Agl(NMe,2naphthoyl) residue was formed on the resin. In short: After Fmoc-D/LAgl(NMe,Boc)-OH was coupled, the Boc protecting group was removed with TFA, 3-equivalent 2naphthoyl chloride and 3-equivalent DIPEA were used to acylate the free secondary amino group of the side chain. Removal of the Nα-Fmoc protecting group with 20% piperidine in DMF in two successive 5 and 15 min treatments was followed by the standard elongation protocol until the completion of the peptide.
All of the peptides were cleaved from the resin support with simultaneous side chain deprotection by anhydrous HF containing the scavengers anisole (10% v/v) and methyl sulfide (5% v/v) for 60 min at 0 °C. The diethyl ether precipitated crude peptides were cyclized in 75% acetic acid (200 mL) by addition of iodine (10% solution in methanol) until the appearance of a stable orange color. Forty minutes later, ascorbic acid was added to quench the excess iodine.
Purification and characterization of the analogues
The crude, lyophilized peptides were purified by preparative RP-HPLC49 on a 5 cm × 30 cm cartridge, packed in the laboratory with reversed-phase Vydac C18 silica (15-20 μM particle size, 300 Å) using a Waters Prep LC 4000 preparative chromatograph system, with a Waters 486 tunable absorbance UV detector and Huston Instruments Omni Scribe chart recorder. The peptides eluted with a flow rate of 100 mL/min using a linear gradient of 1% B per 3 min increase from the baseline % B. Eluent A = 0.25 N TEAP pH 2.25, eluent B = 60% CH3CN, 40% A. As a final step, all peptides were rechromatographed in a 0.1% TFA solution and CH3CN on the same cartridge at 100 mL/min (gradient of 1% CH3CN/min). The collected fractions were screened by analytical RP-HPLC on a system using two Waters 501 HPLC pumps, Schimadzu SPD-6A UV detector, Rheodyne Model 7125 injector, Huston Instruments Omni Scribe chart recorder and a Vydac C18 column (0.46 cm × 25 cm, 5 μm particle size, 300 Å pore size). The fractions containing the product were pooled and subjected to lyophilization. The purity of the final peptide was determined by analytical RP-HPLC performed with a linear gradient using 0.1 M TEAP pH 2.5 as eluent A and 60% CH3CN/40% A as eluent B on a Hewlett-Packard Series II 1090 Liquid Chromatograph connected to a Vydac C18 column (0.21 × 15 cm, 5 μm particle size, 300 Å pore size). The capillary zone electrophoresis (CZE) analysis of the peptides was performed on a Beckman P/ACE System 2050; field strength of 15 kV at 30 °C on an Agilent μSil bare fused-silica capillary (75 μm i.d. × 40 cm length).51 Mass spectra (MALDI-TOF-MS) were measured on an ABI-Perseptive DE-STR instrument. The instrument employs a nitrogen laser (337 nm) at a repetition rate of 20 Hz. The applied accelerating voltage was 20 kV. Spectra were recorded in delayed extraction mode (300 ns delay). All spectra were recorded in the positive reflector mode. Spectra were sums of 100 laser shots. Matrix alpha-cyano-4-hydroxycinnamic acid was prepared as saturated solutions in 0.3% TFA in 50% CH3CN. The observed monoisotopic (M + H)+ values of each peptide corresponded with the calculated (M + H)+ values (Table 1). The diastereomers of the Agl containing peptides could be separated by preparative RP-HPLC.
Determination of the stereochemistry of Agl in the peptides
Since the L and D enantiomers of Fmoc-D/LAgl(NMe,Boc)46 used for the synthesis of peptides (21, des-AA1,4-6,10,12,13-[Cpa2,DCys3,LAgl(NMe,2naphthoyl)8]-SRIF-2Nal-NH2 (22), 23-25, 27, des-AA1,2,4-6,10,12,13-[LAgl(NMe,2naphthoyl)8,IAmp9,Tyr11]-SRIF-NH2 (28), 29, des-AA1,2,4-6,10,12,13-[DCys3,LAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-2Nal-NH2 (30), 31, des-AA1,4-6,10,12,13-[Tyr2,DCys3,LAgl(NMe,2naphthoyl)8,IAmp9]-SRIF-2Nal-NH2 (32)) were not resolved initially, two diastereomers were generated, isolated, characterized, and tested. Separation of the L- from the DAgl-containing peptides was achieved using RP-HPLC. The absolute configuration of the Agl was deduced from enzymatic hydrolysis studies with Trypsin, Aminopeptidase M and Proteinase K, (Roche Diagnostics Corp., Indianapolis, IN) respectively as we published earlier.50 All enzymatic hydrolyses were carried out according to the protocols suggested by the manufacturers.
Trypsin, which is highly specific toward positively charged side chains with lysine and arginine, was used to determine the stereochemistry of 21 and 22. Trypsin (Roche, 5 μg) in 0.05% TFA (20 μL) was added to peptide (0.02 μmol) dissolved in 0.046 M Tris buffer (200 μL, pH = 8.1) containing 0.01M CaCl2. The hydrolysis was followed by RP-HPLC for 6 days. Trypsin was able to open the ring structure of 21 and 22 but the rate of reactions was very much different. We suggest that 22 contains the L-isomer of the Agl derivative and 21 contains the D-isomer of the Agl derivative because 55% of 22 versus only 28% of 21 were hydrolyzed in 6 days.
Peptides with L-amino acids at their N-terminus and IAmp at position 9 instead of Lys (23 versus 24 and 27 versus 28) were digested with Aminopeptidase M, which is a metalloprotease, and can hydrolyze peptides at a free α-amino group of L-amino acids (except X-Pro bonds and the amino groups of Asp, Gln or βAla), to determine the absolute configuration of Agl. The treatment of 24 and 28 with Aminopeptidase M at room temperature for 48 hours resulted in many very hydrophilic products followed by RP-HPLC, indicating that these peptides have been completely hydrolyzed, providing evidence that these analogues contained the L-enantiomer of Agl in their sequence. Compounds 23 and 27 were hydrolyzed only into two products suggesting that these two peptides contained the D-enantiomer of Agl in their sequence resulting in more resistance to the enzymatic hydrolysis. To confirm this conclusion, these analogues were also digested with Proteinase K, which is a serine protease that exhibits very broad cleavage specificity. The predominant site of cleavage is the peptide bond adjacent to the carboxyl group of hydrophobic aliphatic and aromatic amino acids with blocked alpha amino groups. Peptides were dissolved (20 μg) at a concentration of 0.3 mg/mL in 10 mM Tris buffer pH 7.8. Proteinase K (5 μL, 6 U) was added and reacted for 16 hours at 37 °C. An aliquot (5 μL) was quenched with 45 μL 0.1 N HCl prior to analysis by HPLC for determination of degradation products. The aliquots were screened using microbore RP-HPLC under buffer system A; 0.1%TFA/H2O, buffer system B, 70% CH3CN in A at a flow rate of 0.05 mL/min under gradient conditions 10-80% B over 30 min. Fragments of 23 identified as H-Phe-Agl(NMe,2naphthoyl)-IAmp-Phe-Cys-Thr-NH2 and H-Phe-Agl(NMe,2naphthoyl)-IAmp-Phe-OH were confirmed by MALDI-MS. These observations verified that 23 contained the DAgl. The mass of fragments of 24 were found to be <500 Da indicating the presence of LAgl.
Analogues 25, 26, 29, 30 and 31, 32 contain a D-residue at position 2 (dTyr) or 3 (dCys), respectively, therefore enzymatic hydrolysis by Aminopeptidase M could not be expected for any of these compounds. Instead, these analogues were digested with Proteinase K as described above.
The digestion of 25 with Proteinase K for 4 days resulted in four products, which were identified as H-cyclo[Cys-Phe-Agl(NMe,2naphthoyl)-IAmp-Phe-Cys]-Thr-NH2, H-Phe-Agl(NMe,2naphthoyl)-IAmp-Phe-Cys-Thr-NH2, H-Phe-Agl(NMe,2naphthoyl)-IAmp-Phe-OH and the starting material H-dTyr-cyclo[Cys-Phe-Agl(NMe,2naphthoyl)-IAmp-Phe-Cys]-Thr-NH2 confirmed by MALDI-MS. After the digestion of 26 with Proteinase K for 4 days no starting material was detected by HPLC, one of the fragments was identified as H-dTyr-Cys-Phe-Agl(NMe,2naphthoyl)-IAmp-Phe-Cys-OH and all of the other detected fragments had a mass of less than 500 suggesting that this peptide contains the L-isomer of Agl.
The digestion of 29 with Proteinase K for 7 days resulted in three products, which were identified as H-cyclo[dCys-Phe-Agl(NMe,2-naphthoyl)-IAmp-Phe-Cys]-OH, H-dCys-Phe-Agl(NMe,2naphthoyl)-IAmp-Phe-OH and the starting material H-cyclo[dCys-Phe-Agl(NMe,2naphthoyl)-IAmp-Phe-Cys]-Nal-NH2 confirmed by MALDI-MS. After the digestion of 30 with Proteinase K for 47 hours, a small amount of the starting material was detected by HPLC, and the fragments were identified as H-Agl(NMe,2naphthoyl)-IAmp-Phe-OH, H-IAmp-Phe-Cys-Nal-NH2 confirmed by MALDI-MS suggesting that this peptide contains the L-isomer of Agl.
The digestion of 31 with Proteinase K for 47 hours resulted in three fragments, which were identified as H-cyclo[dCys-Phe-Agl(NMe,2naphthoyl)-IAmp-Phe-Cys]-Nal-NH2, H-dCys-(Cys-Nal-NH2)-Phe-Agl(NMe,2naphthoyl)-IAmp-Phe-OH, H-dCys-Phe-Agl(NMe,2naphthoyl)-IAmp-Phe-OH and the starting material confirmed by MALDI-MS. After the digestion of 32 with Proteinase K for 47 hours a small amount of the starting material was detected by HPLC, and no other fragments could be identified suggesting that this peptide contains the L-isomer of Agl.
Overall, aminoglycine-containing enantiomers were relatively easy to separate using HPLC under gradient conditions (Table 7).
Table 7. Separation of the D- from the L-Agl-containing peptides by RP-HPLC.
| Compound | HPLC-Column | Gradient | Retention time (min) |
|---|---|---|---|
| 21 | Vydac C18 | 20 to 80%B in 40 min | 32.33 |
| 22 | Vydac C18 | 20 to 80%B in 40 min | 35.26 |
| 23 | Vydac C18 | 30 to 60%B in 30 min | 17.93 |
| 24 | Vydac C18 | 30 to 60%B in 30 min | 23.39 |
| 25 | Gemini C18 | 20 to 60%B in 40 min | 30.56 |
| 26 | Gemini C18 | 20 to 60%B in 40 min | 34.15 |
| 27 | Vydac C18 | 30 to 70%B in 30 min | 11.75 |
| 28 | Vydac C18 | 30 to 70%B in 30 min | 14.91 |
| 29 | Gemini C18 | 40 to 70%B in 30 min | 20.28 |
| 30 | Gemini C18 | 40 to 70%B in 30 min | 25.10 |
| 31 | Vydac C18 | 30 to 70%B in 40 min | 27.68 |
| 32 | Vydac C18 | 30 to 70%B in 40 min | 32.75 |
HPLC buffer system: A = TEAP (pH 2.5) and B = 60% CH3CN/40% A with a gradient slope of 1% B/min, at flow rate of 0.2 mL/min.
NMR Experiments
The 1H NMR spectra were recorded on a Bruker 700 MHz spectrometer operating at proton frequency of 700 MHz. Chemical shifts were measured using DMSO (δ = 2.49 ppm) as an internal standard. The 2D spectra were acquired at 298 K. Resonance assignments of the various proton resonances have been carried out using total correlation spectroscopy (TOCSY);75, 76 double-quantum filtered spectroscopy (DQF-COSY)77 and nuclear Overhauser enhancement spectroscopy (NOESY).78-80 The TOCSY experiments employed the MLEV-17 spin-locking sequence suggested by Davis and Bax,75 applied for a mixing time of 50 or 70 ms. The NOESY experiments were carried out with a mixing time of 100 ms or 150 ms. The TOCSY and NOESY spectra were acquired using 800 complex data points in the ω1 dimension and 1024 complex data points in the ω2 dimension and were subsequently zero-filled to 1024 × 2048 before Fourier transformation. The DQF-COSY spectra were acquired with 1024 × 4096 data points and were zero-filled to 2048 × 4096 before Fourier transformation. The TOCSY, DQF-COSY and NOESY spectra were acquired with 16, 16 and 24 scans, respectively, with a relaxation delay of 1 s. The signal from the residual water of the solvent was suppressed using pre-saturation during the relaxation delay and during the mixing time. The TOCSY and NOESY data were multiplied by 75° shifted sine-function in both dimensions. To differentiate exchange peaks from the NOEs, rotating frame NOESY (ROESY)81, 82 spectra were measured at 150, 250 and 400 msec. All spectra were processed using the software PROSA.83 The spectra were analyzed using the software X-EASY.84
Structure Determination
The chemical shift assignment of the major/minor conformer was obtained by the standard procedure using DQF-COSY and TOCSY spectra for intra-residual assignment and the NOESY spectrum was used for the sequential assignment.85 The collection of structural restraints is based on the NOEs and vicinal 3JNHα couplings. Dihedral angle constraints were obtained from the 3JNHα couplings, which were measured from the 1D 1H NMR spectra and from the intra-residual and sequential NOEs along with the macro GRIDSEARCH in the program CYANA.53 The calibration of NOE intensities versus 1H-1H distance restraints and appropriate pseudo-atom corrections to the non-stereo specifically assigned methylene, methyl and ring protons were performed using the program CYANA. On an average, approximately 70 NOE constraints and 15 angle constraints were utilized while calculating the conformers (Table 3). A total of 100 conformers were initially generated by CYANA and a bundle containing 20 CYANA conformers with the lowest target function values were utilized for further restrained energy minimization, using the program DISCOVER with steepest decent algorithm.54 The resulting energy minimized bundle of 20 conformers was used as a basis for discussing the solution conformation of the different SRIF analogues. The structures were analyzed using the program MOLMOL.86
Biology: Reagents
All reagents were of the best grade available and were purchased from common suppliers. Lactalbumin hydrolysate was from HyClone, ATP from Sigma-Aldrich, D-luciferin from Roche Diagnostics and coenzyme A from Calbiochem.
Cell lines
Chinese hamster lung fibroblasts (CCL39) stably expressing the human sst1 and the luciferase reporter gene under the control of the serum response element (CCL39-sst1-Luci) were from D. Hoyer (Novartis, Basel, Switzerland) and grown in DMEM with GlutaMAX-I / Ham’s F-12 Nut. Mix. with GlutaMAX-I (1:1) supplemented with 10% foetal bovine serum, 100U/ml penicillin and 100 μg/ml streptomycin, and 400 μg/ml Geneticin (G418-sulfate) at 37 °C and 5% CO2. All culture reagents were from Gibco BRL, Life Technologies.
Receptor autoradiography
Cell membrane pellets were prepared as previously described and stored at -80 °C.87 Receptor autoradiography was performed on 20-μm thick cryostat (Microm HM 500, Walldorf, Germany) sections of the membrane pellets, mounted on microscope slides, and then stored at -20 °C. For each of the tested compounds, complete competition experiments with the universal SRIF radioligand [Leu8, D-Trp22, 125I-Tyr25]-SRIF-28 (125I-[LTT]-SRIF-28) (2,000 Ci/mmol; ANAWA, Wangen, Switzerland) using 15,000 cpm/100 μL and increasing concentrations of the unlabelled peptide ranging from 0.1 - 1000 nM were performed. As control, unlabelled SRIF-28 was run in parallel using the same increasing concentrations. The sections were incubated with 125I-[LTT]-SRIF-28 for 2 hours at room temperature in 170 mmol/L Tris-HCl buffer (pH 8.2), containing 1% BSA, 40 mg/L bacitracin, and 10 mmol/L MgCl2 to inhibit endogenous proteases. The incubated sections were washed twice for 5 min in cold 170 mmol/L Tris-HCl (pH 8.2) containing 0.25% BSA. After a brief dip in 170 mmol/L Tris-HCl (pH 8.2), the sections were dried quickly and exposed for 1 week to Kodak BioMax MR film. IC50 values were calculated after quantification of the data using a computer-assisted image processing system as described previously.88 Tissue standards (Autoradiographic [125I] and/or [14C] microscales, GE Healthcare; Little Chalfont, UK) that contain known amounts of isotope, cross-calibrated to tissue-equivalent ligand concentrations were used for quantification.88-91
The analogue 25 was 125I-mono-radiolabeled (2,000 Ci/mmol, ANAWA, Switzerland) and used as radioligand in binding studies on cell membrane pellet sections of cells stably expressing sst1 receptors and on tissue sections, following the same protocol as described above for 125I-[LTT]-SRIF-28.
Luciferase assay
The luciferase reporter gene assay was performed as described previously by Hoyer and colleagues32, 58 with minor modifications. CCL39-sst1-Luci cells were seeded at 25′000 cells per well in 96-well plates. After 24 hours, the cells were washed once with PBS and then serum-deprived for 24 hours in assay medium (DMEM with GlutaMax-I/Ham’s F12 Nut. Mix. with GlutaMax-I (1:1) containing 5 g/l lactalbumin hydrolysate and 20 mM HEPES) at 37 °C and 5% CO2. The cells were then treated in triplicate at 37 °C and 5% CO2 with assay medium alone (basal) or with assay medium containing the compounds to be tested at concentrations between 0.1 nM and 1 μM for 5 h. The cells were then washed with PBS and lysed in lysis buffer (100 mM KPi-buffer containing 0.2% Triton X-100 and 1 mM DL-dithiothreitol). Luciferase activity was measured using a luminometer by injecting first the ATP-reagent (10 mM ATP, 35 mM glycyl-glycine and 20 mM MgCl2, pH 7.8) and then the luciferin-reagent (0.47 mM D-luciferin, 0.27 mM coenzyme A, 35 mM glycyl-glycine and 20 mM MgCl2, pH 7.8). Measuring parameters: delay time: 5 seconds; measurement time: 10 seconds. The 1 μM SRIF-14 effect minus the basal effect was set as 100%. Stimulation of luciferase reporter gene activity by the different compounds is expressed as % stimulation of the 1 μM SRIF-14 effect. As positive control for the luciferase reporter gene activity 10% foetal bovine serum in assay medium was used.
Acknowledgments
The project described was supported in part by Award Number 5R01DK59953-05 from the National Institute of Diabetes and Digestive and Kidney Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Diseases or the National Institutes of Health. We are indebted to R. Kaiser, C. Miller for technical assistance in the synthesis and characterization of the peptides and Dr. W. Fisher, and William Low for mass spectrometric analyses. JR is the Dr. Frederik Paulsen Chair in Neurosciences Professor.
Abbreviations
The abbreviations for the common amino acids are in accordance with the recommendations of the IUPAC-IUB Joint Commission on Biochemical Nomenclature (Eur. J. Biochem. 1984, 138:9-37). The symbols represent the L-isomer except when indicated otherwise. Additional abbreviations:
- AA
amino acid
- Agl
aminoglycine
- ATP
adenosine 5′-triphosphate
- Boc
tert-butoxycarbonyl
- Bzl
benzyl
- Bzl(3Br)
3-bromobenzyl
- cAMP
3′,5′-cyclic adenosine monophosphate
- Cbm
carbamoyl
- CM
chloromethyl
- CZE
capillary zone electrophoresis
- DIC
N,N’-diisopropylcarbodiimide
- DIPEA
diisopropylethylamine
- DMEM
Dulbecco’s modified Eagle’s medium
- DMF
dimethylformamide
- DQF-COSY
double quantum filtered correlation spectroscopy
- Fmoc
9-fluorenylmethoxycarbonyl
- HEPES
4-(2-hydroxyethyl) piperazine-1-ethansulfonic acid
- hhLys
homo-homo-lysine
- HOBt
1-hydroxybenzotriazole
- IAmp
4-(N-isopropyl)-aminomethylphenylalanine
- MBHA
4-methylbenzhydrylamine
- Mob
4-methoxybenzyl
- Nal
3-(2-naphthyl)-alanine
- NMP
N-methylpirrolidinone
- NOESY
nuclear Overhauser enhancement spectroscopy
- PBS
phosphate buffered saline
- RMSD
root mean square deviation
- ROESY
rotating frame nuclear Overhauser enhancement spectroscopy
- SRE
serum response element
- SRIF
somatostatin
- SRIF-28
somatostatin-28
- ssts
SRIF receptors
- TEA
triethylamine
- TEAP
triethylammonium phosphate
- TFA
trifluoroacetic acid
- TOCSY
total correlation spectroscopy
- Z(2Br)
2-bromobenzyloxycarbonyl
- Z(2Cl)
2-chlorobenzyloxycarbonyl
References
- 1.Grace CRR, Durrer L, Koerber SC, Erchegyi J, Reubi JC, Rivier JE, Riek R. Somatostatin receptor 1 selective analogues: 4. Three-dimensional consensus structure by NMR. J. Med. Chem. 2005;48:523–533. doi: 10.1021/jm049518u. [DOI] [PubMed] [Google Scholar]
- 2.Brazeau P, Vale WW, Burgus R, Ling N, Butcher M, Rivier JE, Guillemin R. Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science. 1973;179:77–79. doi: 10.1126/science.179.4068.77. [DOI] [PubMed] [Google Scholar]
- 3.Guillemin R, Gerich JE. Somatostatin: physiological and clinical significance. Ann. Rev. Med. 1976;27:379–388. doi: 10.1146/annurev.me.27.020176.002115. [DOI] [PubMed] [Google Scholar]
- 4.Tannenbaum GS, Epelbaum J. Somatostatin. In: Kostyo JL, Goodman HM, editors. Handbook of physiology - the endocrine system. V. Oxford University Press; New York: 1999. pp. 221–265. Hormonal Control of Growth. [Google Scholar]
- 5.Janecka A, Zubrzycka M, Janecki T. Review: Somatostatin analogs. J. Pept. Res. 2001;58:91–107. doi: 10.1034/j.1399-3011.2001.00873.x. [DOI] [PubMed] [Google Scholar]
- 6.Schally AV, Comaru-Schally AM, Nagy A, Kovacs M, Szepeshazi K, Plonowski A, Varga JL, Halmos G. Hypothalamic hormones and cancer. Front. Neuroendocrinol. 2001;22:248–291. doi: 10.1006/frne.2001.0217. [DOI] [PubMed] [Google Scholar]
- 7.Hannon JP, Nunn C, Stolz B, Bruns C, Weckbecker G, Lewis I, Troxler T, Hurth K, Hoyer D. Drug design at peptide receptors: somatostatin receptor ligands. J. Mol. Neurosci. 2002;18:15–27. doi: 10.1385/JMN:18:1-2:15. [DOI] [PubMed] [Google Scholar]
- 8.Hoyer D, Epelbaum J, Feniuk W, Humphrey PPA, Meyerhof W, O’Caroll AM, Patl Y, Reisine T, Reubi JC, Schindler M, Schonbrunn A, Taylor JE, Vezzani A. Somatostatin receptors. In: Girdlestrom D, editor. The IUPHAR compendium of receptor characterization and classification. 2 ed. IUPHAR Media; London: 2000. pp. 354–364. [Google Scholar]
- 9.Csaba Z, Dournaud P. Cellular biology of somatostatin receptors. Neuropeptides. 2001;35:1–23. doi: 10.1054/npep.2001.0848. [DOI] [PubMed] [Google Scholar]
- 10.Patel YC. Somatostatin and its receptor family. Front. Neuroendocrinol. 1999;20:157–198. doi: 10.1006/frne.1999.0183. [DOI] [PubMed] [Google Scholar]
- 11.Siehler S, Hoyer D. Characterisation of human recombinant somatostatin receptors. 4 Modulation of phospholipase C activity. Naunyn-Schmiedeberg’s Arch Pharmacol. 1999;360:522–532. doi: 10.1007/s002109900144. [DOI] [PubMed] [Google Scholar]
- 12.Cescato R, Schulz S, Waser B, Eltschinger V, Rivier JE, Wester H-J, Culler M, Ginj M, Liu Q, Schonbrunn A, Reubi JC. Internalization of sst2, sst3 and sst5 receptors: Effects of somatostatin agonist and antagonists. J. Nucl. Med. 2006;47:502–511. [PubMed] [Google Scholar]
- 13.Ginj M, Zhang H, Waser B, Cescato R, Wild D, Erchegyi J, Rivier J, Mäcke HR, Reubi JC. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. USA. 2006;103:16436–16441. doi: 10.1073/pnas.0607761103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olias G, Viollet C, Kusserow H, Epelbaum J, Meyerhof W. Regulation and function of somatostatin receptors. J. Neurochem. 2004;89:1057–91. doi: 10.1111/j.1471-4159.2004.02402.x. [DOI] [PubMed] [Google Scholar]
- 15.Weckbecker G, Lewis I, Albert R, Schmid HA, Hoyer D, Bruns C. Opportunities in somatostatin research: biological, chemical and therapeutic aspects. Nat. Rev. Drug Discov. 2003;2:999–1017. doi: 10.1038/nrd1255. [DOI] [PubMed] [Google Scholar]
- 16.Piwko C, Thoss VS, Schupbach E, Kummer J, Langenegger D, Probst A, Hoyer D. Pharmacological characterisation of human cerebral cortex somatostatin SRIF1 and SRIF2 receptors. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:161–167. doi: 10.1007/pl00004927. [DOI] [PubMed] [Google Scholar]
- 17.Thermos K. Functional mapping of somatostatin receptors in the retina: a review. Vision Res. 2003;43:1805–1815. doi: 10.1016/s0042-6989(03)00169-x. [DOI] [PubMed] [Google Scholar]
- 18.Bocci G, Culler MD, Fioravanti A, Orlandi P, Fasciani A, Colucci R, Taylor JE, Sadat D, Danesi R, Del Tacca M. In vitro antiangiogenic activity of selective somatostatin subtype-1 receptor agonists. Eur. J. Clin. Invest. 2007;37:700–708. doi: 10.1111/j.1365-2362.2007.01848.x. [DOI] [PubMed] [Google Scholar]
- 19.Reubi JC, Schaer JC, Waser B, Hoeger C, Rivier J. A selective analog for the somatostatin sst1-receptor subtype expressed by human tumors. Eur. J. Pharmacol. 1998;345:103–110. doi: 10.1016/s0014-2999(97)01618-x. [DOI] [PubMed] [Google Scholar]
- 20.Reubi JC, Waser B, Laissue JA, Gebbers JO. Somatostatin and vasoactive intestinal peptide receptors in human mesenchymal tumors: in vitro identification. Cancer Res. 1996;56:1922–1931. [PubMed] [Google Scholar]
- 21.Reubi JC, Waser B, Schaer J-C, Laissue JA. Somatostatin receptor sst1-sst5 expression in normal and neoplastic human tissues using receptor autoradiography with subtype-selective ligands. European Journal of Nuclear Medicine. 2001;28:836–846. doi: 10.1007/s002590100541. [DOI] [PubMed] [Google Scholar]
- 22.Reubi JC, Schaer JC, Waser B, Mengod G. Expression and localization of somatostatin receptor SSTR1, SSTR2 and SSTR3 mRNAs in primary human tumors using in situ hybridization. Cancer Res. 1994;54:3455–3459. [PubMed] [Google Scholar]
- 23.Reubi JC, Waser B, Schaer JC, Markwalder R. Somatostatin receptors in human prostate and prostate cancer. J. Clin. Endocrinol. Metab. 1995;80:2806–2814. doi: 10.1210/jcem.80.9.7673428. [DOI] [PubMed] [Google Scholar]
- 24.Miller GM, Alexander JM, Bikkal HA, Katznelson L, Zervas NT, Klibanski A. Somatostatin receptor subtype gene expression in pituitary adenomas. J. Clin. Endocrinol. Metab. 1995;80:1386–1392. doi: 10.1210/jcem.80.4.7714115. [DOI] [PubMed] [Google Scholar]
- 25.Lanneau C, Bluet-Pajot MT, Zizzari P, Csaba Z, Dournaud P, Helboe L, Hoyer D, Pellegrini E, Tannenbaum GS, Epelbaum J, Gardette R. Involvement of the Sst1 somatostatin receptor subtype in the intrahypothalamic neuronal network regulating growth hormone secretion: an in vitro and in vivo antisense study. Endocrinology. 2000;141:967–979. doi: 10.1210/endo.141.3.7349. [DOI] [PubMed] [Google Scholar]
- 26.Kreienkamp H-J, Akgün E, Baumeister H, Meyerhof W, Richter D. Somatostatin receptor subtype 1 modulates basal inhibition of growth hormone release in somatotrophs. FEBS Lett. 1999;462:464–466. doi: 10.1016/s0014-5793(99)01582-3. [DOI] [PubMed] [Google Scholar]
- 27.Zatelli MC, Piccin D, Tagliati F, Ambrosio MR, Margutti A, Padovani R, Scanarini M, Culler MD, degli Uberti EC. Somatostatin receptor subtype 1 selective activation in human growth hormone (GH)- and prolactin (PRL)-secreting pituitary adenomas: effects on cell viability, GH, and PRL secretion. J. Clin. Endocrinol. Metab. 2003;88:2797–2802. doi: 10.1210/jc.2002-021825. [DOI] [PubMed] [Google Scholar]
- 28.Vasilaki A, Papasava D, Hoyer D, Thermos K. The somatostatin receptor (sst(1)) modulates the release of somatostatin in the nucleus accumbens of the rat. Neuropharmacology. 2004;47:612–618. doi: 10.1016/j.neuropharm.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Dal Monte MD, Petrucci C, Vasilaki A, Cervia D, Grouselle D, Epelbaum J, Kreienkamp HJ, Richter D, Hoyer D, Bagnoli P. Genetic deletion of somatostatin receptor 1 alters somatostatinergic transmission in the mouse retina. Neuropharmacology. 2003;45:1080–1092. doi: 10.1016/s0028-3908(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 30.Thermos K, Bagnoli P, Epelbaum J, Hoyer D. The somatostatin sst1 receptor: an autoreceptor for somatostatin in brain and retina? Pharmacol. Ther. 2006;110:455–464. doi: 10.1016/j.pharmthera.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Hoyer D, Dixon K, Gentsch C, Vassout A, Enz A, Jaton A, Nunn C, Schoeffter P, Neumann P, Troxler T, Pfaeffli P. NVP-SRA880, a somatostatin sst1 receptor antagonist promotes social interactions, reduces aggressive behaviour and stimulates learning. The Pharmacologist. 2002;44:A254. [Google Scholar]
- 32.Hoyer D, Nunn C, Hannon J, Schoeffter P, Feuerbach D, Schuepbach E, Langenegger D, Bouhelal R, Hurth K, Neumann P, Troxler T, Pfaeffli P. SRA880, in vitro characterization of the first non-peptide somatostatin sst(1) receptor antagonist. Neurosci. Lett. 2004;361:132–5. doi: 10.1016/j.neulet.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 33.Matrone C, Pivonello R, Colao A, Cappabianca P, Cavallo LM, Del Basso De Caro ML, Taylor JE, Culler MD, Lombardi G, Di Renzo GF, Annunziato L. Expression and function of somatostatin receptor subtype 1 in human growth hormone secreting pituitary tumors deriving from patients partially responsive or resistant to long-term treatment with somatostatin analogs. Neuroendocrinology. 2004;79:142–148. doi: 10.1159/000077272. [DOI] [PubMed] [Google Scholar]
- 34.Zatelli MC, Tagliati F, Piccin D, Taylor JE, Culler MD, Bondanelli M, degli Uberti EC. Somatostatin receptor subtype 1-selective activation reduces cell growth and calcitonin secretion in a human medullary thyroid carcinoma cell line. Biochem. Biophys. Res. Commun. 2002;297:828–834. doi: 10.1016/s0006-291x(02)02307-0. [DOI] [PubMed] [Google Scholar]
- 35.Zatelli MC, Piccin D, Tagliati F, Bottoni A, Luchin A, Vignali C, Margutti A, Bondanelli M, Pansini GC, Rosa Pelizzo M, Culler MD, Degli Uberti EC. Selective activation of somatostatin receptor subtypes differentially modulates secretion and viability in human medullary thyroid carcinoma primary cultures: potential clinical perspectives. J. Clin. Endocrinol. Metab. 2006 doi: 10.1210/jc.2006-0334. [DOI] [PubMed] [Google Scholar]
- 36.Liapakis G, Hoeger C, Rivier J, Reisine T. Development of a selective agonist at the somatostatin receptor subtype SSTR1. Journal of Pharmacology and Experimental Therapeutics. 1996;276:1089–1094. [PubMed] [Google Scholar]
- 37.Rivier JE, Hoeger C, Erchegyi J, Gulyas J, DeBoard R, Craig AG, Koerber SC, Wenger S, Waser B, Schaer J-C, Reubi JC. Potent somatostatin undecapeptide agonists selective for somatostatin receptor 1 (sst1) J. Med. Chem. 2001;44:2238–2246. doi: 10.1021/jm010037+. [DOI] [PubMed] [Google Scholar]
- 38.Rivier JE, Kirby DA, Erchegyi J, Waser B, Eltschinger V, Cescato R, Reubi JC. Somatostatin receptor 1 selective analogues: 3. Dicyclic peptides. J. Med. Chem. 2005;48:515–522. doi: 10.1021/jm049519m. [DOI] [PubMed] [Google Scholar]
- 39.Erchegyi J, Hoeger CA, Low W, Hoyer D, Waser B, Eltschinger V, Schaer J-C, Cescato R, Reubi JC, Rivier JE. Somatostatin receptor 1 selective analogues: 2. N-methylated scan. J. Med. Chem. 2005;48:507–514. doi: 10.1021/jm049520l. [DOI] [PubMed] [Google Scholar]
- 40.Erchegyi J, Hoeger C, Wenger S, Waser B, Schaer J-C, Reubi JC, Rivier JE. In: Lebl M, Houghten RA, editors. N-methyl scan of a sst1-selective somatostatin (SRIF) analog; Peptides - The wave of the future: 2nd International Peptide Symposium/17th American Peptide Symposium; San Diego, CA. June 10-14, 2001; 2001. pp. 719–720. American Peptide Symposium: San Diego, CA, 2001. [Google Scholar]
- 41.Jiang G, Miller C, Koerber SC, Porter J, Craig AG, Bhattacharjee S, Kraft P, Burris TP, Campen CA, Rivier CL, Rivier JE. Betidamino acid scan of the GnRH antagonist Acyline. J. Med. Chem. 1997;40:3739–3748. doi: 10.1021/jm970024p. [DOI] [PubMed] [Google Scholar]
- 42.Cervini L, Theobald P, Corrigan A, Craig AG, Rivier C, Vale W, Rivier J. Corticotropin releasing factor (CRF) agonists with reduced amide bonds and Ser7 substitutions. J. Med. Chem. 1999;42:761–768. doi: 10.1021/jm980607e. [DOI] [PubMed] [Google Scholar]
- 43.Reubi JC, Schaer J-C, Wenger S, Hoeger C, Erchegyi J, Waser B, Rivier J. SST3-selective potent peptidic somatostatin receptor antagonists. Proc. Natl. Acad. Sci. USA. 2000;97:13973–13978. doi: 10.1073/pnas.250483897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erchegyi J, Wenger S, Waser B, Eltschinger V, Cescato R, Reubi JC, Koerber SC, Grace RCR, Riek R, Rivier JE. In: Blondelle SE, editor. Use of betidamino acids in drug design; Understanding Biology Using Peptides, The 19th American Peptide Symposium; San Diego, CA. June 18-23, 2005; 2005. pp. 517–518. American Peptide Society: San Diego, CA, 2005. [Google Scholar]
- 45.Qasmi D, René L, Badet B. An α-aminoglycine derivative suitable for solid phase peptide synthesis using Fmoc strategy. Tetrahedron Letters. 1993;34:3861–3862. [Google Scholar]
- 46.Jiang G-C, Simon L, Rivier JE. Orthogonally protected N-methyl-substituted α-aminoglycines. Prot. Pep. Lett. 1996;3:219–224. [Google Scholar]
- 47.Rivier JE, Jiang G-C, Koerber SC, Porter J, Craig AG, Hoeger C. Betidamino acids: Versatile and constrained scaffolds for drug discovery. Proc. Natl. Acad. Sci. USA. 1996;93:2031–2036. doi: 10.1073/pnas.93.5.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoeger CA, Galyean RF, Boublik J, McClintock RA, Rivier JE. Preparative reversed phase high performance liquid chromatography. II. Effects of buffer pH on the purification of synthetic peptides. Biochromatography. 1987;2:134–142. [Google Scholar]
- 49.Miller C, Rivier J. Peptide chemistry: Development of high-performance liquid chromatography and capillary zone electrophoresis. Biopolymers Pept. Sci. 1996;40:265–317. doi: 10.1002/(SICI)1097-0282(1996)40:3%3C265::AID-BIP2%3E3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 50.Rivier J, Erchegyi J, Hoeger C, Miller C, Low W, Wenger S, Waser B, Schaer J-C, Reubi JC. Novel sst4-selective somatostatin (SRIF) agonists. Part I: Lead identification using a betide scan. J. Med. Chem. 2003;46:5579–5586. doi: 10.1021/jm030243c. [DOI] [PubMed] [Google Scholar]
- 51.Miller C, Rivier J. Analysis of synthetic peptides by capillary zone electrophoresis in organic/aqueous buffers. J. Pept. Res. 1998;51:444–451. doi: 10.1111/j.1399-3011.1998.tb00643.x. [DOI] [PubMed] [Google Scholar]
- 52.Gairi M, Saiz P, Madurga S, Roig X, Erchegyi J, Koerber SC, Reubi JC, Rivier JE, Giralt E. Conformational analysis of a potent SSTR3-selective somatostatin analogue by NMR in water solution. J. Peptide Sci. 2006;12:82–91. doi: 10.1002/psc.743. [DOI] [PubMed] [Google Scholar]
- 53.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 54.Hagler AT. Theoretical simulation of conformation, energetics and dynamics of peptides. In: Udenfriend S, Meienhofer J, Hruby VJ, editors. The Peptides: Analysis, Synthesis, Biology. Vol. 7. Academic Press; Orlando, FL: 1985. pp. 213–299. [Google Scholar]
- 55.Cescato R, Erchegyi J, Waser B, Piccand V, Maecke HR, Rivier JE, Reubi JC. Design and in vitro characterization of highly sst2-selective somatostatin antagonists suitable for radio-targeting. J. Med. Chem. 2008;51:4030–4037. doi: 10.1021/jm701618q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hill CS, Treisman R. Differential activation of c-fos promoter elements by serum, lysophosphatidic acid, G proteins and polypeptide growth factors. Embo J. 1995;14:5037–5047. doi: 10.1002/j.1460-2075.1995.tb00186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Treisman R. Journey to the surface of the cell: Fos regulation and the SRE. EMBO J. 1995;14:4905–4913. doi: 10.1002/j.1460-2075.1995.tb00173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nunn C, Cervia D, Langenegger D, Tenaillon L, Bouhelal R, Hoyer D. Comparison of functional profiles at human recombinant somatostatin sst2 receptor: simultaneous determination of intracellular Ca2+ and luciferase expression in CHO-K1 cells. Br. J. Pharmacol. 2004;142:150–160. doi: 10.1038/sj.bjp.0705735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reubi JC, Schaer J-C, Waser B, Hoeger C, Rivier J. A selective analog for the somatostatin sst1-receptor subtype expressed by human tumors. Eur. J. Pharmacol. 1998;345:103–110. doi: 10.1016/s0014-2999(97)01618-x. [DOI] [PubMed] [Google Scholar]
- 60.Hocart SJ, Jain R, Murphy WA, Taylor JE, Coy DH. Highly potent cyclic disulfide antagonists of somatostatin. J. Med. Chem. 1999;42:1863–1871. doi: 10.1021/jm9806289. [DOI] [PubMed] [Google Scholar]
- 61.Hocart SJ, Jain R, Murphy WA, Taylor JE, Morgan B, Coy DH. Potent antagonists of somatostatin: Synthesis and biology. J. Med. Chem. 1998;41:1146–1154. doi: 10.1021/jm970730q. [DOI] [PubMed] [Google Scholar]
- 62.Melacini G, Zhu Q, Osapay G, Goodman M. A refined model for the somatostatin pharmacophore: Conformational analysis of lanthionine-sandostatin analogs. J. Med. Chem. 1997;40:2252–2258. doi: 10.1021/jm960851a. [DOI] [PubMed] [Google Scholar]
- 63.Grace CRR, Erchegyi J, Koerber SC, Reubi JC, Rivier J, Riek R. Novel sst2-selective somatostatin agonists. Three-dimensional consensus structure by NMR. J. Med. Chem. 2006;49:4487–4496. doi: 10.1021/jm060363v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bass RT, Buckwalter BL, Patel BP, Pausch MH, Price LA, Strnad J, Hadcock JR. Identification and characterization of novel somatostatin antagonists. Mol. Pharmacol. 1996;50:709–715. [PubMed] [Google Scholar]
- 65.Bass RT, Buckwalter BL, Patel BP, Pausch MH, Price LA, Strnad J, Hadcock JR. Mol. Pharmacol. Vol. 51. 1997. Identification and characterization of novel somatostatin antagonists; p. 170. Erratum. [PubMed] [Google Scholar]
- 66.Grace CRR, Erchegyi J, Koerber SC, Reubi JC, Rivier J, Riek R. Novel sst4-selective somatostatin (SRIF) agonists. Part IV: Three-dimensional consensus structure by NMR. J. Med. Chem. 2003;46:5606–5618. doi: 10.1021/jm030246p. [DOI] [PubMed] [Google Scholar]
- 67.Grace CRR, Erchegyi J, Reubi JC, Rivier JE, Riek R. Three-dimensional consensus of structure of sst2-selective somatostatin (SRIF) antagonists by NMR. Biopolymers. 2008 doi: 10.1002/bip.21060. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Siehler S, Seuwen K, Hoyer D. [125I]Tyr10-cortistatin14 labels all five somatostatin receptors. Naunyn Schmiedebergs Arch Pharmacol. 1998;357:483–489. doi: 10.1007/pl00005197. [DOI] [PubMed] [Google Scholar]
- 69.Siehler S, Seuwen K, Hoyer D. Characterisation of human recombinant somatostatin receptors. 1. Radioligand binding studies. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:488–499. doi: 10.1007/s002109900141. [DOI] [PubMed] [Google Scholar]
- 70.Fehlmann D, Langenegger D, Schuepbach E, Siehler S, Feuerbach D, Hoyer D. Distribution and characterisation of somatostatin receptor mRNA and binding sites in the brain and periphery. J. Physiol. (Paris) 2000;94:265–281. doi: 10.1016/s0928-4257(00)00208-4. [DOI] [PubMed] [Google Scholar]
- 71.Horiki K, Igano K, Inouye K. Amino acids and peptides. Part 6. Synthesis of the Merrifield resin esters of N-protected amino acids with the aid of hydrogen bonding. Chem. Lett. 1978;2:165–168. [Google Scholar]
- 72.Rivier JE, Jiang G, Porter J, Hoeger C, Craig AG, Corrigan AZ, Vale WW, Rivier CL. GnRH antagonists: novel members of the azaline B family. J. Med. Chem. 1995;38:2649–2662. doi: 10.1021/jm00014a017. [DOI] [PubMed] [Google Scholar]
- 73.Sypniewski M, Penke B, Simon L, Rivier J. (R)-tert-Butoxycarbonylamino-fluorenylmethoxycarbonyl-glycine from (S)-Benzyloxycarbonyl-serine or from papain resolution of the corresponding amide or methyl ester. J. Org. Chem. 2000;65:6595–6600. doi: 10.1021/jo000736e. [DOI] [PubMed] [Google Scholar]
- 74.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 75.Davis DG, Bax A. Assignment of complex 1H NMR spectra via two-dimensional homonuclear Hartmann-Hahn spectroscopy. J. Am. Chem. Soc. 1985;107:2820–2821. [Google Scholar]
- 76.Braunschweiler L, Ernst RR. Coherence transfer by isotropic mixing: Application to proton correlation spectroscopy. J. Magn. Reson. 1983;53:521–528. [Google Scholar]
- 77.Rance M, Sorensen OW, Bodenhausen B, Wagner G, Ernst RR, Wüthrich K. Improved spectral resolution in COSY1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 1983;117:479–485. doi: 10.1016/0006-291x(83)91225-1. [DOI] [PubMed] [Google Scholar]
- 78.Kumar A, Wagner G, Ernst RR, Wüthrich K. Buildup rates of the nuclear Overhauser effect measured by two-dimensional proton magnetic resonance spectroscopy: Implications for studies of protein conformation. J. Am. Chem. Soc. 1981;103:3654–3658. [Google Scholar]
- 79.Macura S, Ernst RR. Elucidation of cross-relaxation in liquids by two-dimensional NMR spectroscopy. Mol. Phys. 1980;41:95–117. [Google Scholar]
- 80.Macura S, Huang Y, Suter D, Ernst RR. Two-dimensional chemical exchange and cross-relaxation spectroscopy of coupled nuclear spins. J. Magn. Reson. 1981;43:259–281. [Google Scholar]
- 81.Bax A, Davis DG. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 1985;65:355–360. [Google Scholar]
- 82.Bothner-By AA, Stephens RL, Lee J, Warren CD, Jeanloz RW. Structure determination of a tetrasaccharide: transient nuclear Overhauser effects in the rotating frame. J. Am. Chem. Soc. 1984;106:811–813. [Google Scholar]
- 83.Güntert P, Dotsch V, Wider G, Wüthrich K. Processing of multi-dimensional NMR data with the new software PROSA. J. Biolmol. NMR. 1992;2:619–629. [Google Scholar]
- 84.Eccles C, Güntert P, Billeter M, Wüthrich K. Efficient analysis of protein 2D NMR spectra using the software package EASY. J. Biomol. NMR. 1991;1:111–130. doi: 10.1007/BF01877224. [DOI] [PubMed] [Google Scholar]
- 85.Wüthrich K. NMR of Proteins and Nucleic Acids. J. Wiley & Sons; New York: 1986. [Google Scholar]
- 86.Koradi R, Billeter M. MOLMOL: a program for display and analysis of macromolecular structures. PDB Newsletter. 1998;84:5–7. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 87.Reubi JC, Schaer JC, Waser B, Wenger S, Heppeler A, Schmitt JS, Mäcke HR. Affinity profiles for human somatostatin receptor sst1-sst5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. European Journal of Nuclear Medicine. 2000;27:273–282. doi: 10.1007/s002590050034. [DOI] [PubMed] [Google Scholar]
- 88.Reubi JC, Kvols LK, Waser B, Nagorney DM, Heitz PU, Charboneau JW, Reading CC, Moertel C. Detection of somatostatin receptors in surgical and percutaneous needle biopsy samples of carcinoids and islet cell carcinomas. Cancer Res. 1990;50:5969–5977. [PubMed] [Google Scholar]
- 89.Reubi JC. In vitro identification of vasoactive intestinal peptide receptors in human tumors: Implications for tumor imaging. J. Nucl. Med. 1995;36:1846–1853. [PubMed] [Google Scholar]
- 90.Baskin DG, Wimpy TH. Calibration of [14C]plastic standards for quantitative autoradiography of [125I]labeled ligands with Amersham Hyperfilm beta-max. Neurosci. Lett. 1989;104:171–177. doi: 10.1016/0304-3940(89)90350-9. [DOI] [PubMed] [Google Scholar]
- 91.Miller JA, Zahniser NR. The use of 14C-labeled tissue paste standards for the calibration of 125I-labeled ligands in quantitative autoradiography. Neurosci. Lett. 1987;81:345–350. doi: 10.1016/0304-3940(87)90408-3. [DOI] [PubMed] [Google Scholar]