

Abstract
Genetic variation in mRNA expression plays a critical role in human phenotypic diversity, but it has proven difficult to detect regulatory polymorphisms - mostly single nucleotide polymorphisms (rSNPs). Additionally, variants in the transcribed region, termed here ‘structural RNA SNPs’ (srSNPs), can affect mRNA processing and turnover. Both rSNPs and srSNPs cause allelic mRNA expression imbalance (AEI) in heterozygous individuals. We have applied a rapid and accurate AEI methodology for testing 42 genes implicated in human diseases and drug response, specifically cardiovascular and CNS diseases, and affecting drug metabolism and transport. Each gene was analyzed in physiologically relevant human autopsy tissues, including brain, heart, liver, intestines, and lymphocytes. Substantial AEI was observed in ∼55% of the surveyed genes. Focusing on cardiovascular candidate genes in human hearts, AEI analysis revealed frequent cis-acting regulatory factors in SOD2 and ACE mRNA expression, having potential clinical significance. SNP scanning to locate regulatory polymorphisms in a number of genes failed to support several previously proposed promoter SNPs discovered with use of reporter gene assays in heterologous tissues, while srSNPs appear more frequent than expected. Computational analysis of mRNA folding indicates that ∼90% of srSNPs affects mRNA folding, and hence potentially function. Our results indicate that both rSNPs and srSNPs represent a still largely untapped reservoir of variants that contribute to human phenotypic diversity.
Introduction
Genetic, epigenetic, and environmental factors determine phenotypic variability, including susceptibility to disease or treatment outcome. Polymorphisms that change the amino acid sequences in coding regions (cSNPs) are readily detectable. However, regulatory polymorphisms (rSNPs) appear to be more prevalent than functional nonsynonymous cSNPs [1-5]. Genome-wide surveys and SNP association analysis with mRNA expression trait mapping [5,6] indicate regulatory polymorphisms as major factors in human phenotypic evolution and variability [5,7]. A third type of functional polymorphism affects mRNA processing (splicing, maturation, stability, transport) and translation [8]. We refer to this class of polymorphisms as ‘structural RNA polymorphisms’ (srSNPs). However, the overall role of rSNPs and srSNP still requires systematic evaluation.
Whereas mRNA levels are subject to both cis- and trans-acting factors, measuring the relative allelic mRNA expression selectively detects only cis-acting factors. Allelic expression imbalance (AEI), i.e., a different number or type of mRNAs generated between alleles, is a robust and quantitative phenotype directly linked to cis-acting polymorphisms [3,5,8-21] and epigenetic regulation, including X-inactivation, imprinting, and gene silencing [4,22,23].
Genome-wide association studies continue to increase the number of candidate genes, while knowledge of the functional genetic variants is lagging. AEI analysis is a powerful tool for finding regulatory polymorphisms, but technical difficulties hamper broad usage. Earlier AEI methods mostly targeted monoallelic expression, while polymorphisms resulting in relatively small changes, although potentially physiologically relevant, are more difficult to measure. Array- and RTPCR-based methods with limited precision or sensitivity have been applied to detect partial regulatory changes, but have mostly been applied to small sets of candidate genes in lymphocytes. Results from these studies suggest that 20-50% of genes show detectable AEI [2,3,24-26]. Yet, because the impact of rSNPs and srSNPs strongly depends on the tissue context, AEI analysis should be performed in physiologically relevant tissues [27,28]. Systematic and accurate surveys of AEI in many genes applied to a variety of human target tissues are lacking. Yet, autopsy tissues present additional difficulties because of partial mRNA degradation.
For rapid detection of regulatory polymorphisms in multiple genes, we have developed a robust and fast methodology applicable to human autopsy tissues, filling an important gap between large-scale candidate gene discovery and resolution of the functional variants. This study surveyed AEI for 42 genes in human autopsy tissues, including brain, heart, liver, intestines, and kidney, as well as peripheral mononuclear cells, revealing frequent AEI in a large fraction of genes. In cardiovascular genes where regulatory polymorphisms had been reported previously, we tested whether the observed AEI ratios were compatible with any effects of these polymorphisms on allelic expression in relevant tissues. We also addressed the question of how srSNPs affect mRNA folding, and point to a number of genes where frequent srSNPs affect mRNA expression. The results provide insight into the prevalence of rSNPs and srSNPs.
Results
Methodology for AEI analysis of multiple genes in human autopsy tissues
We developed a rapid methodology for measuring allelic ratios in genomic DNA and mRNA (as cDNA) (AEI analysis) in human autopsy target tissues. The assay relies on PCR/RT-PCR amplification, followed by a primer extension step with fluorescently labeled dideoxynucleotides, and analysis by capillary electrophoresis. Details of the assays applied to single genes have been published previously by us for several genes included in the present survey [9-16]. To facilitate application to multiple genes in human autopsy tissues, we have introduced several steps for obtaining reproducible allelic gDNA and mRNA ratios, including use of multiple gene-specific primers to maximize cDNA yields for the target genes. Assay throughput is ∼150 samples/hour, or higher with multiplexing, with an error rate in the order of 5% (gDNA) and 10-15% (mRNA).
Application of AEI analysis to candidate genes
AEI analysis was applied to 42 candidate genes in a variety of human tissues (Table 1), divided into genes for cardiovascular and CNS disorders, and drug metabolism and transport. This selection provides information on the frequency of cis-acting factors but was not designed to cover the much larger number of possible candidate genes. We first determined (by RTPCR) all 42 genes were well expressed in the target tissues examined and then determined >4,200 individual genotypes for mRNA marker SNPs in the candidate genes. The 1,008 heterozygous samples suitable for use in AEI assays yielded relative allelic expression for an average of 23 subjects or 46 individual chromosomes per gene (average marker SNP heterozygosity ∼24%). Results for four genes are shown in Fig.1. This study was well powered to detect frequent functional polymorphisms (>5% minor allele frequency), similar to previous AEI studies [2,25,26]. Details on tissue source, number of samples, marker SNPs, and allele frequency are found in Supplemental Table 1. As a conservative detection threshold for the presence of mRNA AEI ratios (major:minor allele), we used ±log2 0.5 (1:1.4 or 1.4:1) corresponding to 3 SD or more relative to DNA ratios, similar to previous studies [24,25].
Table 1. List of candidate genes tested for the presence of AEI.
| Cardiovascular disease and pharmacogenetic candidate genes |
|---|
| ACE, CCL2, SOD2, NNMT, LPL, HMGCR, CSF1, PTGDS, HIF1A, NOS3, FLT1, CACNA1C, ADRB2, KCNMB1, VKORC1, GGCX, CETP, HMOX1 |
| Drug metabolism and drug transporter candidate genes |
| ABCB1, CYP2D6, CYP2C9, SLC15A1, SLC15A2 |
| CNS disorder and pharmacogenetics candidate genes |
| DRD2, CHRNA4, OPRM1, HTR2A, BDNF, SLC6A4, TPH2, SLC6A2, DTNBP1, NRG1, HTR1B, MAOA, DRD3, ESR1, SLC6A3, COMT, DAO, NR3C1, NQO2 |
Details on the number of subjects and the target tissues analyzed are in Supplemental Table 1. Genes showing significant AEI are listed in Table 2.
Figure 1.
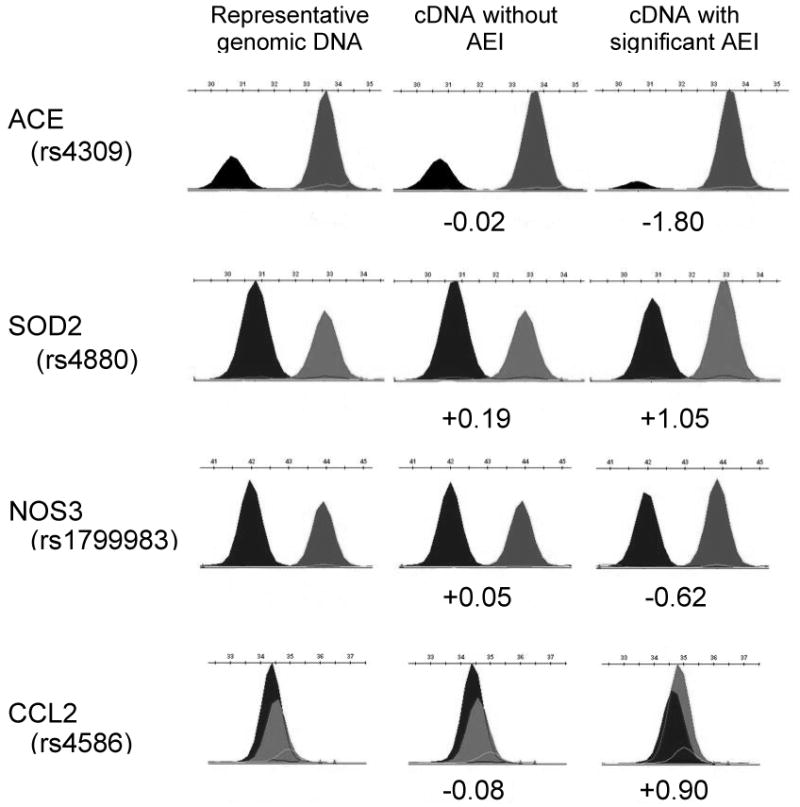
Results of AEI analysis for ACE, SOD2, NOS3 and CCL2, in heart left ventricular tissues. Each peak represents a distinct allele measured in genomic DNA or cDNA from a single heterozygous individual. The selected samples (columns, left to right) represent the typical genomic DNA ratio observed, a cDNA showing insignificant deviation from the expected ratio and a cDNA sample showing highly significant deviation from unity. Normalization to the average genomic DNA is used in the calculation of AEI values (cDNA values listed as major:minor allele on a log2 scale) and accounts for differences in fluorescent dideoxynucleotide incorporation efficiencies and fluorescence yields. See Table 1 for a list of genes reported here and Table 2 for marker SNPs and genes showing significant AEI results.
Table 2 contains results for genes meeting the detection threshold in at least one sample, along with information on the marker SNPs, number of replicate analyses, frequency, magnitude and direction of AEI. If a suspected functional polymorphism is in near complete linkage disequilibrium with the marker SNP, most or all AEI ratios are unidirectional (either <1 or >1), as observed with SOD2 in heart tissues (Fig.2). In contrast, functional polymorphisms unlinked to the marker SNP are revealed by random distribution of ratios <1 and >1 (Fig.2, Table 2), indicating these are located in other haplotype blocks. Lesser AEI ratios may also be of physiological relevance but should be subject to more extensive analytical validation to exclude artifacts. The results reveal AEI above our threshold in 67% of the candidate genes, with AEI in two or more subjects in 55% of genes. Where genes lack significant AEI this argues against the presence of cis-acting factors in the tissues analyzed.
Table 2. Genes showing significant allelic mRNA expression ratios (at least one sample showing minimally ±20.5, or ∼40% AEI in either direction).
| Symbol | Tissue | Marker SNP(s) | AEI ratio range | % samples with significant AEI (total number of samples) | Further publication |
|---|---|---|---|---|---|
| Cardiovascular disease candidate genes | |||||
| ACE | Heart, Liver, Small bowel | rs4309 | 0.2-1.5 | 48% (n=73) | |
| rs4343 | |||||
| MCP1/CCL2 | Heart, Monocytes | rs4586 | 0.66-1.87 | 18% (n=55) | |
| rs13900 | |||||
| SOD2 | Heart | rs4880 | 0.9-2.1 | 83% (n=41) | |
| rs5746092 | |||||
| NNMT | Liver, Small bowel | rs4646335 | 0.58-2.9 | 15% (n=39) | |
| CSF1 | Monocytes | rs333970 | 0.98-1.9 | 11% (n=18) | |
| PTGDS | Heart | rs6926 | 0.7-1.3 | 3% (n=30) | |
| NOS3 | Heart | rs1799983 | 0.65-1.11 | 18% (n=22) | |
| VKORC1 | Liver | rs7294 | 0.5-1 | 68% (n=29) | |
| CETP | Liver | rs5884 | 0.67-1.89 | 77% (n=29) | |
| Drug metabolism candidate genes | |||||
| ABCB1 | Liver | rs1045642 | 1.0-1.6 | 36% (n=22) | [9] |
| rs1128503 | |||||
| rs2032582 | |||||
| CYP2D6 | Liver | rs1065852 | 0.1-10 | 88% (n=26) | |
| rs1058164 | |||||
| rs16947 | |||||
| rs1135840 | |||||
| CYP2C9 | Liver | rs2017319 | 0.6-1.2 | 21% (n=19) | |
| SLC15A2 | Kidney | rs1143670 | 0.6-1.1 | 13% (n=8) | |
| rs1143671 | |||||
| CNS disorder candidate genes | |||||
| DRD2 | Brain | rs6277 | 0.5-2.0 | 25% (n=30) | [16] |
| rs6275 | |||||
| rs6279 | |||||
| CHRNA4 | Brain | rs1044393 | 0.6-2.0 | 85% (n=13) | |
| rs1044397 | |||||
| rs2229959 | |||||
| rs2236196 | |||||
| OPRM1 | Brain | rs1799971 | 1.0-3.0 | 100% (n=8) | [10] |
| BDNF | Brain | rs6265 | 0.9-1.6 | 11% (n=9) | |
| TPH2 | Brain | rs7305115 | 0.92-2.55 | 48% (n=27) | [13] |
| rs4290270 | |||||
| DTNBP1 | Brain | rs1047631 | 0.88-1.45 | 22% (n=9) | |
| MAOA | Brain | rs1137070 | 0.3-4.0 | 89% (n=19) | [12] |
| rs6232 | |||||
| DRD3 | Brain | rs6280 | 0.3-2.7 | 57% (n=21) | |
| ESR1 | Brain | rs3798577 | 0.5-1.7 | 24% (n=46) | |
| SLC6A3 | Brain | rs6347 | 0.1-1.2 | 95% (n=21) | |
| COMT | Brain | rs4633 | 0.9-1.4 | 10% (n=10) | |
| DAO | Brain | rs2070588 | 0.3-4.5 | 57%(n=21) | |
| NR3C1 | Brain | rs6196 | 0.4-1.0 | 5% (n=20) | |
| NQO2 | Coriell sample | rs1143684 | 0.88-2.60 | 33% (n= 9) | |
AEI ratios are defined by mRNA abundance of major over minor allele for a given marker SNP (normalized to genomic allele ratios measured with SNaPshot). Frequency of AEI is given as % of total number of samples heterozygous for the marker SNP(s). Direction of AEI ratios can be determined by the range of values observed (e.g., >1, <1, and <1>), indicating the minor allele is less or more abundant, respectively. For several genes, references provide detailed AEI studies published separately.
Figure 2.

Allelic mRNA expression ratios (major allele over minor allele, normalized to the mean allelic ratio in genomic DNA) measured in heart failure samples for 12 cardiovascular candidate genes. Results for individual samples are displayed with the magnitude and direction of AEI indicated on a log2 scale (y-axis). Potential AEI in individual samples is indicated by ratios >(+0.3) log2 or <(-0.3) log2, a cutoff arrived at by analysis of the extent of variation in genomic DNA ratios. For the present survey study we considered ratios >(+0.5) log2 or <(-0.5) log2 to represent significant AEI.
Several well-studied genes, such as ACE and SOD2, displayed substantial AEI that was unexpected from previous genetic analyses (Table 2). In some cases, our AEI data confirm previous studies, for example, the modest AEI ratios observed for COMT [17], and a similar frequency and extent of AEI for NQO2 in white blood cells [26] and DTNBP1 in the pons region [29]. We also failed to observe significant AEI in 5HT2A, as reported [30]; however, another study suggests the presence of AEI [31] but lacks rigorous validation of the results.
It is possible that AEI may be detectable only in certain ethnogeographic populations where regulatory alleles are sufficiently frequent (see ACE below), or in specific tissues, environmental conditions, and diseases. For example, AEI was observed for VKORC1 only in the liver but was undetectable in heart tissues and B-lymphoblasts (CEPH samples) (Table 2).
Relationship between AEI and mRNA levels
We tested whether the presence of AEI is correlated with total mRNA levels, measured by RTPCR, in a subset of genes (SOD2, CCL2, NOS3, FLT1, HIF1A, LPL, PTGDS, and MAOA). Borderline significant correlations between AEI and mRNA levels were observed for HIF1A (r=-0.45, p<0.06) and PTGDS (r=0.38, p<0.04). These moderate correlations reflect the greater variability of overall mRNA levels compared to allelic ratios.
Cardiovascular disease candidate genes
AEI analysis was applied to 18 cardiovascular candidate genes that serve as drug targets and have roles in inflammation, coagulation, lipid metabolism, vasomotor tone, and heart contractility (Table 1). Target tissues included 65 heart failure explants from transplant recipients, livers, ex vivo monocytes, and peripheral blood monocyte-derived macrophages. AEI was detectable for 15 cardiovascular genes at a 20% imbalance threshold (Table 1), while 9 genes displayed AEI when we set our more stringent threshold based on the typical error rates (±log2 0.5). AEI ratios for genes surveyed in heart tissues are shown in Figure 2. Allelic mRNA expression of ACE, CCL2, SOD2, CACNA1C, and KCNMB1 was validated using a second marker SNP, with cDNA derived from a different primer (Table 2). CCL2, PTGDS, and KCNMB1 showed allelic ratios below and above 1, suggesting multiple functional polymorphisms and/or incomplete linkage disequilibrium between the marker SNPs and functional alleles (Fig.2). In contrast, ACE displayed large unidirectional AEI ratios only in African Americans, suggesting the presence of a cis-acting factor enriched in this population. AEI results for ACE were confirmed with use of a second marker SNP (r2=0.98 in compound heterozygotes). Standard curves were linear, obtained with homozygous DNA representing both alleles (r2=0.99). A detailed analysis of regulatory ACE polymorphisms will be reported separately. Both SOD2 and NOS3 showed AEI largely in a single direction – suggestive of a functional polymorphism in a shared haplotype with the marker SNP, or that the marker SNP itself is functional. Our results on the frequency and extent of NOS3 AEI are consistent with published AEI results in brain tissues [27].
A number of genes did not show any AEI, for example, the L-type channel CACNA1C – a gene featuring >55 exons across ∼250 kB. Subsequent use of several marker SNPs and AEI analysis of splice variants failed to reveal any cis-acting factor that could have caused highly variable splicing observed for CACNA1C in human heart [15].
Relationship between AEI and previously suggested regulatory polymorphisms
The frequency and directionality of AEI ratios enables us to investigate whether previously proposed regulatory polymorphisms in NOS3 (rs2070744), CCL2 (rs1024611), SOD2 (rs5746091), PTGDS (rs6926), and ACE (intron 16 I/D) contribute to this phenotype. We genotyped the proposed regulatory polymorphisms and tested for association between genotype and AEI ratios. We analyzed AEI ratios with two discrete thresholds, and also as a continuous variable. The results in Table 3 indicate that the putative regulatory polymorphisms cannot account for or are only marginally associated with AEI. For example, a proposed promoter SNP (rs1024611) [32] in CCL2 was incompatible with AEI observed in two subjects, or for the absence of AEI in many samples where this SNP is heterozygous, in both heart tissues and macrophages (Table 3). Similarly, a putative regulatory SNP, T-786C (rs2070744) upstream of NOS3, and rs6296 in PTGDS, were not significantly associated with the AEI observed in human target tissues (Table 3). A marginal association between the intensely studied ACE intron 16 I/D was detectable when AEI was analyzed as a continuous variable, but there was no association with the large AEI ratios shown in Figure 2.
Table 3. Genotyping of suspected functional polymorphisms compared with AEI data.
| Gene | Suspected functional polymorphism/marker | Literature reference | Heterozygous with AEI | Heterozygous without AEI | Homozygous with AEI | Homozygous without AEI | Kappa correlation coefficient (0.5 cutoff) | Kappa correlation coefficient (0.2 cutoff) | p-value (AEI as continuous) |
| ACE | Intron 16 I/D | [39] | 2 | 21 | 3 | 6 | -0.15 | -0.43 | 0.04 |
| CCL2 | rs1024611 (macrophages) | [32] | 2 | 24 | 0 | 8 | 0.04 | 0.04 | 0.95 |
| CCL2 | rs1024611 (heart) | [32] | 1 | 26 | 1 | 5 | -0.05 | 0.19 | 0.82 |
| NOS3 | rs2070744 | [51] | 3 | 9 | 0 | 5 | 0.16 | -0.02 | 0.66 |
| PTGDS | rs6296 | n/a | 1 | 29 | n/a | n/a | n/a | n/a | n/a |
| SOD2 | rs4880 | [41] | 27 | 7 | 7 | 0 | -0.21 | -0.11 | 0.67 |
| SOD2 | rs5746092 | n/a | 21 | 8 | 13 | 4 | -0.04 | 0.20 | 0.71 |
| SOD2 | rs5746091 | [33] | 3 | 0 | 4 | 3 | n/a | n/a | n/a |
Association between heterozygosity of the suggested variants and AEI was done with AEI ratios as a categorical variable using the Kappa test with AEI cutoff (±log2 0.5). The number of subjects heterozygous or homozygous with or without AEI at the 0.5 cutoff is given. Alternatively we applied a relaxed cutoff (±log2 0.2) to address the possibility that lower, but detectable AEI may also be significant (counts not shown). We also tested for association with AEI as a continuous measure using HelixTree (taking the absolute value of AEI for each sample). Tests considered significant at p<0.05 are indicated in bold. We did not test rs6296 or rs5746091 for association (listed as “n/a”) given low numbers of heterozygous and/or homozygous samples.
Detailed analysis of AEI observed for SOD2
Allelic mRNA ratios for SOD2 were ∼1.5-fold in 83% of heart tissues heterozygous for marker rs4880, indicating that the ‘major allele’ has ∼50% greater expression (however, since allele frequency is close to 50% assignment of the minor allele is arbitrary). A second marker, rs5746092 in the 5′UTR in modest LD with rs4880, gave similar results (r2=0.73, in 16 compound heterozygotes), supporting the accuracy of the assay. Neither rs5746092 (37% heterozygosity) nor rs4880 (52% heterozygosity) were completely associated with AEI, as several homozygotes or heterozygotes displayed significant or no AEI, respectively. The results suggest one or more regulatory factors within a common haplotype block. Testing a proposed functional promoter SNP, rs5746091 [33] in 10 subjects, we found that 3 homozygous carriers had no AEI and 3 heterozygous carriers did show AEI (allelic ratio >1.4), but 4 homozygous carriers displayed significant AEI, indicating that rs5746091 could not have played a sole role in allelic expression. Because the AEI ratios are substantiated for each individual by multiple replicates, each subject showing discrepancy between AEI and SNP heterozygosity is informative and, thus fails to support a putative functional role for that SNP.
Since epigenetic factors could affect allelic expression, methylation of a CpG island close to rs4880 was measured. Distant CpG islands outside this haplotype block were not expected to preferentially affect alleles marked by rs4880. To test allele-selective methylation, we digested DNA at a Hpa II methylation-sensitive restriction site near rs4880 and measured the DNA allelic ratios, in comparison to a standard curve from mixed ratios of digested and undigested reference DNA. CpG methylation differed detectably between alleles, but allele-specific methylation did not correlate with corresponding allele-specific mRNA expression ratios (Pearson r2=0.03) (Fig. 3), arguing against an effect on allelic mRNA expression.
Figure 3.

Lack of correlation between SOD2 allelic mRNA expression ratios and allelic CpG methylation ratios in 34 heart tissue samples. Allelic methylation ratios were determined from triplicate assays using Hpa II digestion of the genomic DNA region containing rs4880 (only non-methylated DNA is cut), followed by SNaPshot analysis of the allelic ratios for uncut genomic DNA.
Effect of srSNPs on predicted mRNA folding structures
To assess the potential of SNP-induced changes in mRNA folding, we estimated changes in folding energies for all possible transitions (C<>U, G<>A) and transversions (C<>G, C<>A, G<>U, A<>U) in the mRNA coding regions of the μ, κ and δ opioid receptors (OPRM1, OPRK1, OPRD1), using Mfold. We calculated both the minimum free energy structures (MFE) and the ensembles of suboptimal structures in varying sized windows around all nucleotide positions. A majority of SNPs showed the potential to alter mRNA folding, often predicting more profound changes than the known functional A118G SNP in OPRM1 [17] (see arrow in Fig. 4). Approximately 60% of single nucleotide substitutions affected MFE structures, and ∼90% altered the ensemble of suboptimal structures, with the potential to affect mRNA functions [34]. The relevance of a region of the OPRM1 mRNA displaying low susceptibility to structural disturbance by in silico SNP insertion (Fig.4; also present in OPRD and OPRK (data not shown)) remains to be determined.
Figure 4.

Computed changes of mRNA folding (minimum free energy conformations) induced by all transitions (SNP generated by C<>T and G<>A substitutions) in the transcribed exonic domains of OPRM1 mRNA. The arrow indicates the location of the functional SNP A118G, affecting mRNA levels in human brain (18). The x-axis denotes the nucleotide position in the mature OPRM1 mRNA (cDNA), while the y-axis represent a scale of the extent by which predicted mRNA folding is affected by any given transition. Conformations were calculated for wild-type and mutant sequences using Mfold, and then the sum of the differences in the Mfold single-strandedness count measure at each nucleotide was computed both globally (across the full mRNA structure, each point shown here) and in more regional sliding windows of different sizes. Sliding windows and analysis of both types of transversions at each position (pyrimidine<>purine), as well as A>G transitions alone all gave very similar results (data not shown).
Because SOD2 allelic expression was consistently in a single direction in such a high proportion (>80%) of samples, we suspected the SOD2 marker SNPs might have a direct, functional effect on expression. Thus, we further analyzed the predicted allelic effects on mRNA folding for the marker SNPs in SOD2 (rs4880, rs5646092). Both SNPs are in regions that display highly stable structures, with rs5646092 positioned within an 18bp helix near the transcription and translation initiation sites. These results suggest that one or more of these alleles could affect gene expression through a change in mRNA structure.
Discussion
Robust assay of allelic ratios in genomic DNA and mRNA
We have developed broadly applicable methodology for rapid and robust assays of allelic gene expression (AEI) in human autopsy tissues. Measuring allelic ratios circumvents at least in part problems arising from post-mortem mRNA degradation. The AEI analysis can be scaled up to address multiple genes at a time, and thus, represents an intermediate tool for discovering functional polymorphisms affecting gene regulation (rSNPs) and RNA processing (srSNPs) in candidate genes. The effect of rSNPs and srSNPs is expected to vary with the cellular environment, so that studies on human genes in physiologically relevant target tissues are of critical importance, for example the pontine brainstem for SERT and TPH2 mRNA [11,13].
Factors other than rSNPs and srSNPs could contribute to AEI, including variable copy number (CNV) in germline DNA or more frequently as somatic mutations in cancer [35]. We observed deviations of the DNA ratios from unity only with TPH2 in two subjects [13], indicating that gene duplications are rare among the 42 genes studied. On the other hand, complete loss of one allele in germline DNA at the marker SNP locus cannot be assessed with the SNaPshot method as presented because hemizygous carriers would appear as homozygotes, unless the gene dosage is quantitated.
Another possible source of AEI, allele-selective epigenetic regulation of gene expression must be considered where SNP scanning fails to reveal regulatory polymorphisms. The relatively high precision by which the AEI ratios can be measured, facilitated the dissection of genetic and epigenetic regulation of the X-linked MAOA, with both processes contributing to AEI [12].
Prevalence of AEI in the candidate genes
Our study permits an estimation of the prevalence of cis-acting polymorphisms in 42 candidate genes in human target tissues, a larger, more diverse sampling than previous studies. Table 2 provides information on the magnitude, direction, and frequency of AEI, as guides for more detailed studies. Substantial AEI (>log2 0.5) in more than one subject was observed for 55% of the surveyed genes (Table 2), similar to previous studies [2,3,25]; however, the frequency is higher than estimates from other studies performed with a random selection of genes in cell lines and blood cells [24,26]. These differences may be attributable to the selection of strong candidate genes, or differences in methodology, tissue specificity, number of subjects, and stringency of AEI thresholds. The presence of frequent AEI was unexpected for some of the candidate genes that had already been intensely studied for genetic polymorphisms (e.g., SOD2, ACE, TPH2 [13], DRD2 [16]).
Differential post-mortem decay for alleles could represent a confounding factor that can be overcome by molecular genetic studies of the functional polymorphisms. Polymorphisms affecting alternative splicing may not be detectable if the splice isoforms have similar turnover rates. To address this issue, allelic mRNA expression can be performed after specific amplification of each splice variant, as we have demonstrated for DRD2 (intron 5 and 6 SNPs alter formation of D2S and D2L) [16].
Scanning for regulatory polymorphisms using allelic mRNA expression profiles
AEI patterns provide a means of determining the location of the functional polymorphism by SNP scanning or sequencing the gene locus, followed by molecular genetic analysis of the rSNP or srSNPs, as shown for OPRM1, MDR1, MAOA, SERT, TPH2, and DRD2 [9-13,16].
Reporter gene assays in heterologous tissues are commonly used to characterize regulatory polymorphisms. If these polymorphisms are functional in vivo, one expects corresponding changes in the AEI ratios. However, for the five genes tested (Table 3) we have failed to detect significant linkage between the observed AEI ratios and the putative regulatory SNPs. Similarly, our genotype scanning with AEI did not support a role for a putative SERT promoter polymorphism (SERT-LPR), although we cannot rule out that this promoter polymorphism might be active in development, or under stress [11]. Previously suggested regulatory polymorphisms in DRD2 also failed to correlate with AEI ratios [16]. A separate study of 4 genes (MAOA, NOS3, PDYN, NPY) using AEI analysis again yielded results incompatible with reporter gene assays [27], corroborating our results for MAOA and NOS3. Similarly, the AEI observed with CCL2 (MCP1) was not associated with the putative promoter SNP rs1024611 [32]. Therefore, reporter gene assays are not always reliable indicators of regulatory polymorphisms. Combined use of AEI analysis and reporter gene assays can yield more definitive results regarding regulatory polymorphisms [16].
Potential relevance of structural RNA SNPs (srSNPs)
For OPRM1, MDR1, TPH2, and DRD2, we have linked the AEI ratios to SNPs in the transcribed region of the gene, likely involved in mRNA processing, turnover, and splicing [9,10,13,16]. srSNPs have been shown to affect mRNA stability [9,36] and alternative splicing [16,37]. Our AEI analysis of marker SNPs in SOD2 and NQO2 indicates they (or SNPs in tight LD with them) may also affect RNA structures. Taken together, these results support the notion that srSNPs can be at least as prevalent as rSNPs.
srSNPs could alter mRNA function through changed folding dynamics [15,16,34]. Using Mfold to predict mRNA structural changes resulting from systematic nucleotide exchanges in opioid receptor mRNAs (Fig.4), we find that most SNPs affect the likely ensemble of structural conformations. Consistent with this, SNPs can be detected by a physical method based on ‘single-strand conformational polymorphisms’, with a 95% discovery rate.
srSNPs can further affect translation, as suggested for the OPRM1 SNP A118G [16], and COMT haplotypes with altered mRNA folding [38]. Measuring AEI ratios at the protein level with use of nonsynonymous marker SNPs is a future possibility that would allow the determination of quantitative effects of polymorphisms on translation and protein turnover.
Cardiovascular disease candidate genes
Half of the 18 cardiovascular genes studied displayed AEI at a conservative cutoff, with ACE and SOD2 conspicuous examples. An intron 16 I/D polymorphism of ACE had been extensively tested in clinical association studies, but its functional role remained unclear [39]. Our results suggest strong cis-acting factors unrelated to the I/D variant in heart tissues, with high frequency in African Americans. Analysis of regulatory ACE polymorphisms and clinical relevance will be reported separately (A. Johnson et al.).
SOD2 (mitochondrial manganese superoxide dismutase) is a key factor involved in metabolizing superoxide molecules and may have a role in failing human hearts [40]. Previous association studies of two variants in SOD2 with cardiomyopathy [41,42], cancers (e.g., [43], and other disorders have yielded inconsistent results. The nonsynonymous marker SNP used here lies in a leader sequence (rs4880, -9A>V) and was suggested to affect mitochondrial uptake of the mature protein [41], while a promoter region SNP (rs5746091) disrupts binding of AP-2 [33]. Common AEI observed here in failed heart tissues (Fig.2), with allelic mRNA ratios consistently >1, indicates presence of a frequent functional variant(s) in a haplotype block containing the marker SNPs. Limited genotype scanning of the SOD2 locus indicated that the two marker SNPs (rs4880, rs5746092) each taken alone cannot account for the observed AEI, but may interact with each other or merely represent tags for a functional srSNP in this region. The promoter SNP rs5746091 did not appear to play a main role. Previous studies have implicated structural elements in SOD2 expression, including a GC-rich 5′ region upstream of the transcription start site that also extends into the 5′ end of the transcript [44] and regions in the 3′UTR of the mRNA [45]. Highly favorable RNA structures exist in the region of rs5746092 and rs4880 suggesting multiple structural states in SOD2 mRNA could affect functions. Alternatively, epigenetic regulation of SOD2 expression by CpG methylation [46] could have contributed to AEI, but our initial results argue against this possibility. While further study of SOD2 regulation is necessary, the measured AEI ratios clearly demonstrate functional variation of SOD2 mRNA expression.
FLT1, HIF1A, HMOX1, and LPL did not display common and large AEI. However, because the studied candidate genes all have important physiological roles, even relatively small AEI ratios, as observed for CCL2, NOS3, FLT1, HIF1A, HMOX1, HMGCR, and LPL, may be of clinical importance [35]. Even a small activity change of a critical gene such as HMGCR could affect cholesterol production over an individual's lifetime. Moreover, pravastatin response was associated with two intronic SNPs in HMGCR, with frequency >5% in the population [47], and a genome-wide association study for LDL cholesterol also revealed an association with an intronic HMGCR SNP [48].
In summary, we have applied mRNA AEI analysis to the detection of cis-acting variation for 42 candidate genes, revealing many instances of yet unrecognized functional polymorphisms or other cis-acting factors. The AEI methodology can be applied on a fairly large scale while maintaining high accuracy.
Experimental Procedures
Human tissue selection and sources
We obtained autopsy or biopsy tissue samples from liver, kidney, intestines, peripheral white blood cells, and various brain regions (prefrontal cortex, hippocampus, ventral tegmental area (VTA), amygdala, and nucleus accumbens, and pontine nuclei of the brain stem (for SERT and TPH2)). Specimens from up to ∼100 subjects for each cell or tissue were obtained from various sources and tissue banks (OSU tissue procurement division, NIH Cooperative Human Tissue Network, 105 brain sections from the Stanley Foundation, Red Cross blood samples, and tissue banks at the University of Maryland and the National Disease Research Interchange). Left ventricular pieces were collected from the failed hearts of transplant recipients under an IRB-approved protocol at The Ohio State University. Ninety EBV-transformed B-lymphoblast cell lines were obtained from the Coriell cell repository, consisting of 30 Caucasian family trios. A majority of the tissues are from normal subjects, while some tissues included subjects diagnosed with schizophrenia, bipolar disorder, Alzheimer's disease, and cancer. Ethnic distributions varied between tissues repositories; no attempt was made to cover ethnic groups evenly. The objective of this study was to detect functional polymorphisms with allele frequencies of 5% or more.
Sample preparation
Genomic DNA and RNA were prepared from peripheral lymphocytes, or B-lymphocyte pellets, and frozen tissue samples (brain, liver, etc) as described previously [9-16]. Monocytes and monocyte-derived macrophages were cultured as described [49]. For whole blood extractions, the buffy coat was harvested, then red cells were either lysed using ammonium chloride to yield a leukocyte pellet for RNA extraction, or red and white cells were lysed with a sucrose Triton solution, providing a nuclear pellet for DNA purification. Frozen tissue samples were pulverized under liquid nitrogen and portioned into aliquots for DNA and RNA extractions. DNA was prepared by digestion of the pellet or frozen powder with SDS and proteinase K followed by NaCl salting out of proteins. DNA was recovered by ethanol precipitation, and RNA was extracted in Trizol™, chloroform extracted, and recovered by precipitation with isopropanol. RNA precipitates were dissolved in RNase-free water or Qiagen buffer, and then extracted using Qiagen RNeasy columns.
Analysis of allelic mRNA expression ratios for detection of allelic expression imbalance (AEI) Assay design
Allelic ratios of genomic DNA and mRNA were measured with SNaPshot as reported [9-16]. Briefly, DNA or mRNA (after conversion to cDNA) regions containing a marker SNP (Supplemental Table 1) were PCR amplified, followed by SNaPshot primer extension analysis of each allele (Supplementary Table 2). The procedure differs from earlier studies (e.g., [2]) by combining multiple gene-specific primers close to the marker SNP region for cDNA synthesis to compensate for mRNA degradation. Accurate AEI analysis requires robust expression (RT-PCR cycle threshold 27 or less). Selection criteria for a marker SNP were as follows: 1) location in the transcribed region, coding or non-coding, 2) high minor allele frequency (0.15-0.50), 3) position of marker SNPs preferably more than 20 bp from exon boundaries so that the same set of primers for PCR amplification can be used in both DNA and RNA.
Complementary DNA synthesis
cDNA was generated from total RNA (1 ug) by Superscript II reverse transcriptase (Invitrogen). Because oligo-dT priming often fails in autopsy tissues, we used both oligo-dT and gene-specific oligonucleotide primers targeting a region immediately 3′ of the marker SNP (same oligonucleotide used for PCR). We have multiplexed up to 30 primers to permit 30 different AEI assays per cDNA preparation. Comparisons between single and multiple primers showed no significant differences where tested. cDNA was successfully extracted from autopsy tissues to yield reproducible results between independent cDNA preparations [9-16].
Quantitative PCR-based mRNA analysis
We determined the mRNA levels for each candidate gene in each tissue or cell line, using RT-PCR, to assure that expression is sufficient for accurate AEI analysis (cycle thresholds equal to or below 27 cycles). Primers used for RT-PCR were the same as those selected for the AEI analysis, with PCR conditions optimized for each primer pair on an ABI7000 cycler with SYBR-Green. Results were normalized to an internal standard (β-actin or GAPDH).
Computational analysis of mRNA folding
We used Mfold version 3.0 to estimate the effect of SNPs on mRNA folding [50]. Wild-type Refseq mRNA sequences of OPRM1, OPRD1 and OPRK1 were obtained without untranslated regions. A custom Unix program created every possible variant at each base position and fed sequences to Mfold for structure prediction, and subsequent automated analysis. Changes in minimum free energy, as well as pairwise comparisons in structural interactions (paired vs. unpaired) were calculated relative to the wild-type structure using sliding windows around the induced variants, and across the complete mRNA structure.
Supplementary Material
Supplemental Table 1. List of candidate genes analyzed in this study, grouped by indication (disease or pharmacology). Marker SNPs are all located in transcribed regions of the mature mRNA, or a splice variant. For some genes more than one marker SNP and tissue were used.
Supplemental Table 2. Oligonucleotide primers used for PCR amplification and SNaPshot primer extension reactions. The reverse PCR primer also served for gene specific cDNA synthesis.
Acknowledgments
This work was supported by NIH/NHLBI (5R01HL074730-04), an AHA predoctoral fellowship (0515157B), NIDA (DA022199, DA018744, and DA021620), NIGMS (5U01GM061390-07), NIMH (MH081237) and a grant from the HDL Foundation, Pfizer Pharmaceuticals.
Footnotes
Conflict of Interest Statement: None of the authors have any competing interests to report.
Literature Cited
- 1.Rockman MV, Wray GA. Abundant raw material for cis-regulatory evolution in humans. Mol Biol Evol. 2002;19:1991–2004. doi: 10.1093/oxfordjournals.molbev.a004023. [DOI] [PubMed] [Google Scholar]
- 2.Bray NJ, Buckland PR, Owen MJ, O'Donovan MC. Cis-acting variation in the expression of a high proportion of genes in human brain. Hum Genet. 2003;113:149–53. doi: 10.1007/s00439-003-0956-y. [DOI] [PubMed] [Google Scholar]
- 3.Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW. Allelic variation in human gene expression. Science. 2002;297:1143. doi: 10.1126/science.1072545. [DOI] [PubMed] [Google Scholar]
- 4.Yan H, Zhou W. Allelic variations in gene expression. Curr Opin Oncol. 2004;16:39–43. doi: 10.1097/00001622-200401000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Pastinen T, Ge B, Hudson TJ. Influence of human genome polymorphism on gene expression. Hum Mol Genet. 2006;15 Spec No 1:R9–16. doi: 10.1093/hmg/ddl044. [DOI] [PubMed] [Google Scholar]
- 6.Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen KY, Morley M, Spielman RS. Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet. 2003;33:422–5. doi: 10.1038/ng1094. [DOI] [PubMed] [Google Scholar]
- 7.Rockman MV, Hahn MW, Soranzo N, Zimprich F, Goldstein DB, Wray GA. Ancient and recent positive selection transformed opioid cis-regulation in humans. PLoS Biol. 2005;3:e387. doi: 10.1371/journal.pbio.0030387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson AD, Wang D, Sadee W. Polymorphisms affecting gene regulation and mRNA processing: broad implications for pharmacogenetics. Pharmacol Ther. 2005;106:19–38. doi: 10.1016/j.pharmthera.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Wang D, Johnson AD, Papp AC, Kroetz DL, Sadee W. Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant 3435C>T affects mRNA stability. Pharmacogenet Genomics. 2005;15:693–704. [PubMed] [Google Scholar]
- 10.Zhang Y, Wang D, Johnson AD, Papp AC, Sadee W. Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. J Biol Chem. 2005;280:32618–24. doi: 10.1074/jbc.M504942200. [DOI] [PubMed] [Google Scholar]
- 11.Lim JE, Papp A, Pinsonneault J, Sadee W, Saffen D. Allelic expression of serotonin transporter (SERT) mRNA in human pons: lack of correlation with the polymorphism SERTLPR. Mol Psychiatry. 2006;11:649–62. doi: 10.1038/sj.mp.4001797. [DOI] [PubMed] [Google Scholar]
- 12.Pinsonneault JK, Papp AC, Sadee W. Allelic mRNA expression of X-linked monoamine oxidase a (MAOA) in human brain: dissection of epigenetic and genetic factors. Hum Mol Genet. 2006;15:2636–49. doi: 10.1093/hmg/ddl192. [DOI] [PubMed] [Google Scholar]
- 13.Lim JE, Pinsonneault J, Sadee W, Saffen D. Tryptophan hydroxylase 2 (TPH2) haplotypes predict levels of TPH2 mRNA expression in human pons. Mol Psychiatry. 2007;12:491–501. doi: 10.1038/sj.mp.4001923. [DOI] [PubMed] [Google Scholar]
- 14.Pinsonneault J, Nielsen CU, Sadee W. Genetic Variants of the Human H+/Dipeptide Transporter PEPT2: Analysis of Haplotype Functions. J Pharmacol Exp Ther. 2004;311:1088–96. doi: 10.1124/jpet.104.073098. [DOI] [PubMed] [Google Scholar]
- 15.Wang D, Papp AC, Binkley PF, Johnson JA, Sadee W. Highly variable mRNA expression and splicing of L-type voltage-dependent calcium channel alpha subunit 1C in human heart tissues. Pharmacogenet Genomics. 2006;16:735–45. doi: 10.1097/01.fpc.0000230119.34205.8a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Bertolino A, Fazio L, Blasi G, Rampino A, Romano R, Lee MLT, Xiao T, Papp A, Wang D, et al. Novel polymorphisms in the human dopamine D2 receptor gene affect gene expression, splicing, and neuronal activity. PNAS. 2007;104:20552–57. doi: 10.1073/pnas.0707106104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bray NJ, Buckland PR, Williams NM, Williams HJ, Norton N, Owen MJ, O'Donovan MC. A haplotype implicated in schizophrenia susceptibility is associated with reduced COMT expression in human brain. Am J Hum Genet. 2003;73:152–61. doi: 10.1086/376578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knight JC, Keating BJ, Rockett KA, Kwiatkowski DP. In vivo characterization of regulatory polymorphisms by allele-specific quantification of RNA polymerase loading. Nat Genet. 2003;33:469–75. doi: 10.1038/ng1124. [DOI] [PubMed] [Google Scholar]
- 19.Ge B, Gurd S, Gaudin T, Dore C, Lepage P, Harmsen E, Hudson TJ, Pastinen T. Survey of allelic expression using EST mining. Genome Res. 2005;15:1584–91. doi: 10.1101/gr.4023805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin W, Yang HH, Lee MP. Allelic variation in gene expression identified through computational analysis of the dbEST database. Genomics. 2005;86:518–27. doi: 10.1016/j.ygeno.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Cowles CR, Hirschhorn JN, Altshuler D, Lander ES. Detection of regulatory variation in mouse genes. Nat Genet. 2002;32:432–7. doi: 10.1038/ng992. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Chen M, Deng C, Bourc'his D, Nealon JG, Erlichman B, Bestor TH, Weinstein LS. Identification of the control region for tissue-specific imprinting of the stimulatory G protein alpha-subunit. Proc Natl Acad Sci U S A. 2005;102:5513–8. doi: 10.1073/pnas.0408262102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singer-Sam J, Chapman V, LeBon JM, Riggs AD. Parental imprinting studied by allele-specific primer extension after PCR: paternal X chromosome-linked genes are transcribed prior to preferential paternal X chromosome inactivation. Proc Natl Acad Sci U S A. 1992;89:10469–73. doi: 10.1073/pnas.89.21.10469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pastinen T, Sladek R, Gurd S, Sammak A, Ge B, Lepage P, Lavergne K, Villeneuve A, Gaudin T, Brandstrom H, et al. A survey of genetic and epigenetic variation affecting human gene expression. Physiol Genomics. 2004;16:184–93. doi: 10.1152/physiolgenomics.00163.2003. [DOI] [PubMed] [Google Scholar]
- 25.He H, Olesnanik K, Nagy R, Liyanarachchi S, Prasad ML, Stratakis CA, Kloos RT, de la Chapelle A. Allelic variation in gene expression in thyroid tissue. Thyroid. 2005;15:660–7. doi: 10.1089/thy.2005.15.660. [DOI] [PubMed] [Google Scholar]
- 26.Pant PV, Tao H, Beilharz EJ, Ballinger DG, Cox DR, Frazer KA. Analysis of allelic differential expression in human white blood cells. Genome Res. 2006;16:331–9. doi: 10.1101/gr.4559106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cirulli ET, Goldstein DB. In vitro assays fail to predict in vivo effects of regulatory polymorphisms. Hum Mol Genet. 2007;16:1931–9. doi: 10.1093/hmg/ddm140. [DOI] [PubMed] [Google Scholar]
- 28.Wilkins JM, Southam L, Price AJ, Mustafa Z, Carr A, Loughlin J. Extreme context specificity in differential allelic expression. Hum Mol Genet. 2007;16:537–46. doi: 10.1093/hmg/ddl488. [DOI] [PubMed] [Google Scholar]
- 29.Bray NJ, Preece A, Williams NM, Moskvina V, Buckland PR, Owen MJ, O'Donovan MC. Haplotypes at the dystrobrevin binding protein 1 (DTNBP1) gene locus mediates risk for schizophrenia through reduced DTNBP1 expression. Hum Mol Genet. 2005;14:1947–54. doi: 10.1093/hmg/ddi199. [DOI] [PubMed] [Google Scholar]
- 30.Bray NJ, Buckland PR, Hall H, Owen MJ, O'Donovan MC. The serotonin-2A receptor gene locus does not contain common polymorphism affecting mRNA levels in adult brain. Mol Psychiatry. 2004;9:109–14. doi: 10.1038/sj.mp.4001366. [DOI] [PubMed] [Google Scholar]
- 31.Fukuda Y, Koga M, Arai M, Noguchi E, Ohtsuki T, Horiuchi Y, Ishiguro H, Niizato K, Iritani S, Itokawa M, et al. Monoallelic and unequal allelic expression of the HTR2A gene in human brain and peripheral lymphocytes. Biol Psychiatry. 2006;60:1331–5. doi: 10.1016/j.biopsych.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 32.Rovin BH, Lu L, Saxena R. A novel polymorphism in the MCP-1 gene regulatory region that influences MCP-1 expression. Biochem Biophys Res Commun. 1999;259:344–8. doi: 10.1006/bbrc.1999.0796. [DOI] [PubMed] [Google Scholar]
- 33.Xu Y, Porntadavity S, St Clair DK. Transcriptional regulation of the human manganese superoxide dismutase gene: the role of specificity protein 1 (Sp1) and activating protein-2 (AP-2) Biochem J. 2002;362:401–12. doi: 10.1042/0264-6021:3620401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miklos I, Meyer IM, Nagy B. Moments of the Boltzmann distribution for RNA secondary structures. Bull Math Biol. 2005;67:1031–47. doi: 10.1016/j.bulm.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 35.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–9. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 36.Duan J, Wainwright MS, Comeron JM, Saitou N, Sanders AR, Gelernter J, Gejman PV. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet. 2003;12:205–16. doi: 10.1093/hmg/ddg055. [DOI] [PubMed] [Google Scholar]
- 37.Howe D, Lynas C. The cyclin D1 alternative transcripts [a] and [b] are expressed in normal and malignant lymphocytes and their relative levels are influenced by the polymorphism at codon 241. Haematologica. 2001;86:563–9. [PubMed] [Google Scholar]
- 38.Nackley AG, Shabalina SA, Tchivileva IE, Satterfield K, Korchynskyi O, Makarov SS, Maixner W, Diatchenko L. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science. 2006;314:1930–3. doi: 10.1126/science.1131262. [DOI] [PubMed] [Google Scholar]
- 39.Sayed-Tabatabaei FA, Oostra BA, Isaacs A, van Duijn CM, Witteman JC. ACE polymorphisms. Circ Res. 2006;98:1123–33. doi: 10.1161/01.RES.0000223145.74217.e7. [DOI] [PubMed] [Google Scholar]
- 40.Sam F, Kerstetter DL, Pimental DR, Mulukutla S, Tabaee A, Bristow MR, Colucci WS, Sawyer DB. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J Card Fail. 2005;11:473–80. doi: 10.1016/j.cardfail.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 41.Hiroi S, Harada H, Nishi H, Satoh M, Nagai R, Kimura A. Polymorphisms in the SOD2 and HLA-DRB1 genes are associated with nonfamilial idiopathic dilated cardiomyopathy in Japanese. Biochem Biophys Res Commun. 1999;261:332–9. doi: 10.1006/bbrc.1999.1036. [DOI] [PubMed] [Google Scholar]
- 42.Valenti L, Conte D, Piperno A, Dongiovanni P, Fracanzani AL, Fraquelli M, Vergani A, Gianni C, Carmagnola L, Fargion S. The mitochondrial superoxide dismutase A16V polymorphism in the cardiomyopathy associated with hereditary haemochromatosis. J Med Genet. 2004;41:946–50. doi: 10.1136/jmg.2004.019588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hung RJ, Boffetta P, Brennan P, Malaveille C, Gelatti U, Placidi D, Carta A, Hautefeuille A, Porru S. Genetic polymorphisms of MPO, COMT, MnSOD, NQO1, interactions with environmental exposures and bladder cancer risk. Carcinogenesis. 2004;25:973–8. doi: 10.1093/carcin/bgh080. [DOI] [PubMed] [Google Scholar]
- 44.Xu Y, Fang F, Dhar SK, St Clair WH, Kasarskis EJ, St Clair DK. The role of a single-stranded nucleotide loop in transcriptional regulation of the human sod2 gene. J Biol Chem. 2007;282:15981–94. doi: 10.1074/jbc.M608979200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ginsberg MD, Feliciello A, Jones JK, Avvedimento EV, Gottesman ME. PKA-dependent binding of mRNA to the mitochondrial AKAP121 protein. J Mol Biol. 2003;327:885–97. doi: 10.1016/s0022-2836(03)00173-6. [DOI] [PubMed] [Google Scholar]
- 46.Huang Y, He T, Domann FE. Decreased expression of manganese superoxide dismutase in transformed cells is associated with increased cytosine methylation of the SOD2 gene. DNA Cell Biol. 1999;18:643–52. doi: 10.1089/104454999315051. [DOI] [PubMed] [Google Scholar]
- 47.Chasman DI, Posada D, Subrahmanyan L, Cook NR, Stanton VP, Jr, Ridker PM. Pharmacogenetic study of statin therapy and cholesterol reduction. Jama. 2004;291:2821–7. doi: 10.1001/jama.291.23.2821. [DOI] [PubMed] [Google Scholar]
- 48.Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, Rieder MJ, Cooper GM, Roos C, Voight BF, Havulinna AS, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Gen. 2008;40:189–97. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eubank TD, Galloway M, Montague CM, Waldman WJ, Marsh CB. M-CSF induces vascular endothelial growth factor production and angiogenic activity from human monocytes. J Immunol. 2003;171:2637–43. doi: 10.4049/jimmunol.171.5.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakayama M, Yasue H, Yoshimura M, Shimasaki Y, Kugiyama K, Ogawa H, Motoyama T, Saito Y, Ogawa Y, Miyamoto Y, et al. T-786-->C mutation in the 5′-flanking region of the endothelial nitric oxide synthase gene is associated with coronary spasm. Circulation. 1999;99:2864–70. doi: 10.1161/01.cir.99.22.2864. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. List of candidate genes analyzed in this study, grouped by indication (disease or pharmacology). Marker SNPs are all located in transcribed regions of the mature mRNA, or a splice variant. For some genes more than one marker SNP and tissue were used.
Supplemental Table 2. Oligonucleotide primers used for PCR amplification and SNaPshot primer extension reactions. The reverse PCR primer also served for gene specific cDNA synthesis.