

Abstract
CPT-11 is an antitumor prodrug that is hydrolyzed by carboxylesterases (CE) to yield SN-38, a potent topoisomerase I poison. However, the dose limiting toxicity is delayed diarrhea that is thought to arise, in part, from activation of the prodrug by a human intestinal CE (hiCE). Therefore, we have sought to identify selective inhibitors of hiCE that may have utility in modulating drug toxicity. We have evaluated one such class of molecules (benzene sulfonamides), and developed QSAR models for inhibition of this protein. Using these predictive models, we have synthesized a panel of fluorene analogues that are selective for hiCE, demonstrating no cross reactivity to the human liver CE, hCE1, or towards human cholinesterases, and have Ki values as low as 14nM. These compounds prevented hiCE-mediated hydrolysis of the drug and the potency of enzyme inhibition correlated with the clogP of the molecules. These studies will allow the development and application of hiCE-specific inhibitors designed to selectively modulate drug hydrolysis in vivo.
INTRODUCTION
Carboxylesterases (CE) are ubiquitously expressed enzymes that are thought to be responsible for the hydrolysis of xenobiotics.1 They catalyze the conversion of esters to their corresponding alcohols and carboxylic acids. Since numerous clinically used compounds are esterified, an approach used by the pharmaceutical industry to improve the water solubility of molecules, they are substrates for these enzymes. Hence, drugs such as heroin, cocaine, 1 (irinotecan; CPT-112; Figure 1), capecitabine, oseltamivir (Tamiflu), lidocaine, and meperidine (Demerol) are all hydrolyzed by CEs.3–16 Therefore, identifying compounds that modulate the hydrolysis of these agents may be useful in either altering the half-life and/or toxicities associated with these drugs. For example, flestolol, a β-blocker is rapidly hydrolyzed by CEs to an inactive metabolite and hence its biological activity is rapidly lost.17 Inhibition of the enzyme responsible for this hydrolysis would increase the in vivo stability of the molecule and likely improve its therapeutic utility. In contrast, the delayed diarrhea that is associated with 1 treatment is thought to arise, in part, from hydrolysis of the drug in the intestine by the human intestinal CE (hiCE, CES2)12, 13, 18 to yield 2 (7-ethyl-10-hydroxycamptothecin; SN-38; Figure 1). Since this is the dose limiting toxicity for this highly effective anticancer agent, approaches that ameliorate this side effect would improve patient quality of care and potentially allow drug dose intensification. This could potentially be achieved by an inhibitor that targets hiCE within the gut. We have sought therefore to identify compounds that can inhibit CEs without impacting human acetyl- or butyrylcholinesterase (hAChE and hBChE, respectively). Initially, we screened a library of compounds from Telik using their Target-Related Affinity Profiling (TRAP™) technology19 and identified several compounds that were selective inhibitors of CEs.20, 21 Of these, one class demonstrated selectivity towards hiCE versus the human liver CE, hCE1 (CES1).21 The majority of these compounds were benzene sulfonamides and preliminary studies indicated that halogen substitution tended to increase the potency of the inhibitors. However, these studies were based on a series of 9 compounds (4–12 in Table 1) with a disparate set of different chemotypes.21 Here we have considerably expanded these analyses, and now assayed and analyzed 57 benzene sulfonamides for their ability to inhibit hiCE, hCE1, hAChE or hBChE. Using detailed QSAR models, we have designed a series of novel fluorene analogues that are highly potent hiCE inhibitors and can modulate 1 metabolism. Potentially, these molecules would be lead compounds for subsequent drug design.
Figure 1.

The chemical structure and hydrolysis of 1 resulting in the formation of 2.
Table 1.
Ki values for the inhibition of human CEs and cholinesterases by the benzene sulfonamides. For the CEs, 3 was used as a substrate and the respective thiocholines were used for hAChE and hBChE. The general structure of the sulfonamides is indicated. The X in the subfragment represents the point of attachment to the sulfonamide moiety.
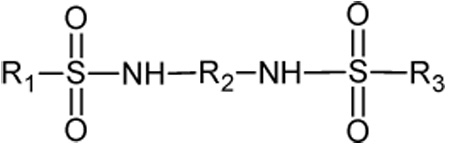 | |||||||
|---|---|---|---|---|---|---|---|
| ID | Compound | R1 | R2 | R3 | clog P | hiCE Ki ± SE (nM) | hCE1 Ki ± SE (nM) |
| 4 | 4,6-dimethyl-N1,N3- diphenylbenzene-1,3- disulfonamide | Bza | 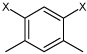 |
Bz | 2.75 | 218 ± 45b | >100,000 |
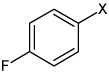
| 5 | 4-chloro-N-(4 ethoxyphenyl) benzene sulfonamide | 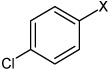 |
None | 3.46 | 1,310 ± 176b | >100,000 | |
| 6 | 4-bromo-N-(4-phenoxyphenyl)benzene sulfonamide | 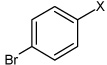 |
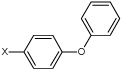 |
None | 5.03 | 165 ± 33b | >100,000 |
| 7 | N,N'-(4,4'-disulfanediylbis(4,1-phenylene)) dimethane sulfonamide | H | 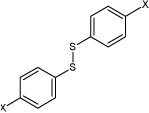 |
H | 2.39 | 767 ± 285b | >100,000 |
| 8 | N,N'-(1,4-phenylene)benzene sulfonamide | Bz | Bz | Bz | 3.63 | 1,060 ± 133b | >100,000 |
| 9 | N,N'-(2-methyl-1,4-phenylene)benzene sulfonamide | Bz | 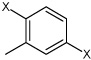 |
Bz | 3.36 | 365 ± 87b | >100,000 |
| 10 | N,N'-(naphthalene-1,4-diyl)bis(4-chloro benzene sulfonamide) | 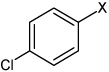 |
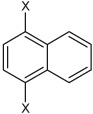 |
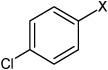 |
5.08 | 194 ± 23b | >100,000 |
| 11 | N,N'-(1,4-phenylene)bis(4-chlorobenzene sulfonamide) | 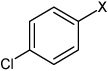 |
Bz | 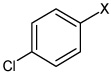 |
4.40 | 53.2 ± 5.5b | 13,700 ± 4,870a |
| 12 | N,N'-(perchloro-1,4-phenylene)dibenzene sulfonamide | Bz | 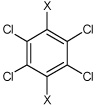 |
Bz | 4.63 | 451 ± 39b | >100,000 |
| 13 | N,N'-(2,3,5,6-fluoro-1,4-phenylene) bis(4-chlorobenzene sulfonamide) | 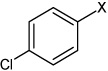 |
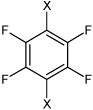 |
 |
4.33 | 41.5 ± 6.5b | >100,000 |
| 14 | N,N'-(1,4-phenylene)bis(2-fluoro benzene sulfonamide) | 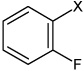 |
Bz | 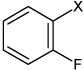 |
3.02 | 1,450 ± 154 | >100,000 |
| 15 | N,N'-(1,4-phenylene)bis(3-fluorobenzene sulfonamide) | 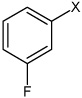 |
Bz | 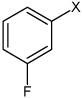 |
3.61 | 248 ± 26 | >100,000 |
| 16 | N,N'-(1,4-phenylene)bis(4-fluorobenzene sulfonamide) | 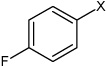 |
Bz | 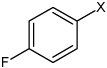 |
3.72 | 355 ± 70 | >100,000 |
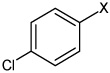
| 17 | N,N'-(1,4-phenylene)bis(3-chlorobenzene sulfonamide) | 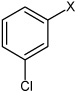 |
Bz | 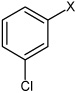 |
4.31 | 74.0 ± 5.5 | >100,000 |
| 18 | N,N'-(1,4-phenylene)bis(3-bromobenzene sulfonamide) | 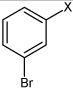 |
Bz | 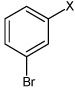 |
4.55 | 67.0 ± 9.6b | >100,000 |
| 19 | N,N'-(1,4-phenylene)bis(4-bromobenzene sulfonamide) | 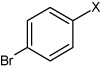 |
Bz | 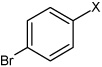 |
4.70 | 85.2 ± 10.1 | >100,000 |
| 20 | N,N'-(1,4-phenylene)bis(2,6-difluoro benzene sulfonamide) | 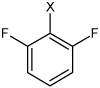 |
Bz | 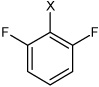 |
2.78 | 5,260 ± 620 | >100,000 |
| 21 | N,N'-(1,4-phenylene)bis(3,4-difluoro benzene sulfonamide) | 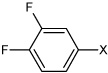 |
Bz | 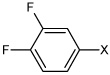 |
3.81 | 160 ± 7.5 | >100,000 |
| 22 | N,N'-(1,4-phenylene)bis(3,5-difluoro benzene sulfonamide) | 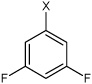 |
Bz | 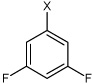 |
3.76 | 246 ± 34 | >100,000 |
| 23 | N,N'-(1,4-phenylene)bis(3,4,5-trifluorobenzene sulfonamide) | 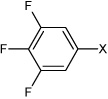 |
Bz | 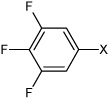 |
3.81 | 210 ± 33 | >100,000 |
| 24 | N,N'-(1,4-phenylene)bis(2,3-dichlorobenzene sulfonamide) | 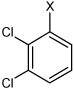 |
Bz | 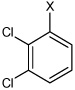 |
4.63 | 268 ± 137 | >100,000 |
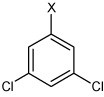
| 25 | N,N'-(1,4-phenylene)bis(2,5-dichloro benzene sulfonamide) | 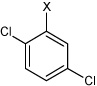 |
Bz | 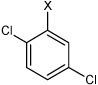 |
4.60 | 119 ± 15 | >100,000 |
| 26 | N,N'-(1,4-phenylene)bis(3,4-dichloro benzene sulfonamide) | 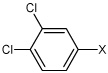 |
Bz | 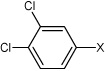 |
4.63 | 91.5 ± 23 | >100,000 |
| 27 | N,N'-(1,4-phenylene)bis(3,5-dichloro benzene sulfonamide) | 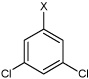 |
Bz | 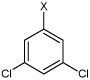 |
4.87 | 385 ± 80 | >100,000 |
| 28 | N,N'-(1,4-phenylene)bis(2,4,5-trichlorobenzene sulfonamide) | 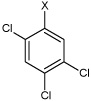 |
Bz | 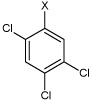 |
4.74 | >100,000 | >100,000 |
| 29 | 4-chloro-N-(4-(4-fluorophenylsulfonamido) phenyl)benzene sulfonamide | 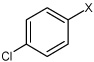 |
Bz | 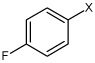 |
4.06 | 200 ± 11 | >100,000 |
| 30 | 3,4-difluoro-N-(4-(phenylsulfonamido) phenyl)benzene sulfonamide | 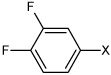 |
Bz | Bz | 3.64 | 488 ± 30 | >100,000 |
| 31 | 4-chloro-N-(4-(4-methylphenyl sulfonamido)phenyl) benzene sulfonamide | 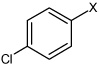 |
Bz | 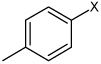 |
4.40 | 161 ± 9.3 | >100,000 |
| 32 | 4-fluoro-N-(4-(4-methylphenyl sulfonamido)phenyl) benzene sulfonamide | 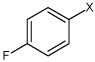 |
Bz | 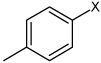 |
4.02 | 281 ± 38 | >100,000 |
| 33 | 2,4-difluoro-N-(4-(4-methylphenyl sulfonamido)phenyl) benzene sulfonamide | 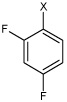 |
Bz | 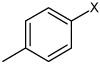 |
3.69 | 385 ± 50 | >100,000 |
| 34 | 2,5-difluoro-N-(4-(4-methylphenyl sulfonamido)phenyl)benz ene sulfonamide | 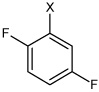 |
Bz | 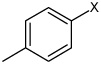 |
3.67 | 467 ± 38 | >100,000 |
| 35 | 3,4-difluoro-N-(4-(4-methylphenyl sulfonamido)phenyl)benz ene sulfonamide | 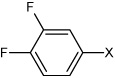 |
Bz | 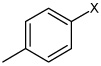 |
4.15 | 194 ± 18 | >100,000 |
| 36 | N,N'-(1,4-phenylene)bis(4-fluoro-2-methyl benzene sulfonamide) | 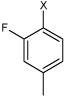 |
Bz | 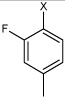 |
3.35 | 273 ± 33 | >100,000 |
| 37 | 4-fluoro-N-(4-(4-methyoxy phenyl sulfonamido)phenyl)benzene sulfonamide | 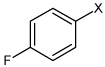 |
Bz | 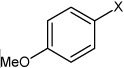 |
4.10 | 568 ± 76 | >100,000 |
| 38 | N-(4-(4-fluorophenylsulfonamido) phenyl)-2,5-dimethylbenzene sulfonamide | 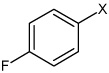 |
Bz | 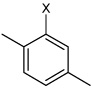 |
3.62 | 240 ± 30 | >100,000 |
| 39 | 4-(N-(4-(4-fluorophenylsulfonamido) phenyl)sulfamoyl) benzoic acid | 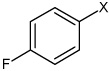 |
Bz | 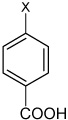 |
3.06 | >100,000 | >100,000 |
| 40 | 3-(N-(4-(4-fluorophenylsulfonamido) phenyl)sulfamoyl)-4-methyl benzoic acid | 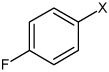 |
Bz | 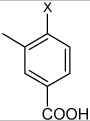 |
3.06 | 693 ± 187 | >100,000 |
| 41 | 4-(N-(4-(4-fluorophenyl sulfonamido) phenyl)sulfamoyl)-N-methyl benzamide | 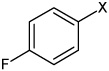 |
Bz | 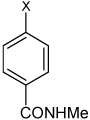 |
3.77 | >100,000 | >100,000 |
| 42 | N,N'-(2-chloro-1,4-phenylene) dibenzene sulfonamide | Bz | 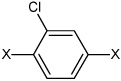 |
Bz | 4.25 | 696 ± 28 | >100,000 |
| 43 | N,N'-(2,5-dichloro-1,4-phenylene) bis(4-chlorobenzene sulfonamide) | 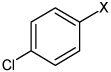 |
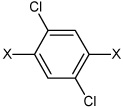 |
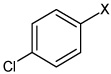 |
4.72 | 455 ± 79 | >100,000 |
| 44 | N,N'-(2,5-dibromo-1,4-phenylene) bisbenzene sulfonamide | Bz | 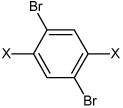 |
Bz | 4.24 | >100,000 | >100,000 |
| 45 | N,N'-(2,5-dibromo-1,4-phenylene) bis(4-chlorobenzene sulfonamide | 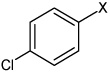 |
 |
 |
4.82 | >100,000 | >100,000 |
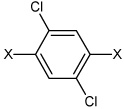
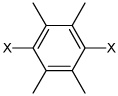
| 46 | N,N'-(2,3,5,6-tetramethyl-1,4-phenylene) bis(4-fluorobenzene sulfonamide) |  |
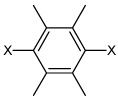 |
 |
2.82 | >100,000 | >100,000 |
| 47 | N,N'-(2,3,5,6-tetramethyl-1,4-phenylene) bis(3-chlorobenzene sulfonamide) | 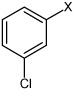 |
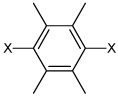 |
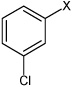 |
3.16 | 2,060 ± 750 | >100,000 |
| 48 | N,N'-(2,3,5,6-tetramethyl-1,4-phenylene) bis(4-chlorobenzene Sulfonamide | 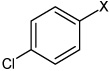 |
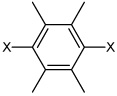 |
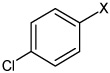 |
3.21 | 2,010 ± 710 | >100,000 |
| 49 | N,N'-(2,3,5,6- tetramethyl-1,4-phenylene) bis(3-bromobenzene sulfonamide) | 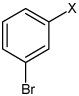 |
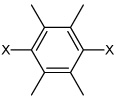 |
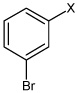 |
3.12 | 1,570 ± 480 | >100,000 |
| 50 | N,N'-(2,3,5,6-tetramethyl-1,4-phenylene) bis(4-bromobenzene sulfonamide) | 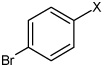 |
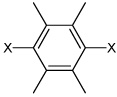 |
 |
3.22 | 3,240 ± 1,780 | >100,000 |
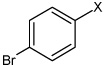
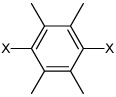
| 51 | N,N'-(2,3,5,6-tetramethyl-1,4-phenylene) bis(3,4-dichlorobenzene sulfonamide) | 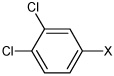 |
 |
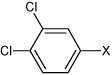 |
3.99 | 344 ± 57 | >100,000 |
| 52 | N,N'-(2,3,5,6-tetramethyl-1,4-phenylene) bis(3,5-dichlorobenzene sulfonamide) | 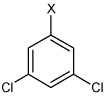 |
 |
 |
4.13 | 2,600 ± 1,010 | >100,000 |
| 53 | N,N'-(2,3,5,6-tetrafluoro-1,4-phenylene) bis(4-fluorobenzene sulfonamide) | 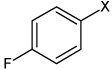 |
 |
 |
4.03 | 314 ± 46 | >100,000 |
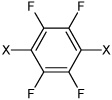
| 54 | N,N'-(2,3,5,6-tetrafluoro-1,4-phenylene) bis(4-bromobenzene sulfonamide) |  |
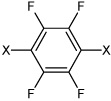 |
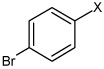 |
4.75 | 23.4 ± 2.7b | >100,000 |
| 55 | N,N'-1,4-phenylenebis-2- naphthalene sulfonamide | 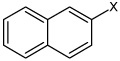 |
Bz | 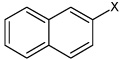 |
4.87 | 1,580 ± 730 | >100,000 |
Bz represents benzene ring.
Data taken from Wadkins et al.21
Data taken from Hatfield et al (manuscript submitted).
RESULTS
Selective inhibition of hiCE by benzene sulfonamides
Based upon our previous work,21 we had identified benzene sulfonamides as selective inhibitors of hiCE. The 3D-QSAR analysis presented in this article indicated that (i) halogenation of the phenyl rings resulted in more potent compounds, and (ii) that the central region of the inhibitor-enzyme complex was hydrophobic and could accommodate a large aromatic structure. Therefore, we synthesized or acquired a total of 57 sulfonamide analogues, mostly containing halogen atoms appended to the benzene rings and assessed their inhibitory potency towards the CEs hiCE and hCE1, as well as hAChE and hBChE. These assays used 3 (o-nitrophenyl acetate) as a substrate for the former enzymes, and acetylthiocholine and butyrylthiocholine for the respective cholinesterases. As indicated in Table 1, the vast majority of the compounds were excellent inhibitors with Ki values ranging from 41nM to 3,240nM. The sulfonamides were selective for hiCE, with only one molecule demonstrating weak activity towards hCE1 (compound 11). However, 11 was still over 250-fold more potent against hiCE (Ki values were 53nM vs 13,700nM for hiCE and hCE1, respectively). None of the sulfonamides inhibited either hAChE or hBChE (data not shown) consistent with our previously reports of these types of compounds.21 Since both CEs and cholinesterases demonstrate very similar crystal structures,4, 22 we presume that the specificity of the sulfonamides for hiCE is due to unique interactions with amino acids within the active site of this protein. Six compounds were inactive towards all enzymes (28, 39, 41, and 44–46). The majority of these compounds contained large bulky atoms or moieties present within either the terminal benzene rings (28, 39, 41), or the central domain of the molecule (44–46). These groups would likely impede access of the inhibitor to that active site gorge, thereby mitigating their biological activity. The mode of enzyme inhibition by the sulfonamides inhibitors was partially competitive, indicating that while their structure resembled the substrate molecule, they were unable to completely inhibit substrate hydrolysis.23 For this series of compounds therefore, the inhibitory potency will be partially dependent upon the structure of the substrate. To confirm that these molecules could indeed inhibit the hydrolysis of other substrates, their effect on the metabolism of 1 was assessed.
Inhibition of hiCE-mediated 1 hydrolysis
Having demonstrated potent inhibition of the hydrolysis of 3 by the sulfonamides, we determined their ability to prevent conversion of 1 to 2. As indicated above, this was necessary since the inhibitors acted in a partially competitive fashion. Table 2 displays the Ki values for the inhibition of 1 hydrolysis by these compounds. As can be seen, very potent inhibitors were obtained, with inhibition constants as low as 23.4nM being observed. A comparison of the inhibition constants for 1 and 3 demonstrated a good linear correlation (r2 = 0.72; Figure 2) as well as an excellent Spearman r value (r = 0.80; p = 0.0003). Again, only compounds that inhibited the hydrolysis of both substrates were included in these analyses. These results indicate that, in general, there is a commonality in the potency of enzyme inhibition and ability of selected compounds to inhibit the hydrolysis of specific substrates. Furthermore, we have also demonstrated that compounds 18 and 54 can also inhibit the metabolism of heroin and cocaine (data not shown; Hatfield et al, manuscript in preparation). In summary, these results demonstrate that while determination of the actual Ki values for specific substrates will always be necessary, it is likely that compounds that can inhibit o-NPA hydrolysis will also be active towards other esters.
Table 2.
Ki values for the inhibition of hiCE-mediated hydrolysis of 1 by selected benzene sulfonamides. Inhibition constants using 3 are included for comparison.
| ID | Ki 1 (nM) | Ki 3 (nM) |
|---|---|---|
| 4 | >100,000a | 218 ± 45a |
| 5 | 3,220 ± 950a | 1,310 ± 176a |
| 6 | >100,000a | 165 ± 33a |
| 7 | >100,000a | 767 ± 285a |
| 8 | 892 ± 67a | 1,060 ± 133a |
| 9 | 238 ± 29a | 365 ± 87a |
| 10 | >100,000 | 194 ± 23a |
| 11 | 141 ± 64a | 53.2 ± 5.5a |
| 12 | >100,000 | 451 ± 39a |
| 13 | 110 ± 23a | 41.5 ± 6.5a |
| 15 | 152 ± 29 | 248 ± 26 |
| 16 | 79 ± 15 | 355 ± 70 |
| 17 | 62 ± 15 | 74.0 ± 5.5 |
| 18 | 121 ± 15b | 67.0 ± 9.6b |
| 19 | 44 ± 10 | 85.2 ± 10.1 |
| 20 | >100,000 | 5,260 ± 620 |
| 23 | 192 ± 44 | 210 ± 33 |
| 46 | >100,000 | >100,000 |
| 50 | >100,000 | 3,240 ± 1,780 |
| 52 | >100,000 | 2,600 ± 1,010 |
| 54 | 28 ± 4b | 23.4 ± 2.7b |
Data taken from Wadkins et al.21
Data taken from Hatfield et al (manuscript in preparation).
Figure 2.

Graph demonstrating the correlation between the Ki values for inhibition of 1 hydrolysis, versus those for 3 hydrolysis, when using hiCE. The linear correlation coefficient (r2) for the curve fit is indicated.
Correlation between clogP and inhibitor potency
We have previously demonstrated that for a series of isatins, the inhibitory potency of the compounds toward CEs was related to their clogP values.24 This is likely due to that fact that the active sites of these proteins are highly hydrophobic gorges that project up to 30Å into the interior of the enzyme.4, 22, 25 These hydrophobic regions are also present in the 3D-QSAR models and are thought to reflect the nature of the amino acids that line the gorge. Therefore, we graphed the inhibition constants for the sulfonamides with hiCE versus the clogP values for the respective compounds. For analyses with 3, we excluded the datasets for compounds 28, 39, 41, and 44–46 since they did not inhibit the enzyme. Similarly with CPT-11 as a substrate, we excluded data obtained from compounds 4, 6, 7, 10, 12, 20, 46, 50 and 52. As indicated in Figure 3, reasonable correlations were observed between the Ki values and clogP for both substrates, with linear correlation coefficients (r2) for the curve fit of 0.49 and 0.43 for 1 and 3, respectively. Analysis of these data however, using a Spearman rank order calculation, demonstrated highly significant results (Table 3). Since this statistical test uses a ranking system for determining significance, these analyses cannot be used to predict the Ki value for novel sulfonamides. Nevertheless, it is clear that the relative hydrophobicity of the molecule is an important factor in determining the biological potency of these compounds.
Figure 3.

Graph demonstrating the correlation between Ki values and clogP for the sulfonamide inhibitors. Assays were run using either 1 (red) or 3 (blue) as substrates. Linear correlation coefficients (r2) for the curve fits are indicated.
Table 3.
Linear correlation coefficients and P values for the statistical analysis of the relationship between the clogP and the Ki values for hiCE inhibition by the sulfonamides. Results are presented using either 1 or 3 as substrates.
| Substrate | Parameter | ||
|---|---|---|---|
| r2 | Spearman r | P | |
| 1 | 0.486 | −0.832 | <0.0001 |
| 3 | 0.432 | −0.615 | <0.0001 |
Inhibition of hiCE by fluorene analogues
Based on the information from our earlier 3D-QSAR studies and the clogP correlations, we hypothesized that a planar hydrophobic ring structure, larger than benzene, in the central core of the molecule might improve inhibitor potency. We excluded compounds that might increase the bulkiness of the molecule since the active site for hiCE is thought to exist as a long deep gorge, that projects into the interior of the protein. Therefore, we synthesized a series of fluorene derivatives (compounds 56–60) and assessed their ability to selectively inhibit hiCE. As can be seen from Table 4, all of these analogues were potent inhibitors of hiCE with Ki values ranging from 14–91nM. In addition, all of these molecules inhibited hiCE in a partially competitive fashion, similar to that seen for the other sulfonamides. Direct comparison of the inhibition constants for compounds 8 and 56, that differ only by the fluorene moiety in the central domain of the molecule, indicate that the latter is ∼11-fold more potent at inhibiting hiCE. Furthermore, addition of halogen atoms to the terminal benzene rings also increased the efficacy of the molecules, with compounds 57, 58 and 60 being among the most potent at inhibiting both the hydrolysis of 1 and 3 (Table 2). Indeed these sulfonamides yielded Ki values of 58nM, 40nM and 38nM, with 1, respectively. These results indicate that the inclusion of a planar, aromatic, hydrophobic domain within the center of the molecule is beneficial for hiCE inhibition.
Table 4.
Predicted and observed Ki values for hiCE with 5 fluorene analogues (compounds 56–60) that were postulated from the QSAR analyses to be excellent CE inhibitors.
| ID | Structure | clogP | Pred Ki 3 (nM) | Exp Ki 3 (± SE, nM) | Pred Ki 1 (nM) | Exp Ki 1 (± SE, nM) | hCE1 Ki 3 (± SE, nM) |
|---|---|---|---|---|---|---|---|
| 56 | 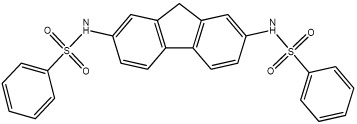 |
4.83 | 98.4 (Train) | 90.8 ± 1.1 | 1,820 | 84.7 ± 14.0 | >100,000 |
| 57 | 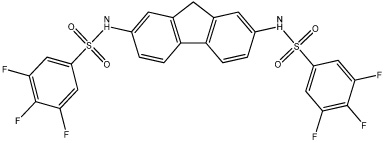 |
4.10 | 7.9 | 18.6 ± 5.3 | 9,350 | 58.1 ± 12.3 | >100,000 |
| 58 | 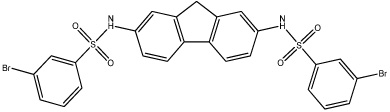 |
5.69 | 570 | 21.2 ± 2.5 | 72.8 | 39.7 ± 7.9 | >100,000 |
| 59 | 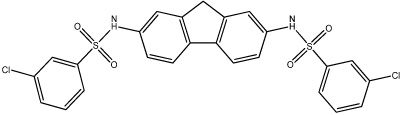 |
5.52 | 100 | 13.9 ± 2.5 | 9,780 | 52.8 ± 20.7 | >100,000 |
| 60 | 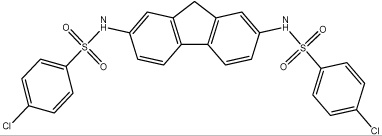 |
5.55 | 25.2 (Train) | 23.5 ± 5.7 | 3,930 | 37.7 ± 7.0 | >100,000 |
3D-QSAR analyses
Having developed relatively simple relationships for enzyme inhibition based upon the clogP value of the inhibitors, we undertook detailed 3D-QSAR analyses with the goal of identifying specific domains within the molecules that might improve (or ablate) inhibitory potency. We have previously performed similar studies and the pseudoreceptor site models that have been generated have significantly enhanced subsequent inhibitor design. Therefore, compounds 5, 8, 8–11, 14–31, 35–37, 47–56 and 60 were used as a training set for the development of the QSAR model. This was then validated using the other molecules (the test set). As can be seen from Table 5 and Figure 4, the linear correlation coefficients (r2) for the observed versus predicted Ki values for the training set were ∼0.9 with similar values for the cross correlation coefficients (q2). Since q2 values greater than 0.4 are considered statistically significant for biological systems,26 these models are likely to have excellent predictive value in the design of novel sulfonamide-based hiCE inhibitors. In addition, the q2/r2 values were close to unity (Table 5), confirming the validity of the models. As indicated in Table 4, the QSAR models were reasonable at predicting the Ki values for the fluorene analogues 56 – 60, with all compounds except 58 being considered excellent inhibitors of hiCE (<100nM) when using 3 as a substrate. Interestingly, the latter molecule had the highest clogP value (5.69), suggesting that for these models, this parameter is not a major determinant of biological potency. Significantly, while the fluorene moiety increased the length of the molecule by 4–5Å as compared to the benzene derivative, this did not significantly impact enzyme inhibition. Also it should be noted (see Figure 3) that the model was very poor at predicting the efficacy of 7. This is potentially due to the fact that this compound contains a disulfide chemotype (unlike all of the other molecules that were assayed) and can potentially adopt alternate conformations that would not be accurately predicted by the model. The QSAR predicted Ki values for the inhibition of hiCE-mediated 1 metabolism were not as good as though seen for 3 (Table 4). Indeed, while the fluorene compounds were considered good inhibitors of hiCE (Ki values in the low µM range), only 58 was predicted with any great accuracy (Table 4 and Figure 4). This is likely due to the fact that only 12 compounds were used as the training set for this model, and this did not include any of the fluorene analogues. However, these QSAR models will allow for rapid screening of analogues for inhibitor potency, prior to the initiation of chemical synthesis.
Table 5.
QSAR validation parameters obtained from Quasar software when using the Ki values for hiCE inhibition with either 1 or 3 as a substrate.
| Substrate | r2 | q2 | q2/r2 |
|---|---|---|---|
| 1 | 0.89 | 0.83 | 0.93 |
| 3 | 0.91 | 0.88 | 0.96 |
Figure 4.

Graphs demonstrating the correlation between the predicted and the observed Ki values obtained from the QSAR models. Panel A represents the graph when using 3 as a substrate, and panel B when using 1. Linear correlation coefficients for the line fits (r2) are indicated on the graphs.
Pseudoreceptor site QSAR models
To provide a graphical representation of the QSAR results, we developed a pseudoreceptor model for hiCE with datasets derived from the inhibition of hydrolysis of 3 (Figure 5A). This figure outlines the interactions that describe receptor-ligand binding in the enzyme. The model has primarily anionic (red areas) regions of charge located in a cluster at the base of the model and a weakly cationic (blue spheres) domain located at the top. This is consistent with all our previous analysis of hiCE inhibitors, where charge asymmetry was observed both in the QSAR models20, 21, 24, 27–29 and in the enzyme structure determined from homology modeling.21 We also present an electrostatic potential map of the model (Figure 5B) oriented to emphasize the charge asymmetry. While the origin of this charge distribution is not completely understood, at least for homology structures of hiCE, it maps well onto the position of charged amino acids present within the active site.21 It should be remembered that these figures represent a description of the interior surface of the active site and not the combined surfaces of the inhibitor molecules. However, in general, the electrostatic potential inside the active site was negative30 and hence it is not clear whether the positive areas required to fit the inhibition data reflect interactions within the active site alone, or represent potential interactions with positively charged residues near the active site opening. The interpretation of these models will be greatly enhanced if a crystal structure of hiCE becomes available.
Figure 5.

3D-QSAR pseudoreceptor site models generated from the enzyme inhibition data for hiCE using the benzene sulfonamides with 3 as a substrate. These models were generated using results obtained from all 57 sulfonamides and include compound 8 for reference. Both figures were generated using Raster3D43 and Molscript.44
A. The model is depicted as colored spheres on a hydrophobic gray grid. Areas that are hydrophobic are indicated in gray, where dark blue spheres represent regions that are positively charged (+0.25e) and light blue spheres correspond to reduced charge (+0.1e). Dark red spheres indicate domains that are negatively charged (−0.25e) and light red spheres represent areas of reduced negative charge (−0.1e). In all cases, e is the charge of the proton.
B. A stereo view of a 3D-QSAR pseudoreceptor site model that describes sulfonamide binding as a molecular surface upon which the electrostatic potential is mapped. The electrostatic potential is calculated from the Quasar software partial charges, which were defined as −0.25e, −0.1e, +0.1e and +0.25e for negative salt bridge, hydrophobic negative, hydrophobic positive, and positive salt bridge characteristics respectively. As above, e is the charge of the proton. The figure is oriented to emphasize the charge asymmetry that appears in all QSAR models that we have observed in previous analyses.20, 21, 24, 27–29
DISCUSSION
In this article, we have demonstrated that potent, selective inhibitors of hiCE based upon the benzene sulfonamide scaffold can be developed. This has resulted in the development of a series of fluorene analogues that have Ki values in the low nM range for both the inhibition of hydrolysis of 1 and 3. These compounds (56–60) were designed and synthesized based upon prior 3D-QSAR pseudoreceptor site models that indicated that a bulky, hydrophobic central domain within the inhibitors improved their potency. The benzene sulfonamide analogues that we assayed fell into 4 broad classes. Compounds 4–13 were originally identified in a small scale library screen21 and they essentially contained 3 domains. This included terminal and central phenyl rings bonded via sulfonamide chemotypes, and substitutions within the rings that altered the chemical properties of the compounds. Since compound 13 demonstrated the lowest Ki for hiCE inhibition, this molecule was used as a scaffold for the design and synthesis of analogues 14–55. Due to the fact that the most potent compounds tended to be halogen substituted,21 we concentrated our efforts on the generation of inhibitors containing these atoms, principally in the terminal benzene rings (molecules 14–41). Amongst this latter series of compounds, we noted that substitution in either the 3- (meta-) or 4-(para-) position with chlorine or bromine (compounds 17–19; Ki values ranging from 6–85nM) resulted in significant lower inhibition constants as compared to the unsubstituted molecule (8; Ki = 1,060nM). Indeed 17–19 were the most potent of the molecules containing substitutions within the terminal benzene rings. These halogens increase the hydrophobicity of the molecules (clog P for 17–19 range from 4.3–4.7, ∼1 log greater than compound 8) and therefore these analogues would be more likely to localize within the hydrophobic active site gorge of the protein.
In contrast, substitution with multiple larger atoms (e.g. compound 28) resulted in loss of biological activity, and is likely due to the fact that this molecule is too large to fit within the active site of hiCE. Furthermore, molecules containing a carboxylic acid or amide group at the 4-position (39 and 41) were inactive. Whether the loss of enzyme inhibition this is due to electronic effects on the other atoms within the sulfonamide, or a steric interaction that forces the inhibitor into a conformation such that it can no longer interact with amino acids that line the active site, or a combination of both, is unclear. However, it is apparent that introducing substitutions that increase the clogP without dramatically increasing the size of the molecules can improve the potency of these analogues.
Compounds 42–54 contained substitutions within the central benzene ring coupled with halogens appended in the terminal phenyl groups. In general, these modifications resulted in reduced potency towards hiCE inhibition. This is exemplified by compounds 44–46, that are inactive in this assay. However, 54 that contains the 2,3,5,6-tetrafluoro substitution, resulted in increased activity as compared to the unsubstituted molecule (19). Since these compounds demonstrate very similar clogP values, the increase in biological activity is likely due to a change in the electron distribution afforded by the very electronegative fluorine atoms. As we have not yet obtained the X-ray structure of hiCE, it has not been possible to identify the specific amino acids with which these sulfonamides interact. Hence, it is unclear how these small molecules demonstrate specificity for hiCE. However, based upon previous observations that sulfonamides can inhibit thrombin, we presume that a similar mechanism of enzyme inhibition occurs.31–34 These studies demonstrated that the oxygen atoms within the sulfonamide group can hydrogen bond with amino acids present within the active site of this protein. Therefore, we believe that the unique arrangement of residues present within the hiCE catalytic gorge, and their ability to form hydrogen bonds with the sulfonamides, represents the key interactions responsible for selective CE inhibition.
Consistent with our previous reports describing CE inhibitors,20, 24, 27 molecules that contained substitutions that increased the bulkiness or width of the compound were generally poor inhibitors. This was exemplified in this series of analogues by 28, 39, 41, and 44–46. All of these sulfonamides contained groups or atoms that significantly increased either the width or length of the molecule that would preclude facile access of the inhibitor to the CE active site. Since the hydrolysis of compounds by CEs is dramatically influenced by their ability to interact with the catalytic amino acids,14, 30 it is highly likely that the same holds true for inhibitor molecules. Therefore, compounds that exceed the dimensions of the active site gorge in these proteins would not be inhibitors of these proteins. Molecules 28, 39, 41, and 44–46 are much larger and bulkier than the other compounds described here, and therefore do not inhibit hiCE.
Based upon the results of the QSAR analyses, we hypothesized that introducing a larger, planar, aromatic core domain within the center of the molecule should increase its potency. This was due to the fact that this moiety would increase the clogP (hydrophobicity) of the compound, without impeding the ability of the sulfonyl groups to interact with the amino acids present within the active site. This is consistent with the 3D-QSAR model depicted in Figure 4. This dataset was generated using both compounds 56 and 60 in the training set. Excluding these compounds resulted in QSAR relationships that predicted 56–60 to be good inhibitors, but the experimental data revealed them to be excellent inhibitors: much better than predicted and some of the most potent compounds we have examined to date. One difficulty with any QSAR analysis is that the fewer members of a class of molecules in the training set, the less likelihood of correct prediction of that series of compounds. Hence, we rebuilt the models, including 56 and 60, to determine whether this would result in improved prediction of the Ki values for 57–59. As expected, the predictive properties of the data were improved by at least 1 order of magnitude by including 56 and 60, suggesting that as we synthesize and test more fluorene-containing molecules, we should be able to produce a more accurate QSAR relationship for other classes of molecules beyond those used in this study. In general, halogen substitution of the distal phenyl rings slightly decreased the Ki value as compared to the unsubstituted analog 11, whereas methyl groups added to the central phenyl ring did not much affect the binding of the analogs. This was somewhat surprising as the 3D-QSAR pseudoreceptor model (Figure 5) is highly hydrophobic in the central ring area, as indicated by gray lines. However, replacement of the central phenyl ring with a naphthyl or fluorene chemotype greatly reduced the inhibition constants of the compounds, i.e. making them more potent hiCE inhibitors. We attribute this to π-π interactions of the small molecules with the tryptophan and phenylalanine rings that are known to line the active site gorge of hiCE.25 Similarly, this would explain the potent inhibitory power of the indole-containing isatins that we have previously characterized.24
In summary, we have synthesized and analyzed the inhibitory properties of a panel of bisbenzene sulfonamides, and demonstrated that these compounds have specificity for hiCE. This has allowed the design of a series of fluorene analogues that are highly potent inhibitors, with Ki values in the low nM range when using either 1 or 3 as substrates. We are currently determining the ability of these compounds to inhibit hiCE intracellularly, and evaluating whether any of these molecules represent valid lead compounds for in vivo use. If so, they may allow the design of clinical candidates, suitable for the modulation of 1-induced toxicity, in patients treated with this drug.
EXPERIMENTAL SECTION
Reagents and chemicals
General reagents and chemicals were obtained from Sigma Aldrich (St. Louis, MO). Sulfonamides were obtained from the following commercial sources: 4–12 from Telik (Palo Alto, CA); 25 and 32 from Akos (Steinen, Germany); 28, 36, 42, 44 and 45 from Princeton (Monmouth Junction, NJ); 29, 30, 37–41 and 55 from Enamine (Monmouth Junction, NJ); 31 and 33–35 from Scientific Exchange (Center Ossipee, NH); and 43 from AsinEx (Winston-Salem, NC). The synthesis of compound 13 has been previously described.21 Compounds were dissolved in DMSO (typically at a concentration of 10mM) immediately prior to biochemical analysis.
Sulfonamide synthesis
Sulfonamides 14–24, 26, 27, 46–54, and 56–60 were synthesized by condensation of the respective diamines and sulfonyl chlorides. Briefly, diamine (5mmol) was dissolved in CH2Cl2 (∼30ml) with stirring and 2ml of pyridine and 0.2g of dimethylaminopyridine were added. The sulfonyl chloride (12mmol), dissolved in 15ml CH2Cl2, was added in one portion and the reaction was allowed to stir for 2–16 hours. During this time, the reaction became dark pink/orange/red and a fine precipitate appeared. The N,N’-bis-arene sulfonamide was recovered by filtration and washed with 100mls of CH2Cl2 and 100ml of ethyl acetate. After drying under vacuum, the material was resuspended in 250mls of 0.1M HCl and stirred at room temperature for 2–3 hours. The mixture was filtered, washed with water and dried to yield the pure product. All compounds were validated by TLC, 1H and 13C NMR, HRMS, melting point and elemental analysis, and were greater than 95% pure. Full details are provided as Supporting Information.
Human enzymes
hCE1 was purified as previously described4, 22, 35 and hiCE was generated in a similar manner, except that a slightly different purification procedure was adopted (Hatfield et al, manuscript in preparation). hAChE and hBChE were purchased from Sigma Aldrich.
Carboxylesterase inhibition assays and determination of Ki values
Parameters for CE inhibition were determined as previously described.20, 21, 24, 28, 29, 36 Briefly, the inhibition of hydrolysis of 3 to o-nitrophenol was measured spectrophotometrically in the presence of increasing concentrations of inhibitor. The fractional inhibition was then graphed versus inhibitor concentration and data fitted to the following equation;23
where [I] = inhibitor concentration, [S] = substrate concentration, i = fractional inhibition, Ks is the dissociation constant for the enzyme substrate complex (assuming negligible commitment to catalysis), α = change in affinity of the substrate for the enzyme in the presence of the inhibitor (where α>0), β = change in the rate of enzyme substrate complex decomposition in the presence of the inhibitor (where 1>β>0), and Ki is the inhibitor constant. Routinely, results were evaluated using Perl Data Language and graphed with GraphPad Prism software with the mode of enzyme inhibition being determined using Akaike’s information criteria.20 Ki values were then calculated using the best fit model described from these analyses.
Inhibition of acetyl- and butyrylcholinesterase
Inhibition of hAChE and hBChE were determined as previously reported,20, 21, 24, 28, 29 using either acetylthiocholine or butyrylthiocholine as substrates, respectively.
Inhibition of hiCE-mediated 1 hydrolysis
The inhibition of the conversion of 1 to 2 by hiCE was determined using the following conditions. Inhibitor concentrations, ranging from 1nM to 100µM, were added to reactions (100µl) containing 25U of hiCE (1U represents the amount of enzyme that catalyzes the conversion of 1nmole of o-nitrophenyl acetate per min), 20µM CPT-11 and 50mM Hepes pH7.4. Reactions were incubated at 37°C for 5 min and terminated by the addition of 100µl of cold acidified methanol. Samples were clarified by centrifugation at 14000g for 10 min and concentrations of SN-38 in the supernatant were then determined by reverse phase HPLC, as previously described.21, 37 Ki values were calculated in an identical fashion to that indicated above.
Calculation of clogP values
clogP values were predicted using ChemSilico Predict software (ChemSilico, Tewksbury, MA).
Data graphing and statistical analyses
Results were graphed and analyzed using GraphPad Prism software. Statistical correlations were determined using the Spearman method (non parametric) to generate two-tailed P values. Spearman correlation coefficients (r) equal to −1 or +1 represent perfect negative or positive correlations, respectively. P values <0.05 were considered statistically significant.
3D-QSAR modeling of inhibitors
Multidimensional-QSAR modeling of carboxylesterase inhibitors was performed using Quasar 5 software38–41 running on a Macintosh G5. The structure for each compound was built using Chem 3D and each structure was minimized with MM2 and PM3 formalisms. The solvation energies and charges were calculated using the AMSOL 7.1 program42 using the SM5.42R solvation method. The gas phase charges obtained were used for all QSAR analyses. A resulting receptor surface was then generated using Quasar 5.0 and analyzed to yield over 200 independent models and subsequently refined to generate ∼7000 pseudoreceptor site models. Repeated analyses of the data sets were then performed until the cross correlation coefficients (q2) were greater than 0.7 for the experimentally determined versus the predicted Ki values. Routinely, this yielded correlation coefficients (r2) of >0.8.
Two independent QSAR models were constructed using two different training sets. The first was constructed using 37 sulfonamides as inhibitors of hiCE using 3 as a substrate. The second was obtained using the Ki values of 12 inhibitors of 1 hydrolysis. In all cases, a range of Ki values spanning 3 orders of magnitude were used to assure a reasonable model. We then included the structures of a number of potential inhibitors of hiCE in the test set, to identify novel chemical scaffolds that might be potential selective inhibitors of hiCE. These included the fluorene-containing compounds 56–60.
Supplementary Material
Physical and spectral parameters for the compounds synthesized as part of these studies are available as Supporting Information.
Acknowledgements
We thank Dr. J.P. McGovren for the gift of CPT-11. This work was supported in part by NIH grants CA108775, a Cancer Center core grant CA21765,a NSF EPSCoR grant EPS-0556308 and by the American Lebanese Syrian Associated Charities.
Abbreviations used
- AChE
acetylcholinesterase
- BChE
butyrylcholinesterase
- CE
carboxylesterase
- hAChE
human AChE
- hBChE
human BChE
- hCE1
CES1, human carboxylesterase 1
- hiCE
CES2, human intestinal carboxylesterase
- HPLC
high performance liquid chromatography
- Ki
inhibition constant
- TRAP
Target Related Affinity Profiling
REFERENCES
- 1.Cashman J, Perroti B, Berkman C, Lin J. Pharmacokinetics and molecular detoxification. Environ. Health Perspect. 1996;104:23–40. doi: 10.1289/ehp.96104s123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunimoto T, Nitta K, Tanaka T, Uehara N, Baba H, Takeuchi M, Yokokura T, Sawada S, Miyasaka T, Mutai M. Antitumor activity of 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxy-camptothecin, a novel water-soluble derivative of camptothecin, against murine tumors. Cancer Res. 1987;47:5944–5947. [PubMed] [Google Scholar]
- 3.Pindel EV, Kedishvili NY, Abraham TL, Brzezinski MR, Zhang J, Dean RA, Bosron WF. Purification and cloning of a broad substrate specificity human liver carboxylesterase that catalyzes the hydrolysis of cocaine and heroin. J. Biol. Chem. 1997;272:14769–14775. doi: 10.1074/jbc.272.23.14769. [DOI] [PubMed] [Google Scholar]
- 4.Bencharit S, Morton CL, Xue Y, Potter PM, Redinbo MR. Structural basis of heroin and cocaine metabolism by a promiscuous human drug-processing enzyme. Nat. Struct. Biol. 2003;10:349–356. doi: 10.1038/nsb919. [DOI] [PubMed] [Google Scholar]
- 5.Kamendulis LM, Brzezinski MR, Pindel EV, Bosron WF, Dean RA. Metabolism of cocaine and heroin is catalyzed by the same human liver carboxylesterases. J. Pharmacol. Expt. Therap. 1996;279:713–717. [PubMed] [Google Scholar]
- 6.Brzezinski MR, Spink BJ, Dean RA, Berkman CE, Cashman JR, Bosron WF. Human liver carboxylesterase hCE-1: binding specificity for cocaine, heroin, and their metabolites and analogs. Drug Metab. Dispos. 1997;25:1089–1096. [PubMed] [Google Scholar]
- 7.Shi D, Yang J, Yang D, LeCluyse EL, Black C, You L, Akhlaghi F, Yan B. Anti-influenza prodrug oseltamivir is activated by carboxylesterase human carboxylesterase 1, and the activation is inhibited by antiplatelet agent clopidogrel. J. Pharmacol. Exp. Ther. 2006;319:1477–1484. doi: 10.1124/jpet.106.111807. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Burnell JC, Dumaual N, Bosron WF. Binding and hydrolysis of meperidine by human liver carboxylesterase hCE-1. J. Pharmacol. Exp. Ther. 1999;290:314–318. [PubMed] [Google Scholar]
- 9.Alexson SE, Diczfalusy M, Halldin M, Swedmark S. Involvement of liver carboxylesterases in the in vitro metabolism of lidocaine. Drug Metab. Dispos. 2002;30:643–647. doi: 10.1124/dmd.30.6.643. [DOI] [PubMed] [Google Scholar]
- 10.Tsuji T, Kaneda N, Kado K, Yokokura T, Yoshimoto T, Tsuru D. CPT-11 converting enzyme from rat serum: purification and some properties. J. Pharmacobiodyn. 1991;14:341–349. doi: 10.1248/bpb1978.14.341. [DOI] [PubMed] [Google Scholar]
- 11.Potter PM, Pawlik CA, Morton CL, Naeve CW, Danks MK. Isolation and partial characterization of a cDNA encoding a rabbit liver carboxylesterase that activates the prodrug Irinotecan (CPT-11) Cancer Res. 1998;52:2646–2651. [PubMed] [Google Scholar]
- 12.Humerickhouse R, Lohrbach K, Li L, Bosron W, Dolan M. Characterization of CPT-11 hydrolysis by human liver carboxylesterase isoforms hCE-1 and hCE-2. Cancer Res. 2000;60:1189–1192. [PubMed] [Google Scholar]
- 13.Khanna R, Morton CL, Danks MK, Potter PM. Proficient metabolism of CPT-11 by a human intestinal carboxylesterase. Cancer Res. 2000;60:4725–4728. [PubMed] [Google Scholar]
- 14.Wierdl M, Tsurkan L, Hyatt JL, Hatfield MJ, Edwards CC, Danks MK, Redinbo MR, Potter PM. An improved human carboxylesterase for use in enzyme/prodrug therapy with CPT-11. Cancer Gene Therap. 2008;15:183–192. doi: 10.1038/sj.cgt.7701112. [DOI] [PubMed] [Google Scholar]
- 15.Tabata T, Katoh M, Tokudome S, Nakajima M, Yokoi T. Identification of the cytosolic carboxylesterase catalyzing the 5'-deoxy-5-fluorocytidine formation from capecitabine in human liver. Drug Metab. Dispos. 2004;32:1103–1110. doi: 10.1124/dmd.104.000554. [DOI] [PubMed] [Google Scholar]
- 16.Quinney SK, Sanghani SP, Davis WI, Hurley TD, Sun Z, Murry DJ, Bosron WF. Hydrolysis of capecitabine to 5'-deoxy-5-fluorocytidine by human carboxylesterases and inhibition by loperamide. J Pharmacol Exp Ther. 2005;313:1011–1016. doi: 10.1124/jpet.104.081265. [DOI] [PubMed] [Google Scholar]
- 17.Stampfli HF, Quon CY. Polymorphic metabolism of flestolol and other ester containing compounds by a carboxylesterase in New Zealand white rabbit blood and cornea. Res. Commun. Mol. Pathol. Pharmacol. 1995;88:87–97. [PubMed] [Google Scholar]
- 18.Morton CL, Iacono L, Hyatt JL, Taylor KR, Cheshire PJ, Houghton PJ, Danks MK, Stewart CF, Potter PM. Metabolism and antitumor activity of CPT-11 in plasma esterase-deficient mice. Cancer Chemother. Pharmacol. 2005;56:629–636. doi: 10.1007/s00280-005-1027-y. [DOI] [PubMed] [Google Scholar]
- 19.Beroza P, Damodaran K, Lum RT. Target-related affinity profiling: Telik's lead discovery technology. Curr. Top. Med. Chem. 2005;5:371–381. doi: 10.2174/1568026053828394. [DOI] [PubMed] [Google Scholar]
- 20.Wadkins RM, Hyatt JL, Wei X, Yoon KJ, Wierdl M, Edwards CC, Morton CL, Obenauer JC, Damodaran K, Beroza P, Danks MK, Potter PM. Identification and characterization of novel benzil (diphenylethane-1,2-dione) analogues as inhibitors of mammalian carboxylesterases. J. Med. Chem. 2005;48:2905–2915. doi: 10.1021/jm049011j. [DOI] [PubMed] [Google Scholar]
- 21.Wadkins RM, Hyatt JL, Yoon KJ, Morton CL, Lee RE, Damodaran K, Beroza P, Danks MK, Potter PM. Identification of novel selective human intestinal carboxylesterase inhibitors for the amelioration of irinotecan-induced diarrhea: Synthesis, quantitative structure-activity relationship analysis, and biological activity. Mol. Pharmacol. 2004;65:1336–1343. doi: 10.1124/mol.65.6.1336. [DOI] [PubMed] [Google Scholar]
- 22.Bencharit S, Morton CL, Hyatt JL, Kuhn P, Danks MK, Potter PM, Redinbo MR. Crystal structure of human carboxylesterase 1 complexed with the Alzheimer's drug tacrine. From binding promiscuity to selective inhibition. Chem. & Biol. 2003;10:341–349. doi: 10.1016/s1074-5521(03)00071-1. [DOI] [PubMed] [Google Scholar]
- 23.Webb JL. Enzyme and Metabolic Inhibitors. Volume 1. General Principles of Inhibition. New York: Academic Press Inc; 1963. [Google Scholar]
- 24.Hyatt JL, Moak T, Hatfield JM, Tsurkan L, Edwards CC, Wierdl M, Danks MK, Wadkins RM, Potter PM. Selective inhibition of carboxylesterases by isatins, indole-2,3-diones. J. Med. Chem. 2007;50:1876–1885. doi: 10.1021/jm061471k. [DOI] [PubMed] [Google Scholar]
- 25.Potter PM, Wadkins RM. Carboxylesterases-detoxifying enzymes and targets for drug therapy. Curr. Med. Chem. 2006;13:1045–1054. doi: 10.2174/092986706776360969. [DOI] [PubMed] [Google Scholar]
- 26.Lundstedt T, Seifert E, Abramo L, Thelin B, Nystrom A, Pettersen J, Bergman B. Experimental design and optimization. Chemom. Intell. Lab. Syst. 1998;42:3–40. [Google Scholar]
- 27.Hicks LD, Hyatt JL, Moak T, Edwards CC, Tsurkan L, Wierdl M, Ferreira AM, Wadkins RM, Potter PM. Analysis of the inhibition of mammalian carboxylesterases by novel fluorobenzoins and fluorobenzils. Bioorg Med Chem. 2007;15:3801–3817. doi: 10.1016/j.bmc.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hyatt JL, Stacy V, Wadkins RM, Yoon KJ, Wierdl M, Edwards CC, Zeller M, Hunter AD, Danks MK, Crundwell G, Potter PM. Inhibition of carboxylesterases by benzil (diphenylethane-1,2-dione) and heterocyclic analogues is dependent upon the aromaticity of the ring and the flexibility of the dione moiety. J. Med. Chem. 2005;48:5543–5550. doi: 10.1021/jm0504196. [DOI] [PubMed] [Google Scholar]
- 29.Wadkins RM, Hyatt JL, Edwards CC, Tsurkan L, Redinbo MR, Wheelock CE, Jones PD, Hammock BD, Potter PM. Analysis of mammalian carboxylesterase inhibition by trifluoromethylketone-containing compounds. Mol. Pharmacol. 2007;71:713–723. doi: 10.1124/mol.105.021683. [DOI] [PubMed] [Google Scholar]
- 30.Wadkins RM, Morton CL, Weeks JK, Oliver L, Wierdl M, Danks MK, Potter PM. Structural constraints affect the metabolism of 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (CPT-11) by carboxylesterases. Mol. Pharmacol. 2001;60:355–362. doi: 10.1124/mol.60.2.355. [DOI] [PubMed] [Google Scholar]
- 31.Brandstetter H, Turk D, Hoeffken HW, Grosse D, Sturzebecher J, Martin PD, Edwards BF, Bode W. Refined 2.3 A X-ray crystal structure of bovine thrombin complexes formed with the benzamidine and arginine-based thrombin inhibitors NAPAP, 4-TAPAP and MQPA. A starting point for improving antithrombotics. J. Mol. Biol. 1992;226:1085–1099. doi: 10.1016/0022-2836(92)91054-s. [DOI] [PubMed] [Google Scholar]
- 32.Sturzebecher J, Hauptmann J, Steinmetzer T, Thrombin . In: Proteinase and peptidase inhibition: recent potential targets for drug development. Smith HJ, Simmons C, editors. London: Taylor & Francis; 2002. pp. 185–201. [Google Scholar]
- 33.Okamoto S, Kinjo K, Hijikata A, Kikumoto R, Tamao Y, Ohkubo K, Tonomura S. Thrombin inhibitors. 1. Ester derivatives of N alpha-(arylsulfonyl)-L-arginine. J. Med. Chem. 1980;23:827–830. doi: 10.1021/jm00182a003. [DOI] [PubMed] [Google Scholar]
- 34.Sturzebecher J, Markwardt F, Voigt B, Wagner G, Walsmann P. Cyclic amides of N alpha-arylsulfonylaminoacylated 4-amidinophenylalanine--tight binding inhibitors of thrombin. Thromb. Res. 1983;29:635–642. doi: 10.1016/0049-3848(83)90218-9. [DOI] [PubMed] [Google Scholar]
- 35.Morton CL, Potter PM. Comparison of Escherichia coli, Saccharomyces cerevisiae, Pichia pastoris, Spodoptera frugiperda and COS7 cells for recombinant gene expression: Application to a rabbit liver carboxylesterase. Mol. Biotechnol. 2000;16:193–202. doi: 10.1385/MB:16:3:193. [DOI] [PubMed] [Google Scholar]
- 36.Hyatt JL, Wadkins RM, Tsurkan L, Hicks LD, Hatfield MJ, Edwards CC, Ii CR, Cantalupo SA, Crundwell G, Danks MK, Guy RK, Potter PM. Planarity and constraint of the carbonyl groups in 1,2-diones are determinants for selective inhibition of human carboxylesterase 1. J. Med. Chem. 2007;50:5727–5734. doi: 10.1021/jm0706867. [DOI] [PubMed] [Google Scholar]
- 37.Guichard S, Morton CL, Krull EJ, Stewart CF, Danks MK, Potter PM. Conversion of the CPT-11 metabolite APC to SN-38 by rabbit liver carboxylesterase. Clin. Cancer Res. 1998;4:3089–3094. [PubMed] [Google Scholar]
- 38.Stewart JJ. MOPAC: a semiempirical molecular orbital program. J. Comput. Aided Mol. Des. 1990;4:1–105. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- 39.Vedani A, Dobler M. Multidimensional QSAR: Moving from three-to five-dimensional concepts. Quant. Struct.-Act. Relat. 2002;21:382–390. [Google Scholar]
- 40.Vedani A, Dobler M. 5D-QSAR: the key for simulating induced fit? J. Med. Chem. 2002;45:2139–2149. doi: 10.1021/jm011005p. [DOI] [PubMed] [Google Scholar]
- 41.Vedani A, Dobler M, Lill MA. Combining protein modeling and 6D-QSAR simulating the binding of structurally diverse ligands to the estrogen receptor. J. Med. Chem. 2005;48:3700–3703. doi: 10.1021/jm050185q. [DOI] [PubMed] [Google Scholar]
- 42.Hawkins GD, Giesen D, Lynch G, Chambers C, Rossi I, Storer J, Li J, Zhu T, Thompson J, Winget P, Lynch B, Rinaldi D, Liotard D, Cramer CJ, Truhlar DG, Amsol V. 7.1. Regents of the University of Minnesota. 2004 [Google Scholar]
- 43.Merritt EA, Bacon DJ. Raster 3D: Photorealistic molecular graphics. Meth. Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 44.Kraulis PJ. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 1991;24:946–950. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Physical and spectral parameters for the compounds synthesized as part of these studies are available as Supporting Information.