

Abstract
At least 119 mutations in the gene encoding copper/zinc superoxide dismutase (SOD1) cause amyotrophic lateral sclerosis by an unidentified toxic gain of function. We compared the dynamic properties of 13 as-isolated, partially metallated, SOD1 variant enzymes using hydrogen-deuterium exchange. We identified a shared property of these familial amyotrophic lateral sclerosis-related SOD1 variants, namely structural and dynamic change affecting the electrostatic loop (loop VII) of SOD1. Furthermore, SOD1 variants that have severely compromised metal binding affinities demonstrated additional structural and dynamic changes to the zinc-binding loop (loop IV) of SOD1. Although the biological consequences of increased loop VII mobility are not fully understood, this common property is consistent with the hypotheses that SOD1 mutations exert toxicity via aggregation or aberrant association with other cellular constituents.
Introduction
At least 119 mutations in the gene encoding copper/zinc superoxide dismutase cause fALS3 by introducing an unknown toxic gain of function. Numerous hypotheses have been proposed to explain mechanisms of SOD1 variant toxicity (reviewed in Refs. 2 and 3). Proposed hypotheses to define the structural or functional alterations shared by SOD1 variants include: 1) decreased stability of apo (metal-deficient) or metallated SOD1 (4), 2) increased hydrophobicity (5), 3) increased self-aggregation propensity (6), 4) increased susceptibility to post-translational modification, 5) decreased metal affinity (7), 6) destabilization of the SOD1 native dimer, and 7) aberrant copper-mediated chemistry (8). The literature, as a whole, supports the notion that the aforementioned hypotheses are in fact related, because SOD1 metal content, native disulfide bond status, hydrophobicity, and stability are interrelated (2) and may thereby affect the specificity of copper-mediated chemistry (9) and aggregation propensity.
Proposed hypotheses to explain the biological consequences of SOD1 toxicity include: 1) impairment of axonal transport (10), 2) excitotoxicity (11), 3) impairment of proteasome (12) or chaperone activity (13), and 4) mitochondrial (14, 15) or endoplasmic reticulum-Golgi dysfunction (15). The relationship of mutation-associated structural changes of SOD1 with these downstream effects is not well established. SOD1 aggregation has been implicated as a contributor to mitochondrial (15, 16), endoplasmic reticulum-Golgi (15, 17, 18), proteasome (19), and chaperone (13) dysfunction and thus bridges the conceptual gap between mutation-related changes to the physicochemical properties of SOD1 and the varied cellular consequences of SOD1 mutations. Indeed, the only property shared by all fALS SOD1 variants thus far studied is an increased propensity to form proteinaceous aggregates of SOD1, as evidenced in fALS patients (20, 21), 21 rodent lines of 13 different SOD1 mutations (22), and at least 13 fALS mutations in cell culture models. Moreover, proteinaceous SOD1-containing inclusions were also found in a subset of sporadic amyotrophic lateral sclerosis patients (20). The sequence of events leading to in vivo aggregation or other abnormal associations involving SOD1 variants, however, is not understood.
Amide hydrogen-deuterium (H/D) exchange reactions coupled with proteolysis and liquid chromatography-mass spectrometry (MS) is an increasingly popular method to study protein dynamics. Upon mixing with deuterated buffer, an amide hydrogen of a protein exchanges with a deuterium in the bulk solvent at a rate depending on the dynamic properties of the site (23). The location of deuteriums can be determined by proteolytic digestion followed by mass analysis. Although x-ray crystallography can give a high resolution structure of a protein, it is usually a snapshot of the energy minimum structure. The H/D exchange results can describe the dynamic properties of a protein, although the resolution is limited by the size of peptic fragments (typically 5–20 residues long). The dynamic information of a protein determined by H/D exchange can complement x-ray crystallographic and NMR data to provide a fuller description of the properties of a protein.
The objective of this study was to use H/D exchange mass spectrometry to correlate the effects of 13 fALS-causing variants upon SOD1 structure and dynamics. We have chosen variants ranging from fully active to completely inactive including SOD1 A4V, L38V, G41S, H46R, G72Y, D76S, G85R, D90A, G93A, D124V, D125H, E133del, and S134N. Herein we report a structural/dynamic change to the SOD1 electrostatic loop (loop VII) that is common to every fALS variant of the 13 we studied. Moreover, we suggest that this common structural change may contribute to abnormal intermolecular interactions involving SOD1.
EXPERIMENTAL PROCEDURES
Materials
Because fALS can be caused by changes as simple as the addition of a methyl group (G93A), we chose to study SOD1 from a eukaryotic preparation, assuring N-terminal acetylation. Wild type (WT) SOD1 purified from human erythrocytes was purchased from Sigma and was fully metallated as indicated by native gel electrophoresis and MS of the intact protein. Metal content of the Sigma protein was analyzed by measuring the intact protein mass by performing electrospray ionization-MS out of pure, high pressure liquid chromatography (HPLC) grade water as described previously (24), affording the ratio of metallated versus apo SOD1. Differences in ionization efficiency of the apo versus metallated SOD1, as well as lower limits for the detection of apo SOD1 were accounted for by converting the metallated SOD1 to apo SOD1 using 0.1% formic acid. By this method, greater than 98% of Sigma SOD1 was judged to contain one copper and one zinc/monomer.
All proteins other than Sigma SOD1 were purified as previously described (25). Protein concentration was determined spectroscopically as well as using the modified Bradford assay (26) (Coomassie PlusTM protein assay reagent; Pierce). Metal content was determined using inductively coupled plasma-MS as previously described (25, 27). Using a Fourier transform ion cyclotron resonance mass spectrometer (apex qe-94; Bruker Daltonics Inc., Billerica, MA), all of the proteins were checked for post-translational modifications via MS of the intact protein as described in Ref. 24. WT SOD1 (Sigma) was judged to consist of less than 12% non-native post-translationally modified SOD1, and the most modified fALS-SOD1 variant consisted of less than 30% non-native post-translationally modified proteins. The most prevalent post-translational modifications were +16, +22, and +32 Da, corresponding to SOD1 with one oxygen, one sodium (a common adduct of proteins analyzed by electrospray MS), and two oxygen atoms, respectively. The sites of modification were not determined, although previous studies showed SOD residues Trp32 (24) and Cys111 (28) to be modified by oxygen in vivo.
H/D Exchange Experiments of WT SOD1 and fALS-SOD1 Variants
More detailed general procedures of H/D exchange experiments are described elsewhere (29). To 3 μl of 1.0 mg/ml SOD1 (in 20 mm potassium phosphate buffer, 150 mm potassium chloride, pH 7.2) was added 17 μl of phosphate-buffered saline (in D2O, pD 7.2), to initiate the exchange reactions, so that H/D exchange took place at 10 μm SOD1 near the in vivo concentration (30, 31). The samples were incubated at 4 °C for a predetermined time of 15, 50, 150, and 500 s. The reaction mixture was quenched by the addition of 30 μl of cold 2 m guanidine hydrochloride, 1 m tris(2-carboxyethyl)phosphine, pH 1.4. The quenched solution was digested by passing the sample over an immobilized pepsin column at a flow rate of 200 μl/min. The digested peptic fragments were temporarily collected on a reverse phase column (4-μl bed volume), separated by C18 HPLC (Magic C18; Michrom BioResources, Inc., Auburn, CA) with a linear gradient of 13–30% buffer B (buffer A, water with 0.05% trifluoroacetic acid, buffer B, 95% acetonitrile, and 5% buffer A) in 23 min and detected by a Finnegan LCQ mass spectrometer (Thermo Fisher Scientific, San Jose, CA).
Optimization of Digestion/Separation Conditions for SOD1
Prior to the H/D exchange experiments, the acidic quenching buffer (the types and concentrations of both denaturant and acid, as well as pH), the flow rate over the pepsin column, and HPLC gradient were optimized to give the best sequence coverage and resolution. Because SOD1 is among the most stable mesotrophic proteins (SOD1 melts at 95 °C and is stable and active in 8 m guanidine HCl or 2% SDS), nonstandard conditions (more acidic quenching and higher capillary temperatures) are required to achieve 100% sequence coverage. These conditions increased back-exchange from our average deuterium recovery of ∼70% to around 57% but are within the range of literature reported values, which vary between 30 and 90%.
During the digestion/separation optimization, to quickly identify pepsin-generated peptides for each digestion/separation condition employed, spectral data were acquired in data-dependent MS/MS mode with dynamic exclusion. The SEQUEST (32) software program (Thermo Fisher Scientific) was used to identify the sequence of the dynamically selected parent peptide ions. This tentative peptide identification was verified by visual confirmation of the parent ion charge state presumed by the SEQUEST program for each peptide. This set of peptides was then further examined to determine whether the quality of the measured isotopic envelope of peptides was sufficient to allow accurate measurement of the geometric centroid of isotopic envelopes on deuterated samples. We observed qualitatively different digestion patterns for variants L38V and S134N, because residues 23–37 were not observed in L38V digests, and residues 119–132 were not observed in S134N digests. The change in L38V and S134N digestion is attributed to the substitution of an amino acid at pepsin cleavage site residues 37 and 38 and near pepsin cleavage site residues 132–133, respectively. In both cases, longer peptides (residues 23–53 for L38V and residues 119–144 for S134N) were observed, allowing coverage of these regions.
Calculation of Deuteration Level
To determine the deuteration level of each peptic fragment, control samples of nondeuterated (run without deuterated buffers) and fully deuterated (incubated in 85 mm tris(2-carboxyethyl)phosphine in 85% D2O for 5 h at 60 °C) SOD1 were also analyzed. The centroids of probe peptide isotopic envelopes were measured using an in-house-developed program in collaboration with Sierra Analytics (Mateo, CA). The corrections for back-exchange were made employing methods described previously (33),
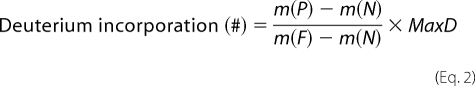 |
where m(P), m(N), and m(F) are the centroid values of partially deuterated (on-exchange) peptide, nondeuterated peptide, and fully deuterated peptide, respectively. MaxD is the maximum deuterium incorporation calculated by subtracting the number of prolines in the third or later amino acid and two from the number of amino acids in the peptide of interest (assuming the first two amino acids cannot retain deuteriums (34)). For SOD1, the deuterium recovery of a fully deuterated sample ((m(F) − m(N))/MaxD) was on average 57%.
The first two amino acids of a peptide lose deuteriums; therefore no information can be gathered from these residues. Therefore, for example, peptide 7–20 can provide the H/D exchange behaviors of segments 9–20. This causes the gaps seen in Table 1 as well as in Figs. 1 and 3. The exceptions are peptides 1–6 and 1–8. These two peptides provide the information on segments 1–6 and 1–8, respectively, because there is no loss of deuteriums in these peptides because of N-terminal acetylation of the protein.
TABLE 1.
Average deuteration level in each segment of WT SOD1 and average increase in deuteration level in each segment of SOD1 variants at 4 °C and pH 7.2
The deuteration levels at four time points, 15, 50, 150, and 500 s, were averaged for each segment. Three different measurements were performed on three different days for WT SOD1. The two N-terminal amino acids of peptides were not analyzed because of the loss of deuteriums during the analysis. For example, peptide 7–20 gives the information on segment 9–20 (“Experimental Procedures”). Charge is the charge state of the corresponding peptide monitored. Charge 0 indicates that the deuteration level of segments is calculated by subtraction. For example, the number of deuteriums attached in segment 38–53 was obtained by the subtraction of the number of deuteriums in segment 23–37 from that in segment 23–53. Average deuteration levels between 10 and 30% versus WT SOD1 are highlighted in yellow, those >30% are highlighted in red.

FIGURE 1.

Pepsin digest coverage map of SOD1. Top panel, bold lines indicate the peptide fragment ions identified. Note the redundant coverage (two or more peptide ions/amino acid) of the entire SOD1 primary sequence. Bottom panel, H/D exchange of WT copper/zinc SOD1 at 4 °C (in triplicate). From top, the results shown were measured in March, April, and January. Each block contains four time points 15, 50, 150, and 500 s. The deuteration level at each time point of each peptide is color-coded as a heat map (shown to the right).
FIGURE 3.

Changes to the structure/dynamics of the electrostatic loop are a shared property of fALS SOD1 mutants. Deuterium incorporations relative to copper/zinc-WT SOD1 (Fig. 2 illustrates absolute rates) at times (from top to bottom in the matrix) 15, 50, 150, and 500 s were compared for 13 fALS SOD1 variants, copper-deficient SOD1, and a duplicate copper/zinc SOD1 control. WT-like variants (upper eight rows) and metal-binding variants (lower five rows) are separated by a larger space. Note that each mutant was found to increase the dynamics of the electrostatic loop. This region of shared perturbation was colored orange and threaded onto the secondary structure (middle panel), where the thickness of orange is proportional to the number of mutants affected in a particular region, and onto human copper/zinc-SOD crystal structure (bottom panel; Protein Data Bank code 1SPD) (1) with mutated residues labeled. Note that the dimer interface is predominantly the N terminus, residues 49–54, 113–115, and 148–153 and is shown as black bars. In triplicate experiments, no difference greater than 5% was observed between WT SOD1 controls.
H/D Exchange Theoretical Considerations Experiments at 4 °C versus 37 °C
Our H/D exchange mass spectrometry experiments took place at 4 °C, whereas the human physiological temperature is 37 °C, and the temperatures for many of the spectroscopic methods performed by other groups (see discussion) vary from below 0 °C to 37 °C. Formalisms to relate the observed rates of amide H/D exchange to thermodynamic stabilization of proteins have been developed (23). Amide hydrogens of proteins in the native, folded state are proposed to exchange according to the following equation,
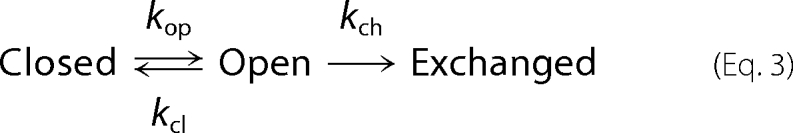 |
where kop is the rate of “opening,” kcl is the rate of “closing,” and kch is the “intrinsic” or chemical exchange rate as discussed above. In this scheme, the observable exchange rate is given by Equation 4.
For most proteins at or below neutral pH, amide H/D exchange occurs by EX2 mechanism (35), where kcl ≫ kch, and Equation 4 becomes Equation 5.
Effects of Temperature on Intrinsic Amide Hydrogen Exchange Rates
Assuming that the protein dynamics do not change upon increasing temperature (Kop, constant), kex will be proportional to kch,
where Ea = 17.4 kcal/mol for base catalyzed amide hydrogen exchange reactions (34, 36). This means
For example, measuring 15, 50, 150, and 500 s at 4 °C is the equivalent of measuring 0.53, 1.8, 5.3, and 18 s at 37 °C. In preliminary experiments we compared the H/D exchange mass spectrometry of SOD1 at 4 °C and at 25 °C and observed no indication of differences in stability at these two temperatures. The deuterium buildup curves at two temperatures overlaid well after using the formula above to normalize the intrinsic exchange.
RESULTS
Pepsin Digestion of SOD1
Prior to H/D exchange experiments, the digestion and separation conditions for SOD1 were optimized to generate a set of peptic fragments that cover the entire protein sequence using WT SOD1. The best results were obtained when two parts of diluted SOD1 were quenched with three parts of 2 m guanidine hydrochloride, 1 m tris(2-carboxyethyl)phosphine, pH 1.4, and digested with an immobilized pepsin column (104 μl, the flow rate over the pepsin column at 200 μl/min) at 1 °C. The separation of digested fragments by reverse phase HPLC and detection by MS generated 58 peptides covering the entire amino acid sequence of SOD1 (Fig. 1). Twenty of the identified peptides, still spanning the entire SOD1 sequence, were of high quality (high signal/noise) and were used for this H/D exchange analysis (Table 1).
As shown in Fig. 1 and Table 1, multiple peptides were often observed for the same region. There are benefits to having peptides from the same regions: 1) a pair of analogous peptides behaving similarly can increase the confidence level of H/D exchange behaviors of the region and 2) a pair of analogous peptides enables the sublocalization of deuteriums. For example, the subtraction of the number of deuteriums in segment 23–37 from that in segment 23–53 provide the number of deuteriums attached in segment 38–53.
H/D Exchange of WT copper/zinc SOD1
H/D exchange perturbations of WT SOD1 were determined at 4 °C at pH 7.2 (Figs. 1 and 2, supplemental Fig. S1, and Table 1). A concentration of 10 μm SOD1 was used throughout the H/D exchange experiments to mimic the in vivo concentration of SOD1 (30, 31). The average deuteration level at each segment was obtained using the deuteration levels at 15, 50, 150, and 500 s. The H/D exchange experiments of both fully metallated and partially metallated copper/zinc WT SOD1 were repeated on three different days, and the standard deviation was typically 2% or less with the exception of the segment containing residues 133–144, whose standard deviation was 5%, suggesting that variation in electrostatic loop structure and dynamics is an intrinsic property of SOD1.
FIGURE 2.

WT (left panel) versus fALS mutant G85R (right panel) superoxide dismutase showing percentage deuteration levels. Percentage deuteration levels following 15 s of exchange were threaded onto a human copper/zinc-SOD crystal structure, Protein Data Bank code 1SPD (1). The region comprising the dimeric interfaces is the vertical blue area in the center of the structure. Red indicates the regions which exchange fast (therefore dynamic), and blue indicates the regions which exchange slowly (therefore rigid). The data demonstrate that the region corresponding to the dimer interface (labeled in structure) affords the greatest protection from amide H/D exchange, whereas the electrostatic loop (labeled in structure) affords the least protection.
None of the segments showed complete deuteration even after 500 s of exchange time, consistent with WT SOD1 being a well folded protein with no disordered regions. The most deuterated segment was residues 133–144 at the electrostatic loop, the structural element that is most distal from the SOD1 dimer interface. Other highly deuterated segments were residues 56–96, which includes the zinc loop, and residues 99–103, which are residues in the β5 strand. In general, the deuteration levels near the dimer interface are relatively low, and those at the edges distal to the dimer interface are relatively high (Fig. 2).
H/D Exchange of SOD1 Variants
Previous studies (25) have characterized SOD1 variants in two classes, those with substantial metal binding and superoxide dismutase activities, termed WT-like variants, and those with minimal metal binding and superoxide dismutase activity, termed metal-binding site variants. The H/D exchange perturbations of 13 fALS-related variants including eight WT-like and five metal-binding variants, as well as native (fully metallated) copper/zinc SOD1 and copper-deficient WT SOD1, are illustrated in Fig. 3. Each protein was analyzed at four different time points (15, 50, 150, and 500 s), and the differences shown are relative to fully metallated WT SOD1 purified from human erythrocytes (Table 1, Fig. 3, and supplemental Fig. S2). Based upon the maximum standard deviations among the three independent WT SOD1 deuteration levels of 5% (see above), our cut-off for significant difference among variants was set at a conservative 10%. As a result of this conservative cut-off, we decreased sensitivity to eliminate false positive results.
All of the fALS SOD1 variants showed perturbations at residues 133–144 (electrostatic loop) in at least one time point, and with the exception of G93A (which was 9%) the average difference in deuteration levels for all time points was greater than 10% (Table 1). It is striking that the H/D exchange perturbations of the disease-associated variants are similar despite the fact that the mutation sites are distributed across the entire primary structure (Fig. 3 and Table 1). Disruption of the dimer interface elements was only observed for A4V and G85R.
WT-like Variants Perturbed Primarily the SOD1 Electrostatic Loop
The H/D exchange patterns of eight WT-like variants, A4V, L38V, G41S, G72S, D76Y, D90A, G93A, and E133del, were determined (Fig. 3 and Table 1). Most of the WT-like variants showed primarily perturbations at residues 133–144 (electrostatic loop), and some also showed additional perturbations at the adjacent residues 119–132 (electrostatic loop). The exception is A4V, which showed the most pronounced perturbations at the N- and C-terminal residues, 1–8 (β1) and 147–153 (β8), as well as minor perturbations in the electrostatic loop residues 133–144, residues 21–37 (loop II) and 56–96 (the zinc loop and part of β5).
Metal-binding Variants Showed Perturbations in the Electrostatic Loop and Additional Perturbation in the Zinc Loop
The H/D exchange patterns of five metal-binding variants, H46R, G85R, D124V, D125H, and S134N, were determined (Fig. 3 and Table 1). The segments containing residues 56–96 (zinc loop), 119–132 (electrostatic loop), and 133–144 (electrostatic loop) of these variants exchanged significantly faster than those of WT SOD1 (Fig. 3 and Table 1). These three regions were not observed in H46R. G85R showed extra perturbations at segments containing residues 38–53 and 98–103. Fig. 2 illustrates the difference in the average deuteration levels for WT SOD1 and the fALS variant with the most perturbed structure/dynamics, G85R SOD1. The rates of H/D exchange for both the electrostatic and zinc-binding loops were perturbed in all of the metal-binding site variants tested, including G85R, S134N, H46R, D124V, and D125H (Fig. 3). G85R, however, demonstrated an additional increased exchange in loop 3 and β-strand 4 not observed in other variants (Fig. 3).
Copper Content Has Little Effect on SOD1 Dynamics
Metal contents for all proteins studied are listed in Table 2. Copper-deficient WT SOD1 and all 13 variants contained less than one molecule of copper/dimer, whereas native WT SOD1 contained about two coppers/dimer. There is little apparent correlation between the copper content and the H/D exchange patterns of WT SOD1 (Fig. 4a). The H/D exchange patterns of copper-deficient WT SOD1 are indistinguishable from native WT SOD1 despite carrying less than one quarter of the copper of native WT SOD1. Moreover, whereas G93A contained ∼10% of the copper of WT SOD1, the H/D exchange patterns are very similar to those of native WT SOD1. Although D76Y has the highest copper content among the variants, its H/D exchange perturbations are among the largest observed in the WT-like variants.
TABLE 2.
Metal content in equivalents per dimer
| Name | Copper | Zinc |
|---|---|---|
| Copper/zinc WT | 2.0 | 2.0 |
| Copper-deficient WT | 0.46 | 1.16 |
| A4V | 0.22 ± 0.11a | 1.62 ± 0.23a |
| G93A | 0.23 | 1.47 |
| L38V | 0.23 | 1.32 |
| G41S | 0.52 | 1.35 |
| G72S | 0.74 | 0.78 |
| D76Y | 0.22 | 0.85 |
| E133del | 0.31 | 1.26 |
| D90A | 0.25 | 1.43 |
| G85R | 0 | 0.014 |
| D124V | 0.01 | 0.026 |
| D125H | 0.09 | 0.36 |
| S134N | 0.15 | 0.33 |
| H46R | 0.05 | 0.1 |
a Because of an oversight, metal content analysis of this A4V preparation was not performed. Shown are the averages and standard deviations of the three prior A4V preparations.
FIGURE 4.

Effect of metallation on structure/dynamics of SOD1. Shown is the correlation between metal content and fALS SOD1 variant H/D exchange perturbations with respect to wild type SOD1. a, copper content and H/D exchange perturbations at electrostatic loop residues 119–132; b, correlation between zinc content and H/D exchange perturbations at zinc loop residues 56–96; c, correlation between zinc content and H/D exchange perturbations at electrostatic loop residues 119–132. R2 value of linear fits are 0.25 (a), 0.75 (b), and 0.43 (c).
Zinc Content Effects the H/D Exchange at Segment 119–132
In general, the WT-like variants contained at least one zinc/dimer, whereas metal-binding site variants had a very low amount of bound zinc/dimer (0.4 zinc or less per dimer). Segments containing residues 56–96 (zinc loop, β5, and loop V) and 119–132 (electrostatic loop) are perturbed in all of the metal-binding variants. There are moderate correlations between zinc content and the H/D exchange perturbations at these two segments among the variants; the higher the zinc content, the smaller the H/D exchange perturbations are in these two regions (Fig. 4, b and c).
Electrostatic Loop Structural Perturbation Is the Dominant Structural Perturbation
To determine how electrostatic or zinc loop perturbation were related to the sum of all SOD1 (global) structural perturbation, each was correlated (Fig. 5). Zinc loop and electrostatic loop perturbations were highly correlated with global SOD1 perturbation, with R2 = 0.80 and p < 0.0001, and R2 = 0.84 and p < 0.0001, respectively. Zinc loop perturbation generally represented ∼15% of the total SOD1 perturbation, whereas electrostatic loop perturbation represented ∼ 50% of the global SOD1 perturbation.
FIGURE 5.

Correlation between loop structure/dynamics and overall SOD1 structure/dynamics. a, correlation between fALS SOD1 variant H/D exchange perturbations for zinc loop (average of % deuteration increase compared with WT for the peptide covering residues 56–96) versus sum of total exchange; b, correlation between fALS SOD1 variant H/D exchange perturbations for electrostatic loop (sum of average of % deuteration increase compared with WT for peptides covering residues 119–132, 119–144, and 133–144) versus total exchange. For S134N, peptide 119–132 was not observed, and so deuteration of 119–144 was taken as the value for residues 133–144 and 119–132. R2 and p values of linear fits are 0.80 and p < 0.0001 (a) and 0.84 and p < 0.0001 (b).
DISCUSSION
The present study identifies perturbation of electrostatic loop residues 133–144 with respect to wild type SOD1 (including copper-deficient WT SOD1) as a property common to 13 partially metallated, fALS-associated SOD1 variants. In addition to electrostatic loop disorder, zinc loop disorder was observed in variants with severely compromised metal binding affinity. Perturbation of the zinc and the electrostatic loops appears to result from the combination of fALS-related mutation and metal deficiency (see below). Perturbation of the SOD1 electrostatic loop is the first structural property common to a large set of SOD1 variants and is expected to lead to a gain of SOD1 intermolecular interaction (37).
This study used variants in the electrostatic loop (D124V, D125H, and S134N); zinc loop (G72S and D76Y); β-strands 1, 4, and 5 (A4V, H46R, G85R, respectively); and loop residues clustered at the β-barrel plug (L38V, G41S, D90A, and G93A). These included the most prevalent fALS-associated mutations. Overall, our H/D exchange results (Fig. 3) do not lend support to certain hypotheses of the common gain of fALS variant structure and function, including that: 1) fALS SOD1 mutations perturb the dimer interface (1, 38); 2) fALS mutations at copper-binding histidine residues of SOD1 dramatically reduce the strength of normal dimer interactions (39); and 3) local or framework perturbations are the dominant structural perturbation (40). For example, no differences in exchange rates at the dimer interface were observed for eleven of 13 variants; only A4V and G85R had perturbed dimer interfaces (Fig. 3), and only seven of the 13 fALS-associated variants showed detectable structural perturbation in the vicinity of the mutations (Fig. 3). Although perturbation of the dimer interface was not observed to be a general phenomenon, we did observe perturbation of the A4V dimer interface, consistent with previous studies (38, 41, 42) (specifically residues Ile106, Ile151, and Ala4).
One caveat of our studies is that they can be confounded by inter- and intramolecular interactions. For example, a structural change that first results in increased solvent accessibility of a given residue (and thus increased H/D exchange) could give rise to intermolecular interactions that decrease the solvent accessibility of the same region. The relatively low protein 10 μm concentrations of SOD1 should, however, minimize such intermolecular interactions. Another caveat of our study is that our 13 variants do not contain a large number of mutations near the dimer interface and occur in only three of the eight β strands. Thus, whereas our results may exclude certain properties as being common to all SOD1 mutations, they do not exclude the possibility that certain properties (e.g. dimer dissociation) occur in a different subset of SOD1 mutations. Finally, our WT-like variants do not have the same degree of metal occupancy compared with purifications from mouse or human tissues (24).
Structural Perturbation of the SOD1 Electrostatic Loop
The electrostatic loop showed the fastest exchange in our data, consistent with reports of disorder in previous x-ray crystallography (43) and NMR studies (44). Proteins characterized included both apo and WT SOD1 (9, 43), both apo and metallated H46R and S134N (45, 46), G85R (47), G93A (48), H43R (40), A4V, I113T (41, 42), D125H (49), and G37R SOD1 (50). For example, our H/D exchange results with G85R, S134N, and H46R (Fig. 3) are consistent with the x-ray crystallographic results, demonstrating that the β-barrel elements of SOD1 were relatively unperturbed, whereas the electrostatic and zinc loops were more affected (they were so disordered as to not be observed in some crystal structures) (37, 45–47). Disorder in the electrostatic loop is associated with the increased intermolecular interactions responsible for fibril formation (45). Moreover, solution NMR studies of fully metallated S134N indicated that major structural and dynamic changes occurred only in the electrostatic loop (37) and that these changes result in non-native interactions that lead to aggregation.
Comparison of many of our H/D exchange results for partially metallated A4V, which showed changes in the SOD1 N and C termini, with the previously published H/D exchange results for apo A4V (51) was not possible because of incomplete sequence coverage in the previous report. We did not observe the change observed for apo A4V at residues 50–53 and did note changes in the electrostatic loop not observed in the first report. We did not observe any bimodal peak distribution indicative of EX1 kinetics, as was observed previously in amino acids 21–53 for apo A4V. This indicates there are substantial differences between the structure/dynamics of apo and metallated A4V, as previous studies of their relative stabilities would indicate (27, 52). Both the previous H/D exchange study and the current study did not detect changes in A4V residues 9–20.
Structural Perturbation Caused by Partial Metal Binding
Our preferred explanation for perturbations in the electrostatic loop is that they are the result of both variant-related structural change and metal deficiency. We are unaware of any finding that rules out decreased metal affinity as a common gain of function associated with ALS mutations, and the loss of metals universally leads to variants that are less stable than wild type SOD1. Previous studies indicate that the loss of native post-translational modifications, including copper, zinc, and the intrasubunit disulfide, destabilize fALS-variant SOD1 structures to a greater extent than wild type SOD1 structure (2, 53). Our metal analysis indicated that our WT-like samples contain roughly one zinc/dimer and less than one copper/dimer, and therefore all of the variants presented in Fig. 3 are indeed copper-deficient.
A number of lines of evidence support a role for metal deficiency in ALS, including: 1) five of the variants studied, G85R, S134N, D14V, D125H, and H46R, the so called metal-binding site variants, are metal-deficient in vivo (25, 54); 2) copper-deficient SOD1 is of potential physiological relevance, considering that roughly 35% of human copper/zinc SOD1 is copper-deficient in vivo (55–57); and 3) copper-deficient SOD1 can cause fALS in mice (39, 58). Our results demonstrate that zinc deficiency, more so than copper deficiency, is related to disorder in the electrostatic loop (Fig. 4). Our H/D exchange data (Fig. 3) are also consistent with the existing hypothesis that fALS variants may perturb copper-mediated chemistry (7, 59), because the electrostatic loop contains copper ligands and normally prohibits access of small molecules to the active site. The finding that proteins that do not bind active site copper still cause ALS, however, argues against copper-mediated chemistry as a universal gain of toxic SOD1 function (39, 58, 60).
Studies of fALS SOD1 variants demonstrate an inverse correlation between the global rate of H/D exchange and apo protein stability, i.e. less stable proteins had faster rates of exchange (52). Some of the variants used here overlap with the variants from this previous study, including S134N and D125H, which were apo in both studies, and A4V and L38V, which were partially metallated in the current study and apo in the previous study. In the current study partially metallated A4V and L38V demonstrated slow H/D exchange compared with other variants; conversely the apo forms of the same proteins demonstrated the fastest rates of H/D exchange of any variants (52). This illustrates the phenomenon pointed out by Valentine and co-workers (52, 53) that the WT-like variants, which have relatively stable metal-bound structures, become the least stable variants upon the loss of metals. Thus, variants with low metal binding affinity are usually metal-free, but as metal-free proteins are relatively stable. Conversely, WT-like variants are usually metallated, but as metal-free proteins are relatively unstable.
In summary, we demonstrate that changes to the structure/dynamics of the SOD1 electrostatic loop are a common property of 13 diverse, fALS-associated SOD1 mutations. Taken together with previous studies indicating that structural changes in the electrostatic loop lead to increased intermolecular interactions and fibril formation (37, 45), our results provide a mechanism for the aggregation of SOD1 from a single, partially folded state as well as for gain-of-interaction with other biomolecules. Gain-of-intermolecular-interactions, including self-association (37) and fibril formation (45), interaction with dynein (61, 62), and interaction with mitochondria (14, 16, 63, 64), are properties shared by all fALS mutations thus far tested. Considering our recent result that aggregation propensity and loss of stability of SOD1 are synergistic risk factors for fALS patient disease severity (65) and the observation that the one shared property of fALS variants in vitro and in vivo is their propensity to form aggregates, we propose that this finding is of potential relevance to ALS etiology.
Supplementary Material
Acknowledgments
We thank the entire Agar laboratory for thoughtful comments and revisions and N. Y. R. Agar, Greg Petsko, and Dagmar Ringe for thoughtful discussion.
This work was supported, in whole or in part, by National Institutes of Health Grant R01NS065263.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- fALS
- familial ALS
- ALS
- amyotrophic lateral sclerosis
- SOD1
- copper/zinc superoxide dismutase
- WT
- wild type
- H/D
- hydrogen-deuterium
- MS
- mass spectrometry
- HPLC
- high pressure liquid chromatography.
REFERENCES
- 1.Deng H. X., Hentati A., Tainer J. A., Iqbal Z., Cayabyab A., Hung W. Y., Getzoff E. D., Hu P., Herzfeldt B., Roos R. P., et al. (1993) Science 261, 1047–1051 [DOI] [PubMed] [Google Scholar]
- 2.Tiwari A., Hayward L. J. (2005) Neurodegener. Dis. 2, 115–127 [DOI] [PubMed] [Google Scholar]
- 3.Valentine J. S., Doucette P. A., Zittin Potter S. (2005) Annu. Rev. Biochem. 74, 563–593 [DOI] [PubMed] [Google Scholar]
- 4.Lindberg M. J., Tibell L., Oliveberg M. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 16607–16612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tiwari A., Xu Z., Hayward L. J. (2005) J. Biol. Chem. 280, 29771–29779 [DOI] [PubMed] [Google Scholar]
- 6.Durham H. D., Roy J., Dong L., Figlewicz D. A. (1997) J. Neuropathol. Exp. Neurol. 56, 523–530 [DOI] [PubMed] [Google Scholar]
- 7.Crow J. P., Sampson J. B., Zhuang Y., Thompson J. A., Beckman J. S. (1997) J. Neurochem. 69, 1936–1944 [DOI] [PubMed] [Google Scholar]
- 8.Beckman J. S., Carson M., Smith C. D., Koppenol W. H. (1993) Nature 364, 584. [DOI] [PubMed] [Google Scholar]
- 9.Roberts B. R., Tainer J. A., Getzoff E. D., Malencik D. A., Anderson S. R., Bomben V. C., Meyers K. R., Karplus P. A., Beckman J. S. (2007) J. Mol. Biol. 373, 877–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collard J. F., Côté F., Julien J. P. (1995) Nature 375, 61–64 [DOI] [PubMed] [Google Scholar]
- 11.Rothstein J. D., Jin L., Dykes-Hoberg M., Kuncl R. W. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 6591–6595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kabashi E., Agar J. N., Taylor D. M., Minotti S., Durham H. D. (2004) J. Neurochem. 89, 1325–1335 [DOI] [PubMed] [Google Scholar]
- 13.Bruening W., Roy J., Giasson B., Figlewicz D. A., Mushynski W. E., Durham H. D. (1999) J. Neurochem. 72, 693–699 [DOI] [PubMed] [Google Scholar]
- 14.Kong J., Xu Z. (1998) J. Neurosci. 18, 3241–3250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dal Canto M. C., Gurney M. E. (1994) Am. J. Pathol. 145, 1271–1279 [PMC free article] [PubMed] [Google Scholar]
- 16.Pasinelli P., Belford M. E., Lennon N., Bacskai B. J., Hyman B. T., Trotti D., Brown R. H., Jr. (2004) Neuron 43, 19–30 [DOI] [PubMed] [Google Scholar]
- 17.Mourelatos Z., Gonatas N. K., Stieber A., Gurney M. E., Dal Canto M. C. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 5472–5477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tobisawa S., Hozumi Y., Arawaka S., Koyama S., Wada M., Nagai M., Aoki M., Itoyama Y., Goto K., Kato T. (2003) Biochem. Biophys. Res. Commun. 303, 496–503 [DOI] [PubMed] [Google Scholar]
- 19.Hyun D. H., Lee M., Halliwell B., Jenner P. (2003) J. Neurochem. 86, 363–373 [DOI] [PubMed] [Google Scholar]
- 20.Shibata N., Hirano A., Kobayashi M., Siddique T., Deng H. X., Hung W. Y., Kato T., Asayama K. (1996) J. Neuropathol. Exp. Neurol. 55, 481–490 [DOI] [PubMed] [Google Scholar]
- 21.Shibata N., Asayama K., Hirano A., Kobayashi M. (1996) Dev. Neurosci. 18, 492–498 [DOI] [PubMed] [Google Scholar]
- 22.Turner B. J., Talbot K. (2008) Prog. Neurobiol. 85, 94–134 [DOI] [PubMed] [Google Scholar]
- 23.Englander S. W., Kallenbach N. R. (1983) Q. Rev. Biophys. 16, 521–655 [DOI] [PubMed] [Google Scholar]
- 24.Taylor D. M., Gibbs B. F., Kabashi E., Minotti S., Durham H. D., Agar J. N. (2007) J. Biol. Chem. 282, 16329–16335 [DOI] [PubMed] [Google Scholar]
- 25.Hayward L. J., Rodriguez J. A., Kim J. W., Tiwari A., Goto J. J., Cabelli D. E., Valentine J. S., Brown R. H., Jr. (2002) J. Biol. Chem. 277, 15923–15931 [DOI] [PubMed] [Google Scholar]
- 26.Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez J. A., Valentine J. S., Eggers D. K., Roe J. A., Tiwari A., Brown R. H., Jr., Hayward L. J. (2002) J. Biol. Chem. 277, 15932–15937 [DOI] [PubMed] [Google Scholar]
- 28.Fujiwara N., Nakano M., Kato S., Yoshihara D., Ookawara T., Eguchi H., Taniguchi N., Suzuki K. (2007) J. Biol. Chem. 282, 35933–35944 [DOI] [PubMed] [Google Scholar]
- 29.Hamuro Y., Coales S. J., Southern M. R., Nemeth-Cawley J. F., Stranz D. D., Griffin P. R. (2003) J. Biomol. Tech. 14, 171–182 [PMC free article] [PubMed] [Google Scholar]
- 30.Kurobe N., Suzuki F., Okajima K., Kato K. (1990) Clin. Chim. Acta 187, 11–20 [DOI] [PubMed] [Google Scholar]
- 31.Yoshida E., Mokuno K., Aoki S., Takahashi A., Riku S., Murayama T., Yanagi T., Kato K. (1994) J. Neurol. Sci. 124, 25–31 [DOI] [PubMed] [Google Scholar]
- 32.Yates J. R., Eng J. K., Clauser K. R., Burlingame A. L. (1996) J. Am. Soc. Mass Spectrom. 7, 1089–1098 [DOI] [PubMed] [Google Scholar]
- 33.Zhang Z., Smith D. L. (1993) Protein Sci. 2, 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bai Y., Milne J. S., Mayne L., Englander S. W. (1993) Proteins 17, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sivaraman T., Arrington C. B., Robertson A. D. (2001) Nat. Struct. Biol. 8, 331–333 [DOI] [PubMed] [Google Scholar]
- 36.Hamuro Y., Burns L., Canaves J., Hoffman R., Taylor S., Woods V. (2002) J. Mol. Biol. 321, 703–714 [DOI] [PubMed] [Google Scholar]
- 37.Banci L., Bertini I., D'Amelio N., Gaggelli E., Libralesso E., Matecko I., Turano P., Valentine J. S. (2005) J. Biol. Chem. 280, 35815–35821 [DOI] [PubMed] [Google Scholar]
- 38.Ray S. S., Nowak R. J., Strokovich K., Brown R. H., Jr., Walz T., Lansbury P. T., Jr. (2004) Biochemistry 43, 4899–4905 [DOI] [PubMed] [Google Scholar]
- 39.Wang J., Caruano-Yzermans A., Rodriguez A., Scheurmann J. P., Slunt H. H., Cao X., Gitlin J., Hart P. J., Borchelt D. R. (2007) J. Biol. Chem. 282, 345–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DiDonato M., Craig L., Huff M. E., Thayer M. M., Cardoso R. M., Kassmann C. J., Lo T. P., Bruns C. K., Powers E. T., Kelly J. W., Getzoff E. D., Tainer J. A. (2003) J. Mol. Biol. 332, 601–615 [DOI] [PubMed] [Google Scholar]
- 41.Cardoso R. M., Thayer M. M., DiDonato M., Lo T. P., Bruns C. K., Getzoff E. D., Tainer J. A. (2002) J. Mol. Biol. 324, 247–256 [DOI] [PubMed] [Google Scholar]
- 42.Hough M. A., Grossmann J. G., Antonyuk S. V., Strange R. W., Doucette P. A., Rodriguez J. A., Whitson L. J., Hart P. J., Hayward L. J., Valentine J. S., Hasnain S. S. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 5976–5981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strange R. W., Antonyuk S., Hough M. A., Doucette P. A., Rodriguez J. A., Hart P. J., Hayward L. J., Valentine J. S., Hasnain S. S. (2003) J. Mol. Biol. 328, 877–891 [DOI] [PubMed] [Google Scholar]
- 44.Banci L., Bertini I., Cramaro F., Del Conte R., Viezzoli M. S. (2003) Biochemistry 42, 9543–9553 [DOI] [PubMed] [Google Scholar]
- 45.Elam J. S., Taylor A. B., Strange R., Antonyuk S., Doucette P. A., Rodriguez J. A., Hasnain S. S., Hayward L. J., Valentine J. S., Yeates T. O., Hart P. J. (2003) Nat. Struct. Biol. 10, 461–467 [DOI] [PubMed] [Google Scholar]
- 46.Antonyuk S., Elam J. S., Hough M. A., Strange R. W., Doucette P. A., Rodriguez J. A., Hayward L. J., Valentine J. S., Hart P. J., Hasnain S. S. (2005) Protein Sci. 14, 1201–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao X., Antonyuk S. V., Seetharaman S. V., Whitson L. J., Taylor A. B., Holloway S. P., Strange R. W., Doucette P. A., Valentine J. S., Tiwari A., Hayward L. J., Padua S., Cohlberg J. A., Hasnain S. S., Hart P. J. (2008) J. Biol. Chem. 283, 16169–16177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shipp E. L., Cantini F., Bertini I., Valentine J. S., Banci L. (2003) Biochemistry 42, 1890–1899 [DOI] [PubMed] [Google Scholar]
- 49.Elam J. S., Malek K., Rodriguez J. A., Doucette P. A., Taylor A. B., Hayward L. J., Cabelli D. E., Valentine J. S., Hart P. J. (2003) J. Biol. Chem. 278, 21032–21039 [DOI] [PubMed] [Google Scholar]
- 50.Hart P. J., Liu H., Pellegrini M., Nersissian A. M., Gralla E. B., Valentine J. S., Eisenberg D. (1998) Protein Sci. 7, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shaw B. F., Durazo A., Nersissian A. M., Whitelegge J. P., Faull K. F., Valentine J. S. (2006) J. Biol. Chem. 281, 18167–18176 [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez J. A., Shaw B. F., Durazo A., Sohn S. H., Doucette P. A., Nersissian A. M., Faull K. F., Eggers D. K., Tiwari A., Hayward L. J., Valentine J. S. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 10516–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shaw B. F., Valentine J. S. (2007) Trends Biochem. Sci. 32, 78–85 [DOI] [PubMed] [Google Scholar]
- 54.Nishida C. R., Gralla E. B., Valentine J. S. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 9906–9910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bartnikas T. B., Gitlin J. D. (2003) J. Biol. Chem. 278, 33602–33608 [DOI] [PubMed] [Google Scholar]
- 56.Petrovic N., Comi A., Ettinger M. J. (1996) J. Biol. Chem. 271, 28335–28340 [DOI] [PubMed] [Google Scholar]
- 57.Petrovic N., Comi A., Ettinger M. J. (1996) J. Biol. Chem. 271, 28331–28334 [DOI] [PubMed] [Google Scholar]
- 58.Wang J., Slunt H., Gonzales V., Fromholt D., Coonfield M., Copeland N. G., Jenkins N. A., Borchelt D. R. (2003) Hum. Mol. Genet. 12, 2753–2764 [DOI] [PubMed] [Google Scholar]
- 59.Crow J. P., Ye Y. Z., Strong M., Kirk M., Barnes S., Beckman J. S. (1997) J. Neurochem. 69, 1945–1953 [DOI] [PubMed] [Google Scholar]
- 60.Wang J., Xu G., Gonzales V., Coonfield M., Fromholt D., Copeland N. G., Jenkins N. A., Borchelt D. R. (2002) Neurobiol. Dis. 10, 128–138 [DOI] [PubMed] [Google Scholar]
- 61.Ström A. L., Gal J., Shi P., Kasarskis E. J., Hayward L. J., Zhu H. (2008) J. Neurochem. 106, 495–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang F., Strom A. L., Fukada K., Lee S., Hayward L. J., Zhu H. (2007) J. Biol. Chem. 282, 16691–16699 [DOI] [PubMed] [Google Scholar]
- 63.Jaarsma D., Rognoni F., van Duijn W., Verspaget H. W., Haasdijk E. D., Holstege J. C. (2001) Acta Neuropathol. 102, 293–305 [DOI] [PubMed] [Google Scholar]
- 64.Liu J., Lillo C., Jonsson P. A., Vande Velde C., Ward C. M., Miller T. M., Subramaniam J. R., Rothstein J. D., Marklund S., Andersen P. M., Brännström T., Gredal O., Wong P. C., Williams D. S., Cleveland D. W. (2004) Neuron 43, 5–17 [DOI] [PubMed] [Google Scholar]
- 65.Wang Q., Johnson J. L., Agar N. Y., Agar J. N. (2008) PLoS Biol. 6, e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.