

Abstract
To understand the role of a crowded physiological environment in the pathogenesis of neurodegenerative diseases, we report the following. 1) The formation of fibrous aggregates of the human Tau fragment Tau-(244–441), when hyperphosphorylated by glycogen synthase kinase-3β, is dramatically facilitated by the addition of crowding agents. 2) Fibril formation of nonphosphorylated Tau-(244–441) is only promoted moderately by macromolecular crowding. 3) Macromolecular crowding dramatically accelerates amyloid formation by human prion protein. A sigmoidal equation has been used to fit these kinetic data, including published data of human α-synuclein, yielding lag times and apparent rate constants for the growth of fibrils for these amyloidogenic proteins. These biochemical data indicate that crowded cell-like environments significantly accelerate the nucleation step of fibril formation of human Tau fragment/human prion protein/human α-synuclein (a significant decrease in the lag time). These results can in principle be predicted based on some known data concerning protein concentration effects on fibril formation both in vitro and in vivo. Furthermore, macromolecular crowding causes human prion protein to form short fibrils and nonfibrillar particles with lower conformational stability and higher protease resistance activity, compared with those formed in dilute solutions. Our data demonstrate that a crowded physiological environment could play an important role in the pathogenesis of neurodegenerative diseases by accelerating amyloidogenic protein misfolding and inducing human prion fibril fragmentation, which is considered to be an essential step in prion replication.
Amyloid fibrils associated with neurodegenerative diseases such as Alzheimer disease, Parkinson disease, Huntington disease, and transmissible spongiform encephalopathy (TSE)3 (1–5) can be considered biologically relevant failures of the cellular protein quality control mechanisms (6) consisting of molecular chaperones and proteases (7). Up to now, about 20 different proteins with unrelated sequences and tertiary structures are known to form fibrous aggregates associated with various neurodegenerative diseases. These amyloidogenic proteins include both natively unfolded proteins, such as human Tau protein (3) and human α-synuclein (8), and folded globular proteins such as human prion protein (4). There are two faces of protein misfolding in neurodegeneration as follows: a gain of toxic function and a loss of physiological function, which can even occur in combination (9).
Human Tau protein, a marker for Alzheimer disease, forms filaments in the brains of patients with Alzheimer disease (3, 10, 11). It has been found that hyperphosphorylation of Tau reduces the binding affinity between Tau and tubulin and contributes to the self-association of Tau and the formation of Tau paired helical filaments (3, 11–13). It has been proposed that glycogen synthase kinase-3β (GSK-3β) hyperphosphorylation of Tau plays an important role in Alzheimer disease (14, 15), and GSK-3β induces an Alzheimer disease-like hyperphosphorylation of Tau when overexpressed in cultured human neurons (16).
A large body of data strongly suggests Creutzfeldt-Jakob disease, bovine spongiform encephalopathy, and other TSEs are caused by prions (4). Prions are infectious proteins that can transmit biological information by propagating protein misfolding and aggregation (17). The infectious agent is believed to consist entirely of the prion protein (PrP) and is devoid of nucleic acid (4, 17). Prion biogenesis is associated with the normal protease-sensitive form of the protein (cellular PrP molecule, PrPC) undergoing structural change into an abnormal, protease-resistant, disease-causing isoform of prion protein (PrPSc) (4, 17). Although the mechanism by which PrPC is converted to PrPSc in TSE-infected cells and in vivo is not clear, data from cell-free reactions suggest this process is akin to autocatalytic polymerization (18).
Misfolding of Tau and prion proteins has been traditionally and widely studied in dilute solutions (10, 19–21). However, the physiological environment is poorly modeled by such dilute solutions, and biochemical reactions in vivo differ greatly from those in dilute solutions (22). The proteins associated with neurodegenerative diseases form fibrils in a physiological environment crowded with other background macromolecules (22–26), such as proteins, glycosaminoglycans, and proteoglycans (23). Crowding is not confined to cellular interiors but also occurs in the extracellular matrix of tissues (24) and takes place at membrane surfaces (27). For example, blood plasma contains ∼80 g/liter protein, a concentration sufficient to cause significant crowding effects (24). Polysaccharides also contribute to crowding, especially in the extracellular matrix of tissues such as collagen (23, 26). The conversion of PrP from a normal soluble conformation PrPC to its pathogenic conformation PrPSc is believed to occur on the cell surface, in the endocytic vesicles, or in the crowded extracellular matrix (18). Thus, macromolecular crowding on the cell surface and in the extracellular matrix may play an important role in the conformational transition and amyloid formation of PrP in vivo, which have not been fully characterized yet. In vitro, such a crowded environment can be achieved experimentally by adding high concentrations of single or mixed nonspecific crowding agents to the system (23–31). Recently, it has been demonstrated that macromolecular crowding significantly enhances the rate of amyloid formation of α-synuclein (32, 33), amyloid-β peptides (27), and human apolipoprotein C-II (34). However, the role of the crowded physiological environment in the pathogenesis of neurodegenerative diseases is poorly understood so far.
To address the contributions of crowded physiological environments on the pathogenesis of neurodegenerative diseases, we report here that macromolecular crowding dramatically accelerates fibril formation by human Tau fragment and by human prion protein under physiological conditions. Our results indicate that macromolecular crowding significantly accelerates the nucleation step of fibril formation of human Tau fragment/human prion protein/human α-synuclein by fitting the data to a sigmoidal equation (35, 36). Furthermore, macromolecular crowding causes human prion protein to form short fibrils and nonfibrillar particles with lower conformational stability and higher protease resistance activity, compared with those formed in dilute solutions.
EXPERIMENTAL PROCEDURES
Materials
The crowding agents, Ficoll 70 and dextran 70, were purchased from Sigma. Heparin (average molecular mass = 6 kDa) and thioflavin T (ThT) were also obtained from Sigma. Guanidine hydrochloride, guanidine thiocyanate (GdnSCN), and trypsin were obtained from Promega Corp. (Madison, WI). Dithiothreitol (DTT), proteinase K, and Triton X-100 were purchased from Amresco Chemical Co. (Solon, OH). All other chemicals used were made in China and were of analytical grade.
Plasmids and Proteins
The cDNA encoding human Tau fragment Tau-(244–441) was amplified using the plasmid for human Tau40 (kindly provided by Dr. Michel Goedert) as a template. The PCR-amplified Tau-(244–441) was subcloned into pRK172 vector. Recombinant Tau-(244–441) was expressed in Escherichia coli and purified to homogeneity by SP-Sepharose chromatography as described previously (37). His-tagged GSK-3β of cDNA was amplified using human GSK-3β plasmid (kindly provided by Dr. Thilo Hagen) as a template. The PCR-amplified enzyme was subcloned into pEa vector. Recombinant GSK-3β was expressed in E. coli and purified to homogeneity by Ni-NTA-Sepharose and SP-Sepharose chromatography sequentially. The cDNA encoding human PrP-(23–231) was constructed into pET30a. Recombinant full-length human prion protein was expressed in E. coli and purified on a Ni-NTA-agarose column as described (38). Purified Tau protein and GSK-3β were analyzed by SDS-PAGE with one band. Purified human PrP was confirmed by SDS-PAGE and mass spectrometry to be a single species with an intact disulfide bond. The concentration of human Tau fragment was determined according to its absorbance at 214 nm with a standard calibration curve drawn by bovine serum albumin, and the concentration of full-length human prion protein was determined by its absorbance at 280 nm using the molar extinction coefficient value of 57,995 m−1 cm−1 deduced from the composition of the protein online.
Phosphorylation of Tau-(244–441)
A pure recombinant Tau fragment (0.50 mg/ml) was hyperphosphorylated by GSK-3β (20 μg/ml) in the phosphorylation solution containing 2 mm ATP, 8 mm MgCl2, 5 mm EGTA, 1 mm phenylmethylsulfonyl fluoride, 2 mm DTT, and 60 mm HEPES (pH 7.4) at 37 °C for 20 h with a re-addition of ATP and DTT after incubation for 12 h and terminated by heating the reaction solutions at 95 °C for 5 min. The cooled hyperphosphorylated Tau was centrifuged at 10,000 × g for 10 min to remove protein aggregates, and the protein was concentrated and stored at −20 °C.
Fibril Formation and Thioflavin T Binding Assays
A 2.5 mm ThT stock solution was freshly prepared in 10 mm Tris-HCl buffer (pH 7.5) and passed through a 0.22-μm pore size filter before use to remove insoluble particles. Under standard conditions, 12 μm Tau-(244–441) was incubated without agitation in the assembly buffer at pH 7.5 (10 mm Tris-HCl buffer containing 1 mm DTT and 20 μm ThT with or without a crowding agent) and at 37 °C for up to 2 h in the presence of fibrillization inducer heparin used in a Tau:heparin molar ratio of 4:1. The fluorescence of ThT was excited at 440 nm with a slit width of 7.5 nm, and the emission was measured at 480 nm with a slit width of 7.5 nm on an LS-55 luminescence spectrometer (PerkinElmer Life Sciences). A similar method was used for the assembly of hyperphosphorylated Tau fragments in the presence of heparin with a Tau:heparin molar ratio of 4:1, except for the addition of 17 mm NaF. The preparation of the samples before the first measurement took 1 min.
Under standard conditions, a stock solution of human PrP in 6 m guanidine hydrochloride was diluted to a final concentration of 22,11, or 5.5 μm and incubated at 37 °C in PBS buffer (pH 7.0) containing 1 m guanidine hydrochloride and 3 m urea in the absence and presence of different concentrations of crowding agents with continuous shaking at 220 rpm, and samples (50 μl) were diluted into PBS buffer containing 12.5 μm ThT, giving a final volume of 2.5 ml. The fluorescence intensity at 480 nm was averaged over 60 s to increase the signal-to-noise ratio of the measurements, and both excitation and emission slits were 5 nm. Control experiments were performed to ensure that the crowding agents had no influence on the ThT binding assays for both human Tau fragments and human PrP.
Mass Spectrometry
In vitro, GSK-3β hyperphosphorylated human Tau fragment Tau-(244–441), on the one hand, was isolated using SDS-PAGE under reducing conditions by 15% (w/v) polyacrylamide gels. Protein bands were stained by Coomassie Blue. Gel bands were manually excised, reduced, alkylated, and digested with trypsin. On the other hand, hyperphosphorylated Tau-(244–441) was directly reduced and alkylated in phosphorylation reaction buffer and then digested with trypsin in a molar ratio of 1:50. Hyperphosphorylated Tau-(244–441) peptides obtained by in-solution proteolytic digestion were performed by phosphopeptide enrichment on immobilized metal-affinity chromatography according to available protocols (Pierce). Peptides obtained by proteolytic digestion and immobilized metal-affinity chromatography were separated by reversed-phase chromatography using a TempoTM nano multidimensional liquid chromatography system (Eksigent Technologies) and sequenced by MS/MS using a hybrid triple quadrupole-linear ion trap mass spectrometer (QTRAP 3200, Applied Biosystems, Foster City, CA) in information- dependent acquisition mode. The mass spectral data were searched against a data base containing the sequences of all the human Tau isoforms using the Mascot searching algorithm (Matrix Science). Phosphopeptides and nonphosphorylated peptides of Tau were identified based on the following search criteria: trypsin used with up to two missed cleavages; carbamidomethylation of cysteine in fixed modification; oxidized methionine and phosphorylation of Ser, Thr, or Tyr in variable modification; peptide tolerance 2.0 Da; and MS/MS tolerance to 0.8 Da. Phosphorylated residues were determined by manual inspection of the mass spectral MS/MS data.
For selective detection of phosphopeptides from the N terminus of the hyperphosphorylated Tau-(244–441), a list of multiple reaction monitoring transitions of potential phosphopeptides was generated by a software script developed by Applied Biosystems. In general, transitions were included for N-terminal tryptic peptides (maximum of one missed cleavage) containing Ser, Thr, or Tyr residues with either one or two modifications and for doubly and triply charged species for the Q1 mass range 400–1600 m/z. The mass spectrometer was instructed to switch from multiple reaction monitoring to enhanced product ion-scanning mode when an individual multiple reaction monitoring signal exceeded 30 counts. Data were initially analyzed by submitting the MS/MS data to Mascot with a peptide tolerance setting of 0.03 Da. This proves successful as the exact parent mass is known and defined in the multiple reaction monitoring transition. Mascot searches were performed against a data base containing the sequences of all the human Tau isoforms with trypsin plus one missed cleavage, carbamidomethylation of cysteine as a fixed modification, phosphorylated STY and oxidized methionine as a variable modification, and an MS/MS tolerance of 0.8 Da. In addition, data were examined manually.
ANS Binding Measurements
The fluorescence spectra of hydrophobic dye ANS binding were recorded between 400 and 650 nm at 25 °C with excitation at 380 nm (an excitation slit of 5 nm and an emission slit of 6.5 nm) on the LS-55 luminescence spectrometer. Human prion protein samples at various time points during amyloid formation were diluted 10-fold by PBS buffer and then incubated with 100 μm ANS for 15 min at room temperature in the dark before monitoring fluorescence intensity. Assays in the absence of the protein were performed to correct for unbound ANS emission fluorescence intensities.
CD Measurements
Circular dichroism spectra were obtained by using a Jasco J-810 spectropolarimeter (Jasco Corp., Tokyo, Japan) with a thermostated cell holder. A quartz cell with a 1-mm light path was used for measurements in the far-UV region. Spectra were recorded from 200 to 250 nm for far-UV CD. The final concentration of human PrP was kept at 13 μm. The averaged spectra of several scans were corrected relative to the buffer blank or the buffer containing crowding agents. Measurements were made at 25 °C.
PK Digestion Assays
Human PrP fibril samples were prepared in 100 mm Tris-HCl (pH 7.5) with 0.5% Triton X-100 or without Triton X-100 and heated in water bath for 15 min at 80 °C, cooled down (20), and then incubated with PK at a PK:PrP molar ratio of 1:1000 to 1:100 for 1 h at 37 °C. After PK digestion, samples were analyzed in 15% SDS-PAGE and detected by silver staining.
Kinetic Models
Kinetic parameters were determined by fitting ThT fluorescence intensity versus time to sigmoidal Equation 1 (35, 36),
where F is the fluorescence intensity; k is the apparent rate constant for the growth of fibrils, and tm is the time to 50% of maximal fluorescence. The initial base line during the lag time is described by F0. The final base line after the growth phase has ended is described by A + ct. The lag time is the time when the growth in fibrils becomes detectable and is calculated as tm − 2/k.
The time-dependent appearance of ThT fluorescence intensity was also found to be well described by the empirical Hill function Equation 2 (34),
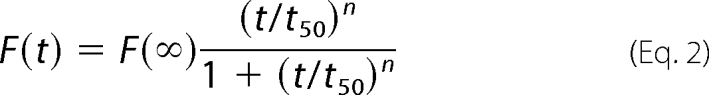 |
where F(∞) is the fluorescence intensity in the long time limit; t50 is the elapsed time when F is equal to one-half of F(∞), and n is a cooperativity parameter.
Transmission Electron Microscopy
The formation of filaments by human Tau fragment and by human prion protein was confirmed by electron microscopy of negatively stained samples. Sample aliquots of 10 μl were placed on copper grids and left at room temperature for 1–2 min, rinsed twice with H2O, and then stained with 2% (w/v) uranyl acetate for another 1–2 min. The stained samples were examined using an H-8100 transmission electron microscope (Hitachi, Tokyo, Japan) operating at 100 kV or an FEI Morgagni transmission electron microscope operating at 80 kV.
Determining Conformational Stability of Human PrP Fibrils
Amyloid fibrils, produced from human PrP incubated in the absence and presence of crowding agents (100 or 150 g/liter Ficoll 70) for 12 h, were incubated for 1 h at 25 °C in the presence of different concentrations of GdnSCN (0–3.5 m). The concentration of GdnSCN was then adjusted to 0.35 m by the addition of PBS buffer containing 12.5 μm ThT, followed by a ThT binding assay as described earlier. The above amyloid fibrils were also incubated with 2% SDS for 5 min at different temperatures (25–100 °C) and then analyzed in 15% SDS-PAGE and detected by Coomassie Blue staining.
RESULTS
Effects of Macromolecular Crowding on Kinetics of Fibril Formation by Human Tau and Prion Proteins
The amino acid sequence of Tau-(244–441) contains two parts as follows: the four repeat microtubule binding domain, which is the core domain related to filament formation; and the C terminus, which has a slight inhibition on the rate and extent of filament formation by the core domain in vitro (19). Because Tau-(244–441) consists of the repeat domain forming the core of paired helical filaments in Alzheimer disease and the C terminus containing some pathologically relevant phosphorylation sites by GSK-3β (39), and also assembles more readily than full-length Tau protein into filaments in vitro, we employed such a Tau fragment for studying the kinetics of Tau fibril formation. The differences of fibrous aggregation kinetics, monitored by ThT fluorescence in the absence and presence of crowding agents, were clearly shown in Fig. 1, A and B, and the kinetic parameters of fibril formation were determined by fitting these kinetic data sets to different models.
FIGURE 1.

Effects of macromolecular crowding on filament formation of Tau-(244–441). Filament formation of nonphosphorylated Tau-(244–441) induced by heparin in the absence and presence of Ficoll 70 (A) and dextran 70 (B), respectively, was monitored by ThT fluorescence. The crowding agent concentrations were 0 (open square), 50 g/liter (solid circle), 100 g/liter (solid triangle), and 200 g/liter (inverted solid triangle). Filament formation of GSK-3β hyperphosphorylated Tau-(244–441) in the absence and presence of Ficoll 70 (C) and dextran 70 (D), respectively, was monitored by ThT fluorescence. The long time incubation of GSK-3β-hyperphosphorylated Tau-(244–441) is shown in the inset of C. The data were fitted to a sigmoidal equation, and the solid lines represent the best fit. The corresponding parameters are summarized in supplemental Table S1.
To compare the overall rates of fibril formation, these kinetic data sets were fitted to the Hill function (34), and the corresponding kinetic parameters are summarized in supplemental Table S1. As shown in Table S1, the value of t50 of the reaction rate for Tau-(244–441) monitored by ThT fluorescence was 1.36 h in the absence of a crowding agent, whereas the addition of crowding agents significantly decreased the value of t50, especially with higher concentrations of crowding agents. The value of t50 of the reaction monitored by ThT fluorescence is 0.492 and 0.440 h in the presence of 200 g/liter Ficoll 70 and 200 g/liter dextran 70, respectively, remarkably shorter than that in the absence of a crowding agent. The above results indicated that the addition of these crowding agents at 100–200 g/liter moderately accelerated filament formation of nonphosphorylated human Tau-(244–441) on the investigated time scale.
To uncover the detailed mechanisms of the enhancing effects of crowding agents on fibrous aggregation by Tau-(244–441), a sigmoidal equation (35, 36) was used to fit the kinetic data, yielding kinetic parameters k, tm, and the lag time, which are also summarized in supplemental Table S1. We found that the addition of a crowding agent such as Ficoll 70 at 200 g/liter moderately accelerated both steps of nucleation and elongation of nonphosphorylated human Tau-(244–441) fibrillization (supplemental Table S1), resulting in a lag time of 0.07 h in the presence of 200 g/liter Ficoll 70, which is 3.4-fold decreased compared with that in the absence of a crowding agent (0.24 h), and an apparent rate constant for the growth of fibrils of 5.29 h−1, which is 1.7-fold larger than that in the absence of a crowding agent (3.07 h−1). These results again indicated that the fibrous aggregation of nonphosphorylated Tau-(244–441) was promoted moderately by macromolecular crowding.
It is interesting to measure the effects of such crowding agents on the fibrous aggregation by hyperphosphorylated Tau-(244–441), due to the complexity of the aggregation of Tau protein, which is modulated by abnormal phosphorylation (39–41), and the substantial role of hyperphosphorylation in the regulation of Tau function (42). In this study, human Tau fragment Tau-(244–441) hyperphosphorylated by GSK-3β was explored. As shown in Fig. 1, C and D, human Tau fragment Tau-(244–441), when hyperphosphorylated by GSK-3β, was unable to form fibrous aggregates in the absence of a crowding agent on the investigated time scale of 2 h; in contrast, the hyperphosphorylated Tau-(244–441) allowed efficient filament formation when a crowding agent (Ficoll 70 or dextran 70) at 200–300 g/liter was used in the same time scale. After a long-time incubation, hyperphosphorylated Tau-(244–441) in the absence of a crowding agent had a slight enhancement in ThT fluorescence intensity, as shown in Fig. 1C, inset, whereas the value of t50 of the reaction was 20.0 h. The lag time (6.10 h) was much longer, and the apparent rate for the growth of fibrils (0.23 h−1) was much slower than nonphosphorylated Tau-(244–441) in the absence of a crowding agent (supplemental Table S1), indicating an inhibition of hyperphosphorylation on fibrous aggregation of Tau-(244–441). However, the addition of crowding agents at 300 g/liter dramatically accelerated both steps, resulting in a lag time of 0.02 h in the presence of Ficoll 70, which is 305-fold shorter than that in the absence of a crowding agent, and an apparent rate constant for the growth of fibrils of 2.65 h−1, which is 12-fold larger than that in the absence of a crowding agent (supplemental Table S1). Clearly, the nucleation of hyperphosphorylated Tau-(244–441) was strongly accelerated by Ficoll 70. Similar results were obtained in the presence of dextran 70 (Fig. 1D and supplemental Table S1). The above data indicate that a crowded cell-like environment significantly accelerates the nucleation step of hyperphosphorylated human Tau fragment misfolding.
SDS-PAGE profiles of Tau-(244–441) and Tau-(244–441) hyperphosphorylated by GSK-3β are shown in supplemental Fig. S1A. Nonphosphorylated Tau-(244–441) (supplemental Fig. S1A, lane 2) migrated as a single band, and hyperphosphorylated Tau-(244–441) (lane 3) migrated slower. Meanwhile, mass spectrometry was used to determine the phosphorylated sites of Tau. The MS/MS spectra generated by peptides phosphorylated on the pathologically relevant site Ser-400 (38, 39) are shown in supplemental Fig. S1B. The y ion series (y7, y8, y9, and y10) here demonstrated a constant neutral loss of phosphoric acid (98 Da) from the precursor ion, a feature often observed when Ser residues are phosphorylated by GSK-3β. Meanwhile, phosphorylation at sites Ser-305, Ser-352, Ser-396, Thr-403, Ser-404, Ser-409, Ser-412, Ser-413, Thr-414, and Ser-416 was also detected by tandem MS/MS (data not shown), indicating that Tau-(244–441) was hyperphosphorylated by GSK-3β. Here, Ser-396, Thr-403, Ser-404, Ser-409, Ser-412, Ser-413, and Thr-414/Ser-416 are pathologically relevant phosphorylation sites on Tau (39, 40, 43).
The above results suggest macromolecular crowding offsets the lost capability of fibril formation caused by the hyperphosphorylation of Tau and partially answers the question of why the assembly of Tau protein is more readily inhibited by hyperphosphorylation in test tubes compared with those in cells (42).
Next, another amyloidogenic protein human PrP was explored (Fig. 2 and supplemental Fig. S2). High concentrations of macromolecular crowding agents used to mimic the crowded physiological conditions did alter the process of amyloid formation of human PrP. As shown in Fig. 2A, the presence of Ficoll 70 at concentrations of 50, 100, 150, and 200 g/liter in the reaction system rotated at 220 rpm all significantly decreased the lag phase and increased the elongation rates of 0.50 mg/ml human PrP. Fitting human prion protein aggregation kinetic data with the Hill function gave t50 and F(∞) values that reflect the lag phase and the final quantity of PrP amyloid formation, respectively. The corresponding kinetic parameters are summarized in supplemental Table S2. As shown in supplemental Table S2, t50 was strongly decreased with the increase of crowding agent concentration, compared with amyloid formation of human PrP in dilute solutions. The above results indicate that macromolecular crowding dramatically accelerates fibril formation by human prion protein on the investigated time scale.
FIGURE 2.

Effects of macromolecular crowding on amyloid formation of human prion protein. Human prion protein in the absence and presence of Ficoll 70 was monitored by ThT fluorescence (A) and ANS fluorescence (B). The concentration of PrP was 0.50 mg/ml. The crowding agent concentrations were 0 (open square), 50 g/liter (solid circle), 100 g/liter (solid triangle), 150 g/liter (inverted solid triangle), and 200 g/liter (solid square). The data were fitted to a sigmoidal equation, and the solid lines represent the best fit. The corresponding parameters from A are summarized in supplemental Table S2.
To elucidate the mechanism of the enhancing effect of macromolecular crowding on amyloid formation of human prion protein, the sigmoidal equation was used to fit the kinetic data yielding three kinetic parameters, which are also summarized in supplemental Table S2. The addition of Ficoll 70 at 200 g/liter dramatically accelerated both steps (Fig. 2A), resulting in a lag time of 0.81 h in the presence of Ficoll 70, which is decreased 7.5-fold compared with that in the absence of a crowding agent (6.06 h), and an apparent rate constant for the growth of fibrils of 6.64 h−1, which is 4.4-fold larger than that in the absence of a crowding agent (1.51 h−1). Clearly, the nucleation of human PrP was strongly accelerated by Ficoll 70 (supplemental Table S2). The kinetics of amyloid formation as a function of human PrP protein concentration was analyzed (Fig. 2A and supplemental Fig. S2A). A similar conclusion was reached for 0.25 or 0.125 mg/ml human PrP in the presence of Ficoll 70 (supplemental Fig. S2A). A similar conclusion was also reached for 0.50 mg/ml human PrP in the presence of Ficoll 70 when the reaction system was rotated at 110 rpm, a modest agitation (data not shown). These results indicated that a crowded cell-like environment significantly accelerated the nucleation step of human prion protein misfolding. As shown in supplemental Fig. S2A and supplemental Table S2, a 4-fold increase in concentration (0.125 to 0.50 mg/ml) resulted in reduction of the lag phase from 7.21 to 6.06 h and an increase of the apparent rate constant for the growth of fibrils from 0.92 to 1.51 h−1 in dilute solution. The kinetics of fibril formation by human PrP in the presence of Ficoll 70 at the same concentration varied with protein concentration in a very similar manner, displaying only modest concentration dependence (supplemental Fig. S2A and supplemental Table S2).
The effects of added crowding agents on the rate of fibril formation of human PrP were further monitored via measurement of time-dependent ANS fluorescence (Fig. 2B) and turbidity (supplemental Fig. S2B). Changes in ANS fluorescence are frequently used to detect the solvent-exposed hydrophobic clusters (44). Fig. 2B shows the changes in ANS binding fluorescence intensity during amyloid formation by human PrP in dilute solutions and in the presence of 50–150 g/liter Ficoll 70. Amyloid formation by human PrP under such different conditions all induced a pronounced blue shift of ANS emission maximum from 510 to 480 nm together with a remarkable increase in ANS fluorescence intensity. As shown in Fig. 2B, a strong enhancement of the ANS fluorescence intensity was observed when samples were incubated for 6 h in 100 g/liter Ficoll 70; however, the ANS fluorescence intensity only began to grow when samples were incubated for 6 h in dilute solutions. The results from turbidity measurements were even more pronounced. As shown in supplemental Fig. S2B, a strong enhancement of the absorbance at 400 nm was observed when samples were incubated for 4 h in 100 g/liter Ficoll 70; however, the absorbance at 400 nm only began to grow when samples were incubated for 6 h in dilute solutions. Here, control experiments were performed to ensure that the crowding agents had no influence on the turbidity assays. Clearly, macromolecular crowding dramatically accelerated the aggregation of human PrP, and the nucleation of human PrP was strongly accelerated by Ficoll 70 (Fig. 2B and supplemental Fig. S2B).
Finally, we used the same kinetic analysis to examine the published results of human α-synuclein fiber formation from Uversky et al. (32). As shown in supplemental Table S3, the addition of crowding agents significantly accelerates amyloid formation by human α-synuclein. The kinetic study of crowding agents on amyloid formation by human α-synuclein (supplemental Table S3) is in perfect agreement with the idea that the nucleation step of amyloidogenic protein misfolding is significantly accelerated by crowded cell-like environments.
Effects of Macromolecular Crowding on Morphology of Human Tau and PrP Samples
Transmission electron microscopy (TEM) was used to study the morphology of human Tau samples and human PrP samples incubated under different conditions. Fig. 3 shows TEM images of Tau fragment samples incubated in the solution of Ficoll 70. In the presence of 300 g/liter Ficoll 70, the majority of hyperphosphorylated Tau-(244–441) was observed as separate small fibrils (Fig. 3B), but no fibrils were observed for the hyperphosphorylated fragment in the absence of a crowding agent after incubation for 2 h (Fig. 3A). For nonphosphorylated Tau-(244–441), the addition of crowding agents had no significant effect on the morphology of Tau samples, and long and branched fibrils were observed in both samples (Fig. 3, C and D). Fig. 4 shows TEM images of human PrP samples incubated in dilute solutions or in the solution of 150 g/liter Ficoll 70 at different time points. In the absence of a crowding agent, the fibrils appear as a long, twisted, and branched structure after incubation for 8 h (Fig. 4B), and the fibrils became shorter after incubation for 12 h (Fig. 4C), but no fibrils were observed when human PrP samples were incubated for 6 h (Fig. 4A). As shown in Fig. 4, D–F, the addition of crowding agents significantly accelerates amyloid formation by human PrP. In the presence of 150 g/liter Ficoll 70, long and branched fibrils (length 1–2 μm), accompanied by some smaller fibrils, were observed when human PrP samples were incubated for only 1.5 h (Fig. 4D). When human PrP samples were incubated for 3 and 6 h, short fibrillar fragments and spherical or ellipsoidal particles (∼15–30 nm in diameter) became more abundant (Fig. 4, E and F). In other words, with the increase of the incubation time, the crowded cell-like environment produced more fragmented fibrils and nonfibrillar particles, which could have implications for prion replication because smaller species are generally considered more infectious. Furthermore, the morphology of hyperphosphorylated Tau protein formed under crowded conditions is similar to that of Tau filaments formed in the brains of patients with Alzheimer disease (45), and the morphology of human PrP fibrils formed in crowded solutions is similar to that of PrPSc purified from scrapie-infected hamster brain (46, 47).
FIGURE 3.

Transmission electron micrographs of human Tau fragment samples at physiological pH after incubation under different conditions. Hyperphosphorylated Tau-(244–441) (A, 2-h incubation) and nonphosphorylated Tau-(244–441) (C, 2-h incubation) were incubated in the absence of a crowding agent. Hyperphosphorylated Tau-(244–441) was incubated in the presence of 300 g/liter Ficoll 70 (B, 2-h incubation), and nonphosphorylated Tau-(244–441) was incubated in the presence of Ficoll 70 at 150 g/liter (D, 2-h incubation). A 2% (w/v) uranyl acetate solution was used to negatively stain the fibrils. The scale bars represent 200 nm for A and B and 100 nm for C and D, respectively.
FIGURE 4.

Transmission electron micrographs of human prion protein samples at physiological pH after incubation under different conditions. Prion protein samples were incubated in the absence of a crowding agent for 6 (A), 8 (B), and 12 h (C) or in the presence of 150 g/liter Ficoll 70 for 1.5 (D), 3 (E), and 6 h (F). A 2% (w/v) uranyl acetate solution was used to negatively stain the fibrils. The scale bars represent 300 nm.
Effects of Macromolecular Crowding on Conformational Stability and Protease Resistance of Human PrP Fibrils
The physical properties of human PrP fibrils such as conformational stability appear to determine to a large extent their biological effects (48). To evaluate the conformational stability, the fibrils produced from human prion protein in the absence and presence of crowding agents were incubated in the presence of increasing concentrations of guanidine thiocyanate (from 0 to 3.5 m) for 1 h, and the amount of intact amyloid structure was analyzed using the ThT binding assay. As shown in Fig. 5A, human PrP fibrils formed in dilute solutions showed a denaturation profile with a C½ of ∼2.7 m, whereas those formed in 150 g/liter Ficoll 70 displayed a denaturation profile with a much smaller C½ value (∼1.2 m). Here, C½ is the half-concentration at which the ThT fluorescence intensity of human PrP fibrils is decreased by 50%. Different conformations of human PrP fibrils were formed at different temperatures. As shown in Fig. 5, B and C, human PrP fibrils formed in 150 g/liter Ficoll 70 showed a much lower melting temperature (∼55 °C) in the presence of SDS than those formed in dilute solutions (∼72 °C). The above results indicated that macromolecular crowding significantly decreased the conformation stability of human PrP fibrils.
FIGURE 5.

Global denaturation of human PrP fibrils analyzed by ThT fluorescence and SDS-PAGE. Guanidine thiocyanate-induced denaturation profiles were monitored for human PrP fibrils in the absence of a crowding agent (open squares) and in the presence of 100 g/liter Ficoll 70 (solid circles) or 150 g/liter Ficoll 70 (solid triangles) (A). Amyloid fibrils, produced from human PrP incubated in the absence and presence of crowding agents for 12 h, were incubated for 1 h at 25 °C in the presence of different concentrations of GdnSCN. The concentration of GdnSCN was then adjusted to 0.35 m, followed by a ThT binding assay. The slight increase in ThT fluorescence observed in the absence of Ficoll 70 at low concentrations of denaturant was presumably due to GdnSCN-induced dissociation of co-aggregated amyloid fibrils. Stability of human PrP fibrils in the absence of a crowding agent (B) and in the presence of 150 g/liter Ficoll 70 (C) under different temperatures was determined by SDS-PAGE. Amyloid fibrils, produced from human PrP incubated in the absence and presence of crowding agents for 12 h, were incubated with 2% SDS for 5 min under each temperature used. Lane M, protein molecular weight marker, restriction endonuclease Bsp98I (25.0 kDa) and β-lactoglobulin (18.4 kDa). Proteins in the gel were visualized by Coomassie Blue staining. The band intensities reflected susceptibility of aggregates to thermal solubilization.
Prion propagation involves the conversion of PrPC into PrPSc, shifting from a predominantly α-helical to β-sheet structure (49). Fig. 6, A and B, shows the CD spectra of native human PrP and human PrP fibrils formed in the absence and presence of 150 g/liter Ficoll 70. Under both conditions, human PrP formed amyloid fibrils with β- sheet-rich conformation from the native state, which has predominant α-helix conformation.
FIGURE 6.

Secondary structural changes of human PrP isoforms monitored by far-UV CD (A and B) and proteinase K digestion assays of human PrP fibrils (C and D) formed in the absence of a crowding agent (A and C) and in the presence of 150 g/liter Ficoll 70 (B and D). Curve a, native human PrP. Curve b, human PrP fibrils. Samples were treated with PK for 1 h at 37 °C at PK:PrP molar ratios as follows: 1:1000 (lanes 2 and 6), 1:500 (lanes 3 and 7), and 1:100 (lanes 4 and 8). The controls with zero protease in the absence of a crowding agent and in the presence of 150 g/liter Ficoll 70 were loaded on lanes 1 and 5, respectively. Amyloid fibrils were produced from human PrP incubated in the absence and presence of crowding agents for 12 h. Protein fragments were separated by SDS-PAGE and detected by silver staining.
PK resistance activity has been widely used to distinguish PrPC from PrPSc since the pioneering studies of Prusiner and co-workers (50). Limited exposure of PrPSc to PK digestion generates a core fragment of 19–21 kDa encompassing residues ∼90–231, named PrP-(27–30) (50–56). Here, an annealing PK digestion assay (20), in which the conformational rearrangement was induced in the presence of 0.5% Triton X-100 by a 15-min exposure to 80 °C, was used to further characterize human PrP fibrils under different conditions. As shown in Fig. 6, C and D, human PrP fibrils produced in the presence of Triton X-100 and in the absence and presence of crowding agents generated a PK-resistant core fragment of 17 kDa encompassing residues 97–231 after PK digestion for 1 h, which shares the primary N-terminal sequence with PrP-(27–30) type 2 (57) but lacks a few amino acids at the very end of the C terminus together with the glycosylphosphatidylinositol anchor (56). Also generated were three abnormally short PK-resistant fragments (12-, 10-, and 8-kDa bands), similar to those reported previously (20, 53). Furthermore, the presence of 150 g/liter Ficoll 70 caused a remarkable increase in optical density of the 17-kDa band, compared with that in dilute solutions, and a small amount of intact human PrP (23.6-kDa band) was observed on the SDS-polyacrylamide gel under such crowded conditions (Fig. 6, C and D). As shown in supplemental Fig. S3, A and B, when PK digestion assays were preformed in the absence of Triton X-100, no 17-kDa band was detected at PK:PrP molar ratios of 1:500 and 1:100 in the absence of crowding agents; however, both the 17- and 23.6-kDa band appeared in the presence of 150 g/liter Ficoll 70. These results indicated that the addition of crowding agents had an enhancing effect on protease resistance activity of human PrP fibrils.
DISCUSSION
For most amyloidogenic proteins, including human Tau protein, human PrP, and human α-synuclein, the fibril formation can be described by the “nucleation-elongation” reaction (8, 58, 59). Taking Tau protein as an example, the pathological aggregation of Tau is a nucleation-elongation reaction that involves the formation of β-structure containing some hexapeptide motifs of the repeat domain (37). The “driving force” for filament formation by the Tau repeat domain involves both the formation of an intermolecular disulfide bond (58) and noncovalent interactions such as hydrogen bonds, electrostatic interactions, and hydrophobic interactions. The elongation can then proceed from the nuclei, which have been reported previously to be as small as 8–14 Tau monomers (58). The nucleation is usually a rate-limiting step, and factors that affect the nucleation rate are likely important to the rate and extent of fibril formation.
Macromolecular crowding has become a greater concern to biochemists, especially in the fields of protein folding/misfolding and association (27–34, 60). For its role in protein misfolding, some widely accepted generalizations are as follows. (i) Amyloid fibrils can form when the native structure of a protein is destabilized, which favors formation of partially folded conformations (1, 61, 62), and such partially folded states are destabilized by macromolecular crowding (60). (ii) Simple excluded volume theory predicts that high concentrations of “inert” macromolecules should stabilize either the compact native state or the compact filaments relative to any less compact unfolded or partially folded state of the polypeptide (22–25). This work explores the mechanisms of the enhancing effects of macromolecular crowding on protein misfolding and addresses the physiological question why and how macromolecular crowding will affect fibril formation of proteins associated with neurodegenerative diseases. Our biochemical data indicate that the lag time of hyperphosphorylated human Tau fragments, human PrP, and human α-synuclein is 305-, 7.5-, and 10.5-fold shorter, respectively, under optimal crowding conditions than that under dilute solutions. Furthermore, no lag time for a conformational transition to β-sheet structures has been observed for amyloid-β peptide samples added to membranes in the presence of 200 g/liter Ficoll 70, whereas in the crowding-free case only minor changes can be seen up to the 6th day, indicating a lag time (27). Combining the results from hyperphosphorylated human Tau fragments, human PrP, human α-synuclein, and amyloid β, we conclude that the nucleation step of amyloidogenic protein misfolding is significantly accelerated by the crowded medium. Therefore, the crowded physiological environment might play an important role in the onset of neurodegenerative diseases by significantly accelerating the nucleation step of amyloidogenic protein misfolding. Our study suggests the crowdedness of cellular environments is an important factor that should be taken into consideration in kinetic analyses of physiological events such as amyloidogenic protein misfolding.
Crowding is expected to shift equilibria toward a state of the system in which the excluded volume is minimized and to affect the reaction accompanied by a significant change in the volume excluded to background solutes (22–25). The extent of this effect is dependent on the relative sizes and shapes of the reactants and products and also those of background molecules (22–25). The most favored situation is the reaction that will generate a greater excluded volume change. According to the nucleation-elongation model, nuclei formation from the partially unfolded/native disordered monomeric proteins is thought to be the initial step and the rate-limiting step (the slowest step) in most amyloid formation processes. As predicted by the excluded volume theory, macromolecular crowding agents should strongly favor the nuclei formation from such monomeric proteins and significantly accelerate the nucleation step of amyloidogenic protein misfolding. Based on our biochemical data and the recently reported results (27–30, 32–34), we propose a valuable hypothesis relating to the tendency of natively unfolded proteins to form fibrils under crowded physiological conditions and the tendency of those proteins that have a stable native state to refold under crowded physiological conditions.
The experiments on human PrP (this work) and human α-synuclein (32) were performed under conditions of agitation. This is usually performed as a convenient way of speeding aggregation (35, 63–65). It also causes fragmentation of amyloid fibrils of prion protein, which occurs in parallel with elongation (63, 64) and affects the kinetics of fibril formation by prion protein (66). Because the experiments on hyperphosphorylated human Tau fragments were performed without agitation, it is easy to understand that the enhancement of the nucleation step in filament formation of hyperphosphorylated Tau fragments by macromolecular crowding is stronger than that of human PrP and human α-synuclein. For human PrP and human α-synuclein, amyloid formation is accelerated by both macromolecular crowding and agitation, but for hyperphosphorylated Tau, filament formation is only accelerated by macromolecular crowding.
In this study, we demonstrate that macromolecular crowding significantly accelerates the nucleation step of amyloidogenic protein misfolding. These results are consistent with known data concerning protein concentration effects on fibril formation both in vitro (67) and in vivo (68). Specifically the papers showing protein concentration effects on amyloid formation by yeast prion proteins in vitro (63, 66) as well as in vivo (prion induction by protein overproduction) (69–71) appear to be relevant to our study. Recent data have shown that pre-existing aggregates (72–74) or intracellular cytoskeletal structures (75) may promote nucleation of amyloid-like complexes by other proteins in the yeast cell. The kinetics of fibril formation by human PrP display modest concentration dependence, acceleration by agitation, and sigmoidal shape of the polymerization time course either in dilute solutions or in crowded media, in perfect agreement with that reported for yeast prion protein (63) and truncated human PrP (64).
Besides the enhancing effect of macromolecular crowding on fibril formation of human PrP, the addition of crowding agents also induced the fragmentation of human PrP fibrils, which will affect the kinetics of fibril formation by prion protein. A three-step model (nucleation, monomer addition, and fiber fragmentation), accurately accounting for the kinetic features of amyloid formation by a yeast prion protein, is proposed by a recent paper (63). The extension of this study of the kinetic data of human PrP modeled by the sigmoidal equation to the modeling of these data by such a complicated three-step model should lead to a better understanding of how fibril fragmentation affects the kinetics of amyloid formation by human PrP in a crowded environment.
Fibril fragmentation is considered to be an essential step in prion replication (76). Recently, Sun et al. (76) have demonstrated that conformational stability of PrP amyloid fibrils controls their smallest possible fragment size. Silveira et al. (47) have reported that nonfibrillar particles fragmented from large PrPSc aggregates, with masses equivalent to 14–28 PrP molecules, are the most efficient initiators of TSE disease. Saborio et al. (77) have reported a procedure involving cyclic amplification of protein misfolding that allows a rapid conversion of large excess PrPC into PrPSc in vitro. In this procedure, aggregates formed when PrPSc is incubated with PrPC are fragmented by sonication to generate multiple smaller units for the continued formation of new PrPSc. The experimental work of Tanaka et al. (78) has revealed a strong link between conformational stability of the yeast prion protein Sup35 and infectivity of amyloid fibrils generated from Sup35 in vitro. The fibrils that display low conformational stability represented by a relatively low melting temperature have been found to exhibit a high efficiency of infection, whereas fibrils with high conformational stability have lower infectivity (78). The question of whether the same rule applies to mammalian prion fibrils is of great interest (48). This study also demonstrates that macromolecular crowding results in human prion protein forming short fibrils and nonfibrillar particles with lower conformational stability and higher protease resistance compared with those formed in noncrowded solutions. A crowded physiological environment may affect the types of fibrillar species that form and accumulate, which may in turn affect pathogenesis of prions and perhaps other amyloidogenic proteins.
Supplementary Material
Acknowledgments
We sincerely thank Prof. Michel Goedert (Laboratory of Molecular Biology, Medical Research Council, Cambridge, UK) for the kind gift of the Tau plasmids and Prof. Thilo Hagen (University of Nottingham, Nottingham, UK) for the kind gift of the GSK-3β plasmids. We thank Prof. Robert L. Baldwin (Stanford University School of Medicine, Stanford, CA) and Prof. Allen P. Minton (NIDDK, National Institutes of Health, Bethesda) for their helpful suggestions. We gratefully acknowledge Prof. Vladimir N. Uversky (Indiana University School of Medicine, Indianapolis) for kindly providing the primary experimental data of human α-synuclein and Dr. Li Li (College of Life Sciences, Wuhan University, Wuhan, China) for technical assistance on TEM.
This work was authored, in part, by National Institutes of Health staff. This work was supported by National Key Basic Research Foundation of China Grant 2006CB910301 and National Natural Science Foundation of China Grants 30770421 and 30970599.
 The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3 and Tables S1–S3.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3 and Tables S1–S3.
- TSE
- transmissible spongiform encephalopathy
- ANS
- 8-anilino-1-naphthalene sulfonic acid
- DTT
- dithiothreitol
- GdnSCN
- guanidine thiocyanate
- GSK-3β
- glycogen synthase kinase-3β
- MS/MS
- tandem mass spectrometry
- Ni-NTA
- nickel-nitrilotriacetic acid
- PK
- proteinase K
- PrP
- prion protein
- PrPC
- the normal cellular PrP molecule
- PrPSc
- an abnormal disease-causing isoform of prion protein
- TEM
- transmission electron microscopy
- ThT
- thioflavin T
- PBS
- phosphate-buffered saline.
REFERENCES
- 1.Dobson C. M. (1999) Trends Biochem. Sci. 24, 329–332 [DOI] [PubMed] [Google Scholar]
- 2.Dobson C. M. (2003) Nature 426, 884–890 [DOI] [PubMed] [Google Scholar]
- 3.Goedert M. (1993) Trends Neurosci. 16, 460–465 [DOI] [PubMed] [Google Scholar]
- 4.Prusiner S. B. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soto C. (2003) Nat. Rev. Neurosci. 4, 49–60 [DOI] [PubMed] [Google Scholar]
- 6.Kaganovich D., Kopito R., Frydman J. (2008) Nature 454, 1088–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wickner S., Maurizi M. R., Gottesman S. (1999) Science 286, 1888–1893 [DOI] [PubMed] [Google Scholar]
- 8.Fink A. L. (2006) Acc. Chem. Res. 39, 628–634 [DOI] [PubMed] [Google Scholar]
- 9.Winklhofer K. F., Tatzelt J., Haass C. (2008) EMBO J. 27, 336–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goedert M., Jakes R., Spillantini M. G., Hasegawa M., Smith M. J., Crowther R. A. (1996) Nature 383, 550–553 [DOI] [PubMed] [Google Scholar]
- 11.Mandelkow E. M., Mandelkow E. (1993) Trends Biochem. Sci. 18, 480–483 [DOI] [PubMed] [Google Scholar]
- 12.Alonso A., Zaidi T., Novak M., Grundke-Iqbal I., Iqbal K. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 6923–6928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grundke-Iqbal I., Iqbal K., Tung Y. C., Quinlan M., Wisniewski H. M., Binder L. I. (1986) Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geschwind D. H. (2003) Neuron 40, 457–460 [DOI] [PubMed] [Google Scholar]
- 15.Lovestone S., Reynolds C. H., Latimer D., Davis D. R., Anderton B. H., Gallo J. M., Hanger D., Mulot S., Marquardt B., Stabel S., Woodgett J. R., Miller C. C. (1994) Curr. Biol. 4, 1077–1086 [DOI] [PubMed] [Google Scholar]
- 16.Hong M., Chen D. C., Klein P. S., Lee V. M. (1997) J. Biol. Chem. 272, 25326–25332 [DOI] [PubMed] [Google Scholar]
- 17.Soto C., Estrada L., Castilla J. (2006) Trends Biochem. Sci. 31, 150–155 [DOI] [PubMed] [Google Scholar]
- 18.Baron G. S., Wehrly K., Dorward D. W., Chesebro B., Caughey B. (2002) EMBO J. 21, 1031–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abraha A., Ghoshal N., Gamblin T. C., Cryns V., Berry R. W., Kuret J., Binder L. I. (2000) J. Cell Sci. 113, 3737–3745 [DOI] [PubMed] [Google Scholar]
- 20.Bocharova O. V., Makarava N., Breydo L., Anderson M., Salnikov V. V., Baskakov I. V. (2006) J. Biol. Chem. 281, 2373–2379 [DOI] [PubMed] [Google Scholar]
- 21.Stöhr J., Weinmann N., Wille H., Kaimann T., Nagel-Steger L., Birkmann E., Panza G., Prusiner S. B., Eigen M., Riesner D. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 2409–2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minton A. P. (2006) J. Cell Sci. 119, 2863–2869 [DOI] [PubMed] [Google Scholar]
- 23.Ellis R. J. (2001) Curr. Opin. Struct. Biol. 11, 114–119 [DOI] [PubMed] [Google Scholar]
- 24.Ellis R. J. (2001) Trends Biochem. Sci. 26, 597–604 [DOI] [PubMed] [Google Scholar]
- 25.Zhou H. X., Rivas G., Minton A. P. (2008) Annu. Rev. Biophys. 37, 375–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bellotti V., Chiti F. (2008) Curr. Opin. Struct. Biol. 18, 771–779 [DOI] [PubMed] [Google Scholar]
- 27.Bokvist M., Gröbner G. (2007) J. Am. Chem. Soc. 129, 14848–14849 [DOI] [PubMed] [Google Scholar]
- 28.van den Berg B., Wain R., Dobson C. M., Ellis R. J. (2000) EMBO J. 19, 3870–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou B. R., Liang Y., Du F., Zhou Z., Chen J. (2004) J. Biol. Chem. 279, 55109–55116 [DOI] [PubMed] [Google Scholar]
- 30.Du F., Zhou Z., Mo Z. Y., Shi J. Z., Chen J., Liang Y. (2006) J. Mol. Biol. 364, 469–482 [DOI] [PubMed] [Google Scholar]
- 31.Homouz D., Perham M., Samiotakis A., Cheung M. S., Wittung-Stafshede P. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 11754–11759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uversky V. N., Cooper E. M., Bower K. S., Li J., Fink A. L. (2002) FEBS Lett. 515, 99–103 [DOI] [PubMed] [Google Scholar]
- 33.Shtilerman M. D., Ding T. T., Lansbury P. T., Jr. (2002) Biochemistry 41, 3855–3860 [DOI] [PubMed] [Google Scholar]
- 34.Hatters D. M., Minton A. P., Howlett G. J. (2002) J. Biol. Chem. 277, 7824–7830 [DOI] [PubMed] [Google Scholar]
- 35.Cohlberg J. A., Li J., Uversky V. N., Fink A. L. (2002) Biochemistry 41, 1502–1511 [DOI] [PubMed] [Google Scholar]
- 36.Chattopadhyay M., Durazo A., Sohn S. H., Strong C. D., Gralla E. B., Whitelegge J. P., Valentine J. S. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 18663–18668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barghorn S., Biernat J., Mandelkow E. (2005) Methods Mol. Biol. 299, 35–51 [DOI] [PubMed] [Google Scholar]
- 38.Yin S. M., Zheng Y., Tien P. (2003) Protein Expr. Purif. 32, 104–109 [DOI] [PubMed] [Google Scholar]
- 39.Morishima-Kawashima M., Hasegawa M., Takio K., Suzuki M., Yoshida H., Titani K., Ihara Y. (1995) J. Biol. Chem. 270, 823–829 [DOI] [PubMed] [Google Scholar]
- 40.Hanger D. P., Byers H. L., Wray S., Leung K. Y., Saxton M. J., Seereeram A., Reynolds C. H., Ward M. A., Anderton B. H. (2007) J. Biol. Chem. 282, 23645–23654 [DOI] [PubMed] [Google Scholar]
- 41.Wang Y. P., Biernat J., Pickhardt M., Mandelkow E., Mandelkow E. M. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 10252–10257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ballatore C., Lee V. M., Trojanowski J. Q. (2007) Nat. Rev. Neurosci. 8, 663–672 [DOI] [PubMed] [Google Scholar]
- 43.Yuzwa S. A., Macauley M. S., Heinonen J. E., Shan X., Dennis R. J., He Y., Whitworth G. E., Stubbs K. A., McEachern E. J., Davies G. J., Vocadlo D. J. (2008) Nat. Chem. Biol. 4, 483–490 [DOI] [PubMed] [Google Scholar]
- 44.Liang Y., Du F., Sanglier S., Zhou B. R., Xia Y., Van Dorsselaer A., Maechling C., Kilhoffer M. C., Haiech J. (2003) J. Biol. Chem. 278, 30098–30105 [DOI] [PubMed] [Google Scholar]
- 45.Berriman J., Serpell L. C., Oberg K. A., Fink A. L., Goedert M., Crowther R. A. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 9034–9038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prusiner S. B., McKinley M. P., Bowman K. A., Bolton D. C., Bendheim P. E., Groth D. F., Glenner G. G. (1983) Cell 35, 349–358 [DOI] [PubMed] [Google Scholar]
- 47.Silveira J. R., Raymond G. J., Hughson A. G., Race R. E., Sim V. L., Hayes S. F., Caughey B. (2005) Nature 437, 257–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun Y., Breydo L., Makarava N., Yang Q., Bocharova O. V., Baskakov I. V. (2007) J. Biol. Chem. 282, 9090–9097 [DOI] [PubMed] [Google Scholar]
- 49.Jackson G. S., Hosszu L. L., Power A., Hill A. F., Kenney J., Saibil H., Craven C. J., Waltho J. P., Clarke A. R., Collinge J. (1999) Science 283, 1935–1937 [DOI] [PubMed] [Google Scholar]
- 50.McKinley M. P., Bolton D. C., Prusiner S. B. (1983) Cell 35, 57–62 [DOI] [PubMed] [Google Scholar]
- 51.Oesch B., Westaway D., Wälchli M., McKinley M. P., Kent S. B., Aebersold R., Barry R. A., Tempst P., Teplow D. B., Hood L. E., Prusiner S. B., Weissmann C. (1985) Cell 40, 735–746 [DOI] [PubMed] [Google Scholar]
- 52.Baskakov I. V., Legname G., Baldwin M. A., Prusiner S. B., Cohen F. E. (2002) J. Biol. Chem. 277, 21140–21148 [DOI] [PubMed] [Google Scholar]
- 53.Bocharova O. V., Breydo L., Salnikov V. V., Gill A. C., Baskakov I. V. (2005) Protein Sci. 14, 1222–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bocharova O. V., Breydo L., Parfenov A. S., Salnikov V. V., Baskakov I. V. (2005) J. Mol. Biol. 346, 645–659 [DOI] [PubMed] [Google Scholar]
- 55.Castilla J., Saá P., Hetz C., Soto C. (2005) Cell 121, 195–206 [DOI] [PubMed] [Google Scholar]
- 56.Notari S., Strammiello R., Capellari S., Giese A., Cescatti M., Grassi J., Ghetti B., Langeveld J. P., Zou W. Q., Gambetti P., Kretzschmar H. A., Parchi P. (2008) J. Biol. Chem. 283, 30557–30565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parchi P., Zou W., Wang W., Brown P., Capellari S., Ghetti B., Kopp N., Schulz-Schaeffer W. J., Kretzschmar H. A., Head M. W., Ironside J. W., Gambetti P., Chen S. G. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 10168–10172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Friedhoff P., von Bergen M., Mandelkow E. M., Davies P., Mandelkow E. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 15712–15717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harper J. D., Lansbury P. T., Jr. (1997) Annu. Rev. Biochem. 66, 385–407 [DOI] [PubMed] [Google Scholar]
- 60.Engel R., Westphal A. H., Huberts D. H., Nabuurs S. M., Lindhoud S., Visser A. J., van Mierlo C. P. (2008) J. Biol. Chem. 283, 27383–27394 [DOI] [PubMed] [Google Scholar]
- 61.Bellotti V., Mangione P., Stoppini M. (1999) Cell. Mol. Life Sci. 55, 977–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kelly J. W. (1998) Curr. Opin. Struct. Biol. 8, 101–106 [DOI] [PubMed] [Google Scholar]
- 63.Collins S. R., Douglass A., Vale R. D., Weissman J. S. (2004) PLoS Biol. 2, e321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baskakov I. V., Bocharova O. V. (2005) Biochemistry 44, 2339–2348 [DOI] [PubMed] [Google Scholar]
- 65.Xue W. F., Homans S. W., Radford S. E. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 8926–8931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Serio T. R., Cashikar A. G., Kowal A. S., Sawicki G. J., Moslehi J. J., Serpell L., Arnsdorf M. F., Lindquist S. L. (2000) Science 289, 1317–1321 [DOI] [PubMed] [Google Scholar]
- 67.Scherzinger E., Sittler A., Schweiger K., Heiser V., Lurz R., Hasenbank R., Bates G. P., Lehrach H., Wanker E. E. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 4604–4609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Colby D. W., Cassady J. P., Lin G. C., Ingram V. M., Wittrup K. D. (2006) Nat. Chem. Biol. 2, 319–323 [DOI] [PubMed] [Google Scholar]
- 69.Ter-Avanesyan M. D., Kushnirov V. V., Dagkesamanskaya A. R., Didichenko S. A., Chernoff Y. O., Inge-Vechtomov S. G., Smirnov V. N. (1993) Mol. Microbiol. 7, 683–692 [DOI] [PubMed] [Google Scholar]
- 70.Wickner R. B. (1994) Science 264, 566–569 [DOI] [PubMed] [Google Scholar]
- 71.Derkatch I. L., Chernoff Y. O., Kushnirov V. V., Inge-Vechtomov S. G., Liebman S. W. (1996) Genetics 144, 1375–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Osherovich L. Z., Weissman J. S. (2001) Cell 106, 183–194 [DOI] [PubMed] [Google Scholar]
- 73.Derkatch I. L., Bradley M. E., Hong J. Y., Liebman S. W. (2001) Cell 106, 171–182 [DOI] [PubMed] [Google Scholar]
- 74.Meriin A. B., Zhang X., He X., Newnam G. P., Chernoff Y. O., Sherman M. Y. (2002) J. Cell Biol. 157, 997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ganusova E. E., Ozolins L. N., Bhagat S., Newnam G. P., Wegrzyn R. D., Sherman M. Y., Chernoff Y. O. (2006) Mol. Cell. Biol. 26, 617–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sun Y., Makarava N., Lee C. I., Laksanalamai P., Robb F. T., Baskakov I. V. (2008) J. Mol. Biol. 376, 1155–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saborio G. P., Permanne B., Soto C. (2001) Nature 411, 810–813 [DOI] [PubMed] [Google Scholar]
- 78.Tanaka M., Chien P., Naber N., Cooke R., Weissman J. S. (2004) Nature 428, 323–328 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.