

Abstract
Seryl-tRNA synthetase (SerRS) from methanogenic archaeon Methanosarcina barkeri, contains an idiosyncratic N-terminal domain, composed of an antiparallel β-sheet capped by a helical bundle, connected to the catalytic core by a short linker peptide. It is very different from the coiled-coil tRNA binding domain in bacterial-type SerRS. Because the crystal structure of the methanogenic-type SerRS·tRNA complex has not been obtained, a docking model was produced, which indicated that highly conserved helices H2 and H3 of the N-terminal domain may be important for recognition of the extra arm of tRNASer. Based on structural information and the docking model, we have mutated various positions within the N-terminal region and probed their involvement in tRNA binding and serylation. Total loss of activity and inability of the R76A variant to form the complex with cognate tRNA identifies Arg76 located in helix H2 as a crucial tRNA-interacting residue. Alteration of Lys79 positioned in helix H2 and Arg94 in the loop between helix H2 and β-strand A4 have a pronounced effect on SerRS·tRNASer complex formation and dissociation constants (KD) determined by surface plasmon resonance. The replacement of residues Arg38 (located in the loop between helix H1 and β-strand A2), Lys141 and Asn142 (from H3), and Arg143 (between H3 and H4) moderately affect both the serylation activity and the KD values. Furthermore, we have obtained a striking correlation between these results and in vivo effects of these mutations by quantifying the efficiency of suppression of bacterial amber mutations, after coexpression of the genes for M. barkeri suppressor tRNASer and a set of mMbSerRS variants in Escherichia coli.
The aminoacyl-tRNA synthetases (aaRSs)2 catalyze the activation of cognate amino acids and their transfer to the 3′-end of corresponding tRNA molecules. The aaRSs are a highly conserved family of enzymes comprised of two distinct structural groups referred to as classes I and II (1–3), with a notable exception of LysRS representatives, which belong to both classes (4). Although the catalytic mechanisms of various aaRSs are broadly similar (5), each enzyme has developed a high specificity in recognizing its cognate amino acid and tRNA, which is pivotal for accurate translation of the genetic code (1). The discrimination of the amino acids is based on recognizing the differences in the size and charge of the molecules (6). The specificity of tRNA selection depends on a set of identity determinants that are mostly located at two distal extremities: the anticodon loop and the amino acid accepting stem. In a few instances, identity elements are also found in the D-arm, T-arm, and variable loop. They can either act as positive determinants that enhance aminoacylation or negative ones that prevent aminoacylation. The recognition of tRNAs by synthetases can also be affected by the modification of particular nucleotides (7, 8). AaRSs show divergent strategies for tRNA recognition. Most notably, class I and class II aaRSs (including pyrrolysyl-tRNA synthetase, see Ref. 9) approach tRNAs from the minor and major groove sides of the acceptor stem, respectively (10). Although the majority of determinants are in direct contact with cognate synthetases (8), the aminoacylation fidelity is controlled by kinetic differences more than by binding affinities (11).
Seryl-tRNA synthetases (SerRSs), which catalyze the aminoacylation of several tRNASer isoacceptors and tRNASec with serine, can be divided into two structurally different groups: bacterial-type SerRSs function in a variety of archaeal, bacterial, and eukaryotic organisms, whereas the methanogenic-type was found only in methanogenic archaea (12, 13). Furthermore, based on sequence comparison (14, 15) and x-ray analyses, two subgroups of bacterial-type SerRSs were identified: one consists of the enzymes from bacterial sources, best represented by those from Thermus thermophilus (16) and Escherichia coli (3), and an archaeal/eukaryal-type, structurally related to SerRS from archaeon Pyrococcus horikoshii (17).
All SerRSs are functional homodimers with a C-terminal active site domain typical for class II aaRSs and an N-terminal domain that is responsible for binding of the long variable arm of tRNASer isoacceptors (reviewed in Ref. 18), with exception of the mammalian mitochondrial enzyme (19). The long variable arm of tRNASer categorizes it as one of the type 2 tRNAs, including the tRNASer and tRNALeu species (from all organisms or domains of life) and bacterial tRNATyr species (20). In bacterial-type SerRS, the N-terminal domain forms an antiparallel α-helical coiled-coil structure (3, 16), whereas in the methanogenic-type counterpart it is significantly larger and composed of a six-stranded antiparallel β-sheet capped by a bundle of three helices (H1, H2, and H4) with up-down topology and an additional short helix (H3) that runs almost perpendicular to helix H4 (21) (see Fig. 1). Despite pronounced structural differences between the tRNA-binding domains in two SerRS types, in each case the N-terminal domain of one subunit interacts with the extra arm of tRNASer, to position the 3′-end of tRNA into the C-terminal active site of another subunit (23–25). The recent crystal structure of the first archaeal/eukaryal SerRS from the archaeon P. horikoshii, and the structure-based model of the enzyme bound with the T. thermophilus and P. horikoshii tRNAsSer, suggested that the helical N-terminal domain of P. horikoshii SerRS is also involved in the binding of the extra arm of tRNA (17).
FIGURE 1.

Design of mMbSerRS variants based on the conserved amino acid residues in the N-terminal domain of methanogenic-type SerRSs and their proximity to tRNA in mMbSerRS·tRNASer docking model. A, the structure-based sequence alignment of the mMbSerRS N-terminal domain with selected SerRS sequences derived from methanogenic archaea (Mk, Methanopyrus kandleri; Mt, Methanothermobacter thermoautotrophicus; Mj, Methanococcus jannaschii; Mm, Methanococcus maripaludis; mMb, Methanosarcina barkeri). The sequence alignment was generated using the program MUSCLE (22). Amino acids that are completely conserved are in red, whereas those with 80% conservation are in yellow. Secondary structural elements are indicated above the alignment with gray cylinders and arrows for helices and β-sheets, respectively. Mutated residues are marked with dots. B, mMbSerRS·tRNASer docking model. The SerRS dimer is shown in gray and tRNASer is colored cyan. Conserved amino acids are indicated by the same color code as in A.
The recognition of tRNA by SerRS relies, besides on the long extra arm, on the identity elements in tRNASer acceptor arms, achieved by the motif 2 residues. Unlike the majority of aaRSs systems, the anticodon triplet is not recognized by SerRS. The first four base pairs in the tRNA acceptor arm (G1:C72, G2:C71, A/U3:U/A70, and R4:Y69) are identity elements for bacterial SerRS, and among them, the second G2:C71 base pair is the most significant (reviewed in Ref. 18). Consistently, the crystal structure of the T. thermophilus SerRS·tRNASer complex revealed that SerRS interacts with the tRNASer acceptor stem, and Ser261 is responsible for the base-specific interaction with G2 (23, 26). Although the acceptor stem sequences are not well conserved among the eukaryal tRNASer isoacceptors, the discriminator base G73 is an essential identity requirement for human tRNASer and serves as an anti-determinant in lower eukaryotes (reviewed in Ref. 18). Unlike eukaryal tRNAsSer, archaeal tRNAsSer conserve the G1:C72, C2:G71, C3:G70, and G4:C69 base pairs in the acceptor stem. However, the tRNA specificity of the archaeal/eukaryal SerRS from P. horikoshii seems to depend mainly on the extra arm, but not on the acceptor stem. Indeed, this enzyme exhibits quite relaxed specificity for tRNASer recognition (17).
Archaeon Methanosarcina barkeri, which possesses two dissimilar SerRSs, one of a bacterial- and the other of methanogenic-type, provides an excellent system for studying the evolution of tRNASer determinants. Two enzymes recognize the same set of tRNA isoacceptors in vitro (27). We have undertaken several approaches to elucidate the basis for their different serylation mechanisms. Kinetic analysis of variant tRNASer transcripts by the two archaeal SerRS enzymes (27) revealed that the length of the variable arm is a critical recognition element for both enzymes, as is the identity of the discriminator base (G73) and base pair G30:C40 in the anticodon stem. However, additional determinants were identified as being required for specific serylation by the unusual methanogenic-type enzyme, which relies on G1:C72 identity and on the number of unpaired nucleotides at the base of the variable loop. The tRNA recognition pattern by two M. barkeri SerRS may differ in vivo, because only bacterial-type SerRS complements the function of thermolabile E. coli SerRS (28).
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis and Purification of Proteins
The seryl-tRNA synthetase expression vector (pET15bmMbSerRS) has been reported previously (27). Primers listed in supplemental Table S1 and the QuikChange mutagenesis kit (Stratagene) were used for site-directed mutagenesis. Point mutations were confirmed by DNA sequencing.
Wild type and mutated SerRS proteins were expressed in E. coli, as described previously (21). Because WT mMbSerRS and its variants are His-tagged, they were first purified by affinity chromatography on nickel-nitrilotriacetic acid-agarose. Greater purity of proteins was achieved using cation exchange chromatography. Proteins were loaded on a Resource S 6-ml column and eluted with a linear KCl gradient (100–500 mm) in buffer containing 25 mm Mes, pH 6.2, 5 mm dithiothreitol (DTT), and 10 mm MgCl2. Fractions enriched in SerRS were pooled and desalted on gel-filtration columns (PD-10), concentrated by ultrafiltration, and stored at −80 °C in a buffer containing 25 mm Hepes, pH 7.0, 200 mm KCl, 5 mm MgCl2, 5 mm DTT, and 10% glycerol.
tRNA Cloning and in Vitro Transcription
The gene for MbtRNAGGASer was constructed from synthetic oligomers according to the published sequence (27). In vitro transcription was performed as reported previously (29). The tRNA transcript was carefully renatured prior to use in the kinetic assay, gel mobility shift assay, and surface plasmon resonance spectroscopy by heating for 5 min at 70 °C in 10 mm Tris/HCl, pH 7.5, followed by addition of MgCl2 to the final concentration of 5 mm and placing on ice. MbtRNAGGASer was maximally serylated to 80% as determined in the standard reaction mixture (described below).
Aminoacylation Assay
Aminoacylation was carried out at 37 °C in the reaction mixture containing 50 mm Hepes/HCl, pH 7.0, 15 mm MgCl2, 4 mm DTT, 5 mm ATP, 125 μm [14C]serine, 0.25 μm SerRS, and 1.5 μm tRNA. Quantification of synthesized radioactive seryl-tRNASer was done as described (29). Relative serylation rates represent the average of at least three independent experiments.
Electrophoretic Mobility Shift Assay
To check for complex formation between cognate tRNA and wild type or mutated mMbSerRS a constant amount of purified protein (8.3 pmol) was mixed with tRNA (14.8 pmol) and incubated for 15 min at 37 °C in a 13.5-μl volume containing 30 mm Hepes, pH 7.0, and 6 mm MgCl2 followed by cooling on ice. To test salt influence on non-covalent complex formation between protein and nucleic acid, salt (25 or 250 mm KCl) at different amounts was added in the reaction mixture prior to incubation for 15 min at 37 °C. Samples were subjected to electrophoresis on a native 9% acrylamide (w/v) gel of acrylamide:bis-acrylamide (19:1) containing 5% glycerol in electrophoresis buffer (25 mm Mes, 25 mm Tris, pH 7.6). Electrophoresis was performed at 4 °C for 2.5 h at 120 V, and gels were stained with silver.
Surface Plasmon Resonance
Kinetic studies were performed at 25 °C using a BIACORE T100 surface plasmon resonance (SPR) instrument. Wild type protein and mutants were covalently attached to a carboxymethyl dextran-coated gold surface (CM5 sensor chip). The carboxymethyl groups of dextran were activated with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride and N-hydroxysuccinimide, and seryl-tRNA synthetase, or its mutants, were attached at pH 5.0 in 10 mm sodium acetate. Any remaining reactive sites were blocked by reaction with ethanolamine and the surface was washed with 50 mm NaOH to remove any non-covalently bound ligand. Proteins were immobilized at levels of 1000 response units in one flow cell. The kinetics of association and dissociation were monitored at a flow rate of 60 μl/min. The renatured tRNA analyte was diluted in a running buffer (30 mm Hepes, pH 7.0, 6 mm MgCl2, and 5 mm DTT). Binding was measured at concentration ranges of 19.5 nm to 8 μm tRNA. After the end of each injection, tRNA was allowed to dissociate for 500 s and then the chip was regenerated with 3 m KCl at a flow of 100 μl/min for 5 min. Data reported are the differences in SPR signal between the flow cell containing the wild type or mutant mMbSerRS and the reference cell without enzyme immobilized. Duplicate injections were made for each tRNA concentration in one round of measurement and each experiment was repeated twice. The data were analyzed using Biacore T100 Evaluation Software.
Suppression of E. coli Amber Mutations
The assay is based on monitoring the suppression efficiency of bacterial amber mutations in E. coli strain XAC-A24 (F′ ara argE(UAG) rpoB gyrA Δlac pro/F′ lacI(UAG)-Z proAB) (28), after co-expression of the M. barkeri SerRS gene, encoding methanogenic-type SerRS, with the gene for cognate archaeal suppressor tRNA (supMb). The synthetic gene for supMb, bearing the CGA anticodon, was inserted into the pTech plasmid, behind the lpp promoter, as described (28). To obtain higher expression of genes of some SerRS mutants, NcoI–XhoI fragments containing synthetase genes with the N-terminal His6 tag from plasmid pET15b were then recloned into pBAD24 vector (30), where SerRS expression is under control of the arabinose inducible promoter. Suppression of the argE amber mutation was tested by plating E. coli cells on selective M9 minimal glucose plates. The efficiency of suppression was determined by measuring the β-galactosidase activity produced from lacI-lacZ fusion harboring a nonsense mutation in the lacI portion (31).
RESULTS
Selection of Target Sites for Site-directed Mutagenesis
Our previously published structure-based model of the mMbSerRS· tRNASer complex (Fig. 1) implies that tRNASer binds across two subunits of dimeric enzyme, as observed in the bacterial synthetase·tRNA complex structure. Experimental verification of predicted cross-dimer binding was provided by testing the activity of constructed SerRS heterodimers (25). In the model the long variable arm of the tRNA is positioned to interact with the N-terminal domain of mMbSerRS, in accordance with our previous biochemical experiments that identified the long variable arm of archaeal tRNASer as a major tRNA recognition determinant (27, 29, 32). Furthermore, electrostatic potential calculations of the mMbSerRS dimer show an extended area of positive surface potential on the inner bow of the N-terminal domain, supporting the idea of N-terminal domain involvement in recognition of the negatively charged tRNA backbone (21). As target sites for mutagenesis we have chosen positions encoding highly conserved amino acids in the N-terminal domain of mMbSerRS (marked by dots in Fig. 1A), that are according to the docking model, in the proximity of tRNA (Fig. 1B). In all constructed mMbSerRS variants (listed in supplemental Table S1) a single amino acid was replaced with alanine: arginine 38, arginine 76, arginine 78, lysine 79, lysine 87, lysine 88, tyrosine 89, lysine 90, arginine 94, lysine 141, asparagine 142, and arginine 143. Most altered residues carry basic side chains expected to interact with negatively charged tRNA. Functional characterization of mMbSerRS mutants was performed in vitro and in vivo.
A Single Amino Acid Change (R76A) Causes Complete Loss of Aminoacylation Activity
Serylation propensity of all SerRS variants was tested in a standard aminoacylation assay with in vitro transcribed MbtRNAGGASer as a substrate (Fig. 2). Mutant R76A completely failed to serylate tRNASer, a notable drop in aminoacylation activity was detected for mutants K79A and R94A, whereas mMbSerRS variants carrying R38A, K141A, N142A, and R143A replacements revealed moderately decreased initial serylation rates in comparison with WT MbSerRS. Structural integrity of the variant carrying the R76A replacement was probed by circular dichroism, which confirmed that the observed lack of tRNA charging capacity was not caused by protein misfolding (data not shown). This is also supported by the ability of the R76A, K79A, R94A, and R143A mutants to catalyze amino acid activation in the pyrophosphate exchange reaction (data not shown).
FIGURE 2.

Aminoacylation activity of mMbSerRS variants. Relative serylation rates of in vitro transcribed MbtRNASer by mMbSerRS variants are presented by horizontal bars. Mutated enzymes that retain the serylation ability comparable with the WT enzyme are colored black, mutants with the serylation rate lowered to 59–80% of the WT are designated with traverse lines, variants with a significant drop in the serylation rate (33% of the WT serylation rate) are marked with a black and white square pattern, and one which completely loses the serylation ability is highlighted white.
Gel Retardation Assay Reveals Amino Acids Crucial for Non-covalent Complex Formation with Cognate tRNA
All mMbSerRS variants were tested for the ability to participate in the non-covalent complex formation with in vitro transcribed MbtRNAGGASer (Fig. 3). Mutants R38A, R78A, K87A, K88A, Y89A, K90A, K141A, N142A, and R143A formed complexes stable enough to be detected on the gel (Fig. 3, lanes c, e, g–j, and l–n, respectively), whereas the variant R76A did not form a complex, as expected. Because the interactions between synthetase and tRNA are in general electrostatic, raising the salt concentration may affect the stability of the complexes. Gel mobility shift assay was thus performed in the presence of 25 and 250 mm KCl (Fig. 4) for the wild type enzyme and mutants with moderately decreased serylation propensity (R38A, K141A, and R143A). As shown in Fig. 4A the complexes involving mutants R38A and R143A are much weaker in the presence of 10-fold higher salt (lanes g and i, respectively, in comparison with lanes c and e), whereas the K141A·tRNA complex vanishes completely (lane h in comparison with lane d). Thus, amino acids Arg38, Lys141, and Arg143 seem to be involved in non-covalent complex formation. Moreover, diminished serylation activities of these mutants in the presence of a higher salt concentration emphasize their involvement in ionic interactions (Fig. 4B). The activity of the WT enzyme at 25 mm KCl was used as the reference point (100% activity) and the activities of WT SerRS, R38A and K141A enzymes were determined at salt concentrations of 125 and 250 mm. Evidently, the activity of mutants drops more rapidly than the enzyme activity of the WT. More precisely, there is a 60% drop in activity of mutated enzymes when the concentration of KCl was increased from 25 to 125 mm, whereas in the case of the wild type enzyme activity drops only 20% (and the enzyme is still around 80% active in the presence of 125 mm KCl). Although inactive in tRNA complex formation, the capacity of mutants K79A and R94A to catalyze the formation of seryl-adenylate remained comparable with the wild type enzyme, as revealed by a PPi-exchange assay (not shown).
FIGURE 3.

Gel mobility shift assay of the non-covalent complexes between MbtRNASer and various mMbSerRS mutants. MbtRNASer was incubated with different mMbSerRS variants (final concentration of tRNA and enzymes was 1.1 and 0.6 μm, respectively) and subjected to PAGE under native conditions: WT, lane b; R38A, lane c; R76A, lane d; R78A, lane e; K79A, lane f; K87A, lane g; K88A, lane h; Y89A, lane i; K90A, lane j; R94A, lane k; K141A, lane l; N142A, lane m; and R143A, lane n. Non-complexed mMbSerRS (8.3 pmol) and MbtRNASer (14.8 pmol) were loaded on the gel as electrophoretic mobility markers (lanes a and o, respectively). Non-covalent complexes and non-complexed tRNA are marked with black and white arrows, respectively.
FIGURE 4.

Effect of different salt concentrations on non-covalent complex formation and serylation propensity. A, non-covalent complexes were made as described under “Experimental Procedures” and in the legend to Fig. 3, except that in the reaction mixture the concentration of KCl was varied as indicated, and then subjected to PAGE under native conditions. Non-complexed WT mMbSerRS is visible in lane a, whereas non-covalent complexes between tRNA and mMbSerRS variants in the presence of 25 mm KCl were loaded into following lanes: WT, lane b; R38A, lane c; K141A, lane d; and R143A, lane e; non-covalent complexes between tRNA and mMbSerRS variants in the presence of 250 mm KCl were loaded into following lanes: WT, lane f; R38A, lane g; K141A, lane h; and R143A, lane i; non-complexed tRNA is in lane j. Non-covalent complexes and non-complexed tRNA are marked with black and white arrows, respectively. B, relative serylation rate of the enzymes was tested at three different KCl concentrations (25, 125, and 250 mm). Activity of the WT enzyme at 25 mm KCl was taken as the reference point (100% activity). Contrary to the WT enzyme, a significant drop in the serylation rate was detected for both mutants after raising the KCl concentration from 25 to 125 mm.
Determination of Dissociation Constants for tRNA·mMbSerRS Complexes by SPR Reveals the Importance of Arg94 in tRNA Binding
We used SPR to compare the interaction of 12 mMbSerRS proteins (WT enzyme and 11 mutants) and tRNA. The study was performed on a CM5 sensor chip on which the WT SerRS and the variants were independently immobilized. Different concentrations of tRNA were injected at a flow rate of 60 μl/min for 210 s. The sensorgram obtained for the WT enzyme agreed best with a two-state binding model with a conformational change, described by the following equation,
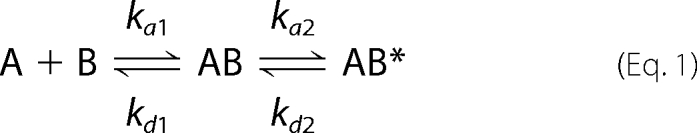 |
where KA = (ka1/kd1)(1 + ka2/kd2) and KD = 1/KA. In this model, the analyte A (tRNA) binds to the ligand B (mMbSerRS) to form an initial complex AB. The complex then undergoes a change in conformation to form a more stable complex AB* (33). The sensorgrams of all other mutants agreed best with the two-state binding model with conformational change as well, except for mutant R94A. Curves obtained for this mutant could have been interpreted only by using the steady state affinity model. In that sense, the KD value (without ka1, kd1, ka2, and kd2 constants) was solely determined for the R94A variant. An example of the sensorgram, denoting tRNA binding to the R143A variant, and the kinetic constants calculated from the fitted curves, are given in Fig. 5 and Table 1, respectively. SPR analysis for mutant R76A was not performed because that particular enzyme was inactive in aminoacylation and did not form a non-covalent complex detectable on the gel. According to the data presented in Table 1 the largest effect on binding the tRNA was achieved by altering arginine 94 to alanine. The KD value for the R94A mutant was almost 50-fold higher than for the wild type enzyme (122 nm for the WT enzyme and 5.81 μm for the R94A variant). A significant effect on interaction with tRNA have mutations of lysines 79 and 141 and arginine 143. Their respective mutants K79A, K141A, and R143A have between 4- and 7-fold higher KD values than the wild type enzyme. Variants R38A and N142A gave similar KD values (422 nm and 386 nm, respectively) that were ∼3.5-fold higher than the wild type KD. Dissociation constants for all other mutants (R78A, K87A, K88A, Y89A, and K90A) ranged from 151 to 301 nm.
FIGURE 5.

Kinetic analysis of tRNA binding to immobilized mMbSerRS R143A monitored by a biosensor. Sensorgrams (red) were obtained for the binding of different concentrations of tRNASer (70.3–2250 nm) in 30 mm Hepes, pH 7.0, 6 mm MgCl2, and 5 mm DTT to R143A mMbSerRS. Data were fit to the two-state conformational change model (black).
TABLE 1.
Kinetic constants obtained from the fit to the conformational change model of sensorgrams acquired for the binding of different concentrations of tRNA (19.5 nm to 8 μm) to various SerRS mutants
Only for mutant R94A steady state affinity model was used (text).
| SerRS variant | ka1 | kd1 | ka2 | kd2 | KD | Relative KDa | χ2b |
|---|---|---|---|---|---|---|---|
| m−1s−1/105 | s−1/10−1 | s−1/10−2 | s−1/10−3 | m/10−7 | |||
| WT | 1.471 ± 0.010 | 1.239 ± 0.009 | 1.482 ± 0.006 | 2.513 ± 0.003 | 1.22 | 1 | 2.71 |
| R38A | 0.442 ± 0.001 | 0.342 ± 0.001 | 0.832 ± 0.004 | 9.949 ± 0.017 | 4.22 | 3.5 | 0.20 |
| R78A | 0.558 ± 0.002 | 0.417 ± 0.002 | 1.204 ± 0.004 | 4.192 ± 0.006 | 1.93 | 1.6 | 0.28 |
| K79A | 0.299 ± 0.001 | 0.231 ± 0.001 | 0.283 ± 0.002 | 7.543 ± 0.029 | 5.61 | 4.6 | 0.18 |
| K87A | 0.983 ± 0.013 | 0.714 ± 0.011 | 1.345 ± 0.014 | 3.552 ± 0.011 | 1.52 | 1.2 | 1.95 |
| K88A | 1.175 ± 0.004 | 0.862 ± 0.003 | 0.993 ± 0.002 | 2.703 ± 0.002 | 1.57 | 1.3 | 0.53 |
| Y89A | 0.568 ± 0.002 | 0.498 ± 0.003 | 1.048 ± 0.004 | 5.494 ± 0.008 | 3.01 | 2.5 | 0.20 |
| K90A | 0.572 ± 0.005 | 0.547 ± 0.006 | 1.606 ± 0.005 | 4.568 ± 0.006 | 2.12 | 1.7 | 0.77 |
| R94A | 58.10 ± 1.70 | 47.6 | 0.07 | ||||
| K141A | 0.264 ± 0.001 | 0.237 ± 0.001 | 0.515 ± 0.003 | 6.169 ± 0.013 | 4.89 | 4.0 | 1.34 |
| N142A | 1.423 ± 0.005 | 1.212 ± 0.004 | 0.673 ± 0.002 | 5.578 ± 0.005 | 3.86 | 3.2 | 0.77 |
| R143A | 0.324 ± 0.001 | 0.444 ± 0.002 | 0.605 ± 0.004 | 11.27 ± 0.03 | 8.90 | 7.3 | 0.09 |
Relative KD is determined as KD(mutant)/KD(WT).
Statistical value describing the closeness of fit. Values <10 are acceptable (34).
The first association constant, ka1 (constant of initial complex formation), is in the range of 104 m−1 s−1 for most of the mutants. Only the wild type enzyme and variants K88A and N142A have 1.175 × 105 m−1 s−1 ≤ ka1 ≤ 1.471 × 105 m−1 s−1 (Table 1), which is a value between 2- and 6-fold higher ka1 than for other mutated mMbSerRSs. The first-order rate constants (ka2) for WT, R78A, K87A, K88A, Y89A, and K90A were 2–6-fold larger than kd2; these mutants have KD values similar to the dissociation constant of the wild type (1.2–2.5 KD of the wild type enzyme). In the case of the N142A, ka2 is just slightly higher than kd2, whereas mutants R38A, K79A, K141A, and R143A have second dissociation rate constants (kd2) even greater than ka2. Their KD values are at least 3.5-fold larger than KD for the WT enzyme. The SPR experiment also allowed the estimation of so called “KD1” (KD of the first reaction of the two-step mechanism; KD1 = kd1/ka1), which is similar for all the mutants. Taken together, our results suggest that the second step of the reaction predominantly determines the observed KD values.
Effects of Engineered mMbSerRS Amino Acid Alterations on Serylation of tRNA in Vivo
We have recently shown that expression of the gene encoding M. barkeri bacterial-type SerRS (bMbSerRS) in E. coli complements the function of thermolabile SerRS at the nonpermissive temperature, whereas expression of the mMbSerRS gene does not (28). However, co-expression of the M. barkeri seryl-tRNA synthetase gene, encoding either bacterial- or methanogenic-type SerRS, with the gene for the cognate archaeal suppressor tRNA leads to suppression of bacterial amber mutations, implying that the E. coli translation machinery can use serylated tRNA from methanogenic archaea as a substrate in protein synthesis (28). Bacterial strain XAC-A24 carries two amber mutations XAC-A24 (F′ ara argE(UAG) rpoB gyrA Δlac pro/F′ lacI(UAG)-Z proAB), suppression of which reflects recognition and aminoacylation levels of suppressor tRNA by selected aminoacyl-tRNA synthetases. The argE(UAG) mutation is suppressible by any amino acid. The other suppressible marker in strain XAC-A24, in which the UAG in-frame codon has been inserted in the lacI part of a lacI-lacZ fusion gene, was used for quantification of suppression efficiency (35, 36). Because serylation of M. barkeri serine-specific tRNA by endogenous E. coli SerRS is negligible (12, 37), suppression is entirely dependent on recognition between archaeal partners (mMbSerRS/suppressor tRNASer). Serine-specific tRNAs are especially suitable to be used in such assays, because the anticodon is not a recognition element for interaction with the cognate synthetase, and its alteration does not change the tRNA identity (38). We have converted the tRNACGAMSer isoacceptor sequence into the tRNASer suppressor sequence (supSMb) and placed it behind the lpp promoter in the pTech plasmid (pTechsupSMb, see Ref. 28). Next, strain XAC-A24 was co-transformed with a pair of compatible plasmids, one of which carried the gene for a methanogenic-type synthetase variant, whereas the second carried the M. barkeri suppressor tRNASer sequence. To analyze the contribution of individual amino acids to tRNA binding and catalysis, enzyme variants were characterized by the ability to serylate the suppressor tRNA in vivo. Expression of WT synthetase or its mutated variants was either from the pET15b plasmid, which enables constitutive expression by inefficient recognition of T7 promoter by bacterial RNA polymerase (28), or from pBAD24, where it is dependent on induction with arabinose (30). Expression of all mMbSerRS variants in the XAC-A24 strain has been verified by Western blot. No suppression was obtained when mMbSerRS variants, R76A and R94A, were co-expressed with archaeal suppressor tRNA (0 and 1.65% activity, respectively; Fig. 6). To exclude the possibility that the lack of suppression was caused by too low expression of two synthetase variants from the pET15b plasmid, their genes were recloned to pBAD24 vector. Higher expression of the mutated synthetase R94A led to production of slightly higher quantities of serylated suppressor tRNA (from 1.65 to 7% suppression efficiency relative to the WT enzyme). This increment of the suppression level shows that in the case of the R94A mutant a weak suppression is possible. Alteration of residue Arg76 resulted in an enzyme that was completely inactive in vivo (0% activity) regardless of which plasmid, pET15b or pBAD24, was used (see Fig. 6). These results are fully in agreement with our in vitro experiments performed with both mutants. Mutants K79A, K90A, K141A, N142A, and R143A displayed reduced suppression efficiency (Fig. 6) compared with the wild type mMbSerRS, confirming the contribution of these side chains in the interactions with tRNA. All other tested variants (R38A, R78A, K87A, K88A, and Y89A) showed more than 40% of the original suppression efficiency detected by the WT enzyme.
FIGURE 6.

Suppression efficiency of M. barkeri SerRS variants. Suppression efficiency was determined by measuring the β-galactosidase activity in E. coli strain XAC-A24. 100% corresponds to the β-galactosidase activity of strain XAC-A24 co-transformed with the pET15b plasmid, carrying the gene for a wild type mMbSerRS, and pTech vector, carrying the M. barkeri suppressor tRNASer sequence. Results were reported as the percentage of mutant enzyme suppression activity relative to that of the wild type enzyme.
DISCUSSION
Idiosyncratic N-terminal Domains of SerRS Enzymes Provide tRNASer Binding Capacity
The canonical tRNASer is characterized by a long variable arm (B20 bases) between the anticodon stem and the T-arm, which SerRS employs as a major tRNA identity element to discriminate its cognate tRNAs from all other species. This elongated variable arm is well conserved throughout the evolutionary process from prokaryotes to eukaryotes (with the only exception being metazoan mitochondria). Accordingly, SerRS enzymes have acquired a unique N-terminal domain, mostly structured as a coiled-coil (3, 16, 17), for recognizing the variable arm. The N-terminal coiled-coil of P. horikoshii SerRS (17) comprises additional basic residues as compared with bacterial SerRSs (3, 16) and a Trp residue (Trp40) found in other archaeal/eukaryal serine-specific synthetases (17). An insertion of 20 amino acids into the N-terminal sequences of metazoans and trypanosomatid SerRS sequences, at the center of the predicted coiled-coil motif, suggests that this region may extend beyond the length seen in the structures solved so far (39). On the other hand, although the coiled-coiled N-terminal domain exists in the metazoan mitochondrial SerRS, it does not bind the tRNA extra arm, because its cognate tRNA structures markedly deviate from the canonical cloverleaf secondary structure with highly truncated and/or intrinsically missing arms (40). Consequently, biochemical (41) and recent structural studies (19) have revealed a truly distinctive mode of tRNA binding by mammalian mitochondrial SerRS. The SerRSs from methanogenic archaea also differ markedly from their bacterial-type counterparts, most notably through the absence of the N-terminal coiled-coil (21). Besides its role in tRNA binding, the N-terminal domain of mMbSerRS is required to assist proper folding of the catalytic domain (25). Interestingly, archaeal SerRSs, either of archaeal/eukaryal- (17) or methanogenic-type, do not conserve the residues of T. thermophilus SerRS that interact with the tRNA extra arm (23, 26), although they can aminoacylate bacterial tRNAsSer, besides their homologous archaeal tRNA substrates (17, 29, 32). Therefore, it is possible that archaeal SerRSs recognize not only the tRNA sequence, but also its overall three-dimensional structure that enables recognition of different tRNAsSer (32).
Identification of Amino Acids in the N-terminal Domain of mMbSerRS Critical for tRNA Recognition
We have demonstrated the contribution of individual amino acid residues toward tRNA binding, because their replacement leads to clear functional defects (see Table 1 and Figs. 2, 3, 5, and 7). The altered residues can have direct influence on tRNA binding (side chains that are in direct contact with tRNA) or indirectly affect interaction between the synthetase and tRNA (side chains that ensure proper positioning of amino acids involved in direct contact with tRNA). We demonstrate, by a range of methods, that the individual substitutions of Arg76, Lys79, and Arg94 have a pronounced effect on the ability of the enzyme to serylate cognate tRNA. The gel mobility shift assay showed that individual substitutions of these residues decrease the stability of SerRS·tRNASer complexes (see Fig. 3). Binding analysis using SPR revealed that wild type archaeal mMbSerRS binds in vitro transcribed MbtRNASer with high affinity (see Table 1). The determined dissociation constant (KD = 0.12 μm) is similar to other synthetase·tRNA complexes (involving CysRS (42), GlnRS (43, 44), and AspRS (45)). The interaction between M. barkeri SerRS and tRNA is entirely lost after replacement of Arg76, located in the short H2 helix, with alanine (see Figs. 1, 2, 3, and 6), preventing the estimation of the KD. The most affected measurable tRNA binding affinity was for mutant R94A, the KD value of which was increased about 50-fold relative to WT mMbSerRS. Arginine 94 is located in the loop between helix H2 and β-strand A4 and according to the docking model positioned to interact with the T-loop of the tRNASer. The crystal structure of the SerRS·tRNASer complex from T. thermophilus (23, 26, 46) revealed that the tRNA-binding coiled-coil of bacterial SerRS is buried between the TψC arm and the long extra arm of tRNASer. Likewise, biochemical studies on the mammalian mitochondrial system pointed to the importance of the T-loop, which is the main identity element for two unusual tRNASer isoacceptors. Previous kinetic analysis of variant tRNASer transcripts by the mMbSerRS showed reduced serylation efficiency after abolishing interactions between the D- and T-loops, as these alterations, presumably, affected the tertiary structure of tRNASer (27). The experiments presented here with the R94A mMbSerRS variant reveal the importance of the T-loop for interactions with methanogenic-type synthetase.
FIGURE 7.

Contribution of the individual amino acids in tRNA binding. One subunit of the mMbSerRS dimer is shown. The size and the brightness of the spheres designate the significance of the particular residue in tRNA binding according to the cumulative influence on biochemical properties of mMbSerRS. The largest and the brightest sphere (arginine 76) annotates the most important side chain in tRNA recognition.
Alteration of Lys79 (positioned in H2) strongly influences tRNA binding affinity, increasing the KD 5-fold. Variants R94A and K79A aminoacylated tRNASer with approximately one-third the velocity of the wild type enzyme, whereas the mutants carrying alanine at positions Arg38, Lys141, Asn142, and Arg143 remained moderately active, but also exhibited diminished affinity for tRNA, as expected. Arg38, with a proline and two glycines in the vicinity (see Fig. 1A), is located in the loop between helix H1 and β-sheet A2 (see Fig. 1A). The flexibility of this region may be required for correct positioning of this mMbSerRS region toward tRNA. All other conserved amino acids in that area are hydrophobic and oriented to the interior of the enzyme, implicating their involvement in maintaining the structure of the N-terminal domain. Arg143 has a dual role: besides direct involvement in tRNA binding, its main chain also interacts with Arg147, located in helix 4 that participates in helix-turn-helix-mediated positioning of the N-terminal domain relative to the catalytic core (25). Accordingly, the R143A variant binds cognate tRNA with 7-fold lower affinity (Fig. 7).
To assess whether selected N-terminal residues are important for aminoacylation in vivo, we tested the ability of corresponding mutant proteins to promote suppression of bacterial amber mutations. When the wild type mMbSerRS gene was introduced with MbtRNASer to the E. coli strain bearing the lacI-lacZ fusion reporter system, robust β-galactosidase activity was produced (Fig. 6). In contrast, mutated SerRS genes promoted production of detectable, but less active reporter enzymes, in agreement with the KD values estimated by SPR. Importantly, variant R76A was inactive in the suppression assay, even when coexpressed from the pBAD24 plasmid behind a stronger promoter. This confirms that the mutated enzyme(s) did not productively interact with tRNASer, not only in vitro, but also in the cellular context, when competing noncognate tRNAs are present.
mMbSerRS Serylation Mechanism and the Importance of Substrate-induced Conformational Changes
Our previous crystallographic studies revealed that the binding of serine to wild type mMbSerRS causes a significant localized conformational change in the serine ordering loop (residues 394–410) of the enzyme (21). Next, positioning of the cognate tRNA in the active site of mMbSerRS is facilitated upon the conformational change of the motif 2 loop, which participates in ATP binding and mediates interactions with the tRNA acceptor stem (37). Our biochemical experiments point to the importance of the flexibility of the tRNA 3′-end binding region (37) for avoiding a steric clash as seen between the acceptor end of tRNA and the motif 2 loop Ile342 in the tRNA·mMbSerRS complex model, and allowing hydrogen bonding of the first base pair.
The structure of the mMbSerRS enzyme shows that the orientation of N-terminal domains in two monomers differs by a rotation of ∼20° indicating that a conformational change likely accompanies tRNA binding (21). Accordingly, the SPR sensorgrams obtained for tRNA binding to immobilized WT mMbSerRS and the majority of constructed variants with mutations in the N-terminal domain agreed best with a two-state binding model implying that a conformational change occurs in the enzyme upon binding of the tRNA substrate (see Table 1 and Fig. 5). A two-step mechanism for the complex formation of synthetase and tRNA in the serine system from yeast was postulated in the mid-seventies (47, 48).
Our model suggests that the first step of binding will involve formation of a bimolecular complex between the tRNA and the flexibly disposed N-terminal domain of the SerRS. In the second step of the binding reaction, accommodation of the tRNA into the active site will occur, accompanied by conformational changes of the enzyme and the acceptor end of the tRNA. Nevertheless, our SPR binding experiments indicate that the second step of the binding is the rate-limiting step for the reaction and that this step is directly affected for all N-terminal domain mutants. These results suggest that the initial binding of the tRNA to the N-terminal domain is fast compared with the overall binding reaction. The slow step of the binding reaction involves the delivery of the tRNA to the active site of the enzyme, accompanied by the loss of entropy due to the organization of the flexibly disposed domains of the enzyme, and the associated conformational changes in the active site. During this second stage of the binding reaction the reduction in the affinity between the tRNA and the N-terminal domain, as indicated by the kinetic data obtained here, will increase the dissociation rate of the complex, perhaps by reducing the precision with which the tRNA is accommodated into the active site. Consequently, this would reduce the likelihood for the associated conformational changes in the active site of the enzyme that stabilize the interaction with the tRNA resulting in an increased kd2 of the binding reaction. A similar kinetic phenomenon was observed by Tsai and Johnson (49) of how T7 DNA polymerase discourages the incorporation of non-cognate nucleotides.
The different relative orientations of the tRNA-binding and catalytic domains were also shown to be associated with tRNA binding in yeast (50), E. coli AspRS (51), and human mitochondrial PheRS (52). Therefore, although tRNA-binding domains in the two SerRS types are non-homologous and evolutionarily unrelated (21), the requirement for a closing movement of the N-terminal domain upon tRNA binding has been observed in the T. thermophilus SerRS·tRNA co-crystal structure (23) and shown here for mMbSerRS.
Supplementary Material
Acknowledgments
We are grateful to Eilika Weber-Ban for useful suggestions regarding experimental procedures and for valuable discussions. Mike Scott and Stefan Schauer from the Functional Genomics Center, Zurich, are greatly acknowledged for assistance in SPR experiments. We are indebted to Rouven Bingel-Erlenmeyer and Martina Trokter for help with protein purification.
This work was supported by Ministry of Science, Education and Sports of the Republic of Croatia Project (119-0982913-1358), grants from the Swiss National Science Foundation, the Scientific Cooperation between Eastern Europe and Switzerland (SCOPES) program of the Swiss National Science Foundation, and Unity through Knowledge Fund project 10/07.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1.
- aaRS
- aminoacyl-tRNA synthetase (standard amino acid abbreviations precede RS throughout)
- SerRS
- seryl-tRNA synthetase
- mMbSerRS
- methanogenic-type Methanosarcina barkeri SerRS
- MbtRNAGGASer
- M. barkeri tRNASer with anticodon GGA
- SPR
- surface plasmon resonance
- WT
- wild type
- DTT
- dithiothreitol
- Mes
- 2-(N-morpholino)ethanesulfonic acid.
REFERENCES
- 1.Ibba M., Soll D. (2000) Annu. Rev. Biochem. 69, 617–650 [DOI] [PubMed] [Google Scholar]
- 2.Eriani G., Delarue M., Poch O., Gangloff J., Moras D. (1990) Nature 347, 203–206 [DOI] [PubMed] [Google Scholar]
- 3.Cusack S., Berthet-Colominas C., Härtlein M., Nassar N., Leberman R. (1990) Nature 347, 249–255 [DOI] [PubMed] [Google Scholar]
- 4.Ibba M., Morgan S., Curnow A. W., Pridmore D. R., Vothknecht U. C., Gardner W., Lin W., Woese C. R., Söll D. (1997) Science 278, 1119–1122 [DOI] [PubMed] [Google Scholar]
- 5.Zhang C. M., Perona J. J., Ryu K., Francklyn C., Hou Y. M. (2006) J. Mol. Biol. 361, 300–311 [DOI] [PubMed] [Google Scholar]
- 6.Ataide S. F., Ibba M. (2006) ACS Chem. Biol. 1, 285–297 [DOI] [PubMed] [Google Scholar]
- 7.Giegé R., Puglisi J. D., Florentz C. (1993) Prog. Nucleic Acid Res. Mol. Biol. 45, 129–206 [DOI] [PubMed] [Google Scholar]
- 8.Giegé R., Sissler M., Florentz C. (1998) Nucleic Acids Res. 26, 5017–5035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nozawa K., O'Donoghue P., Gundllapalli S., Araiso Y., Ishitani R., Umehara T., Söll D., Nureki O. (2009) Nature 457, 1163–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ribas de Pouplana L., Schimmel P. (2001) Cell 104, 191–193 [DOI] [PubMed] [Google Scholar]
- 11.Ebel J. P., Giegé R., Bonnet J., Kern D., Befort N., Bollack C., Fasiolo F., Gangloff J., Dirheimer G. (1973) Biochimie 55, 547–557 [DOI] [PubMed] [Google Scholar]
- 12.Kim H. S., Vothknecht U. C., Hedderich R., Celic I., Söll D. (1998) J. Bacteriol. 180, 6446–6449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tumbula D., Vothknecht U. C., Kim H. S., Ibba M., Min B., Li T., Pelaschier J., Stathopoulos C., Becker H., Söll D. (1999) Genetics 152, 1269–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lenhard B., Orellana O., Ibba M., Weygand-Duraseviæ I. (1999) Nucleic Acids Res. 27, 721–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woese C. R., Olsen G. J., Ibba M., Söll D. (2000) Microbiol. Mol. Biol. Rev. 64, 202–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujinaga M., Berthet-Colominas C., Yaremchuk A. D., Tukalo M. A., Cusack S. (1993) J. Mol. Biol. 234, 222–233 [DOI] [PubMed] [Google Scholar]
- 17.Itoh Y., Sekine S., Kuroishi C., Terada T., Shirouzu M., Kuramitsu S., Yokoyama S. (2008) RNA Biol. 5, 169–177 [DOI] [PubMed] [Google Scholar]
- 18.Weygand-Durasevic I., Cusack S. (2005) in The Aminoacyl-tRNA Synthetases ( Ibba M., Francklyn C., Cusack S. eds) pp. 177–192, Landes Bioscience, Georgetown, TX [Google Scholar]
- 19.Chimnaronk S., Gravers Jeppesen M., Suzuki T., Nyborg J., Watanabe K. (2005) EMBO J. 24, 3369–3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sprinzl M., Vassilenko K. S. (2005) Nucleic Acids Res. 33, D139–D140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bilokapic S., Maier T., Ahel D., Gruic-Sovulj I., Söll D., Weygand- Durasevic I., Ban N. (2006) EMBO J. 25, 2498–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. (1997) Nucleic Acids Res. 25, 4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biou V., Yaremchuk A., Tukalo M., Cusack S. (1994) Science 263, 1404–1410 [DOI] [PubMed] [Google Scholar]
- 24.Vincent C., Borel F., Willison J. C., Leberman R., Härtlein M. (1995) Nucleic Acids Res. 23, 1113–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bilokapic S., Ivic N., Godinic-Mikulcic V., Piantanida I., Ban N., Weygand-Durasevic I. (2009) J. Biol. Chem. 284, 10706–10713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cusack S., Yaremchuk A., Tukalo M. (1996) EMBO J. 15, 2834–2842 [PMC free article] [PubMed] [Google Scholar]
- 27.Korencic D., Polycarpo C., Weygand-Durasevic I., Söll D. (2004) J. Biol. Chem. 279, 48780–48786 [DOI] [PubMed] [Google Scholar]
- 28.Lesjak S., Weygand-Durasevic I. (2009) FEMS Microbiol. Lett. 294, 111–118 [DOI] [PubMed] [Google Scholar]
- 29.Gruic-Sovulj I., Jaric J., Dulic M., Cindric M., Weygand-Durasevic I. (2006) J. Mol. Biol. 361, 128–139 [DOI] [PubMed] [Google Scholar]
- 30.Guzman L. M., Belin D., Carson M. J., Beckwith J. (1995) J. Bacteriol. 177, 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coulondre C., Miller J. H. (1977) J. Mol. Biol. 117, 577–606 [DOI] [PubMed] [Google Scholar]
- 32.Bilokapic S., Korencic D., Söll D., Weygand-Durasevic I. (2004) Eur. J. Biochem. 271, 694–702 [DOI] [PubMed] [Google Scholar]
- 33.Yowler B. C., Schengrund C. L. (2004) Biochemistry 43, 9725–9731 [DOI] [PubMed] [Google Scholar]
- 34.Chenal A., Nizard P., Forge V., Pugnière M., Roy M. O., Mani J. C., Guillain F., Gillet D. (2002) Protein Eng. 15, 383–391 [DOI] [PubMed] [Google Scholar]
- 35.Normanly J., Ogden R. C., Horvath S. J., Abelson J. (1986) Nature 321, 213–219 [DOI] [PubMed] [Google Scholar]
- 36.Polycarpo C. R., Herring S., Bérubé A., Wood J. L., Söll D., Ambrogelly A. (2006) FEBS Lett. 580, 6695–6700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bilokapic S., Rokov Plavec J., Ban N., Weygand-Durasevic I. (2008) FEBS J. 275, 2831–2844 [DOI] [PubMed] [Google Scholar]
- 38.Rogers M. J., Söll D. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 6627–6631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geslain R., Aeby E., Guitart T., Jones T. E., Castro de Moura M., Charrière F., Schneider A., Ribas de Pouplana L. (2006) J. Biol. Chem. 281, 38217–38225 [DOI] [PubMed] [Google Scholar]
- 40.Helm M., Brulé H., Friede D., Giegé R., Pütz D., Florentz C. (2000) RNA 6, 1356–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimada N., Suzuki T., Watanabe K. (2001) J. Biol. Chem. 276, 46770–46778 [DOI] [PubMed] [Google Scholar]
- 42.Zhang C. M., Perona J. J., Hou Y. M. (2003) Biochemistry 42, 10931–10937 [DOI] [PubMed] [Google Scholar]
- 43.Weygand-Duraseviæ I., Schwob E., Söll D. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 2010–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bullock T. L., Sherlin L. D., Perona J. J. (2000) Nat. Struct. Biol. 7, 497–504 [DOI] [PubMed] [Google Scholar]
- 45.Frugier M., Moulinier L., Giegé R. (2000) EMBO J. 19, 2371–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yaremchuk A. D., Tukalo M. A., Krikliviy I., Malchenko N., Biou V., Berthet-Colominas C., Cusack S. (1992) FEBS Lett. 310, 157–161 [DOI] [PubMed] [Google Scholar]
- 47.Rigler R., Pachmann U., Hirsch R., Zachau H. G. (1976) Eur. J. Biochem. 65, 307–315 [DOI] [PubMed] [Google Scholar]
- 48.Riesner D., Pingoud A., Boehme D., Peters F., Maass G. (1976) Eur. J. Biochem. 68, 71–80 [DOI] [PubMed] [Google Scholar]
- 49.Tsai Y. C., Johnson K. A. (2006) Biochemistry 45, 9675–9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sauter C., Lorber B., Cavarelli J., Moras D., Giegé R. (2000) J. Mol. Biol. 299, 1313–1324 [DOI] [PubMed] [Google Scholar]
- 51.Rees B., Webster G., Delarue M., Boeglin M., Moras D. (2000) J. Mol. Biol. 299, 1157–1164 [DOI] [PubMed] [Google Scholar]
- 52.Klipcan L., Levin I., Kessler N., Moor N., Finarov I., Safro M. (2008) Structure 16, 1095–1104 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.