

Abstract

A triazolinylidine carbene catalyzed intermolecular Stetter reaction of glyoxamide and alkylidene ketoamides has been developed. 1,4-dicarbonyl products are afforded in good to excellent yields, enantioselectivities and diastereoselectivities. Further derivatization of the products affords useful intermediates for organic synthesis.
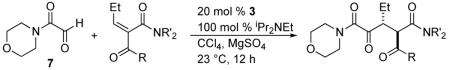
The Stetter reaction,1 the N-heterocyclic carbene (NHC) catalyzed addition of aldehydes to Michael acceptors, is a prototypical example of the emerging class of catalyzed umpolung reactions.2 Following a seminal early report by Enders and Teles,3 we4 and others5 have extensively investigated the asymmetric intramolecular Stetter reaction. The asymmetric intermolecular Stetter, on the other hand, has remained a much more significant challenge.6 In 2008, Enders reported an asymmetric intermolecular Stetter reaction of aromatic aldehydes and chalcones proceeding in good yield and modest modest selectivities.7 Concurrently, we reported the enantioselective intermolecular Stetter reaction of glyoxamides 1 with alkylidene malonates 2 (eq 1).8
 |
(1) |
 |
(2) |
A shortcoming of the use of alkylidene malonates 2 is the need for the second ester group. We considered that the use of alkylidene ketoacid derivatives would provide an opportunity to incorporate synthetically useful substituents on the second carbonyl (eq 2). However, the reaction would generate mixtures of diastereomers, a situation that could be rectified through the use of alkylidene ketoamides. We have already demonstrated that the protonation event in the asymmetric Stetter is highly diastereoselective4d and it has been welldocumented that tertiary β-ketoamides, bearing a stereocenter between the carbonyls, are configurationally stable due to strong A1,3 strain in the enolate.9 Interestingly, catalytic asymmetric transformations that generate ketoamide stereocenters are surprisingly rare.10
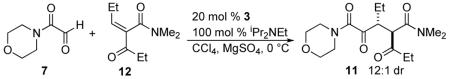
A Knoevenagel reaction of various ketoamides and aldehydes generates the requisite substrates 5 as single olefin isomers.11 Adducts were subjected to our previously developed reaction conditions, Table 1. At ambient temperature, the carbene derived from triazolium salt 3 catalyzes the reaction of glyoxamide 7 with β-ketoamide-derived Michael acceptors in good to excellent yield and high diastereoselectivities. As shown in Table 1, with a dimethylamide Michael acceptor, the product 8 is isolated in 68% yield, 82% ee and 6:1 dr (Table 1), entry 1. When the diethylamide is employed, the product 9 is obtained in similar yield, high dr but lower ee (Table 1, entry 2). The use of a phenylketone on the Michael acceptor results in a nearly racemic product 10 (Table 1, entry 3). With longer alkyl ketone substituents, the product 11 is formed in 92% yield, 89% ee and 5:1 dr (Table 1, entry 4). Lastly, we found that optimal conditions involved conducting the reaction at 0 °C (Table 1, entry 5).
Table 1.
Screen of Michael Acceptors
| entry | R | R' | product | yield (%)a | ee (%)b | drc |
|---|---|---|---|---|---|---|
| 1 | Me | Me | 8 | 68 | 82 | 6:1 |
| 2 | Et | 9 | 66 | 77 | 14:1 | |
| 3 | Ph | Me | 10 | 60 | 7 | 14:1 |
| 4 | Et | Me | 11 | 92 | 89 | 5:1 |
| 5d | Me | 11 | 90 | 92 | 12:1 | |
Reaction conducted with 1 equiv of 7 and 2 equiv of Michael acceptor at 23 °C.
Enantiomeric excess determined by HPLC analysis on a chiral stationary phase.
Diastereomer ratio determined by 1H NMR.
Reaction conducted at 0 °C.
A primary concern at the outset of this study was the configurational stability of the newly formed stereocenters. A control experiment using 20 mol% precatalyst 3 and one equivalent Hünig's base was performed in carbon tetrachloride at 0 °C, shown in Table 2. It was found that the conversion of 11 gradually increases with reaction time with the reaction complete in 12 hours. Fortunately, no epimerization was observed under these basic conditions, consistent with our hypothesis.
Table 2.
Control Experiment to Test for Epimerization
Reaction conducted with 1 equiv of 1 and 2 equiv of 12 at 0 °C.
See Table 1.
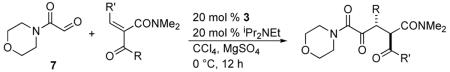
A series of Michael acceptors with different substitution were then synthesized and tested using the optimized reaction conditions, with the results shown in Table 3. When the alkylidene substituent is a methyl group, the product 14 is obtained in excellent yield and 89% ee, with 7:1 dr (Table 2, entry 1). Similar results are observed with other alkyl substituents (Table 2, entry 3-5). The reaction also tolerates a variety of functional groups; substrates with tethered benzyl ether, olefin, and alkyne give desired products in excellent enantioselectivities and good diastereoselectivities (Table 2, entry 7, 9, 10). Compounds with tethered halogen or protected aldehyde are also obtained in good yield and good stereoselectivities (Table 2, entry 8, 11). Substrates with different ketone R groups were also made and subjected to the optimized reaction conditions. When R is propyl, the Stetter adduct 34 is generated in 92% yield, 92% ee and 11:1 dr (Table 2, entry 12). Finally, substrate 37 with a tethered olefin on the ketone leads to product 38 in 94% yield, 90% ee and 9:1 dr.
Table 3.
Substrate Scope
| entry | R | R' | substrate | product | yield (%)a | ee(%)b | drc |
|---|---|---|---|---|---|---|---|
| 1 | Et | Me | 13 | 14 | 25 | 89 | 7:1 |
| 2 | Et | 26 | 11 | 90 | 26 | 12:1 | |
| 3 | Pr | 15 | 16 | 81 | 90 | 6:1 | |
| 4 | Bu | 17 | 18 | 71 | 92 | 12:1 | |
| 5d | Bu | 19 | 20 | 44 | 87 | 11:1 | |
| 6 | CH2CH2Ph | 21 | 22 | 65 | 31 | 19:1 | |
| 7 | CH2CH2OBn | 23 | 24 | 87 | 98 | 11:1 | |
| 8 | CH2CH2CH2CI | 25 | 26 | 83 | 81 | 10:1 | |
| 9 | 27 | 28 | 83 | 90 | 14:1 | ||
| 10 | 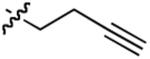 |
29 | 30 | 78 | 92 | 4:1 | |
| 11 | 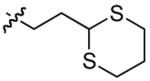 |
31 | 32 | 77 | 86 | 9:1 | |
| 12 | Pr | Et | 33 | 34 | 92 | 92 | 11:1 |
| 13 | 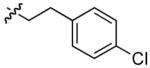 |
35 | 36 | 64 | 94 | 5:1 | |
| 14 | Et | 37 | 38 | 94 | 90 | 9:1 | |
In order to explain the stereochemistry of this transformation, a plausible mechanism is proposed in Scheme 1. Reaction of glyoxamide 7 with carbene derived from 3 will generate a nucleophilic olefin intermediate, which may be one of two different isomers (I or II). Conjugate addition of the favored intermediate I12 to 12 would transiently generate stabilized carbanion IV, which may not even be a local minimum on the energy surface should the conjugate addition/protonation event be concerted.13 The Michael acceptor 12 will approach I from the bottom face to avoid interaction with the benzyl group in the catalyst, as shown in III. The H-bond between the enol and the amide could also play a directing role. An intramolecular proton transfer4d will lead to the desired product 11.14
Scheme 1.

The obtained 1,4-dicarbonyl compounds could be further functionalized to useful building blocks for synthesis (Scheme 2). Reduction of 11 with Super Hydride at -78 °C affords hemiacetal 39 in 72% yield as a 1:1 mixture of diastereomers at the acetal carbon. Treatment of 39 with dry HCl in methanol at 65 °C gives tetra-substituted dihydrofuran 40, which is a versatile intermediate15 and also is a common substructure found in many natural products.16 During this transformation, no epimerization is observed. When 39 is treated with TFA in toluene at 110 °C for 48 hours, chemoselective cleavage of the dimethylamide occurs to provide lactone 4117 with three contiguous stereocenters in 72 % yield.
Scheme 2.

In conclusion, a highly enantio- and diastereoselective intermolecular Stetter reaction was developed. Catalyzed by chiral triazolinylidine carbenes at 0 °C, reactions of glyoxamide and β-keto-amide derived Michael acceptors afford 1,4-dicarbonyl compounds in good to excellent yields, enantioselectivities, and diastereoselectivities. A stereochemical model is proposed to account for the absolute and relative stereochemistry. The obtained product was further functionalized to useful building blocks for organic synthesis.
Supplementary Material
Acknowledgment
We thank NIGMS (GM72586) for support of this research. We thank Johnson & Johnson and Eli Lilly for unrestricted support, and Susie Miller, Derek Dalton and Kevin Oberg (CSU) for solving the crystal structures of 11 and 22.
Footnotes
Supporting Information Available Detailed experimental procedures and spectral data for all new compounds, nOe of 12, and X-ray structures of 11 and 22. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).(a) Stetter H, Schrecke M. Angew. Chem. Int. Ed. Engl. 1973;12:81. [Google Scholar]; (b) Stetter H, Kuhlmann H. Org. React. 1991;40:407–496. [Google Scholar]; (c) Enders D, Balensiefer T. Acc. Chem. Res. 2004;37:534–541. doi: 10.1021/ar030050j. [DOI] [PubMed] [Google Scholar]; (d) Christmann M. Angew. Chem. Int. Ed. 2005;44:2632–2634. doi: 10.1002/anie.200500761. [DOI] [PubMed] [Google Scholar]; (e) Enders D, Niemeier O, Henseler A. Chem. Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; (f) Rovis T. Chem. Lett. 2008;37:2–7. [Google Scholar]
- (2).(a) Corey EJ, Seebach D. Angew. Chem. Int. Ed. Engl. 1965;4:1075–1077. [Google Scholar]; (b) Seebach D. Angew. Chem. Int. Ed. Engl. 1979;18:239–258. [Google Scholar]
- (3).Enders D, Breuer K, Runsink J, Teles JH. Helv. Chim. Acta. 1996;79:1899–1902. [Google Scholar]
- (4).(a) Kerr MS, Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2002;124:10298–10299. doi: 10.1021/ja027411v. [DOI] [PubMed] [Google Scholar]; (b) Kerr MS, Rovis T. Synlett. 2003:1934–1936. [Google Scholar]; (c) Kerr MS, Rovis T. J. Am. Chem. Soc. 2004;126:8876–8877. doi: 10.1021/ja047644h. [DOI] [PubMed] [Google Scholar]; (d) Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2005;127:6284–6289. doi: 10.1021/ja0425132. [DOI] [PubMed] [Google Scholar]; (e) Kerr MS, Read de Alaniz J, Rovis T. J. Org. Chem. 2005;70:5725–5728. doi: 10.1021/jo050645n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Reynolds NT, Rovis T. Tetrahedron. 2005;61:6368–6378. [Google Scholar]; (g) Moore JL, Kerr MS, Rovis T. Tetrahedron. 2006;62:11477–11482. [Google Scholar]; (h) Liu Q, Rovis T. J. Am. Chem. Soc. 2006;128:2552–2553. doi: 10.1021/ja058337u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Liu Q, Rovis T. Org. Proc. Res. Dev. 2007;11:598–604. doi: 10.1021/op600278f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Read de Alaniz J, Kerr MS, Moore JL, Rovis T. J. Org. Chem. 2008;73:2033–2040. doi: 10.1021/jo702313f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Orellana A, Rovis T. Chem. Commun. 2008:730–732. doi: 10.1039/b716445a. [DOI] [PubMed] [Google Scholar]
- (5).(a) Pesch J, Harms K, Bach T. Eur. J. Org. Chem. 2004:2025–2035. [Google Scholar]; (b) Mennen SM, Blank JT, Tran-Dube MB, Imbriglio JE, Miller SJ. Chem. Commun. 2005:195–197. doi: 10.1039/b414574g. [DOI] [PubMed] [Google Scholar]; (c) Matsumoto Y, Tomioka K. Tetrahedron Lett. 2006;47:5843–5846. [Google Scholar]
- (6).(a) Enders D. Stereoselective Synthesis. Springer-Verlag; Heidelberg, Germany: 1993. [Google Scholar]; (b) Enders D, Breuer K. Comp. Asym. Cat. 1999:1093–1104. [Google Scholar]; (c) Nahm MR, Linghu X, Potnick JR, Yates CM, White PS, Johnson JS. Angew. Chem. Int. Ed. 2005;44:2377–2379. doi: 10.1002/anie.200462795. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nahm MR, Potnick JR, White PS, Johnson JS. J. Am. Chem. Soc. 2006;128:2751–2756. doi: 10.1021/ja056018x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Enders D, Han J, Henseler A. Chem. Commun. 2008:3989–3991. doi: 10.1039/b809913h. [DOI] [PubMed] [Google Scholar]
- (8).Liu Q, Perreault S, Rovis T. J. Am. Chem. Soc. 2008;130:14066–14067. doi: 10.1021/ja805680z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Evans DA, Ennis MD, Le T, Mandel N, Mandel G. J. Am. Chem. Soc. 1984;106:1154–1156. [Google Scholar]; (b) Ogata M, Yoshimura T, Fujii H, Ito Y, Katsuki T. Synlett. 1993:728–730. [Google Scholar]; (c) Bartroli J, Turmo E, Belloc J, Forn J. J. Org. Chem. 1995;60:3000–3012. [Google Scholar]; (d) Duan M, Paquette LA. Tetrahedron Lett. 2000;41:3789–3792. [Google Scholar]; (e) Smith AB, III, Brandt BM. Org. Lett. 2001;3:1685–1688. doi: 10.1021/ol0158922. [DOI] [PubMed] [Google Scholar]; (f) Flores-Morales V, Fernandez-Zertuche M, Ordonez M. Tetrahedron: Asymmetry. 2003;14:2693–2698. [Google Scholar]; (g) Sharma KK, Boddy CN. Bioorg. Med. Chem. Lett. 2007;17:3034–3037. doi: 10.1016/j.bmcl.2007.03.060. [DOI] [PubMed] [Google Scholar]
- (10).Kim M-H, Choi S-H, Lee Y-J, Lee J, Nahm K, Jeong B-S, Park H-G, Jew S-S. Chem. Commun. 2009:782–784. doi: 10.1039/b821468a. [DOI] [PubMed] [Google Scholar]
- (11).(a) Hensel MJ, Palmer JT, Learn KS, Fuchs PL. Synth. Commun. 1986;16:1297–1314. [Google Scholar]; (b) Inokuchi T, Kawafuchi H. J. Org. Chem. 2006;71:947–953. doi: 10.1021/jo051952w. [DOI] [PubMed] [Google Scholar]; (c) Ryabukhin SV, Plaskon AS, Volochnyuk DM, Pipko SE, Shivanyuk AN, Tolmachev AA. J. Comb. Chem. 2007;9:1073–1078. doi: 10.1021/cc070073f. [DOI] [PubMed] [Google Scholar]
- (12).Dudding T, Houk KN. Proc. Natl. Acad. Sci. 2004;101:5770–5775. doi: 10.1073/pnas.0307256101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Analogous to the reverse Cope elimination, known to be concerted; see: Niu D, Zhao K. J. Am. Chem. Soc. 1999;121:2456–2459.. Sibi MP, Liu M. Org. Lett. 2000;21:3393–3396. doi: 10.1021/ol006500e.. Sibi MP, Prabagaran N, Ghorpade SG, Jasperse CP. J. Am. Chem. Soc. 2003;125:11796–11797. doi: 10.1021/ja0372309..
- (14).The absolute configuration was confirmed by single crystal XRD of 22; see Supporting Information.
- (15).(a) Pettigrew JD, Bexrud JA, Freeman RP, Wilson PD. Heterocycles. 2004;62:445–452. [Google Scholar]; (b) Zhou G, Corey EJ. J. Am. Chem. Soc. 2005;127:11958–11959. doi: 10.1021/ja054503m. [DOI] [PubMed] [Google Scholar]; (c) Pettigrew JD, Wilson PD. Org. Lett. 2006;8:1427–1429. doi: 10.1021/ol060266w. [DOI] [PubMed] [Google Scholar]; (d) Toth F, Kalaus G, Greiner I, Kajtar-Peredy M, Gomory A, Hazai L, Szantay C. Heterocycles. 2006;68:2301–2317. [Google Scholar]; (e) Feducia JA, Gagne MR. J. Am. Chem. Soc. 2008;130:592–599. doi: 10.1021/ja075518i. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Hayakawa I, Ueda M, Yamaura M, Ikeda Y, Suzuki Y, Yoshizato K, Kigoshi H. Org. Lett. 2008;10:1859–1862. doi: 10.1021/ol800554f. [DOI] [PubMed] [Google Scholar]
- (16).(a) Boeckman RK, Yoon SK, Heckendorn DK. J. Am. Chem. Soc. 1991;113:9682–9684. [Google Scholar]; (b) Harada N, Sugioka T, Uda H, Kuriki T, Kobayashi M, Kitagawa I. J. Org. Chem. 1994;59:6606–6613. [Google Scholar]; (c) Pirrung MC, Lee YR. J. Am. Chem. Soc. 1995;117:4814–4821. [Google Scholar]; (d) Takao K.-i., Ochiai H, Hashizuka T, Koshimura H, Tadano K.-i., Ogawa S. Tetrahedron Lett. 1995;36:1487–1490. [Google Scholar]; (e) Aungst RA, Funk RL. J. Am. Chem. Soc. 2001;123:9455–9456. doi: 10.1021/ja011470b. [DOI] [PubMed] [Google Scholar]
- (17).(a) Rainka MP, Milne JE, Buchwald SL. Angew. Chem., Int. Ed. 2005;44:6177–6180. doi: 10.1002/anie.200501890. [DOI] [PubMed] [Google Scholar]; (b) Howell GP, Fletcher SP, Geurts K, ter Horst B, Feringa BL. J. Am. Chem. Soc. 2006;128:14977–14985. doi: 10.1021/ja0651862. [DOI] [PubMed] [Google Scholar]; (c) Ramazonov NS, Syrov VN. Chem. Nat. Compd. 2006;42:558–561. [Google Scholar]; (d) Akiyama K, Maruyama M, Yamauchi S, Nakashima Y, Nakato T, Tago R, Sugahara T, Kishida T, Koba Y. Biosci., Biotechnol., Biochem. 2007;71:1745–1751. doi: 10.1271/bbb.70168. [DOI] [PubMed] [Google Scholar]; (e) Kim H, Wooten CM, Park Y, Hong J. Org. Lett. 2007;9:3965–3968. doi: 10.1021/ol7016388. [DOI] [PubMed] [Google Scholar]; (f) Koerner M, Hiersemann M. Org. Lett. 2007;9:4979–4982. doi: 10.1021/ol702092h. [DOI] [PubMed] [Google Scholar]; (g) Li Y, Hale KJ. Org. Lett. 2007;9:1267–1270. doi: 10.1021/ol0700862. [DOI] [PubMed] [Google Scholar]; (h) Mitra S, Gurrala SR, Coleman RS. J. Org. Chem. 2007;72:8724–8736. doi: 10.1021/jo701415m. [DOI] [PubMed] [Google Scholar]; (i) Nakato T, Yamauchi S. J. Nat. Prod. 2007;70:1588–1592. doi: 10.1021/np070300v. [DOI] [PubMed] [Google Scholar]; (j) Fillion E, Carret S, Mercier LG, Trepanier VE. Org. Lett. 2008;10:437–440. doi: 10.1021/ol702613f. [DOI] [PubMed] [Google Scholar]; (k) Jacobine AM, Lin W, Walls B, Zercher CK. J. Org. Chem. 2008;73:7409–7412. doi: 10.1021/jo801258h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.