

Abstract
Mitochondria play a major role in cellular function, not only as a major site of ATP production, but also by regulating energy expenditure, apoptosis signaling, and production of reactive oxygen species. Altered mitochondrial function is reported to be a key underlying mechanism of many pathological states and in the aging process. Measurements conducted using intact mitochondria isolated from fresh tissue provides distinct information regarding the function of these organelles that complements conventional mitochondrial assays using previously frozen tissue as well as in vivo assessment using techniques such as magnetic resonance spectroscopy. This chapter describes the process by which mitochondria are isolated from small amounts of human skeletal muscle obtained by needle biopsy and two approaches used to assess mitochondrial oxidative capacity and other key components of mitochondrial physiology. We first describe a bioluminescent approach for measuring the rates of mitochondrial ATP production. Firefly luciferase catalyzes a light-emitting reaction whereby the substrate luciferin is oxidized in an ATP-dependent manner. A luminometer is used to quantify the light signal which is proportional to ATP concentration. We also review a method involving polarographic measurement of oxygen consumption. Measurements of oxygen consumption, which previously required large amounts of tissue, are now feasible with very small amounts of sample obtained by needle biopsy due to recent advances in the field of high-resolution respirometry. We illustrate how careful attention to substrate combinations and inhibitors allows an abundance of unique functional information to be obtained from isolated mitochondria, including function at various energetic states, oxidative capacity with electron flow through distinct complexes, coupling of oxygen consumption to ATP production, and membrane integrity. These measurements, together with studies of mitochondrial DNA abundance, mRNA and protein expression, and synthesis rates of mitochondrial proteins provide insightful mechanistic information about mitochondria in a variety of tissue types.
Introduction
Mitochondria are believed to have originated in eukaryotic cells by endosymbiosis approximately 2 billion years ago (Gray et al., 1999). Although most mitochondrial proteins are encoded by nuclear DNA and imported into the organelle, mitochondria contain ribosomes and between 2 and 10 copies of circular DNA containing 13 protein-encoding regions and 22 tRNA-encoding genes. Mitochondria are an essential part of normal cellular function, particularly in their role in oxidizing carbon substrates to generate energy currency for the cell. As illustrated in Figure 1, mitochondrial ATP synthesis is driven by a proton gradient across the inner mitochondrial membrane as a result of oxidation of carbon substrates in the tricarboxylic acid cycle. These organelles not only produce chemical energy in the form of ATP, but also liberate energy through thermogenic uncoupling, regulate apoptosis, and are a major source of reactive oxygen species (ROS). While ROS production plays an important role in cell signaling, increased ROS levels can result in oxidative damage to mitochondrial DNA, particularly when ROS production exceeds the capacity of antioxidant defense systems (glutathione peroxidase, catalase, and superoxide dismutase) and DNA repair. Indeed, accumulated oxidative damage has been proposed as a mechanism by which the aging process is accelerated (Harman, 1956). The importance of mitochondria in cellular function and energy balance has spawned much interest in their role in the aging process and metabolic diseases such as type 2 diabetes. Accurate, precise assessment of tissue mitochondrial function is necessary to understand the aging process and the underlying mechanisms of many diseases. Various tools, each with distinct advantages and caveats, have been used to probe mitochondrial properties in vitro and in vivo. For example, magnetic resonance spectroscopy permits non-invasive assessment of muscle oxidative capacity (Argov et al., 1987;Kent-Braun and Ng, 2000;Lanza et al., 2007), steady-state mitochondrial ATP synthesis rate (Lebon et al., 2001;Petersen et al., 2003;Befroy et al., 2008), and tricarboxylic acid cycle flux (Lebon et al., 2001;Petersen et al., 2004;Befroy et al., 2008). The ability to assess mitochondrial function under conditions where circulatory and regulatory systems are intact is a strength of these techniques. Experiments performed ex vivo permit a reductionist approach to probe mitochondrial function at specific molecular and cellular levels; information which is crucial to understanding mechanisms by which mitochondrial function is altered with aging and disease and pathways by which exercise and pharmacological interventions exert beneficial effects. Moreover, the ability to quantify mitochondrial DNA copy numbers (Short et al., 2005;Asmann et al., 2006;Chow et al., 2007), expression (mRNA and protein) of various mitochondrial proteins (Short et al., 2005;Lanza et al., 2008), mitochondrial enzyme activities (Rooyackers et al., 1996;Short et al., 2003), and post-translational protein modifications (Jaleel et al., 2005) allows an enormous amount of complementary data to be obtained from the same biopsy sample that is used for measurements of mitochondrial function. In addition, we have recently described a novel methodology to measure synthesis rates of individual skeletal muscle mitochondrial proteins that offers an opportunity to determine the translational efficiency of gene transcripts (Jaleel et al., 2008). Together, these methods permit comprehensive investigation of mitochondrial function and various molecular and cellular mechanisms that underlie mitochondrial changes with aging, disease, and physical activity.
Figure 1. Overview of mitochondrial oxidative phosphorylation. P.

hosphorylation of ADP to ATP at complex V (ATP synthase) is driven by a proton gradient across the inner mitochondrial membrane. Oxidation of carbon substrates in the tricarboxylic acid (TCA) cycle generates reducing equivalents that subsequently provide electron flow to the electron transport system. Electrons are transferred from NADH through complex I (NADH dehydrogenase) and oxidation of succinate by complex II (succinate dehydrogenase). Electron flow from other sources such as electron transferring flavoprotein is not shown.
Historically, the maximal activities of key mitochondrial enzymes have been widely used as indices of mitochondrial oxidative capacity (Wicks and Hood, 1991;Rooyackers et al., 1996;Houmard et al., 1998). Citrate synthase is a common matrix enzyme marker, while succinate dehydrogenase and cytochrome c oxidase are frequently measured as representative enzymes from the inner mitochondrial membrane. Since only small amounts of previously frozen tissue are required, spectrophotometric-based enzyme activity assays are well-suited for human studies where tissue quantities are limited. Although it is not unreasonable to relate maximal activities of these marker enzymes to mitochondrial oxidative capacity, it is unlikely that a single enzyme can accurately reflect the collective function of an organelle as complex as the mitochondrion. Thus, there is a critical need to implement more direct functional measurements of mitochondrial function and oxidative capacity. Measurement of oxygen consumption in isolated mitochondria, pioneered by Britton Chance over 50 years ago (Chance and Williams, 1956), has long been used to assess function of freshly isolated mitochondria. Accurate respiration measurements using conventional respirometers necessitated large quantities of tissue, limiting its practicality for routine human studies. However, recent technological advances in the field of high-resolution respirometry allow these types of measurements using very small amounts of sample (Haller et al., 1994;Gnaiger, 2001), permitting investigators to obtain a wealth of information regarding mitochondrial function from small amounts of human biopsy tissue. In addition to respiration-based measurements, it is also possible to assess mitochondrial function by measuring synthesis of ATP, the ultimate end-product of oxidative phosphorylation. The ability to measure ATP production in freshly isolated intact mitochondria offers an alternative approach for assessing mitochondrial function that provides distinct, complementary information to traditional measurements of oxygen consumption. By exploiting a photon-emitting reaction involving ATP, luciferin, and firefly luciferase, it is possible to quantify the rates of ATP production under various conditions.
For many years luciferase-based measurements of skeletal muscle mitochondrial ATP production rates (MAPR) have become an integral part of studies in our laboratory. We have applied this method to demonstrate an age-related decline in mitochondrial oxidative capacity (Short et al., 2005) and the absence of this trend in individuals who engage in chronic endurance exercise (Lanza et al., 2008). Using this technique, we have observed paradoxically elevated MAPR in Asian Indian individuals in spite of stark insulin resistance compared to people of northern European descent (Nair et al., 2008). To help elucidate the controversial relationship between insulin resistance and mitochondrial function, we have demonstrated that insulin stimulates MAPR in healthy controls but not in insulin resistant individuals (Stump et al., 2003). These studies point to insulin as a key regulator of mitochondrial function and impaired insulin signaling as a potential mechanism by which mitochondrial dysfunction manifests in insulin resistant people (Asmann et al., 2006). This chapter describes methodology for assessing the function of mitochondria isolated from skeletal muscle, with particular attention to the procedures for isolating mitochondria from skeletal muscle samples, a luciferase-based bioluminescent measurement of mitochondrial ATP production, and mitochondrial oxygen consumption measurements using high-resolution respirometry. Although we describe the detailed procedures for functional measurements in isolated mitochondria from skeletal muscle, similar approaches with relatively minor modifications could be applied to investigate other tissues such as liver, adipose, kidney, heart, brain, permeabilized fibers, and cell culture.
Mitochondrial isolation procedures
Skeletal muscle contains two distinct populations of mitochondria (Cogswell et al., 1993). Subsarcolemmal mitochondria (∼20% of total mitochondrial content) are defined as those within 2 micron from the sarcolemma, often densely clustered in the proximity of nuclei and easily liberated by gentle, mechanical homogenization (Elander et al., 1985). Intermyofibrillar mitochondria (∼80% of total mitochondria) are imbedded amongst the contractile machinery and are best liberated by softening the tissue with proteolytic enzymes prior to homogenization (Elander et al., 1985). Improper use of proteolytic enzymes may potentially damage mitochondria, however careful restriction of proteolytic action prevents damage to the organelles while effectively liberating the organelles from contractile machinery. The two populations appear to supply ATP exclusively to their respective subcellular regions with subsacolemmal mitochondria supplying ATP to the membrane ion and substrate transporters and intramyofibrillar mitochondria supplying ATP primarily to myosin ATPases (Hood, 2001). Furthermore, subsarcolemmal mitochondria have been shown to be more sensitive to changes induced by exercise and disuse (Hood, 2001) and more susceptible to the detrimental effects of aging, type 2 diabetes and obesity (Ritov et al., 2005;Menshikova et al., 2005). The possibility that distinct mitochondrial populations may be affected in different ways by aging and physical activity has prompted some groups to develop methods for isolating distinct mitochondrial fractions (Krieger et al., 1980;Ritov et al., 2005;Menshikova et al., 2006). Muscle tissue is crudely homogenized and to liberate subsarcolemmal mitochondria, which are separated by centrifugation. The remaining intermyofibrillar components are liberated by a more aggressive procedure involving protease or a potassium chloride buffer, which partially dissolves myosin. This approach is complicated by uncertain purity of each isolated fraction and the potential for compromised integrity of the mitochondria. Since our group is interested in characterizing the entire population of skeletal muscle mitochondria regardless of cellular location, we employ an isolation procedure to maximize the yield of both subsarcolemmal and intramyofibrillar mitochondria. A high fractional yield typically requires harsh homogenization methods that often damage the organelles. In contrast, gentle homogenizing procedures preserves the integrity of the outer membrane at the expense of lower yield. To optimize both yield and integrity, our isolation methods are largely based on the careful work of Rasmussen and colleagues (Rasmussen et al., 1997a;Rasmussen and Rasmussen, 1997b) who have extensively published on this topic.
Tissue homogenization
Human muscle tissue is obtained from the vastus lateralis muscle using a modified Bergstrom needle under suction (Edwards et al., 1980;Nair et al., 1988). 50-100 mg of muscle tissue is immediately transferred to gauze soaked in ice-cold saline for the short transport (<3 min) from the patient room to the laboratory. An illustration of the homogenization procedure is shown in figure 2 to accompany the following description. The tissue is trimmed of any obvious fat and connective tissue, blotted with gauze, and weighed before transfer to an ice-cold glass Petri dish with 1-2 ml of cold homogenization buffer. The homogenization buffer (buffer A) contains 100 mM KCl, 50 mM tris, 5 mM MgCl2, 1.8 mM ATP, and 1 mM EDTA, which is mixed in large batches, adjusted to pH 7.2, and frozen in 10 ml aliquots. Muscle tissue is cut into small pieces (∼1 mm3) using a scalpel and sharp forceps. A glass Pasteur pipette is used to carefully draw off buffer, and the minced tissue is incubated for 2 minutes in 1 ml of protease medium (1 ml buffer A + 5.66 mg protease from Bacillus licheniformis, 10.6 U/mg) to effectively disrupt the muscle cells with negligible damage to mitochondria (Rasmussen et al., 1997a). Following the protease treatment, the tissue is immediately washed twice with 3 ml of buffer A to dilute and rinse away the protease to avoid excessive tissue breakdown and damage to mitochondria. Any excess buffer is drawn off before transferring tissue to an ice-cold 5 ml homogenizing vessel with 4 ml of buffer A. Custom-made glass Potter-Elvehjem tissue grinders (Kimble / Kontes, Vineland, NJ) with 0.3 mm clearance between the outer diameter of the pestle and inner diameter of the vessel are ideal for gentle yet effective liberation of both subsarcolemmal and intramyofibrillar mitochondria (Rasmussen et al., 1997a). Although various custom-built motor-driven homogenizer units have been described in the literature, we favor a commercially available homogenizer (Potter S, Sartorius AG, Goettingen, Germany) that is well-suited for mitochondrial isolation. Precise alignment between the plunger and homogenizer vessel minimizes grinding. An integrated cooling vessel allows chilled water to circulate without losing the ability to visually monitor the tissue. Variable rotation speed and careful manual control of the vertical action of the plunger allows the homogenization procedure to be carefully controlled. As extensively discussed (Rasmussen et al., 1997a), these are all key factors that are essential to successful isolation of mitochondria with high yield and integrity. The tissue is homogenized at 150 rpm for 10 minutes with slow vertical plunger movements (∼5 cm in 30 seconds) to minimize the annular flow shear forces that are known to damage the mitochondrial membranes (Rasmussen et al., 1997a). The tissue is carefully monitored through the clear walls of the cooling vessel during the intitial 30-60 seconds of homogenizing where short pestle strokes are used to fragment the bulk of the tissue. Thereafter, the pestle is only momentarily paused at the bottom of the vessel with each vertical plunge to break up any remaining pieces of tissue that settle to the bottom.
Figure 2. Tissue homogenization and isolation of mitochondria.
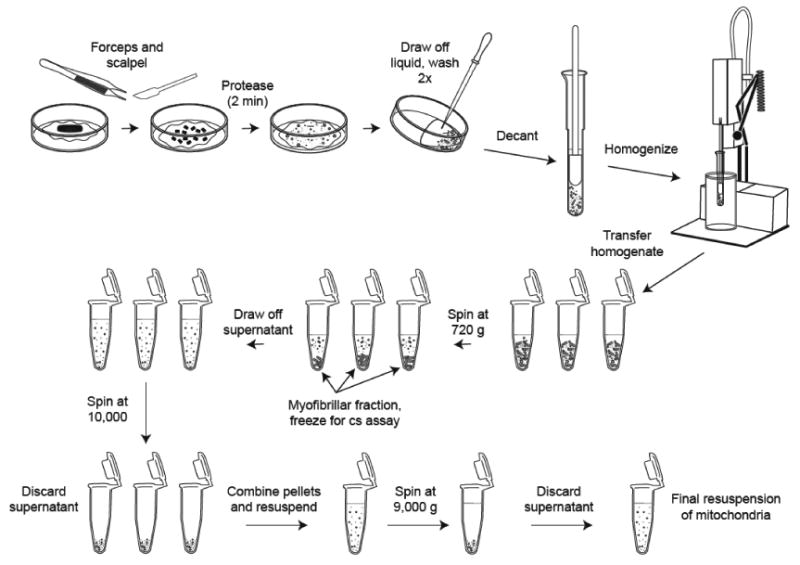
40-100 mg of muscle tissue is placed on an ice-cooled Petri dish and finely minced with forceps and scalpel. A 2-minute protease incubation is used to soften the tissue to help liberate mitochondria that are tightly bound to the contractile filaments. The protease is removed by diluting and washing with buffer. An all glass Potter-Elvehjem tissue grinder (0.3 mm clearance) is used to gently homogenize the tissue in an ice bath. The mitochondria are isolated by differential centrifugation. A low speed spin first removed the myofibrillar portion. A series of two high speed spins generates a mitochondrial pellet, which is resuspended in buffer for functional measurements.
Separation of mitochondria by differential centrifugation
The small size of mitochondria compared to other components of the muscle homogenate allow separation by differential centrifugation. Myofibrillar components form a pellet at low speed centrifugation, whereas sedemintation of mitochondria requires greater centrifugal force. The homogenate is transferred from the homogenizer vessel into 1.5 ml microcentrifuge tubes that have been chilled in an ice bath. Conical, tapered tubes are preferable as they are well-suited to forming pellets that are easily separated from the supernatant. All centrifugation steps are performed at 3-4°C using a microcentrifuge (Eppendorf 5417, Westbury, NY). In the absence of a temperature-controlled environment for the centrifuge, Rasmussen and colleagues describe a clever method for centrifugation of “tip-tubes” in individual ice baths (Rasmussen et al., 1997a). The tubes are centrifuged at low speed (720 g) for 5 minutes to pellet the myofibrillar components, which are frozen for later analysis to determine the homogenization efficiency (described below). The supernatant containing the mitochondrial fraction is transferred to clean, chilled microcentrifuge tubes and centrifuged at 10,000g for 5 minutes to pellet mitochondria. The supernatant is carefully drawn away using a glass Pasteur pipette to avoid disturbing the mitochondrial pellet. The pellets from each tube are combined and resuspended in 1 ml of buffer A by gentle stirring and pipetting. A second high-speed centrifugation is performed at 9,000 g for 5 minutes. The supernatant is discarded and the pellet is gently resuspended in a buffer containing 225 mM sucrose, 44 mM KH2PO4, 12.5 mM Mg acetate, and 6 mM EDTA (buffer B) (Wibom and Hultman, 1990) at a volume of 4 ul of buffer per milligram of muscle tissue, which can be stored on ice for up to 1 hour without any noticeable loss of function. For longer-term storage, mitochondria are suspended in a buffer containing 0.5 mM EGTA, 3mM MgCl2*6H2O, 60 mM potassium lactobionate, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM Sucrose, 1 g/l fatty acid free BSA, 20 mM histidine, 20 μM vitamin E succinate, 3 mM glutathione, 1 μM leupeptine, 2 mM glutamate, 2 mM malate, and 2 mM Mg-ATP (MiPO2, Oroboros, Innsbruck, Austria). Experiments in our lab have found that the function of isolated mitochondria remains unchanged up to 18 hours following homogenization when stored in this buffer at 4°C.
Mitochondrial yield and integrity
The fractional yield (i.e., efficiency) of the homogenization procedure is determined by measuring the mitochondrial marker enzyme citrate synthase remaining in the myofibrillar fraction following the homogenization procedure (Rasmussen et al., 1997a). The previously frozen myofibrillar pellet from the initial low-speed centrifugation is thawed and vigorously homogenized using a tight-fitting tissue grinder to liberate any mitochondria that were not released during the initial homogenization. Homogenization is performed in a buffer containing 20 mM HEPES, 1 mM EDTA, 250 mM sucrose, and 0.1% Triton X-100 to liberate enzymes. Citrate synthase activity is measured spectrophotometrically based on the absorption at 412 nm by the product thionitrobenzoic acid (TNB), which, in the presence of saturating concentrations of substrates acetyl-CoA, oxaloacetate, and dithionitrobenzoic acid (DNTB), is a function of the activity of citrate synthase according to the following 2 step reaction (Sere, 1969):
Acetyl-CoA + oxaloacetate + H2O → citrate + CoA
CoA + DTNB → TNB + CoA-TNB
The homogenization yield is determined by expressing citrate synthase measured in the mitochondrial fraction as a percentage of the total homogenate (myofibrillar fraction + mitochondrial fraction) (Rasmussen et al., 1997a). This homogenization method results in yields of 40-50% (Rasmussen et al., 1997a;Rasmussen et al., 1997b).
Mitochondrial integrity is an important consideration when assessing the function of isolated mitochondria. The outer membrane is easily damaged during homogenization procedures, in which case cytochrome c would exit into the buffer and become rate limiting to oxygen consumption and ATP synthesis. We have confirmed the integrity and function of our mitochondrial preparations in two ways. First a polarographic oxygen electrode (Oxygraph 2K, Oroboros Instruments, Innsbruck, Austria) is often used to measure respiration with serial additions of 10 mM glutamate and 2 mM malate (state 2), 2.5 mM ADP (state 3), and 10 μM cytochrome c, as shown in Figure 3. Respiratory control ratios (state 3:state 4) greater than 8 have been routinely observed, characteristic of high mitochondrial integrity. Furthermore, exogenous cytochrome c does not enhance mitochondrial respiration in these preparations, indicating minimal damage to the outer mitochondrial membrane during isolation procedures. A typical example of a preparation with compromised mitochondrial integrity, reflected by enhanced respiration with exogenous cytochrome c is shown in Panel A of Figure 3. Panel B illustrates the absence of cytochrome c-stimulated respiration in a mitochondrial preparation with high integrity. The inner mitochondrial membrane is impermeable to NADH, allowing inner membrane integrity to be assessed from the increment in the rate of oxygen consumption in the presense of 2.8 mM NADH (Puchowicz et al., 2004). Alternatively, mitochondrial integrity may be assessed by measuring citrate synthase activity in isolated mitochondria before and after membrane disruption by freeze-thaw cycles and extraction of enzyme by Triton X-100. Citrate synthase activity is measured using standard spectrophotometric techniques, as described above. Consistent with the polarographic-based cytochrome c test, we typically find that mitochondrial preparations are 90-95% intact (Asmann et al., 2006).
Figure 3. Testing the integrity of the outer mitochondrial membrane.

Oxygen concentration (grey line) and respiration rate (black line) measured using a polarographic oxygen electrode. Respiration is stimulated by the addition of substrates glutamate and malate (GM) and ADP to stimulate state 3 respiration. 10 μM cytochrome c is added to assess outer membrane integrity. An increment in respiration with exogenous cytochrome c is apparent if the outer mitochondrial membrane is damaged (panel A). Panel B illustrates a preparation where membrane integrity is high.
Mitochondrial ATP production
Principles of the bioluminescent approach
Luciferase from the firefly Photinus pyralis catalyzes a 2-step reaction that oxidizes luciferin in an ATP-dependent reaction that generates a light signal in proportion to ATP concentration (DeLuca and McElroy, 1974):
luciferin + ATP → luciferyl adenylate + PPi
luciferyl adenylate + O2 → oxyluciferin + AMP + light
In nature, these insects use bioluminescence to attract mates. In the laboratory, recombinant luciferase is a powerful analytical tool that can be used to quantify ATP using a luminometer, provided that ATP is the limiting substrate in the reaction (Lundin and Thore, 1975). Since isolated mitochondria respire and generate ATP when provided with appropriate substrates in vitro, it is possible to use a luciferase-based bioluminescent approach to measure the rates of ATP synthesis. Our technique is built on the pioneering methods of Wibom and colleagues who first demonstrated that mitochondrial ATP production could be measured in small amounts of human muscle biopsy tissue (Wibom et al., 1990).
A microplate luminimeter (Veritas, Turner Biosystems) with a highly sensitive photon-counting photomultiplier tube is used to measure bioluminescence from samples in a 96-well plate format (Costar 3912, Corning Life Sciences). A 50 mg muscle sample will yield 200μl of mitochondrial suspension with protein concentrations ranging from 2-5 μg/μl, depending on species and muscle phenotype. A working dilution of 40 μl mitochondrial suspension to 1000 μl buffer B is used for measuring ATP production rates. Since reliable measurements can be achieved with very small amounts of mitochondrial protein (<4μg), it is possible to perform measurements under a variety of conditions in triplicate to take full advantage of the 96-well plate format. Different combinations of substrates and inhibitors permit assessment of the capacity of distinct respiratory chain complexes and different pathways that generate electron flow into the mitochondrial electron transport system, as illustrated in figure 1. The substrate combination of glutamate and malate (GM) provides carbon sources for dehydrogenase reactions in the tricarboxylic acid cycle that generate NADH which is subsequently oxidized by complex I (NADH dehydrogenase). Furthermore, since malate is in equilibrium with fumarate, high malate concentrations will result in product-inhibition of the succinate dehydrogenase reaction, which would otherwise provide direct electron flow into the quinone pool through FADH2. Thus addition of high concentrations of glutamate and malate provides an opportunity to assess the capacity for ATP production with electron flow exclusively through complex I. Similarly, it is possible to assess complex II (succinate dehydrogenase) function in the presence of the substrate succinate and rotenone (SR). By selectively inhibiting complex I, rotenone induces a redox shift that effectively inhibits all of the NADH-linked dehydrogenases in the TCA cycle. Thus, in the presence of rotenone, succinate selectively stimulates electron flow through complex II. ATP production as a result of fatty acid β-oxidation can be assessed using the substrates palmitoyl carnitine and malate (PCM), which provide electron flow through electron-transferring flavoprotein. Finally, a combination of pyruvate, palmitoyl-l-carnitine, α-ketoglutarate and malate (PPKM) provides an index of the additive effects of convergent electron flow from multiple sites along the electron transport system. ADP is a potent regulator of mitochondrial ATP synthesis, both as a substrate and through allosteric enzyme regulation. When ADP is added in saturating concentrations in combination with other substrates, it becomes possible to assess oxidative capacity (state 3) through distinct complexes that shuttle electrons into the Q junction.
Preparation of substrates and ADP
Stock solutions of substrate/inhibitor combinations of GM, SR, PCM, and PPKM are prepared in advance and stored at -20° in small aliquots. With the exception of rotenone, which is dissolved in 50% ethanol, all substrate solutions are made using ultra pure water (Milli-Q, Millipore). The final volume of each well containing substrates (25 μl), ADP (25 μl), sample (25 μl), and luciferin-luciferase reagent (175 μl) is 250 μl. Final concentrations of substrates in each well are as follows:
GM: 10mM glutamate + 5 mM malate
SR: 20 mM succinate + 0.1 mM rotenone
PCM: 0.05 mM palmitoyl-L-carnitine + 2 mM malate
PPKM: 1 mM pyruvate + 0.05 mM palmitoyl-L-carnitine
+ 10 mM α-ketoglutarate + 1 mM malate
A 10mM stock solution of ADP gives a final concentration of 1mM in each well. Since the ADP concentration required to elicit half-maximal respiration (Km) is 10-30 μM in isolated mitochondria (Slater and Holton, 1953;Chance and Williams, 1955), 1 mM ADP is sufficiently high to induce state 3 in the presence of substrates and oxygen. Commercially available ADP is approximately 95% pure with up to 3% contamination with ATP. Even this small ATP contamination substantially increases the background bioluminescence signal and confounds the measurement of mitochondrial ATP levels that are orders of magnitude lower. Therefore, ADP stock solutions must be depleted of ATP, which can be done using high-performance liquid chromatography or enzymatically. The hexokinase reaction can be exploited to enzymatically deplete ATP from ADP stock (Mahaut-Smith et al., 2000) according to the following reaction:
A 10 mM solution of ADP in tris acetate buffer (20 mM tris acetate, 0.2 mM Mg acetate) is incubated for 1 hour at 37°C with 22 mM glucose and 3 U/ml hexokinase. By phosphorylating glucose at the expense of ATP, this reaction effectively depletes ATP from the solution. The solution is then heated to 99°C for 3 minutes to deactivate hexokinase. Lyophilized firefly luciferin-luciferase reagent is commercially available (BioThema 11-501-M) and reconstituted according to the manufacturer's instructions.
Running the experiment and quantitation of ATP
Substrates and sample are added to the wells in triplicate, and the plate is loaded into the luminometer. Luciferase reagent and ADP are added to the wells using programmable injectors that are integrated into the luminometer system. A basic macro controls the volume, timing, and which wells receive the injections. Immediately following the injections, a light reading is taken from each well with a 2 s integration time. Measurements are repeated 7 times on each well with 6 minutes between subsequent readings. Light units are converted to ATP concentration using second-order polynomial equations derived from a series of ATP standard curves that are run on the same plate (Figure 4). Eight ATP standards ranging from 0.1 to 50 μM are measured in duplicate for each combination of substrates. It is necessary to generate an independent standard curve for each substrate condition as some substrate combinations may interfere with the activity of luciferase or block a portion of the bioluminescence signal from being detected by the luminometer. Since the standard curves are being measured under conditions identical to wells containing functional mitochondria, there is no need to account for background signal.
Figure 4. ATP standard curves.

A series of known ATP standards from 0.1 to 50 μM are used to quantify of ATP synthesis rates in isolated mitochondria. Individual standard curves for each substrate combination (GM: mlutamate + malate, SR: succinate + rotenone, PCM: palmitoyl carnitine + malate, PPKM: pyruvate + palmitoyl carnitine + α-ketoglutarate + malate) are generated by measuring light production (relative light units) following the addition of the luciferin-luciferase reagent.
ATP synthesis rates are calculated for each substrate combination using values obtained over the seven measurement cycles. ATP concentration is plotted as a function of time and the overall rate of ATP synthesis is determined as the linear slope through all seven data points (Figure 5). The ATP synthesis rates are commonly normalized using a denominator that allows direct comparison across groups or conditions. For example expressing rates per tissue wet weight is ideal if an investigator wishes to assess oxidative capacity per unit muscle mass, regardless of potential differences in mitochondrial content. Normalizing to citrate synthase activity, mitochondrial protein content, or mitochondrial DNA copy number accounts for differences in mitochondrial content and provides an index of the inherent mitochondrial properties, independent of differences in tissue mitochondrial content.
Figure 5. ATP synthesis rates.

Light units are measured at 7 time points for each substrate combination. Standard curves are used to convert light units into ATP concentration. ATP concentration is plotted as a function of time, of which the linear slope represents the rate of ATP production.
Mitochondrial DNA abundance is measured using real-time quantitative PCR using primer-probe sequences for two marker genes in mitochondrial DNA. A QIAamp DNA mini kit (QIAGEN, Chatworth, CA) is used to extract DNA from frozen muscle samples. The abundance of two marker genes NADH dehydrogenase subunits 1 and 4 are measured using a PCR system (PE Biosystems, Foster City, CA). Samples are run in duplicate and normalized to 28S ribosomal DNA (Short et al., 2005;Lanza et al., 2008). Mitochondrial protein concentration is measured using a colorometric assay (Bio-Rad DC Protein Assay) using a microplate spectrophotometer. There are numerous other measurements that can be performed using frozen tissue that will complement the information obtained from freshly isolated mitochondria. These methods include, but are certainly not limited to, histological analysis, maximal enzyme activities, mRNA expression of nuclear-encoded mitochondrial proteins, protein expression of both nuclear and mitochondrial-encoded proteins, and electron microscopy-based mitochondrial determinations. When combined with functional assessment of freshly isolated mitochondria described below, these methods provide a wealth of complementary mechanistic information.
Mitochondrial respiration
Overview
Oxygen in solution can be measured polarographically with a Clark-type oxygen electrode. Clark electrodes have gold or platinum cathodes and silver or silver/silver chloride anodes, which are connected by a salt bridge and covered by an oxygen-permeable membrane. As oxygen diffuses across the membrane, it is reduced by a fixed voltage between the cathode and anode which generates current in proportion to the concentration of oxygen in solution. By calibrating the voltage with known oxygen concentrations, it is possible to measure the rate of oxygen consumption in a medium containing actively respiring mitochondria. Since reduction of oxygen is a critical step in the process of mitochondrial electron transport and ATP synthesis, measurement of mitochondrial oxygen consumption provides a convenient way to assess mitochondrial function. Although numerous Clark electrode systems are commercially available, our group favors the system manufactured by Oroboros Instruments (Innsbruck, Austria). The combination of twin-2 ml chambers with large (2 mm diameter) cathodes results in high signal-to-noise and very low signal drift. Furthermore, careful choice of inert materials for the chambers, stoppers, and stir bars results in residual oxygen diffusion of less than 2 pmol/s/ml at a PO2 of zero. The sample chambers are housed in an insulated copper block with precise temperature control by Peltier themopiles. This system enables high-resolution measurements of oxygen consumption with very small amounts of tissue (1 mg permeabilized muscle fibers, 0.01mg mitochondrial protein) with a limit of detection of 0.5 pmol/s/ml at steady-state over 5 minutes (Gnaiger, 2001).
An oxygraph protocol for mitochondrial assessment
A 2 ml oxygraph (Oxygraph-2k; Oroboros) chamber is washed with 70% ethanol, rinsed 3 times with distilled water, then filled with respiration medium containing 0.5 mM EGTA, 3 mM MgCl2*6H2O, 60 mM potassium lactobionate, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM Sucrose, and 1 g/l fatty acid free BSA. (MiR05; Oroboros, Innsbruck, Austria). The chamber is allowed to equilibrate with an ambient gas phase at 37°C with a stirrer speed of 750 rpm for >30 minutes to allow air saturation of the respiration medium. Mitochondria are isolated as described above, a 90 μl aliquot of the mitochondrial suspension containing approximately 300 μg mitochondrial protein is added to the chamber. The polyvinylidene fluoride stopper is inserted to generate a closed system with a final volume of 2 ml. Oxygen concentration is recorded at 0.5hz and converted from voltage to oxygen concentration using a two-point calibration. Respiration rates (O2 flux) are calculated as the negative time derivative of oxygen concentration (Datlab Version 4.2.1.50, Oroboros Instruments). The O2 flux values are corrected for the small amount of back-diffusion of oxygen from materials within the chamber, any leak of oxygen from outside of the vessel, and oxygen consumed by the polarographic electrode. Detailed description of standardized instrumental and chemical calibrations is beyond the scope of this chapter and is discussed in detail elsewhere (Gnaiger, 2001). A protocol involving serial additions of various substrates, inhibitors, and uncouplers allows comprehensive assessment of mitochondrial function as shown in Figure 6 and the corresponding stepwise description below:
Figure 6. High-resolution respirometry.

Representative plots of oxygen concentration (top panel) and oxygen flux rate (bottom panel) during a stepwise protocol designed for functional assessment of isolated mitochondria. 1) gas phase equilibration, 2) baseline mitochondrial respiration, 3) substrates glutamate and malate (state 2, complex I), 4) submaximal ADP pulse for assessment of P:O, 5) saturating ADP (state 3, complex I), 6) cytochrome c (membrane integrity), 7) succinate (state 3 complex I + II), 8) rotenone (state 3 complex II), 9) oligomycin (state 4), 10) FCCP (uncoupled respiration), 11) antimycin A (background). Average rates of oxygen consumption for each condition are shown in the bottom panel.
Equilibration with ambient oxygen. Oxygen concentration is measured while in equilibration with a gas phase for air saturation
Baseline respiration. An aliquot of mitochondrial suspension is added to the chamber, the stopper is closed, and respiration is measured in the absence of exogenous substrates.
State 2 respiration. Glutamate (10 mM) and malate (2 mM) are injected through a small capillary tube in the chamber stopper using Hamilton syringes. In the absence of adanylates, oxygen consumption with GM reflects state 2 respiration specific to complex I.
ADP pulse titration. Small pulses of ADP below saturation levels (15 μM) are used to transiently stimulate respiration above state 2. Integration of oxygen flux over the duration of the peak allows quantitation of oxygen consumed per unit of ADP phosphorylated (i.e., ATP/O flux ratio). This approach for measuring mitochondrial phosphorylation efficiency, as well as the use of ADP-injection respirometry is described in detail by Gnaiger and colleagues (Gnaiger et al., 2000).
State 3 respiration (complex I). ADP is added at a saturating level (2.5 mM) to maximally stimulate respiration in the presence of glutamate and malate.
Outer mitochondrial membrane integrity. Mitochondrial integrity may become compromised during isolation procedures, resulting in damage to the outer membrane and release of cytochrome c, which then becomes a limiting factor in mitochondrial respiration. Since cytochrome c cannot penetrate an intact outer mitochondrial membrane, addition of exogenous cytochrome c is a convenient qualitative confirmation of mitochondrial integrity as a quality control measure (Gnaiger and Kuznetsov, 2002;Puchowicz et al., 2004). The absence of a stimulatory effect of cytochrome c on O2 flux rates is indicative of a high quality preparation.
State 3 respiration (complex I + II). The addition of 10 mM succinate stimulates respiration above GM-stimulated state 3 since succinate provides additional electron flow through complex II.
State 3 respiration (complex II). Subsequent addition of 0.5 μM rotenone inhibits complex I. Under these conditions, electron input to complex I is eliminated, providing an index of state 3 respiration through complex II exclusively.
State 4 respiration. The addition of oligomycin (2 μg/μl) inhibits the Fo unit of ATP synthase and induces state 4 respiration by blocking the proton channel and effectively eliminating ATP synthesis. The residual oxygen consumption in the absence of ADP phosphorylation is attributable to proton leak across the inner mitochondrial membrane. Thus, oligomycin inhibited respiration serves as an indicator of the degree of uncoupled respiration or proton leak under these conditions.
Uncoupled respiration. Stepwise titration of the protonophore carbonylcyanide-4-(trifluoromethoxy)-phenylhydrazone (FCCP; 0.05 mM titrations) induces an uncoupled state by dissipating the proton gradient across the inner mitochondrial membrane. Under these conditions, mitochondrial oxidative capacity through complex II can be determined in the absence of the potential control exerted by ATP synthase, adenine nucleotide translocase, or phosphate transporters. Increments in O2 flux above state 3 respiration with rotenone is indicative of a limitation in respiratory capacity by ATP synthase, adenine nucleotide translocases, or phosphate transporters.
Non-mitochondrial respiration. Antimycin A is added at a concentration of 2.5 μM to inhibit cytochrome bc1 complex (complex III). By inhibiting the reduction of cytochrome c, antimycin A allows non-mitochondrial respiration to be measured.
Respiration rates are measured for each condition by an average value of the oxygen flux within a region of stable, consistent oxygen consumption (Figure 5, bottom panel). Respiration rates are then normalized to the amount of mitochondrial protein to allow comparison across groups or conditions without the confounding influence of differences in the amount of mitochondria loaded into the chamber. A standardized, step-wise protocol such as this allows an enormous amount of information to be obtained from a relatively small sample. Although this chapter is focused on measurements using isolated mitochondria, there are numerous excellent references that describe the application of high-resolution respirometry to cultured cells and permeabilized muscle fibers (Kay et al., 2000;Stadlmann et al., 2006;Boushel et al., 2007).
Summary
The function and capacity of intact mitochondria isolated from small amounts of tissue can be objectively determined using polarographic-based measurements of oxygen consumption and luciferase-based bioluminescent measurements of ATP synthesis. Careful selection of substrate combinations and inhibitors enable quantitative measurement of mitochondrial function in various energetic states, oxidative capacity with electron flow through distinct complexes, coupling of oxygen consumption to ATP production, and membrane integrity. These methods provide direct assessment of the function of the organelle in its entirety, as opposed to inferences made from maximal activities of single enzymes. These techniques have wide applicability in both human and animal research and cell culture experiments.
A limitation of the described bioluminescent and polarographic-based measures of mitochondrial function is that the isolated mitochondria are typically exposed to non-physiological conditions. First, isolated mitochondria in vitro are exposed to oxygen concentrations that are much higher than what is present in the cellular microenvironment in vivo. Furthermore, respiration and ATP synthesis rates are measured in response to substrate and ADP concentrations that are saturating and dramatically exceed levels that are endogenous to the cell. Thus, a strength of measuring mitochondrial function in intact muscle fibers or from organisms in vivo is that all circulatory and regulatory systems are intact. Notwithstanding, in vitro measurements from isolated mitochondria permit detailed assessment of the capacity and function of mitochondria, independent of the influence of external variables such as blood flow, oxygen or substrate delivery, or mitochondrial volume density. Furthermore, measurements conducted in isolated mitochondria have the distinct advantage of providing information relevant to the function of distinct components of the electron transport system. Ideally, both in vitro and in vivo methodologies would be used in concert to assess mitochondrial function at the level of the intact organism as well as the individual constituents of the mitochondrial machinery responsible for generating ATP
Reference List
- 1.Argov Z, Bank WJ, Maris J, Peterson P, Chance B. Bioenergetic heterogeneity of human mitochondrial myopathies: phosphorus magnetic resonance spectroscopy study. Neurology. 1987;37:257–262. doi: 10.1212/wnl.37.2.257. [DOI] [PubMed] [Google Scholar]
- 2.Asmann YW, Stump CS, Short KR, Coenen-Schimke JM, Guo Z, Bigelow ML, Nair KS. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes. 2006;55:3309–3319. doi: 10.2337/db05-1230. [DOI] [PubMed] [Google Scholar]
- 3.Befroy DE, Falk PK, Dufour S, Mason GF, Rothman DL, Shulman GI. Increased substrate oxidation and mitochondrial uncoupling in skeletal muscle of endurance-trained individuals. Proc Natl Acad Sci U S A. 2008;%20 doi: 10.1073/pnas.0808889105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boushel R, Gnaiger E, Schjerling P, Skovbro M, Kraunsoe R, Dela F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia. 2007;50:790–796. doi: 10.1007/s00125-007-0594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chance B, Williams G. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem. 1955;217:383–393. [PubMed] [Google Scholar]
- 6.Chance B, Williams G. Respiratory enzymes in oxidative phosphorylation. VI. The effects of adenosine diphosphate on azide-treated mitochondria. J Biol Chem. 1956;221:477–489. [PubMed] [Google Scholar]
- 7.Chow LS, Greenlund LJ, Asmann YW, Short KR, McCrady SK, Levine JA, Nair KS. Impact of endurance training on murine spontaneous activity, muscle mitochondrial DNA abundance, gene transcripts, and function. J Appl Physiol. 2007;102:1078–1089. doi: 10.1152/japplphysiol.00791.2006. [DOI] [PubMed] [Google Scholar]
- 8.Cogswell AM, Stevens RJ, Hood DA. Properties of skeletal muscle mitochondria isolated from subsarcolemmal and intermyofibrillar regions. Am J Physiol. 1993;264:C383–C389. doi: 10.1152/ajpcell.1993.264.2.C383. [DOI] [PubMed] [Google Scholar]
- 9.DeLuca M, McElroy WD. Kinetics of the firefly luciferase catalyzed reactions. Biochemistry. 1974;13:921–925. doi: 10.1021/bi00702a015. [DOI] [PubMed] [Google Scholar]
- 10.Edwards R, Young A, Wiles M. Needle biopsy of skeletal muscle in the diagnosis of myopathy and the clinical study of muscle function and repair. N Engl J Med. 1980;302:261–271. doi: 10.1056/NEJM198001313020504. [DOI] [PubMed] [Google Scholar]
- 11.Elander A, Sjostrom M, Lundgren F, Schersten T, Bylund-Fellenius AC. Biochemical and morphometric properties of mitochondrial populations in human muscle fibres. Clin Sci (Lond) 1985;69:153–164. doi: 10.1042/cs0690153. [DOI] [PubMed] [Google Scholar]
- 12.Gnaiger E. Bioenergetics at low oxygen: dependence of respiration and phosphorylation on oxygen and adenosine diphosphate supply. Respir Physiol. 2001;128:277–297. doi: 10.1016/s0034-5687(01)00307-3. [DOI] [PubMed] [Google Scholar]
- 13.Gnaiger E, Kuznetsov AV. Mitochondrial respiration at low levels of oxygen and cytochrome c. Biochem Soc Trans. 2002;30:252–258. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 14.Gnaiger E, Mendez G, Hand SC. High phosphorylation efficiency and depression of uncoupled respiration in mitochondria under hypoxia. Proc Natl Acad Sci U S A. 2000;97:11080–11085. doi: 10.1073/pnas.97.20.11080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283:1476–1481. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- 16.Haller T, Ortner M, Gnaiger E. A respirometer for investigating oxidative cell metabolism: toward optimization of respiratory studies. Anal Biochem. 1994;218:338–342. doi: 10.1006/abio.1994.1188. [DOI] [PubMed] [Google Scholar]
- 17.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 18.Hood DA. Invited Review: contractile activity-induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol. 2001;90:1137–1157. doi: 10.1152/jappl.2001.90.3.1137. [DOI] [PubMed] [Google Scholar]
- 19.Houmard JA, Weidner ML, Gavigan KE, Tyndall GL, Hickey MS, Alshami A. Fiber type and citrate synthase activity in the human gastrocnemius and vastus lateralis with aging. J Appl Physiol. 1998;85:1337–1341. doi: 10.1152/jappl.1998.85.4.1337. [DOI] [PubMed] [Google Scholar]
- 20.Jaleel A, Halvatsiotis P, Williamson B, Juhasz P, Martin S, Nair KS. Identification of Amadori-modified plasma proteins in type 2 diabetes and the effect of short-term intensive insulin treatment. Diabetes Care. 2005;28:645–652. doi: 10.2337/diacare.28.3.645. [DOI] [PubMed] [Google Scholar]
- 21.Jaleel A, Short KR, Asmann YW, Klaus K, Morse D, Ford GC, Nair KS. In Vivo Measurement of Synthesis Rate of Individual Skeletal Muscle Mitochondrial Proteins. Am J Physiol Endocrinol Metab. 2008 doi: 10.1152/ajpendo.90586.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kay L, Nicolay K, Wieringa B, Saks V, Wallimann T. Direct evidence for the control of mitochondrial respiration by mitochondrial creatine kinase in oxidative muscle cells in situ. J Biol Chem. 2000;275:6937–6944. doi: 10.1074/jbc.275.10.6937. [DOI] [PubMed] [Google Scholar]
- 23.Kent-Braun JA, Ng AV. Skeletal muscle oxidative capacity in young and older women and men. J Appl Physiol. 2000;89:1072–1078. doi: 10.1152/jappl.2000.89.3.1072. [DOI] [PubMed] [Google Scholar]
- 24.Krieger DA, Tate CA, McMillin-Wood J, Booth FW. Populations of rat skeletal muscle mitochondria after exercise and immobilization. J Appl Physiol. 1980;48:23–28. doi: 10.1152/jappl.1980.48.1.23. [DOI] [PubMed] [Google Scholar]
- 25.Lanza IR, Larsen RG, Kent-Braun JA. Effects of old age on human skeletal muscle energetics during fatiguing contractions with and without blood flow. J Physiol. 2007;583:1093–1105. doi: 10.1113/jphysiol.2007.138362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP, Nair KS. Endurance Exercise as a Countermeasure for Aging. Diabetes. 2008 doi: 10.2337/db08-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lebon V, Dufour S, Petersen KF, Ren J, Jucker BM, Slezak LA, Cline GW, Rothman DL, Shulman GI. Effect of triiodothyronine on mitochondrial energy coupling in human skeletal muscle. J Clin Invest. 2001;108:733–737. doi: 10.1172/JCI11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lundin A, Thore A. Analytical information obtainable by evaluation of the time course of firefly bioluminescence in the assay of ATP. Anal Biochem. 1975;66:47–63. doi: 10.1016/0003-2697(75)90723-x. [DOI] [PubMed] [Google Scholar]
- 29.Mahaut-Smith MP, Ennion SJ, Rolf MG, Evans RJ. ADP is not an agonist at P2X(1) receptors: evidence for separate receptors stimulated by ATP and ADP on human platelets. Br J Pharmacol. 2000;131:108–114. doi: 10.1038/sj.bjp.0703517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menshikova EV, Ritov VB, Fairfull L, Ferrell RE, Kelley DE, Goodpaster BH. Effects of exercise on mitochondrial content and function in aging human skeletal muscle. J Gerontol A Biol Sci Med Sci. 2006;61:534–540. doi: 10.1093/gerona/61.6.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Menshikova EV, Ritov VB, Toledo FG, Ferrell RE, Goodpaster BH, Kelley DE. Effects of weight loss and physical activity on skeletal muscle mitochondrial function in obesity. Am J Physiol Endocrinol Metab. 2005;288:E818–E825. doi: 10.1152/ajpendo.00322.2004. [DOI] [PubMed] [Google Scholar]
- 32.Nair KS, Bigelow ML, Asmann YW, Chow LS, Coenen-Schimke JM, Klaus KA, Guo ZK, Sreekumar R, Irving BA. Asian Indians have enhanced skeletal muscle mitochondrial capacity to produce ATP in association with severe insulin resistance. Diabetes. 2008;57:1166–1175. doi: 10.2337/db07-1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nair KS, Halliday D, Griggs RC. Leucine incorporation into mixed skeletal muscle protein in humans. Am J Physiol. 1988;254:E208–E213. doi: 10.1152/ajpendo.1988.254.2.E208. [DOI] [PubMed] [Google Scholar]
- 34.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puchowicz MA, Varnes ME, Cohen BH, Friedman NR, Kerr DS, Hoppel CL. Oxidative phosphorylation analysis: assessing the integrated functional activity of human skeletal muscle mitochondria--case studies. Mitochondrion. 2004;4:377–385. doi: 10.1016/j.mito.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 37.Rasmussen HN, Andersen AJ, Rasmussen UF. Optimization of preparation of mitochondria from 25-100 mg skeletal muscle. Anal Biochem. 1997a;252:153–159. doi: 10.1006/abio.1997.2304. [DOI] [PubMed] [Google Scholar]
- 38.Rasmussen HN, Rasmussen UF. Small scale preparation of skeletal muscle mitochondria, criteria of integrity, and assays with reference to tissue function. Mol Cell Biochem. 1997b;174:55–60. [PubMed] [Google Scholar]
- 39.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 40.Rooyackers OE, Adey DB, Ades PA, Nair KS. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci U S A. 1996;93:15364–15369. doi: 10.1073/pnas.93.26.15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sere PA. Citrate synthase. Methods in Enzymology. 1969:3–5. [Google Scholar]
- 42.Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Short KR, Vittone JL, Bigelow ML, Proctor DN, Rizza RA, Coenen-Schimke JM, Nair KS. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes. 2003;52:1888–1896. doi: 10.2337/diabetes.52.8.1888. [DOI] [PubMed] [Google Scholar]
- 44.Slater E, Holton F. The adeninenucleotide specificity of oxidative phosphorylation. Biochem J. 1953;53:ii. [PubMed] [Google Scholar]
- 45.Stadlmann S, Renner K, Pollheimer J, Moser PL, Zeimet AG, Offner FA, Gnaiger E. Preserved coupling of oxidative phosphorylation but decreased mitochondrial respiratory capacity in IL-1beta-treated human peritoneal mesothelial cells. Cell Biochem Biophys. 2006;44:179–186. doi: 10.1385/CBB:44:2:179. [DOI] [PubMed] [Google Scholar]
- 46.Stump CS, Short KR, Bigelow ML, Schimke JM, Nair KS. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc Natl Acad Sci U S A. 2003;100:7996–8001. doi: 10.1073/pnas.1332551100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wibom R, Hultman E. ATP production rate in mitochondria isolated from microsamples of human muscle. Am J Physiol. 1990;259:E204–E209. doi: 10.1152/ajpendo.1990.259.2.E204. [DOI] [PubMed] [Google Scholar]
- 48.Wicks KL, Hood DA. Mitochondrial adaptations in denervated muscle: relationship to muscle performance. Am J Physiol. 1991;260:C841–C850. doi: 10.1152/ajpcell.1991.260.4.C841. [DOI] [PubMed] [Google Scholar]