

Abstract
Anabolic androgenic steroids (AAS) can promote detrimental effects on social behaviors for which GABA type A (GABAA) receptor-mediated circuits in the forebrain play a critical role. While all AAS bind to androgen receptors (AR), they may also be aromatized to estrogens and thus potentially impart effects via estrogen receptors (ER). Chronic exposure of wild-type male mice to a combination of chemically distinct AAS increased action potential (AP) frequency, selective GABAA receptor subunit mRNAs, and GABAergic synaptic current decay in the medial preoptic area (mPOA). Experiments performed with pharmacological agents and in AR-deficient Tfm mutant mice suggest that the AAS-dependent enhancement of GABAergic transmission in wild-type mice is AR-mediated. In AR-deficient mice, the AAS elicited dramatically different effects, decreasing AP frequency, spontaneous IPSC amplitude and frequency and the expression of selective GABAA receptor subunit mRNAs. Surprisingly, in the absence of AR signaling, the data indicate that the AAS do not act as ER agonists, but rather suggest a novel in vivo action in which the AAS inhibit aromatase and impair endogenous ER signaling. These results show that the AAS have the capacity to alter neuronal function in the forebrain via multiple steroid signaling mechanisms and suggest that effects of these steroids in the brain will depend not only on the balance of AR- versus ER-mediated regulation for different target genes, but also on the ability of these drugs to alter steroid metabolism and thus the endogenous steroid milieu.
Introduction
Anabolic androgenic steroids (AAS) are synthetic derivatives of testosterone designed for therapeutic uses, but now predominantly self-administered to enhance performance or body image (Kochakian and Yesalis, 2000; Llewellyn, 2007). Humans administer AAS in complex regimes characterized by concurrent and prolonged use of multiple AAS at doses that result in supraphysiological levels of these steroids, and >100 AAS are available that vary widely in their chemical properties and their metabolic fates (Llewellyn, 2007). Importantly, although all AAS and AAS metabolites bind to the classical androgen receptor (AR), many, upon aromatization, may also exert physiological effects via classical estrogen receptor (ERα and β) pathways (Basaria et al., 2001; Shahidi, 2001; Clark et al., 2006).
While administered for their anabolic actions, AAS use has also been strongly correlated with untoward actions on social behaviors. Specific effects vary with the regime of AAS administration (duration, dose and drug combinations) and with sex and age of the subject. Depending on these variables, AAS have been shown to both advance and retard pubertal onset in females, enhance and diminish libido in both sexes, and suppress reproductive competence in both sexes (Franke and Berendonk, 1997; Clark and Henderson, 2003; Clark et al., 2006). AAS use is also associated with both enhanced and diminished aggression and anxiety (Clark and Henderson, 2003; Trenton and Currier, 2005). Both AR- and ER-mediated signaling are critically important with respect to the actions of gonadal steroids on the production and complexity of these behaviors. For example, in male rodents, the full repertoire of sexual behaviors reflects the actions of both androgens and estrogens (Hull et al., 2002). Similarly both androgens and estrogens can activate offensive aggression in adult male mice (Simon, 2002; Matsumoto et al., 2003; Scordalakes and Rissman, 2003; Sato et al., 2004).
Expression of these steroid-sensitive behaviors is also dependent on GABAA receptor-mediated transmission in regions of the forebrain including the mPOA (Blaustein and Erskine, 2002; Hull et al., 2002; Miczek et al., 2002; Sewards and Sewards, 2002; Goodson, 2005; Veening et al., 2005). The central portion of the mPOA is characterized by a dense population of neurons that are nearly all GABAergic (Gao and Moore, 1996; Sagrillo and Selmanoff, 1997) and GABA-responsive (Penatti et al., 2005; this study). These cells also express high levels of AR (Lu et al., 1998; Shah et al., 2004) and ERα, lower but appreciable levels of ERβ (Mitra et al., 2003; Nomura et al., 2003; Kudwa et al., 2004), and high levels of aromatase (Foidart et al., 1995). GABAergic transmission in the mPOA is regulated by gonadal steroids (Herbison, 1997) and the AAS (Henderson et al., 2006). The diversity of AAS effects on social behaviors is likely to reflect a complex balance and interplay between AR- and ER-mediated actions on GABAergic and other neural signaling pathways in mPOA and connected regions. Here, we have taken advantage of mutant mice and pharmacological manipulations to assess the roles of AR versus ER in mediating the effects of the AAS within this key forebrain region.
Materials and Methods
Animals.
A single point mutation in the N terminus of the AR gene in the testicular feminization (Tfm) mouse results in the production of a truncated and nonfunctional receptor devoid of both DNA- and steroid-binding domains (Charest et al., 1991; Gaspar et al., 1991; Olsen, 1992; Couse and Korach, 1998). While recognizing that Tfm mice make a truncated AR, we have called these animals functionally “AR-deficient” rather than “androgen-insensitive,” since it is not known how androgen signaling mechanisms at nonclassical (e.g., membrane) AR are affected in Tfm mice and allosteric modulation of ion channels by androgens occurs in these animals (e.g., see Jorge et al., 2002). Because the mutation is X-linked, all Tfm mice generated by conventional breeding methods are infertile and genetically male (Couse and Korach, 1998). Matings of original breeder pairs of heterozygous Aw−J/Aw−J EdaTa−6J +/+ArTfm females and wild-type Tabby Aw−J/Aw−J EdaTa−6J +/Y males obtained from Jackson Laboratories (stock # 001809) have generated a long-standing breeding colony at our facility that has provided both the AR-deficient Tfm (Aw−J/Aw−J +ArTfm/Y) males and wild-type Tabby male littermates used in this study. Offspring were genotyped by PCR according to Scordalakes et al. (2002). All mice were housed in a temperature-controlled and 12/12 h on/off light cycle facility with lights on starting at 7:00 A.M. All animal care procedures and treatment paradigms were approved by the Institutional Animal Care and Use Committee at Dartmouth and are in agreement with the guidelines and recommendations of the National Institutes of Health and American Veterinary Medical Association.
Drug treatment paradigms.
Adult (∼56 d old) male mice were injected intraperitoneally with a combination of three AAS that represent the three major chemical groups of AAS (Clark and Henderson, 2003). (1) Testosterone esters, which are derived from esterification of the 17β-hydroxyl group of testosterone, can be hydrolyzed into free testosterone, reduced to 5α-dihydrotestosterone (DHT) (Martini, 1982; Winters, 1990; Kochakian and Yesalis, 2000) or aromatized to estrogens, including 17β-estradiol (Winters, 1990; Kochakian and Yesalis, 2000). Molecules that have been 5α-reduced cannot be metabolized into estrogens, but may be metabolized into other androgens, such as 3α-androstanediol (3α-diol). (2) 19-nor-testosterone derivatives. These compounds have, in conjunction with the addition of long side chain moieties, substitution of a hydrogen for the methyl group at C19. Like the testosterone esters, 19-nor-testosterone can be aromatized to 17β-estradiol and other estrogens, albeit with less efficiency than free testosterone (Ryan, 1959; Winters, 1990). (3) C17-alkylated derivatives. The 17-methyl moiety precludes aromatization to 17β-estradiol or reduction to DHT (Ryan, 1959; Quincey and Gray, 1967; Winters, 1990), although production of other weak estrogens has been reported (Papaconstantinou et al., 2002; de Gooyer et al., 2003).
Each animal received a mixture containing equal doses of one AAS from each class: 2.5 mg/kg testosterone cypionate (TC), 2.5 mg/kg nandrolone decanoate (ND), and 2.5 mg/kg 17α-Methyltestosterone (17α-MeT) for a total of 7.5 mg AAS/kg/d in sesame oil. Animals were injected for 6 of 7 d for a period of 6 weeks. This total daily AAS concentration is comparable to high doses in human abusers (Kibble and Ross, 1987; Pope and Katz, 1988; Perry et al., 1990). Control subjects were administered with the same injection paradigm and the same volume (∼30 μl; based on body weight) with sesame oil alone. For each cohort studied, 8–10 age-matched wild-type Tabby and AR-deficient Tfm littermates were injected in parallel. For electrophysiological experiments, tamoxifen (2 mg/kg/d) (Rudick and Woolley, 2003; Maguire and Mody, 2007), formestane (4-hydroxyandrost-4-ene-3,17-dione or 4-OHA; 20 mg/kg/d) (Yue et al., 1995), or 17β-estradiol (5 μg per mouse) (Bakker et al., 2004) (for review, see Cornil et al., 2006) was injected into mice for 6 d per week for 6 weeks either alone or in conjunction with the AAS mixture at concentrations indicated above. For physiological experiments in which multiple cohorts were examined, if there was not a significant difference within a treatment group, the data from multiple cohorts were combined. Different cohorts of animals were used for each assay (i.e., electrophysiology, immunocytochemistry, real time PCR, ELISAs, and Western blots). For ERα Westerns, mPOA samples from one Tfm and one wild-type mouse were lost at the time of assay (thus, n = 7 subjects). For ELISAs, mPOA tissue from individual animals was pooled in some cases, and n values reflect the number of determinations, rather than individual animals (see below).
Tissue analyses.
All electrophysiological analyses were made from the central region of the mPOA corresponding to the dorsal aspect of the medial preoptic nucleus (MPN), MPN-medial and encompassing the MPN-central, as defined by Franklin and Paxinos (1997) (Penatti et al., 2005). For mRNA, steroid and protein assessments, dissections were made from a region approximating the MPN, but likely to contain surrounding cells in the mPOA region. Throughout the text, we refer to electrophysiological recordings as made from the MPN and cell biological analyses as made from the mPOA to reflect the fact that tissue samples were likely to include this more extensive region.
RNA extraction and reverse transcription coupled with quantitative real time PCR.
Tissue dissected from coronal slices as described above was stored in the RNA stabilization solution, RNAlater (Ambion) at −20°C. Total RNA was extracted according to manufacturer's protocol for RNAqueous-4PCR kit (Ambion). Briefly, tissue was added to 200 μl lysis/binding buffer and homogenized. An equal volume of 64% EtOH was added and the sample vortexed. The lysate/ethanol mix was applied to an RNAqueous filter (supplied in the kit) and centrifuged for 1 min at 12,000 rpm. The flow through was discarded, the cartridge washed several times and RNA eluted with 50 μl of hot elution buffer. The RNA was treated with 2 units DNase1 (supplied in the kit) for 30 min at 37°C to remove any contaminating genomic DNA. DNase was inactivated by the addition of 0.1 volume DNase Inactivation Reagent, incubated at room temperature (20−22°C) for 2 min, centrifuged, and the DNase-free RNA supernatant collected. The concentration of the RNA was determined by measuring the optical density at 260 nm. Fifty nanograms of total RNA was then reverse transcribed using RETROscript First-Strand Synthesis Kit for real time PCR (RT-PCR) (Ambion) in a total reaction volume of 20 μl. RNA was denatured for 3 min at 75°C with 2 mm dNTPs and 5 μm random decamers. Ten units of RNase inhibitor, RT buffer to 1×, and 100 units of M-MLV reverse transcriptase were added to this mixture, and the reaction was incubated for 1 h at 42°C, followed by inactivation at 92°C for 10 min.
PCR primers and TaqMan MGB probes specific for mouse GABAA receptor α subunit mRNAs were designed using the oligo primer design programs Primer Express Software [Applied Biosystems (ABI)] and OLIGO 6 (Molecular Biology Insights) (Penatti et al., 2005). The specificity of each primer/probe set sequence, for individual GABAA receptor subunits was confirmed by using the National Center for Biotechnology Information sequence alignment algorithm. Primers and probes for the β, γ, δ, and ε subunit mRNAs were obtained from ABI: Mm00433461_m1 (β1), Mm00549788_s1 (β2), Mm00433473-m1 (β3), Mm00433476_m1 (δ), Mm00489932_m1 (ε), Mm00439047_m1 (γ1), and Mm00433489_m1 (γ2), as were primer/probes for ERα (Mm00433149_m1), ERβ (Mm00599821_m1), aromatase (Cyp19) (Mm00484049_m1), progesterone receptor A/B (Mm00435625_m1), KCC2 (Mm00803929_m1), GAD65 (Mm00484923_m1), and GAD67 (Mm00725661_s1). Amplification plots for designed primers corresponding to α1, α2 and α5 GABAA receptor subunit mRNAs had slopes of −3.36 (for α1 and α2) and −3.19 (for α5), giving efficiencies of 0.9844 for α1 and α2 and of 1.0581 for α5.
Real time PCR was performed using an ABI 7500 Sequence Detection System. Validation experiments were conducted to show that subunit-specific primer sets amplified with equal efficiency as reported previously (Penatti et al., 2005). Each mPOA sample was analyzed in triplicate for each assessed cDNA and the 18S rRNA as an internal standard. For each mRNA assessed, a PCR master mix was prepared, containing final concentrations of: 1× TaqMan Universal Master Mix (containing AmpliTaq Gold DNA Polymerase, AmpErase UNG, dNTPs with dUTP, Passive Reference 1, and optimized buffer components), 900 nm forward primer, 900 nm reverse primer, 250 nm probe in a total reaction volume of 25 μl. PCR for 18S rRNA was performed in a 25 μl of reaction containing 1× TaqMan Universal Master Mix, 1× Eukaryotic 18S rRNA Endogenous Control (VIC/MGB Probe, Primer Limited; ABI), to which 1 μl of cDNA was added. Thermocycling conditions included initial steps of 2 min at 50°C, 10 min at 95°C and 40 cycles of PCR at 95°C for 15 s to denature cDNA and 60°C for 1 min for primer/probe annealing and extension. Samples with reverse transcriptase omitted were used to control for genomic DNA contamination and samples with template omitted to control for any reagent contamination. The 2−ΔΔCT method (Livak and Schmittgen, 2001; Peirson et al., 2003) was used for determination of mRNA levels.
Immunocytochemistry.
Immunoreactivity was assessed under confocal microscopy as described previously (Penatti et al., 2005). Expression of the α2 subunit of the GABAA receptor was assessed using an anti-guinea pig polyclonal primary antibody (1:15,000), generously provided by Dr. Jean-Marc Fritschy (University of Zurich, Zurich, Switzerland) and visualized with Alexa Fluor 488 goat anti-guinea pig IgG (Invitrogen; 1:1000) (Penatti et al., 2005). Immunoreactivity for ERα was assessed using a rabbit anti-ERα antiserum (C1355; Upstate Millipore Bioscience Research Reagents; 1:1000) and visualized with anti-goat Alexa 594 (1:1000 dilution).
Steroid immunoassays.
For each assay, heads from age-matched Tabby or Tfm male mice were rapidly removed and placed in PBS (4°C; pH 7.4) to wash away the excess of blood. Brains were quickly removed and the mPOAs were dissected, separated from the underlying meninges and placed in a 1.5 ml Eppendorf tube. For testosterone measurements, one mPOA was assessed in duplicate from each control and AAS-treated animal of each genotype. Levels of 17β-estradiol were lower than those for testosterone and determinations were not reliable for assays from individual mPOAs. Therefore, determinations of 17β-estradiol were made from samples for which mPOA tissue from two to four animals was pooled. Thus, 4–10 animals per treatment condition and per genotype were analyzed for steroid determinations. Each tube was supplemented with PBS/5% ethanol; 70 μl for each testosterone sample or 100 μl for each 17β-estradiol sample (pH 7.4; room temperature). Tissue samples were then hand-homogenized for 30 s followed by centrifugation (7000 rpm; 15 s). The supernatant, devoid of any contaminating tissue, was then carefully collected so as to not disturb the pellet or agitate and was stored overnight at −20°C. Small aliquots (6 μl) from each supernatant were collected for protein assay (BCA Protein Assay Reagent, Pierce). Supernatants clear of any contaminating particulate matter allowed the resulting samples to be assayed directly with Expanded Range Testosterone and High Sensitivity Estradiol Salivary Immunoassay Kits from Salimetrics. These two ELISA kits have low detection thresholds and small volume requirements (6 pg/ml; 25 μl and 1 pg/ml; 100 μl for testosterone and 17β-estradiol, respectively). Optical densities were determined at 450 nm according to the kit's instructions and steroid concentrations were converted from pg/ml to pg/mg mPOA tissue. For each assay, recovery rates (∼60%) were determined by supplementation of five known concentrations of each steroid to samples containing mPOA homogenates of equivalent protein concentration and recovery for each known concentration was determined in duplicate. The recovery rates for each of the five known concentrations were averaged to provide an average recovery rate for each steroid, and raw determinations of the concentrations of testosterone and 17β-estradiol were corrected for recovery. Analysis of intra- and inter-assay variability were estimated from 10 determinations of two sets of three samples of mPOA homogenates containing 5 concentrations of each steroid as the %CV (CV*100) for the highest variation obtained (Steiner et al., 2003; van Geel et al., 2008): for testosterone: %CV(intra-assay): <0.43% and %CV(inter-assay): <1.5%; for 17β-estradiol: %CV(intra-assay): <1.0% and %CV(inter-assay): 1.8%.
Western blot analysis.
For protein extraction, mPOA tissue was homogenized in 25 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 1% NP-40, 1 mm DTT, 5% glycerol, 1 mm PMSF, 10 mm NaF plus a protease inhibitor cocktail, Complete Mini (Roche). Protein concentrations were determined using the BCA Protein Assay Reagent. Forty micrograms lysate for ERα was separated by 7.5% SDS-PAGE gel electrophoresis and transferred to an Immobilon-P PVDF membrane (Millipore). Membranes were blocked in 5% milk/TBST (10 mm Tris-HCl, pH 8.0; 150 mm NaCl, 0.2%Tween-20) for 1 h at room temperature. The ERα antibody, F-10 (s.c.-8002; Santa Cruz Biotechnology) was diluted 1:500 in 5% milk/TBST. The anti-α-tubulin antibody (DM1A, Calbiochem) was diluted 1:20,000 in 5% milk/TBS. Membranes were incubated overnight at 4°C, washed five times for 5 min each in TBST followed by incubation for 1 h with a goat anti-mouse IgG secondary antibody (Pierce) at 1:2000 for ERα and for α-tubulin in 5%milk/TBST. Antibody binding was detected using an enzyme-linked chemiluminescence detection kit, SuperSignal West Femto Maximum Sensitivity Reagent (Pierce), and visualized on autoradiographic film (Kodak). Densitometric measurements (Scion Image, Scion) of the intensity of the signals corresponding to ERα and α-tubulin were corrected for background and the ERα signals were normalized to that for α-tubulin.
Electrophysiological recordings.
Electrophysiological recording and analyses were performed as described previously (Yang et al., 2002; Penatti et al., 2005; Jones et al., 2006). For each experiment, whole brains were quickly removed and placed in ice-cold oxygenated low-sodium sucrose-supplemented dissection solution, in mm: 250 sucrose, 1.2 CaCl2, 10 glucose, 4 KCl, 7 MgSO4, 26 NaHCO3, 1.25 NaH2PO4, and 1 ascorbic acid at pH 7.35. Using an Electron Microscopy Sciences OTS-4000 Vibroslicer, 250 μm coronal sections that included the mPOA were prepared. Slices were superfused with 95% O2/5% CO2-saturated artificial CSF (aCSF), in mm: 125 NaCl, 1.2 CaCl2, 10 glucose, 4 KCl, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, and 1 ascorbic acid at pH 7.35.
Recordings of spontaneous action potentials (APs) were made in the on-cell configuration with aCSF in both the bath and the pipette at room temperature (20−22°C). Data were recorded to tape and subsequently digitized using Acquire 4.0 software (Bruxton Corporation). Action potential frequency was measured and autocorrelograms were constructed with bin-widths of 1–10 ms using software written locally by Brian L. Jones (Oregon Health & Science University, Portland, OR) in MatLab 6.5 R13 (The MathWorks), and rhythmicity was assessed over windows of 0.2–10 s. Larger bin-widths and windows were used for cells with lower firing rates. Classifications of firing patterns from autocorrelograms were made according to Bar-Gad et al. (2001). Categorization of each cell's firing pattern as regular, irregular, or bursty was also made from the raw data. This independent assessment of firing pattern was in universal agreement with autocorrelogram designations made independently by another observer.
For recordings of GABAA receptor-mediated currents, aCSF was supplemented with 2 mm kynurenic acid to block excitatory transmission, and recordings were made in aCSF at room temperature (20−22°C) in the whole-cell configuration of the patch-clamp technique at a holding potential of −70 mV. The internal electrode solution consisted of (in mm): 153 CsCl, 1 MgCl2, 5 EGTA, and 10 HEPES, to which 2 MgATP was added for every experiment. GABAA receptor-mediated spontaneous IPSCs (sIPSCs) were isolated from amino acid-mediated excitatory synapses by addition of 2 mm kynurenic acid to the bath and miniature IPSCs (mIPSCs) isolated by additional perfusion with 1 μm tetrodotoxin (TTX) (Mozrzymas et al., 1999). To confirm that IPSCs were mediated by GABAA receptors, 10 μm bicuculline was used, in some experiments, to reversibly block the events. For analysis of peak current (Ipeak) and current decays (biphasic and fitted with two time constants, τ1 and τ2; Nett et al., 1999; Penatti et al., 2005) sIPSCs or mIPSCs were acquired and averaged from each neuron for a period of 4–12 min per recording condition, with criteria imposed that seal resistance (>1 GΩ), access resistance (<25 MΩ) and holding current did not change more than ±∼10% during the recording. Events were accepted for peak current only if they had rise-times of <2.5 ms and were isolated from other synaptic events (i.e., a synchronous, but concurrent release did not obscure either the rising or the decay phase of the measured response). For analysis of current amplitudes and decay kinetics for which overlapping events were not measured, the total number of events analyzed per cell ranged from >180 to <30 for a minority of cells in which all criteria were met with respect to quality of recording. Coefficients of variation did not vary with the number of events analyzed (e.g., the range for values of Ipeak was 0.34–0.53). All events that exceeded threshold for detection were accepted for assessment of IPSC frequencies. Data were recorded to tape and analyzed using Mini Analysis (Synaptosoft). In addition to estimates of τ1 and τ2, overall synaptic current decay was also described by a single weighted time constant (τw) (Yang et al., 2002, 2005; Jones et al., 2006). The magnitude of tonic GABAA receptor-mediated currents (Itonic) attributed to α5-containing receptors were estimated by minor modifications of previously published procedures (Farrant and Nusser, 2005; Jones et al., 2006) as a change in the baseline holding current (Ibaseline) in the presence of 50 μm (0.1% DMSO) of the α5-selective inverse agonist, L-655,708 (Quirk et al., 1996); a concentration that has been reported to be selective for α5-containing receptors in slice preparations (Caraiscos et al., 2004; Scimemi et al., 2005) and shown to be without effect in α5−/− mice (Caraiscos et al., 2004). Acquisition of data in the presence of L-655,708 was initiated ∼1 min after the drug was perfused into the bath, and acquired data were analyzed for a period of 90–150 s followed by a wash period. All control recordings were performed in 0.1% DMSO for comparison. This concentration of vehicle had no effect on GABAA receptor-mediated responses (i.e., no significant differences between aCSF alone and aCSF/0.1% DMSO), as we have reported previously (Jones et al., 2006).
Drugs.
All chemicals were purchased from Sigma Chemical with the exception of L-655,708 (Tocris Bioscience).
Statistical analyses.
Values for variables are presented as mean ± SE. Shapiro–Wilks or Kolmogorov–Smirnov tests were used to determine if data were distributed normally. For electrophysiological experiments, for non-normally distributed data, values were log-transformed before significance being determined by one- or two-way ANOVA using the general linear model procedure of SAS followed by the Student's t test or means comparison by least significant means, respectively. Significance for autocorrelational analysis of action potential firing was assessed by the X2 test. For real-time PCR analysis, CT values were defined as outliers when ±3 SDs from the mean. Results were qualitatively the same whether or not outliers were included in the final analysis. Differences in the relative abundance of each mRNA for AAS-treated versus control subjects were assessed using Pair-Wise Fixed Reallocation Randomization t test using the excel-based Relative Expression Software Tool (REST) (Pfaffl, 2001; Pfaffl et al., 2002). For mRNA analysis, only positive error bars are depicted in the results; positive and negative error bars differed by ≤15%. For all data, the α level was set at p < 0.05. Throughout the text, n values indicate the number of animals analyzed per condition, except for electrophysiological analyses where n indicates the numbers of cells analyzed per condition. The probability of attaining recordings that met criteria for subsequent analyses (1–3 cells per subject for both control and treated groups) is low in these older animals (>100 d of age at the time of recording), and no one subject within a given group disproportionately contributed cells to the group averages.
Results
I. AAS-induced changes in electrical activity in MPN neurons in wild-type male mice
Electrophysiological recordings were made from the dorsal aspect of the MPN, which is centrally situated within the mPOA. Chronic treatment with the AAS mixture resulted in a significant (p = 0.0010) increase in AP frequency from 1.83 ± 0.33 in MPN neurons from control wild-type mice (n = 32 neurons) to 4.31 ± 0.74 (n = 30 neurons) in AAS-treated subjects (Fig. 1A). This increase was reflected across the entire range of AP frequencies observed. Autocorrelational analysis indicated that firing patterns of MPN neurons could be described as irregular, bursty and regular based on interspike interval (Fig. 1B). AAS treatment resulted in a trend toward a decrease in neurons with bursty firing (p = 0.0550) (Fig. 1C) and a significant decrease in the coefficient of variation in AP frequency (p = 0.0040) in MPN neurons from AAS-treated versus control wild-type mice.
Figure 1.
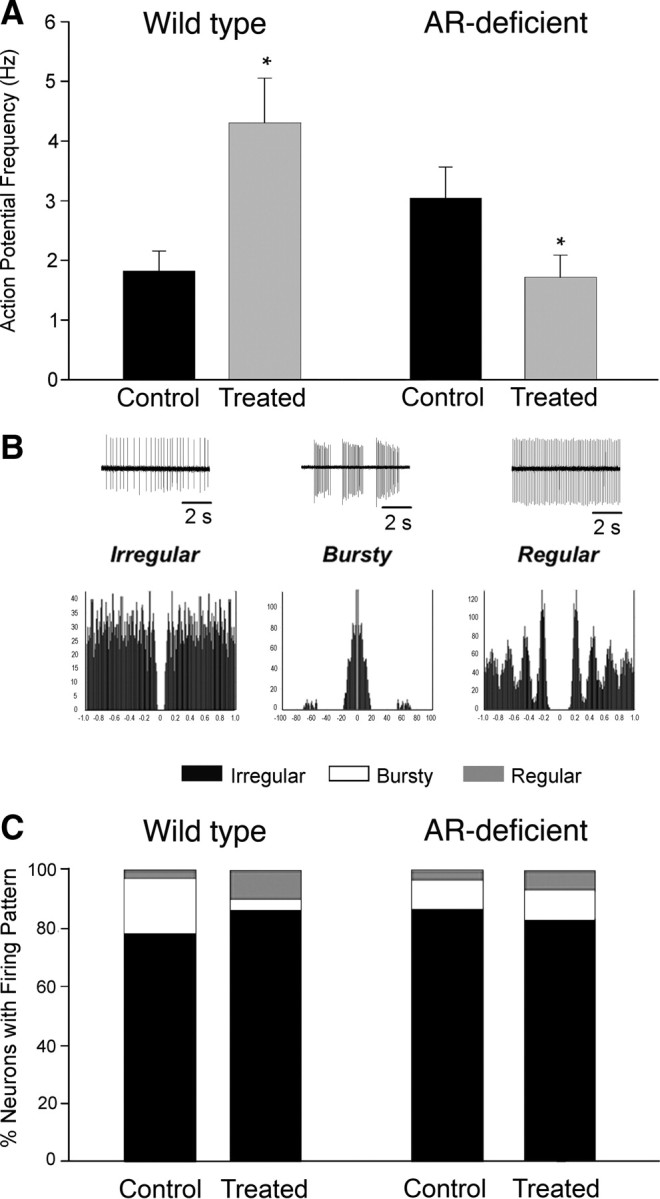
Effects of chronic AAS treatment on AP frequency and patterning in wild-type and AR-deficient male mice. A, Average AP frequency under control (black) and AAS treatment (gray) conditions from wild-type (left) and AR-deficient (right) mice. Asterisks indicate values from AAS-treated subjects were significantly different from their age-matched oil-injected controls in the same genotype. B, Representative examples of action potentials (top) and autocorrelograms (bottom) corresponding to irregular, bursty, and regular firing patterns. C, The percentage of neurons displaying irregular, bursty or regular firing patterns as assessed by autocorrelational analyses of MPN neurons from control or AAS-treated wild-type (left) or AR-deficient (right) mice. Recordings were made from 8 mice for each treatment condition and each genotype.
II. AAS-induced changes in GABAA receptor expression and function in wild-type mice
Changes in GABAA receptor subunit mRNA levels were assessed for the major subunit isoforms of the receptor expressed in the mPOA (α1, α2, α5, β1, β2, β3, γ1, γ2, and ε) (Henderson, 2007), as well as the δ subunit which, while only weakly expressed in the mPOA, may contribute to extrasynaptic tonic conductances in this brain region, as it does in others (Semyanov et al., 2004). Real-time PCR analysis revealed a marked increase in α5 subunit mRNA with AAS treatment in wild-type mice (p = 0.0010; n = 8) (Fig. 2A). A small, but nonetheless significant, increase in β1 subunit mRNA (p = 0.0300; n = 8) was also observed in AAS-treated wild-type subjects relative to this subunit mRNA levels observed in control animals. While the level of α5 mRNA was increased, we observed no concomitant increase in the levels of either δ or ε subunit mRNAs in the mPOA of AAS-treated animals (Fig. 2A); subunits which would likely be present in any new extrasynaptic receptors whose expression was induced by AAS treatment (Henderson, 2007).
Figure 2.

Effects of chronic AAS treatment on GABAA receptor expression and function in the mPOA of wild-type male mice. A, Data indicating average levels of subunit mRNAs in tissue isolated from the mPOA from 8 wild-type mice treated with the AAS mixture for 6 weeks. The mRNA levels for each subunit normalized to 18S rRNA from 8 oil-injected control animals were analyzed and relative subunit mRNA levels for controls were set to 1.00 for each subunit mRNA (horizontal line). Values (2−ΔΔCT) for treated animals were plotted relative to this control value of 1.00. B, Averaged sIPSCs recorded from MPN neurons in slices isolated from control (black line) and AAS-treated (gray line) wild-type mice. C, D, Average Ipeak (left), τw (center) and charge transfer (Qtot; right) (C) and frequency for sIPSCs from these MPN neurons of control (black bars) and AAS-treated (gray bars) mice (D). Asterisks indicate values from AAS-treated subjects that were significantly different from their age-matched oil-injected controls. Recordings were made from 16 mice for each treatment condition.
Receptors containing the α5 subunit contribute to both synaptic and extrasynaptic (tonic) currents in other brain regions (Dunning et al., 1999; Caraiscos et al., 2004; Serwanski et al., 2006; Yamada et al., 2007; Ali and Thomson, 2008). To determine how the AAS-dependent increase in α5 subunit mRNA in wild-type mice may be reflected in changes in GABAergic transmission, GABAA receptor-mediated synaptic and tonic currents were recorded from MPN neurons of control and AAS-treated wild-type animals.
In control wild-type mice, sIPSCs in MPN neurons were characterized by a mean peak current amplitude of 40.7 ± 3.8 pA (Fig. 2B,C), biexponential current decay described by two time constants of 9.96 ± 0.49 and 41.00 ± 4.11 ms (a weighted time constant τw = 21.29 ± 1.63 ms) (Fig. 2B,C), and a mean frequency of 4.7 ± 0.8 Hz (Fig. 2D) (n = 22 cells). AAS treatment of wild-type mice did not significantly increase sIPSC amplitude or frequency (Fig. 2B,D), but did result in an overall slowing of synaptic current decay (τw; p = 0.027) (Fig. 2C), arising from an increase in τ1 (p = 0.0440) and a trend toward prolongation of τ2 in AAS-treated versus control wild-type mice. The overall slowing of synaptic current decay also contributed to a significant (p = 0.0450) increase in total charge transfer (Qtot) in the AAS-treated versus control subjects (Fig. 2C).
As noted above, AAS treatment increased AP firing frequency in MPN neurons of wild-type mice. An AAS-dependent enhancement of GABAergic transmission could directly give rise to an increase in AP firing frequency if GABA acts to depolarize neurons within the mPOA. It has been shown that perturbations in normal neuronal signaling that promote stress can cause reversion to depolarizing actions of GABA in the adult hypothalamus by altering the expression of the anion transporter, K+/Cl– cotransporter 2 (KCC2) (Hewitt et al., 2009). While AAS treatment is known to be associated with changes in the stress response (for review, see Clark and Henderson, 2003), real-time PCR analysis of mPOA tissue indicated that AAS treatment did not result in any significant change in KCC2 mRNA expression in this brain region (p = 0.3245).
The slowing of sIPSC current decay and the increase in total charge transfer observed in AAS-treated wild-type mice would be consistent with an increase in the incorporation of α5 subunits into synaptic receptors, since inclusion of this subunit has been reported to confer more slowly decaying responses relative to receptors containing other α subunits (Burgard et al., 1996; Dunning et al., 1999; Smith and Gong, 2005). Acute application of the α5-selective antagonist, L655,708, to MPN neurons suggested that α5-containing receptors contribute to synaptic responses in ∼50% of MPN neurons tested from both control and AAS-treated mice. For those neurons that were sensitive to the effects of L655,708, τw was significantly (p = 0.0072) longer in neurons from AAS-treated than from control mice (23.07 ± 1.66 ms for control, n = 5 neurons, versus 32.37 ± 1.84 ms for AAS-treated, n = 5 neurons), consistent with the interpretation that there is increased representation of α5-containing receptors in the population of synaptic receptors of AAS-treated wild-type mice. An L655,708-sensitive tonic current (Itonic = 7.7 ± 4.3 pA; n = 8 neurons) was also evident in MPN neurons from wild-type mice, but while AAS treatment augmented the amplitude of this current (16.0 ± 13.1 pA; n = 10 neurons), this difference was not significant.
Chronic exposure to this mixture of commonly abused AAS resulted in significant differences in AP frequency and patterning, GABAA receptor subunit mRNA expression and GABAA receptor-mediated synaptic responses in wild-type mice. As noted previously, the AAS in this mixture not only interact with AR, but also have the capacity to be aromatized to estrogens and to potentially interact with ER. To determine the role of the AR in mediating the effects of this AAS mixture on neuronal activity and GABAA receptor expression and function in the mPOA, recordings were next made from control and AAS-treated AR-deficient Tfm mice.
III. AAS-induced changes in electrical activity in MPN neurons in AR-deficient (Tfm) male mice
In contrast to its effect in wild-type animals, AAS treatment of the AR-deficient mice resulted in a significant decrease in AP frequency (p = 0.0240), from 3.04 ± 0.52 in MPN neurons from control mice (n = 31 neurons) to 1.72 ± 0.37 in MPN neurons from AAS-treated mice (n = 30 neurons) (Fig. 1A), and this decrease was observed across the range of observed frequencies. In contrast to its effects in wild-type mice, AAS treatment did not appreciably alter the patterning (Fig. 1C) or the coefficient of variation for AP firing in the MPN of these AR-deficient mice.
The profile of steady-state levels of GABAA receptor subunit mRNAs in the mPOA of control AR-deficient Tfm mice was comparable to that observed in wild-type Tabby male mice. Of particular note, levels of the α5 subunit mRNA were nearly identical (Fig. 3A). In contrast to the increases in α5 and β1 subunit mRNAs observed in the mPOA with AAS treatment of wild-type mice, AAS treatment of AR-deficient mice did not result in increases (compared with control) in any of the examined GABAA receptor subunit mRNAs in this brain region. Of particular interest, the marked increase in α5 subunit mRNA that was observed in wild-type mice was notably absent in this AR-deficient mutant line. Rather, real-time PCR analysis revealed modest, albeit significant, decreases in β3 (p = 0.0200) and γ2 (p = 0.0060) subunit mRNAs in AAS-treated versus control AR-deficient mice (Fig. 3B).
Figure 3.

GABAA receptor subunit mRNA levels in the mPOA of wild-type and AR-deficient mice. A, Data are presented as the 2−ΔCT values indicating the relative levels of GABAA receptor subunit expression in the mPOA of control wild-type (black) and control AR-deficient (white) mice. B, Data indicating average levels of subunit mRNAs in tissue isolated from the mPOA from AR-deficient mice treated with the AAS mixture for 6 weeks. For data shown in B, values from oil-injected control animals were analyzed and relative subunit mRNA levels for controls were set to 1.00 for each subunit mRNA (horizontal line). The 2−ΔΔCT values for AAS-treated animals were plotted relative to this control value of 1.00. Analyses were made from 8 control and 8 treated animals of each genotype.
For the AR-deficient Tfm mice, consistent with the lack of an AAS-dependent increase in α5 mRNA, AAS treatment promoted neither a prolongation of synaptic current decay nor an increase in total charge transfer in these animals (Fig. 4A,B); changes in current parameters that accompanied the increase in α5 subunit mRNA in the mPOA of wild-type males. To the contrary, although there was no change in sIPSC decay kinetics induced by AAS treatment, the average Ipeak in MPN neurons from AAS-treated mice (n = 28 neurons) was significantly (p = 0.0034) reduced to 71% of that in MPN neurons (n = 25 neurons) from control AR-deficient subjects (Fig. 4A,B). The smaller sIPSC amplitudes in AAS-treated subjects may arise from postsynaptic changes, either a decrease in postsynaptic receptor density or decreased charge transfer through these receptors, or may arise from diminished AP-dependent presynaptic release. Spontaneous IPSC frequency for MPN neurons from AAS-treated AR-deficient mice was significantly (p = 0.0191) lower (47%) than that for neurons from control animals (4.77 ± 0.76 Hz; n = 29 neurons from AAS-treated vs 10.04 ± 2.18 Hz; n = 25 neurons for control) (Fig. 4C). No differences were observed in mIPSC amplitude, decay kinetics, total charge transfer or frequency (Fig. 4D–F) in control versus AAS-treated AR-deficient mice (n = 23 neurons from control and n = 22 neurons from AAS-treated mice), suggesting that neither the decrease in Ipeak nor the decrease in sIPSC frequency in AAS-treated subjects was due to diminished postynaptic sensitivity, which, for analysis of frequency may have resulted in an apparent “loss” of small events in the baseline noise. Rather, the AAS-dependent decrease in Ipeak and frequency of sIPSCs in the MPN observed in the AR-deficient mice (Fig. 4A–C) is consistent with decreased AP firing frequency observed in MPN neurons of AAS-treated AR-deficient mice and concomitant AP-dependent presynaptic release.
Figure 4.

Effects of chronic AAS treatment on GABAA receptor function in the MPN of AR-deficient mice. A, Averaged sIPSCs recorded from MPN neurons in slices isolated from control and AAS-treated AR-deficient mice. B, C, Average Ipeak (left), τw (center) and charge transfer (Qtot; right) (B) and frequency for sIPSCs from these MPN neurons of control and AAS-treated mice (C). D, Average mIPSCs recorded from MPN neurons of control and AAS-treated AR-deficient mice. E, F, Average Ipeak (left), τw (center), and charge transfer (Qtot; right) (E) and frequency for mIPSCs from these MPN neurons (F). Black lines and bars, Control; gray lines and bars, AAS-treated. Asterisks indicate values from AAS-treated subjects that were significantly different from their age-matched oil-injected controls. Recordings were made from 12 to 16 mice for each treatment condition.
As a further assessment of AAS effects on GABAergic tone in the MPN, we also determined the effects of chronic steroid treatment on the levels of the mRNAs encoding the two isoforms of glutamic acid decarboxylase (GAD), the synthetic enzyme for GABA. The 65 kDa isoform of this enzyme, GAD65, is preferentially localized in axon terminals, and levels of GAD65 immunoreactivity strongly correlate with levels of GAD65 mRNA (Esclapez et al., 1994). GAD65 is strongly expressed in the mPOA, and levels of this transcript vary in parallel with the changes in phasic activity in neuroendocrine cells in this and other hypothalamic regions (Feldblum et al., 1993). AAS treatment of AR-deficient mice led to a significant (p = 0.0425) decrease in the levels of GAD65 mRNA in the mPOA to 75% that observed in control animals. AAS treatment did not alter the levels of GAD67 mRNA. These data are also consistent with diminished presynaptic GABAergic tone in the MPN of AAS-treated Tfm mice. AAS treatment did not significantly alter GAD65 (or GAD67) levels in wild-type mice, consistent with an absence of effect of AAS-treatment on sIPSC amplitude or frequency in wild-type subjects.
IV. Effects of ER antagonism on AAS-dependent changes in GABAergic signaling in the Tfm mouse
The significant effects of chronic AAS treatment in the mPOA of Tfm mice suggest that, in the absence of functional AR signaling, significant interactions of the AAS with ER signaling pathways were unmasked. While AAS such as 17α-MeT do not bind to ER (Oosterkamp et al., 1996), all AAS used in this mixture can be metabolized to estrogens (vide supra) and thus have the propensity to act as ER agonists. In addition, in non-neuronal cells assessed in vitro, the AAS have been shown to inhibit aromatase (Mor et al., 2001; de Gooyer et al., 2003) and thus could indirectly act to impair ER signaling by limiting the availability of estrogens in the brain. Experiments were next performed to determine if the ability of the AAS to modulate GABAergic signaling in the MPN in the AR-deficient Tfm mouse could be attributed to these compounds acting by either of these two mechanisms to augment or diminish (respectively) ER-mediated signaling in the mPOA.
To first determine if AAS effects in the AR-deficient mice could be attributed to agonistic actions at the ER, Tfm mice were treated with the selective estrogen receptor modulator (SERM), tamoxifen, or the AAS mixture in conjunction with tamoxifen. Tamoxifen at the dose given here has been shown to be devoid of acute estrogenic activity, but to possess ER antagonistic activity in the rodent brain (Rudick and Woolley, 2003). Surprisingly, concurrent treatment of AR-deficient mice with tamoxifen did not antagonize the actions of AAS treatment on GABAergic transmission in the MPN of the AR-deficient mice, as would have been expected if the AAS were acting as estrogens and tamoxifen as an ER antagonist. Instead, we found that tamoxifen alone also decreased sIPSC amplitudes versus control subjects (p < 0.0001) (Fig. 5) and that there was no difference in Ipeak between AR-deficient animals treated concurrently with the AAS plus tamoxifen versus those treated with tamoxifen alone (Fig. 5). As with AAS treatment, tamoxifen also promoted a significant decrease in the frequency of sIPSCs in MPN neurons of AR-deficient Tfm mice treated with tamoxifen versus control subjects (3.60 ± 0.74 Hz vs 10.04 ± 2.18 Hz; p = 0.0306). There was no difference (p = 1.0000) for sIPSC frequencies between AAS- and tamoxifen-treated Tfm mice. While recognizing that tamoxifen is a SERM with mixed agonistic/antagonistic properties (see for example, Shang et al., 2000; Kressler et al., 2007), these data suggest that the AAS do not act as ER agonists in the AR-deficient mice. Rather, the data suggest that the may be interfering with the actions of estrogens at the ER or may be acting to limit their production via inhibition of aromatase.
Figure 5.

Effects of pharmacological manipulation of estrogens on AAS-dependent modulation of GABAergic sIPSC amplitudes in the MPN of the AR-deficient Tfm mouse. Graphical representation of averaged Ipeak from AR-deficient Tfm mice chronically treated with oil alone (Control), the AAS mixture in oil (AAS), tamoxifen (Tx), the AAS mixture and tamoxifen (Tx+AAS), formestane (Frm), or the AAS mixture and formestane (Frm+AAS). Data for Control and AAS are the same as shown in Figure 5. Identical letters (among the a–d designations) indicate means that were not statistically different from one another as assessed by two-way ANOVA followed by the means comparison by least significant means. In a separate cohort of AR-deficient Tfm mice, experiments were later performed to determine the effects of concurrent treatment with 17β-estradiol and the AAS mixture (E2+AAS) or 17β-estradiol (E2) alone versus control. The identical letter (a') indicates that when concurrently treated with 17β-estradiol and AAS, the AAS did not significantly diminish Ipeak. For both sets of data, numbers in parentheses indicate numbers of cells. Recordings were made from 8 to 12 mice for each treatment condition.
V. Effects of aromatase inhibitors and estrogen on AAS-dependent changes in GABAergic signaling in the Tfm mouse
To directly determine whether inhibition of endogenous estrogen production could mimic the effects of the AAS on GABAergic transmission in AR-deficient mice, Tfm males were treated with formestane or with the AAS mixture in conjunction with formestane. Formestane is a selective and irreversible aromatase inhibitor (Yue and Brodie, 1997; Kohler et al., 2007), which will block production of estrogens from androgens including those AAS that cannot be 5α-reduced (LaMorte et al., 1994). Formestane has been shown to cross the blood brain barrier and to diminish aromatase activity and ER-regulated protein expression in the brain (Foidart et al., 1994; de Fougerolles Nunn et al., 1999).
As in AR-deficient Tfm mice treated with AAS, sIPSC amplitudes in MPN neurons of Tfm mice treated with formestane were significantly lower (p < 0.0001) than those observed in control subjects (Fig. 5). Moreover, there was no difference in sIPSC amplitudes between mice treated with the AAS and those concurrently treated with formestane and the AAS (Fig. 5). The diminution of sIPSC peak amplitudes was, however, significantly (p = 0.0005) greater in animals treated with formestane alone than those treated with the AAS (Fig. 5). These results may reflect the competitive and incomplete inhibition of aromatase activity by the AAS (in vitro assays indicate 17α-MeT and nandrolone inhibit aromatase activity by 53% and 85%, respectively) (de Gooyer et al., 2003) versus the irreversible inhibition of this enzyme imposed by formestane (Yue and Brodie, 1997; Kohler et al., 2007). As with AAS-treatment, the frequency of sIPSCs in MPN neurons of animals treated with formestane was also lower than in control subjects (5.67 ± 0.92 Hz vs 10.04 ± 2.18 Hz), although for formestane alone, this difference did not attain significance. There was no difference in frequency for sIPSCs in MPN neurons from AAS-treated and formestane-treated AR-deficient Tfm mice. Together, these data are consistent with the hypothesis that, in the absence of AR signaling in the Tfm mouse, the AAS diminish GABAergic signaling by limiting the production of estrogens and thus ER-mediated signaling.
If in the absence of functional AR, the ability of the AAS to diminish GABAergic transmission reflects inhibition of aromatase, and thus production of estrogen, supplementing AAS-treated Tfm mice with exogenous estrogen should restore the properties of GABAA receptor-mediated transmission to those observed in control mice. To this end, an additional cohort of AR-deficient mice was treated with oil alone, the AAS mixture in conjunction with 17β-estradiol or 17β-estradiol alone. Consistent with the hypothesis that the AAS are acting as aromatase inhibitors in the Tfm animals, average sIPSC amplitudes were not significantly different between control animals and animals treated with AAS in conjunction with a concentration of 17β-estradiol intended to restore levels of this steroid to a physiological range (Fig. 5). While average Ipeak in animals treated with 17β-estradiol alone was greater than for control subjects, the difference did not attain significance (Fig. 5). As reported above (Section III), AAS treatment of Tfm mice significantly (p = 0.0191) decreased sIPSC frequency to 47% that observed in control mice, but concurrent treatment of the AR-deficient mice with AAS and 17β-estradiol restored the average sIPSC frequency to 95% that observed for controls (p = 0.8848). Treatment of AR-deficient mice with 17β-estradiol alone evoked a nearly significant (p = 0.0539) increase in sIPSC frequency (175% control). As with AAS mixture treatment of Tfm animals, no significant differences were elicited in synaptic current decay by concurrent treatment with AAS mixture and 17β-estradiol or with 17β-estradiol alone.
In contrast to the AR-deficient Tfm mice, formestane treatment of wild-type mice neither mimicked nor enhanced AAS effects on GABAergic transmission in the MPN (i.e., prolongation of synaptic current decay) (Fig. 2C). Specifically, there was no significant difference in τw between control wild-type mice and wild-type mice treated with formestane (21.29 ± 1.63 ms for control vs 20.62 ± 2.04 ms for formestane; p = 0.8048). Similarly, synaptic current decay was equivalently enhanced over control in wild-type animals treated with AAS and in those treated with AAS plus formestane (27.26 ± 2.00 ms for AAS-treated vs 26.62 ± 5.98 ms for AAS plus formestane; p = 0.7933). As with AAS treatment, treatment with formestane alone or concurrent treatment with AAS and formestane had no effect on sIPSC frequency in MPN neurons of wild-type mice. Together, these data indicate that in terms of the actions of these steroids on GABAergic transmission in the MPN, when functional AR are present, AAS actions via the AR predominate over AAS effects as aromatase inhibitors.
VI. Assessment of steroid levels in the brains of AAS-treated mice
Previous studies have shown that adult AR-deficient Tfm mice have markedly lower levels of peripheral testosterone than normal adult male mice (Goldstein and Wilson, 1972; Murphy and O'Shaughnessy, 1991, O'Shaughnessy and Murphy, 1993; Vandenput et al., 2004), but that peripheral levels of estradiol are comparable in Tfm and wild-type Tabby males (Vandenput et al., 2004). Of particular importance, previous studies have also shown that aromatization of testosterone occurs in the Tfm brain (Rosenfeld et al., 1977; Schleicher et al., 1986), and that estradiol derived from testosterone is capable of suppressing follicle stimulating hormone secretion in the Tfm mouse, thus indicating a physiological ER-mediated response of the hypothalamic/pituitary/gonadal axis in this mutant mouse line (Schleicher et al., 1989).
To determine if aromatization of testosterone to estrogen in the mPOA was altered by AAS treatment, steroid immunoassays were performed to assess levels of these steroids in this brain region. Testosterone levels were notably higher for mPOA tissue from male control wild-type than from control AR-deficient animals (Table 1). These data are consistent with previous studies assessing levels of peripheral testosterone in adult male mice (Murphy and O'Shaughnessy, 1991; O'Shaughnessy and Murphy, 1993; Vandenput et al., 2004). As expected, we found marked elevation of testosterone within the mPOA of AAS-treated versus control mice in both wild-type (p = 0.0008) and AR-deficient (p = 0.00004) animals (Table 1), reflecting the significantly higher levels of testosterone derived from the AAS mixture. Assessment of 17β-estradiol in the mPOA indicated that the amounts of this estrogen were comparable in control wild-type and control Tfm animals (p = 0.2621; Table 1), consistent with prior studies examining peripheral estradiol levels (Vandenput et al., 2004).
Table 1.
Levels of testosterone and 17β-estradiol in mPOA tissue
| Genotype/treatment | T (pg/mg protein) | E2 (pg/mg protein) | Aromatization (E2/T) |
|---|---|---|---|
| Wild type (control) | 18.54 ± 9.35 | 1.36 ± 0.21 | 0.074 |
| Wild type (AAS) | 153.30 ± 19.60 | 2.11 ± 0.51 | 0.014 |
| AR-deficient (control) | 4.96a | 0.78 ± 0.08 | 0.157 |
| AR-deficient (AAS) | 135.65 ± 20.4 | 0.33 ± 0.04 | 0.002 |
Mean± SE of the mean levels of testosterone (T) and 17β-estradiol (E2) in mPOA tissue harvested from wild-type control (n = 4 mice for T; n = 8 for E2) or AAS-injected (n = 4 mice for T; n = 10 mice for E2) or AR-deficient control (n = 6 mice for T; n = 8 mice for E2), or AAS-injected (n = 4 mice for T; n = 8 mice for E2) male mice, and the respective ratio of aromatization as determined from the ratio of E2 to T.
aLevels of testosterone were above threshold for detection in only 1 of 6 mPOA samples from AR-deficient animals. For the other 5 animals, the threshold for detection (3.0 pg/mg) was entered for determination of the mean, but no SE was calculated. As noted in Materials and Methods, individual mPOA were assayed for testosterone. As indicated in Materials and Methods, for 17β-estradiol, determinations were made from samples for which mPOA tissue from 2 to 4 animals were pooled.
Previous studies have shown that acute exposure to the AAS for non-neuronal cell lines in vitro inhibits aromatase activity in a dose-dependent manner (Mor et al., 2001; de Gooyer et al., 2003). The Ki for 17α-MeT for inhibition of aromatase was found to be 0.6 μm (Hong et al., 2008), a concentration likely to be attained with the doses of AAS administered here and relevant to human steroid abuse (for review, see Clark et al., 2006). To provide an indication of the level of aromatization (Zhang et al., 2007; Zhao et al., 2007), we determined the ratio of 17β-estradiol to testosterone in the intact mPOA of control animals and animals receiving chronic exposure to the AAS for both wild-type and AR-deficient Tfm mice. In both wild-type and AR-deficient genotypes, the 17β-estradiol to testosterone ratio was markedly lower in AAS-treated than in control subjects. Thus despite dramatically elevated levels of the substrate for aromatase, testosterone, parallel increases in the conversion of testosterone to 17β-estradiol were not observed in either genotype of mice. The lack of enhanced conversion of testosterone to 17β-estradiol in AAS-treated wild-type mice was also notable given that androgens present in the AAS mixture significantly increased levels of aromatase mRNA in the mPOA (vide infra, Fig. 6).
Figure 6.

AAS-dependent changes in Aromatase mRNA in the mPOA. Data are presented as the 2−ΔCT values indicating the relative levels of aromatase (Cyp19) mRNA in tissue isolated from the mPOA from wild-type (left) and AR-deficient Tfm (right) adult male mice injected with oil (control) or treated with the AAS mixture (AAS) for 6 weeks. Identical letters indicate means that were not statistically different from one another as assessed by two-way ANOVA followed by the means comparison by least significant means. Analyses were made from 8 control and 8 treated animals of each genotype.
As a final assessment of testosterone and 17β-estradiol levels in the mPOA of control and AAS-treated mice, we also performed assays for biological endpoints known to be selectively dependent on AR- versus ER-mediated signaling. Specifically, in male rodents, androgens acting at the AR are the major regulators of aromatase expression (Abdelgadir et al., 1994; Foidart et al., 1995; Roselli et al., 1998) (for review, see Roselli and Resko, 1997), while progesterone receptor (PR) mRNA is upregulated in the male mouse mPOA by 17β-estradiol via a mechanism that requires both ERα and ERβ signaling for maximal induction (Kudwa et al., 2004). Aromatase mRNA levels were found to be significantly higher (p < 0.0010) in the mPOA of AAS-treated versus control wild-type mice (Fig. 6), suggesting that the elevated levels of testosterone measured in the mPOA maintained an enhanced level of this AR-regulated transcript over the treatment period. Not surprisingly, levels of aromatase mRNA were not significantly enhanced by AAS treatment in Tfm subjects (p = 0.532) (Fig. 6), consistent with the absence of classical AR-mediated signaling in this mutant mouse line. In contrast to the dramatic effects of the AAS mixture on aromatase mRNA in wild-type mice, no effects of AAS treatment versus control on PR mRNA in the mPOA were evident in either wild-type (p = 0.2915) or Tfm subjects (p = 0.3450). Thus, these data also suggest that androgen, but not estrogen, levels are elevated in AAS-treated mice. Taken in conjunction with the effects of the AAS and agents that alter estrogen signaling on GABAA receptor-mediated sIPSCs (Fig. 5) and the assays of 17β-estradiol to testosterone ratios within the mPOA (Table 1), these data are consistent with a mechanism whereby the AAS interfere with the ability of aromatase to convert androgens to estrogens in the intact mPOA.
VII. Changes in ERα and ERβ expression induced by chronic exposure of AR-deficient Tfm mice to AAS
While the data presented are consistent with an antagonistic action of the AAS on endogenous estrogen production via inhibition of aromatase in the AR-deficient mice, an alternative (or additional) mechanism to account for these effects would be if chronic AAS exposure altered the expression of either ERα or ERβ receptors themselves. Previous studies have indicated that neurons within the mPOA of male mice express both ERα and ERβ, although ERβ was found to be expressed in many fewer cells than ERα (Kudwa et al., 2004). Immunocytochemical assessments for ERα and the predominant somal GABAA receptor α isoform (α2) (Penatti et al., 2005) indicated that there was a high level of coincidence of expression of nuclear ERα in neurons that also express GABAA receptors (data not shown), and that ERα mRNA was ∼26 times more abundant than ERβ mRNA in wild-type mice and ∼22 times more abundant in AR-deficient mice. AAS treatment did not alter the levels of either ERα or ERβ mRNA in either wild-type (treated levels = 105.7 ± 3.2% of control for ERα and 100.0 ± 5.6% control for ERβ) or Tfm (treated levels = 94.2 ± 4.4% of control for ERα and 93.7 ± 4.7% control for ERβ) mice (n = 8 mice for each genotype and each treatment condition). Subsequent Western blot analysis for ERα in mPOA lysates from the AR-deficient Tfm mutants revealed a band of 66 kDa, consistent with the expression of ERα protein, but, as with ERα mRNA levels, averaged densitometric signals, normalized to α-tubulin, revealed no effect of AAS treatment on the level of ERα protein (treated levels = 103.5 ± 13.6% of control; n = 7 control; n = 7 AAS-treated). ERβ protein expression was not assessed due to lack of appropriate antibody. Together, these data suggest that AAS treatment did not elicit marked effects on either ERα or ERβ expression in the mPOA in mice of either genotype.
Discussion
Chronic exposure of adult wild-type male mice to a combination of three chemically distinct AAS resulted in a significant increase in AP firing with a trend toward neurons displaying irregular firing patterns, significant increases in the levels of α5 and β1 subunit mRNAs and a concomitant significant prolongation of GABAA receptor-mediated synaptic current decay and overall charge transfer in neurons of the MPN. Acute exposure to the α5-selective antagonist, L655,708, indicated that receptors containing this subunit are expressed synaptically and extrasynaptically in the MPN and that synaptic currents in L655,708-sensitive neurons are prolonged by AAS treatment. Recombinant receptors containing α5 subunits are predicted to have slower decay kinetics than do receptors containing α1 subunits (Burgard et al., 1996; Dunning et al., 1999; Smith and Gong, 2005; cf. Picton and Fisher, 2007). Moreover, synaptic currents recorded from cortical neurons from α5−/− mice are notably shorter (24.8 ± 2.5 ms) than from wild-type mice (30.3 ± 2.6 ms) (Caraiscos et al., 2004). Our data are thus consistent with the interpretation that the prolongation of synaptic current observed with AAS treatment of wild-type male mice resulted from increased representation of α5-containing receptors at synapses onto MPN neurons, perhaps with α5-containing receptors substituting for α1-containing receptors, which are likely to be the predominant type of α subunit-containing receptors expressed in the MPN of these Tabby (and Tfm) mice. While comparisons with studies assessing effects of acutely blocking GABAergic transmission on AP patterning within the same neuron must be made with great caution, our data demonstrating that long-term AAS treatment resulted in enhanced GABAergic tone and a trend toward more irregular AP firing within the population of MPN neurons are consistent with previous studies on other cells types, demonstrating that acute pharmacological blockade of GABAergic transmission converts irregular to regular AP patterning (Häusser and Clark, 1997; Kononenko and Dudek, 2004; Bright et al., 2007).
AAS treatment of AR-deficient Tfm mice with this AAS mixture resulted in dramatically different results from those obtained in wild-type males. Chronic AAS treatment in the AR-deficient mice resulted in a significant decrease in AP frequency, no increases in either α5 or β1 subunit mRNAs, but small, albeit significant, decreases in β3 and γ2 subunit mRNAs, a significant decrease in sIPSC frequency and amplitude and a significant decrease in GAD65 mRNA levels. While selective GABAA receptors subunit mRNAs were modestly decreased in these AR-deficient mice, we observed no changes in mIPSC amplitude, kinetics, or frequency, and our data are thus consistent with a mechanism by which, in the absence of AR signaling, chronic AAS treatment promoted diminished AP-dependent GABA release onto MPN neurons and thus also the inhibitory drive onto these cells. These data underscore the idea that effects of the AAS are wide-ranging: in both wild-type and AR-deficient mice, chronic treatment with these steroids is likely to have significant effects not only on GABAergic regulation of neurons presynaptic to those studied here, but may also influence electrical activity by altering levels of glutamatergic drive or the function of voltage-gated channels. The diversity of AAS effects on other the molecular components of neural signaling within these critical forebrain regions remains to be explored.
The highly divergent actions on neuronal signaling in the mPOA of wild-type versus AR-deficient Tfm male littermates indicate a critical role for AR signaling in mediating AAS effects in wild-type mice. The importance of AR signaling in mediating AAS-dependent increases in α5 mRNA expression and prolongation of synaptic current decay is highlighted not only by experiments presented here, demonstrating that such changes do not occur in the AR-deficient Tfm line, but also by data indicating that cotreatment with the AR antagonist, flutamide, negates comparable AAS-induced changes in α5 mRNA levels in wild-type C57BL/6J mice (Penatti et al., 2009). We note that while these data with flutamide suggest that some of the actions of the AAS reported here are mediated by activational actions of classical AR, they do not rule out the possibility that there may be inherent differences between the neural templates of wild-type Tabby and Tfm mutants that could also contribute to the divergence in effects of the AAS in the two genotypes. In particular, organizational actions of androgens acting at the AR have been suggested with respect to the acquisition of male-specific behaviors (Sato et al., 2004; Bodo and Rissman, 2007). Such AR-mediated organizational effects may reflect differences in neuronal circuitry within the mPOA, but this has yet been tested. We also note that most of the well described organizational actions of androgens in male rodents are mediated via ER following aromatization of circulating androgens to 17β-estradiol (for review, see Sakuma, 2009). The diminished testosterone levels that have been reported for the Tfm strain are observed only in adult animals. Since synthesis of testosterone (and therefore presumably also 17β-estradiol) is normal in perinatal Tfm mice (Goldstein and Wilson, 1972), many of the early organizational effects attributed to ER activity via aromatized testosterone may thus still occur in this AR-deficient strain.
Experiments in the AR-deficient Tfm line also suggest a novel mechanism by which the AAS may alter neuronal function in vivo. In the absence of classical AR signaling, our data indicate that the AAS do not act, as initially expected, as ER agonists. Rather our data indicate that that abuse-level concentrations of these synthetic steroids interfere with ER signaling by inhibiting aromatase and diminishing production of 17β-estradiol. Specifically, while testosterone levels in the mPOA are dramatically elevated in both wild-type and AR-deficient AAS-treated mice, levels of 17β-estradiol are lower than in controls for both genotypes. Additionally, AAS-treated wild-type mice show significantly elevated levels of the AR-regulated aromatase mRNA, but AAS treatment is without effect on ER-regulated PR mRNA in wild-type or AR-deficient mice. In the AR-deficient Tfm line, the AAS and the aromatase inhibitor, formestane, both significantly decrease sIPSC amplitudes, and the AAS were without further effect on sIPSC amplitudes in animals receiving coincident treatment with formestane. The AAS also promoted a significant decrease in sIPSC frequency versus control in AR-deficient mice, and there were no differences in sIPSC frequency between formestane and AAS-treated Tfm mice or AAS-treated Tfm mice and mice treated concurrently with the AAS and formestane. Formestane has been shown to inhibit estrogen-dependent upregulation of GAD65 and GABA production in hippocampal neurons (Ikeda et al., 2006) and here we show that GAD65 mRNA in the mPOA was decreased in AAS-treated Tfm mice, consistent with a common mechanism of action. Finally, concurrent treatment of AAS-treated Tfm mice with 17β-estradiol negated the effects of AAS treatment on GABAergic transmission. The AAS have been shown to inhibit aromatase activity in vitro in non-neuronal cell lines (Mor et al., 2001; de Gooyer et al., 2003) and aromatase inhibition by the AAS has also been suggested to occur in fish gonads (Zhang et al., 2007). The data presented here extend these findings to suggest that the AAS inhibit aromatase in the mammalian brain. The proposed mechanism of AAS inhibition of aromatase does not preclude additional effects these steroids may have on other key steroid biosynthetic enzymes, such as 5α- and 5β-reductase, that may also affect the endogenous levels of testosterone and estrogens. Such concurrent effects on other enzymes have been demonstrated for the aromatase inhibitor, ATD (Motta et al., 1986; Wozniak et al., 1992; Wade et al., 1994).
In wild-type as well as AR-deficient Tfm mice, AAS treatment did not result in a significant increase in the levels 17β-estradiol or in the induction of PR mRNA, despite dramatically elevated of testosterone as a precursor for 17β-estradiol, consistent with the hypothesis that the AAS may inhibit aromatase in wild-type, as well as AR-deficient animals. Nonetheless, AAS and formestane did not have comparable effects on the GABAergic transmission in wild-type mice, and we conclude that AAS effects mediated via AR predominate on this specific endpoint in wild-type subjects. However, the number of steroid-sensitive molecular targets and steroid-sensitive physiological processes in the brain is extraordinarily expansive and GABAergic signaling in the mPOA is only one component in this repertoire. These many molecular targets and their physiological outcomes are likely to be regulated by equivalently diverse balance between AR- versus ER-mediated signaling. Thus, in the wild-type male mouse, the AAS may upregulate α5 subunit mRNA levels and prolong GABAA receptor-mediated synaptic current decay by acting as AR agonists, but a myriad of steroid-sensitive processes yet to be examined may be more sensitive to an AAS-dependent decrease in estrogen production. Additionally, even the same GABAergic endpoints examined here may be preferentially more sensitive to changes in ER signaling in females or adolescent males where both neural substrates and the endogenous steroid milieu differ significantly.
Humans administer AAS in sophisticated and complex regimes of doses and patterns, often involving self-administration of multiple classes of AAS in a pattern called “stacking” (Llewellyn, 2007). Our data highlight the potential variability of molecular and cellular effects of the AAS in the brain and suggest that the range of actions of these steroids will be highly complex, depending not only upon the chemical signatures and concentrations of the different AAS taken, but also upon region-specific differences in AR, ERα, ERβ, and aromatase expression and changes in the endogenous steroid environment that occur as a function of sex, age, and hormonal state.
Footnotes
This work was supported by the National Institutes of Health (Grants DA18255 and DA14137). We thank Drs. Ann Clark and Joseph Oberlander and Mr. Benjamin Solomon for critical review of this manuscript and Dr. Beth Costine for statistical assistance.
References
- Abdelgadir SE, Resko JA, Ojeda SR, Lephart ED, McPhaul MJ, Roselli CE. Androgens regulate aromatase cytochrome P450 messenger ribonucleic acid in rat brain. Endocrinology. 1994;135:395–401. doi: 10.1210/endo.135.1.8013375. [DOI] [PubMed] [Google Scholar]
- Ali AB, Thomson AM. Synaptic α5 subunit-containing GABAA receptors mediate IPSCs elicited by dendrite-preferring cells in rat neocortex. Cereb Cortex. 2008;18:1260–1271. doi: 10.1093/cercor/bhm160. [DOI] [PubMed] [Google Scholar]
- Bakker J, Honda S, Harada N, Balthazart J. Restoration of male sexual behavior by adult exogenous estrogens in male aromatase knockout mice. Horm Behav. 2004;46:1–10. doi: 10.1016/j.yhbeh.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Bar-Gad I, Ritov Y, Bergman H. The neuronal refractory period causes a short-term peak in the autocorrelation function. J Neurosci Methods. 2001;104:155–163. doi: 10.1016/s0165-0270(00)00335-6. [DOI] [PubMed] [Google Scholar]
- Basaria S, Wahlstrom JT, Dobs AS. Clinical review 138: Anabolic-androgenic steroid therapy in the treatment of chronic diseases. J Clin Endocrinol Metab. 2001;86:5108–5117. doi: 10.1210/jcem.86.11.7983. [DOI] [PubMed] [Google Scholar]
- Blaustein JD, Erskine MS. Feminine sexual behavior: cellular integration of hormonal and afferent information in the rodent forebrain. In: Pfaff DW, Arnold AP, Etgen AM, Fahrbach SE, Rubin RT, editors. Hormones, brain and behavior. Vol 1. Orlando, FL: Academic; 2002. pp. 139–213. [Google Scholar]
- Bodo C, Rissman EF. Androgen receptor is essential for sexual differentiation of responses to olfactory cues in mice. Eur J Neurosci. 2007;25:2182–2190. doi: 10.1111/j.1460-9568.2007.05484.x. [DOI] [PubMed] [Google Scholar]
- Bright DP, Aller MI, Brickley SG. Synaptic release generates a tonic GABAA receptor-mediated conductance that modulates burst precision in thalamic relay neurons. J Neurosci. 2007;27:2560–2569. doi: 10.1523/JNEUROSCI.5100-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgard EC, Tietz EI, Neelands TR, Macdonald RL. Properties of recombinant γ-aminobutyric acidA receptor isoforms containing the α5 subunit subtype. Mol Pharmacol. 1996;50:119–127. [PubMed] [Google Scholar]
- Caraiscos VB, Elliott EM, You-Ten KE, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ, Rosahl TW, Wafford KA, MacDonald JF, Orser BA. Tonic inhibition in mouse hippocampus CA1 pyramidal neurons is mediated by α5 subunit-containing γ-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 2004;101:3662–3667. doi: 10.1073/pnas.0307231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charest NJ, Zhou ZX, Lubahn DB, Olsen KL, Wilson EM, French FS. A frameshift mutation destabilizes androgen receptor messenger RNA in the Tfm mouse. Mol Endocrinol. 1991;5:573–581. doi: 10.1210/mend-5-4-573. [DOI] [PubMed] [Google Scholar]
- Clark AS, Henderson LP. Behavioral and physiological responses to anabolic-androgenic steroids. Neurosci Biobehav Rev. 2003;27:413–436. doi: 10.1016/s0149-7634(03)00064-2. [DOI] [PubMed] [Google Scholar]
- Clark AS, Costine BA, Jones BL, Kelton-Rehkopf MC, Meerts SH, Nutbrown-Greene LL, Penatti CA, Porter DM, Yang P, Henderson LP. Sex- and age-specific effects of anabolic androgenic steroids on reproductive behaviors and on GABAergic transmission in neuroendocrine control regions. Brain Res. 2006;1126:122–138. doi: 10.1016/j.brainres.2006.08.081. [DOI] [PubMed] [Google Scholar]
- Cornil CA, Ball GF, Balthazart J. Functional significance of the rapid regulation of brain estrogen action: where do the estrogens come from? Brain Res. 2006;1126:2–26. doi: 10.1016/j.brainres.2006.07.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couse JF, Korach KS. Exploring the role of sex steroids through studies of receptor deficient mice. J Mol Med. 1998;76:497–511. doi: 10.1007/s001090050244. [DOI] [PubMed] [Google Scholar]
- de Fougerolles Nunn E, Greenstein B, Khamashta M, Hughes GR. Evidence for sexual dimorphism of estrogen receptors in hypothalamus and thymus of neonatal and immature Wistar rats. Int J Immunopharmacol. 1999;21:869–877. doi: 10.1016/s0192-0561(99)00055-7. [DOI] [PubMed] [Google Scholar]
- de Gooyer ME, Oppers-Tiemissen HM, Leysen D, Verheul HA, Kloosterboer HJ. Tibolone is not converted by human aromatase to 7α-methyl-17α-ethynylestradiol (7α-MEE): Analyses with sensitive bioassays for estrogens and androgens and with LC-MSMS. Steroids. 2003;68:235–243. doi: 10.1016/s0039-128x(02)00184-8. [DOI] [PubMed] [Google Scholar]
- Dunning DD, Hoover CL, Soltesz I, Smith MA, O'Dowd DK. GABAA receptor-mediated miniature postsynaptic currents and α-subunit expression in developing cortical neurons. J Neurophysiol. 1999;82:3286–3297. doi: 10.1152/jn.1999.82.6.3286. [DOI] [PubMed] [Google Scholar]
- Esclapez M, Tillakaratne NJ, Kaufman DL, Tobin AJ, Houser CR. Comparative localization of two forms of glutamic acid decarboxylase and their mRNAs in rat brain supports the concept of functional differences between the forms. J Neurosci. 1994;14:1834–1855. doi: 10.1523/JNEUROSCI.14-03-01834.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Feldblum S, Erlander MG, Tobin AJ. Different distributions of GAD65 and GAD67 mRNAs suggest that the two glutamate decarboxylases play distinctive functional roles. J Neurosci Res. 1993;34:689–706. doi: 10.1002/jnr.490340612. [DOI] [PubMed] [Google Scholar]
- Foidart A, Harada N, Balthazart J. Effects of steroidal and non steroidal aromatase inhibitors on sexual behavior and aromatase-immunoreactive cells and fibers in the quail brain. Brain Res. 1994;657:105–123. doi: 10.1016/0006-8993(94)90958-x. [DOI] [PubMed] [Google Scholar]
- Foidart A, Harada N, Balthazart J. Aromatase-immunoreactive cells are present in mouse brain areas that are known to express high levels of aromatase activity. Cell Tissue Res. 1995;280:561–574. doi: 10.1007/BF00318360. [DOI] [PubMed] [Google Scholar]
- Franke WW, Berendonk B. Hormonal doping and androgenization of athletes: a secret program of the German Democratic Republic government. Clin Chem. 1997;43:1262–1279. [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. San Diego: Academic; 1997. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Gao B, Moore RY. The sexually dimorphic nucleus of the hypothalamus contains GABA neurons in rat and man. Brain Res. 1996;742:163–171. doi: 10.1016/s0006-8993(96)01005-0. [DOI] [PubMed] [Google Scholar]
- Gaspar ML, Meo T, Bourgarel P, Guenet JL, Tosi M. A single base pair deletion in the Tfm androgen receptor gene creates a short-lived messenger RNA that directs internal translation initiation. Proc Natl Acad Sci U S A. 1991;88:8606–8610. doi: 10.1073/pnas.88.19.8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Wilson JD. Studies on the pathogenesis of the pseudohermaphroditism in the mouse with testicular feminization. J Clin Invest. 1972;51:1647–1658. doi: 10.1172/JCI106966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson JL. The vertebrate social behavior network: evolutionary themes and variations. Horm Behav. 2005;48:11–22. doi: 10.1016/j.yhbeh.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häusser M, Clark BA. Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron. 1997;19:665–678. doi: 10.1016/s0896-6273(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Henderson LP. Steroid modulation of GABAA receptor-mediated transmission in the hypothalamus: effects on reproductive function. Neuropharmacology. 2007;52:1439–1453. doi: 10.1016/j.neuropharm.2007.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LP, Penatti CA, Jones BL, Yang P, Clark AS. Anabolic androgenic steroids and forebrain GABAergic transmission. Neuroscience. 2006;138:793–799. doi: 10.1016/j.neuroscience.2005.08.039. [DOI] [PubMed] [Google Scholar]
- Herbison AE. Estrogen regulation of GABA transmission in rat preoptic area. Brain Res Bull. 1997;44:321–326. doi: 10.1016/s0361-9230(97)00210-4. [DOI] [PubMed] [Google Scholar]
- Hewitt SA, Wamsteeker JI, Kurz EU, Bains JS. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat Neurosci. 2009;12:438–443. doi: 10.1038/nn.2274. [DOI] [PubMed] [Google Scholar]
- Hong Y, Cho M, Yuan YC, Chen S. Molecular basis for the interaction of four different classes of substrates and inhibitors with human aromatase. Biochem Pharmacol. 2008;75:1161–1169. doi: 10.1016/j.bcp.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Hull EM, Meisel RL, Sachs BD. Male sexual behavior. In: Pfaff DW, Arnold AP, Etgen AM, Fahrbach SE, Rubin RT, editors. Hormones, Brain and Behavior. Vol 1. Orlando, FL: Academic; 2002. pp. 3–137. [Google Scholar]
- Ikeda T, Matsuki N, Yamada MK. Estrogen produced in cultured hippocampal neurons is a functional regulator of a GABAergic machinery. J Neurosci Res. 2006;84:1771–1777. doi: 10.1002/jnr.21083. [DOI] [PubMed] [Google Scholar]
- Jones BL, Whiting PJ, Henderson LP. Mechanisms of anabolic androgenic steroid inhibition of mammalian ε-subunit-containing GABAA receptors. J Physiol. 2006;573:571–593. doi: 10.1113/jphysiol.2006.106534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorge JC, McIntyre KL, Henderson LP. The function and the expression of forebrain GABAA receptors change with hormonal state in the adult mouse. J Neurobiol. 2002;50:137–149. doi: 10.1002/neu.10021. [DOI] [PubMed] [Google Scholar]
- Kibble MW, Ross MB. Adverse effects of anabolic steroids in athletes. Clin Pharm. 1987;6:686–692. [PubMed] [Google Scholar]
- Kochakian C, Yesalis CE. Anabolic-androgenic steroids: a historical perspective and definition. In: Yesalis CE, editor. Anabolic steroids in sport and exercise. Champaign: Human Kinetics; 2000. pp. 4–33. [Google Scholar]
- Kohler M, Parr MK, Opfermann G, Thevis M, Schlörer N, Marner FJ, Schänzer W. Metabolism of 4-hydroxyandrostenedione and 4-hydroxytestosterone: Mass spectrometric identification of urinary metabolites. Steroids. 2007;72:278–286. doi: 10.1016/j.steroids.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Kononenko NI, Dudek FE. Mechanism of irregular firing of suprachiasmatic nucleus neurons in rat hypothalamic slices. J Neurophysiol. 2004;91:267–273. doi: 10.1152/jn.00314.2003. [DOI] [PubMed] [Google Scholar]
- Kressler D, Hock MB, Kralli A. Coactivators PGC-1β and SRC-1 interact functionally to promote agonist activity of the selective estrogen receptor modulator tamoxifen. J Biol Chem. 2007;282:26897–26907. doi: 10.1074/jbc.M705596200. [DOI] [PubMed] [Google Scholar]
- Kudwa AE, Gustafsson JÅ, Rissman EF. Estrogen receptor β modulates estradiol induction of progestin receptor immunoreactivity in male, but not in female, mouse medial preoptic area. Endocrinology. 2004;145:4500–4506. doi: 10.1210/en.2003-1708. [DOI] [PubMed] [Google Scholar]
- LaMorte A, Kumar N, Bardin CW, Sundaram K. Aromatization of 7α-methyl-19-nortestosterone by human placental microsomes in vitro. J Steroid Biochem Mol Biol. 1994;48:297–304. doi: 10.1016/0960-0760(94)90160-0. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Llewellyn W. Ed 6. Jupiter FL: Body of Science; 2007. Anabolics. [Google Scholar]
- Lu SF, McKenna SE, Cologer-Clifford A, Nau EA, Simon NG. Androgen receptor in mouse brain: sex differences and similarities in autoregulation. Endocrinology. 1998;139:1594–1601. doi: 10.1210/endo.139.4.5863. [DOI] [PubMed] [Google Scholar]
- Maguire J, Mody I. Neurosteroid synthesis-mediated regulation of GABAA receptors: relevance to the ovarian cycle and stress. J Neurosci. 2007;27:2155–2162. doi: 10.1523/JNEUROSCI.4945-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini L. The 5 alpha-reduction of testosterone in the neuroendocrine structures. Biochemical and physiological implications. Endocr Rev. 1982;3:1–25. doi: 10.1210/edrv-3-1-1. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Takeyama K, Sato T, Kato S. Androgen receptor functions from reverse genetic models. J Steroid Biochem Mol Biol. 2003;85:95–99. doi: 10.1016/s0960-0760(03)00231-0. [DOI] [PubMed] [Google Scholar]
- Miczek KA, Fish EW, De Bold JF, De Almeida RM. Social and neural determinants of aggressive behavior: pharmacotherapeutic targets at serotonin, dopamine and γ-aminobutyric acid systems. Psychopharmacology. 2002;163:434–458. doi: 10.1007/s00213-002-1139-6. [DOI] [PubMed] [Google Scholar]
- Mitra SW, Hoskin E, Yudkovitz J, Pear L, Wilkinson HA, Hayashi S, Pfaff DW, Ogawa S, Rohrer SP, Schaeffer JM, McEwen BS, Alves SE. Immunolocalization of estrogen receptor β in the mouse brain: comparison with estrogen receptor α. Endocrinology. 2003;144:2055–2067. doi: 10.1210/en.2002-221069. [DOI] [PubMed] [Google Scholar]
- Mor G, Eliza M, Song J, Wiita B, Chen S, Naftolin F. 17α-Methyltestosterone is a competitive inhibitor of aromatase activity in Jar choriocarcinoma cells and macrophage-like THP-1 cells in culture. J Steroid Biochem Mol Biol. 2001;79:239–246. doi: 10.1016/s0960-0760(01)00162-5. [DOI] [PubMed] [Google Scholar]
- Motta M, Zoppi S, Brodie AM, Martini L. Effect of 1,4,6-androstatriene-3,17-dione (ATD), 4-hydroxy-4-androstene-3,17-dione (4-OH-A) and 4-acetoxy-4-androstene-3,17-dione (4-Ac-A) on the 5 alpha-reduction of androgens in the rat prostate. J Steroid Biochem. 1986;25:593–600. doi: 10.1016/0022-4731(86)90408-5. [DOI] [PubMed] [Google Scholar]
- Mozrzymas JW, Barberis A, Michalak K, Cherubini E. Chlorpromazine inhibits miniature GABAergic currents by reducing the binding and by increasing the unbinding rate of GABAA receptors. J Neurosci. 1999;19:2474–2488. doi: 10.1523/JNEUROSCI.19-07-02474.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy L, O'Shaughnessy PJ. Testicular steroidogenesis in the testicular feminized (Tfm) mouse: loss of 17α-hydroxylase activity. J Endocrinol. 1991;131:443–449. doi: 10.1677/joe.0.1310443. [DOI] [PubMed] [Google Scholar]
- Nett ST, Jorge-Rivera JC, Myers M, Clark AS, Henderson LP. Properties and sex-specific differences of GABAA receptors in neurons expressing γ1 subunit mRNA in the preoptic area of the rat. J Neurophysiol. 1999;81:192–203. doi: 10.1152/jn.1999.81.1.192. [DOI] [PubMed] [Google Scholar]
- Nomura M, Korach KS, Pfaff DW, Ogawa S. Estrogen receptor β (ERβ) protein levels in neurons depend on estrogen receptor α (ERα) gene expression and on its ligand in a brain region-specific manner. Brain Res Mol Brain Res. 2003;110:7–14. doi: 10.1016/s0169-328x(02)00544-2. [DOI] [PubMed] [Google Scholar]
- Olsen KL. Genetic influences on sexual behavior differentiation. In: Gerall AA, Moltz H, Ward IL, editors. Handbook of behavioral neurobiology: sexual differentiation. Vol 11. New York: Plenum; 1992. pp. 1–40. [Google Scholar]
- Oosterkamp AJ, Villaverde Herraiz MT, Irth H, Tjaden UR, van der Greef J. Reversed-phase liquid chromatography coupled on-line to receptor affinity detection based on the human estrogen receptor. Anal Chem. 1996;68:1201–1206. doi: 10.1021/ac950736s. [DOI] [PubMed] [Google Scholar]
- O'Shaughnessy PJ, Murphy L. Cytochrome P-450 17α-hydroxylase protein and mRNA in the testis of the testicular feminized (Tfm) mouse. J Mol Endocrinol. 1993;11:77–82. doi: 10.1677/jme.0.0110077. [DOI] [PubMed] [Google Scholar]
- Papaconstantinou AD, Umbreit TH, Goering PL, Brown KM. Effects of 17α-methyltestosterone on uterine morphology and heat shock protein expression are mediated through estrogen and androgen receptors. J Steroid Biochem Mol Biol. 2002;82:305–314. doi: 10.1016/s0960-0760(02)00221-2. [DOI] [PubMed] [Google Scholar]
- Peirson SN, Butler JN, Foster RG. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 2003;31:e73. doi: 10.1093/nar/gng073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penatti CA, Porter DM, Jones BL, Henderson LP. Sex-specific effects of chronic anabolic androgenic steroid treatment on GABAA receptor expression and function in adolescent mice. Neuroscience. 2005;135:533–543. doi: 10.1016/j.neuroscience.2005.06.041. [DOI] [PubMed] [Google Scholar]
- Penatti CA, Costine BA, Porter DM, Henderson LP. Effects of chronic exposure to an anabolic androgenic steroid cocktail on α5-receptor mediated GABAergic transmission and neural signaling in the forebrain of female mice. Neuroscience. 2009;161:526–537. doi: 10.1016/j.neuroscience.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry PJ, Andersen KH, Yates WR. Illicit anabolic steroid use in athletes. A case series analysis. Am J Sports Med. 1990;18:422–428. doi: 10.1177/036354659001800416. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picton AJ, Fisher JL. Effect of the α subunit subtype on the macroscopic kinetic properties of recombinant GABAA receptors. Brain Res. 2007;1165:40–49. doi: 10.1016/j.brainres.2007.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope HG, Jr, Katz DL. Affective and psychotic symptoms associated with anabolic steroid use. Am J Psychiatry. 1988;145:487–490. doi: 10.1176/ajp.145.4.487. [DOI] [PubMed] [Google Scholar]
- Quincey RV, Gray CH. The metabolism of [1,2-3H]17α-methyltestosterone in human subjects. J Endocrinol. 1967;37:37–55. doi: 10.1677/joe.0.0370037. [DOI] [PubMed] [Google Scholar]
- Quirk K, Blurton P, Fletcher S, Leeson P, Tang F, Mellilo D, Ragan CI, McKernan RM. [3H]-L655,708, a novel ligand selective for the benzodiazepine site of GABAA receptors which contain the α5 subunit. Neuropharmacology. 1996;35:1331–1335. doi: 10.1016/s0028-3908(96)00061-5. [DOI] [PubMed] [Google Scholar]
- Roselli CE, Resko JA. Sex differences in androgen-regulated expression of cytochrome P450 aromatase in the rat brain. J Steroid Biochem Mol Biol. 1997;61:365–374. [PubMed] [Google Scholar]
- Roselli CE, Abdelgadir SE, Rønnekleiv OK, Klosterman SA. Anatomic distribution and regulation of aromatase gene expression in the rat brain. Biol Reprod. 1998;58:79–87. doi: 10.1095/biolreprod58.1.79. [DOI] [PubMed] [Google Scholar]
- Rosenfeld JM, Daley JD, Ohno S, YoungLai EV. Central aromatization of testosterone in testicular feminized mice. Experientia. 1977;33:1392–1393. doi: 10.1007/BF01920200. [DOI] [PubMed] [Google Scholar]
- Rudick CN, Woolley CS. Selective estrogen receptor modulators regulate phasic activation of hippocampal CA1 pyramidal cells by estrogen. Endocrinology. 2003;144:179–187. doi: 10.1210/en.2002-220581. [DOI] [PubMed] [Google Scholar]
- Ryan KJ. Biological aromatization of steroids. J Biol Chem. 1959;234:268–272. [PubMed] [Google Scholar]
- Sagrillo CA, Selmanoff M. Castration decreases single cell levels of mRNA encoding glutamic acid decarboxylase in the diagonal band of Broca and the sexually dimorphic nucleus of the preoptic area. J Neuroendocrinol. 1997;9:699–706. doi: 10.1046/j.1365-2826.1997.00630.x. [DOI] [PubMed] [Google Scholar]
- Sakuma Y. Gonadal steroid action and brain sex differentiation in the rat. J Neuroendocrinol. 2009;21:410–414. doi: 10.1111/j.1365-2826.2009.01856.x. [DOI] [PubMed] [Google Scholar]
- Sato T, Matsumoto T, Kawano H, Watanabe T, Uematsu Y, Sekine K, Fukuda T, Aihara K, Krust A, Yamada T, Nakamichi Y, Yamamoto Y, Nakamura T, Yoshimura K, Yoshizawa T, Metzger D, Chambon P, Kato S. Brain masculinization requires androgen receptor function. Proc Natl Acad Sci U S A. 2004;101:1673–1678. doi: 10.1073/pnas.0305303101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleicher G, Stumpf WE, Morin JK, Drews U. Sites of aromatization of [3H]testosterone in forebrain of male, female and androgen receptor-deficient Tfm mice: an autoradiographic study. Brain Res. 1986;397:290–296. doi: 10.1016/0006-8993(86)90630-x. [DOI] [PubMed] [Google Scholar]
- Schleicher G, Khan S, Nieschlag E. Differentiaton between androgen and estrogen receptor mediated effects of testosterone on FSH using androgen receptor deficient (Tfm) and normal mice. J Steroid Biochem. 1989;33:49–51. doi: 10.1016/0022-4731(89)90356-7. [DOI] [PubMed] [Google Scholar]
- Scimemi A, Semyanov A, Sperk G, Kullmann DM, Walker MC. Multiple and plastic receptors mediate tonic GABAA receptor currents in the hippocampus. J Neurosci. 2005;25:10016–10024. doi: 10.1523/JNEUROSCI.2520-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scordalakes EM, Rissman EF. Aggression in male mice lacking estrogen receptor α. Behav Neurosci. 2003;117:38–45. [PubMed] [Google Scholar]
- Scordalakes EM, Shetty SJ, Rissman EF. Roles of estrogen receptor α and androgen receptor in the regulation of neuronal nitric oxide synthase. J Comp Neurol. 2002;453:336–344. doi: 10.1002/cne.10413. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, Silver RA. Tonically active GABAA receptors: modulating gain and maintaining the tone. Trends Neurosci. 2004;27:262–269. doi: 10.1016/j.tins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Serwanski DR, Miralles CP, Christie SB, Mehta AK, Li X, De Blas AL. Synaptic and nonsynaptic localization of GABAA receptors containing the α5 subunit in the rat brain. J Comp Neurol. 2006;499:458–470. doi: 10.1002/cne.21115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewards TV, Sewards MA. Fear and power-dominance drive motivation: neural representations and pathways mediating sensory and mnemonic inputs, and outputs to premotor structures. Neurosci Biobehav Rev. 2002;26:553–579. doi: 10.1016/s0149-7634(02)00020-9. [DOI] [PubMed] [Google Scholar]
- Shah NM, Pisapia DJ, Maniatis S, Mendelsohn MM, Nemes A, Axel R. Visualizing sexual dimorphism in the brain. Neuron. 2004;43:313–319. doi: 10.1016/j.neuron.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Shahidi NT. A review of the chemistry, biological action, and clinical applications of anabolic-androgenic steroids. Clin Ther. 2001;23:1355–1390. doi: 10.1016/s0149-2918(01)80114-4. [DOI] [PubMed] [Google Scholar]
- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- Simon NG. Hormonal processes in the development and expression of aggressive behavior. In: Pfaff DW, Arnold AP, Etgen AM, Farhbach SE, Rubin RT, editors. Hormones, brain and behavior. Vol 1. San Diego: Academic; 2002. pp. 339–392. [Google Scholar]
- Smith SS, Gong QH. Neurosteroid administration and withdrawal alter GABAA receptor kinetics in CA1 hippocampus of female rats. J Physiol 564. 2005;2:421–436. doi: 10.1113/jphysiol.2004.077297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner JM, Teague SR, Williams DA. Development and analytic validation of an enzyme-linked immunosorbent assay for the measurement of canine pancreatic lipase immunoreactivity in serum. Can J Vet Res. 2003;67:175–182. [PMC free article] [PubMed] [Google Scholar]
- Trenton AJ, Currier GW. Behavioural manifestations of anabolic steroid use. CNS Drugs. 2005;19:571–595. doi: 10.2165/00023210-200519070-00002. [DOI] [PubMed] [Google Scholar]
- Vandenput L, Swinnen JV, Boonen S, Van Herck E, Erben RG, Bouillon R, Vanderschueren D. Role of the androgen receptor in skeletal muscle homeostasis: The androgen-resistant testicular feminized male mouse model. J Bone Miner Res. 2004;19:1462–1470. doi: 10.1359/JBMR.040505. [DOI] [PubMed] [Google Scholar]
- van Geel WJA, Abdo WF, Melis R, Williams S, Bloem BR, Verbeek MM. A more efficient enzyme-linked immunosorbent assay for measurement of α-synuclein in cerebrospinal fluid. J Neurosci Methods. 2008;168:182–185. doi: 10.1016/j.jneumeth.2007.09.021. [DOI] [PubMed] [Google Scholar]
- Veening JG, Coolen LM, de Jong TR, Joosten HW, de Boer SF, Koolhaas JM, Olivier B. Do similar neural systems subserve aggressive and sexual behaviour in male rats? Insights from c-Fos and pharmacological studies. Eur J Pharmacol. 2005;526:226–239. doi: 10.1016/j.ejphar.2005.09.041. [DOI] [PubMed] [Google Scholar]
- Wade J, Schlinger BA, Hodges L, Arnold AP. Fadrozole: a potent and specific inhibitor of aromatase in the zebra finch brain. Gen Comp Endocrinol. 1994;94:53–61. doi: 10.1006/gcen.1994.1059. [DOI] [PubMed] [Google Scholar]
- Winters SJ. Androgens: endocrine physiology and pharmacology. NIDA Res Monogr. 1990;102:113–130. [PubMed] [Google Scholar]
- Wozniak A, Holman SD, Hutchison JB. In vitro potency and selectivity of the non-steroidal androgen aromatase inhibitor CGS 16949A compared to steroidal inhibitors in the brain. J Steroid Biochem Mol Biol. 1992;43:281–287. doi: 10.1016/0960-0760(92)90162-c. [DOI] [PubMed] [Google Scholar]
- Yamada J, Furukawa T, Ueno S, Yamamoto S, Fukuda A. Molecular basis for the GABAA receptor-mediated tonic inhibition in rat somatosensory cortex. Cereb Cortex. 2007;17:1782–1787. doi: 10.1093/cercor/bhl087. [DOI] [PubMed] [Google Scholar]
- Yang P, Jones BL, Henderson LP. Mechanisms of anabolic androgenic steroid modulation of α1β3γ2L GABAA receptors. Neuropharmacology. 2002;43:619–633. doi: 10.1016/s0028-3908(02)00155-7. [DOI] [PubMed] [Google Scholar]
- Yang P, Jones BL, Henderson LP. Role of the α subunit in the modulation of GABAA receptors by anabolic androgenic steroids. Neuropharmacology. 2005;49:300–316. doi: 10.1016/j.neuropharm.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Yue W, Brodie AM. Mechanisms of the actions of aromatase inhibitors 4-hydroxyandrostenedione, fadrozole, and aminoglutethimide on aromatase in JEG-3 cell culture. J Steroid Biochem Mol Biol. 1997;63:317–328. doi: 10.1016/s0960-0760(97)00072-1. [DOI] [PubMed] [Google Scholar]
- Yue W, Wang J, Savinov A, Brodie A. Effect of aromatase inhibitors on growth of mammary tumors in a nude mouse model. Cancer Res. 1995;55:3073–3077. [PubMed] [Google Scholar]
- Zhang W, Zhang Y, Zhang L, Zhao H, Li X, Huang H, Lin H. The mRNA expression of P450 aromatase, gonadotropin β-subunits and FTZ-F1 in the orange-spotted grouper (Epinephelus coioides) during 17α-methyltestosterone-induced precocious sex change. Mol Reprod Dev. 2007;74:665–673. doi: 10.1002/mrd.20642. [DOI] [PubMed] [Google Scholar]
- Zhao C, Fujinaga R, Tanaka M, Yanai A, Nakahama K, Shinoda K. Region-specific expression and sex-steroidal regulation on aromatase and its mRNA in the male rat brain: immunohistochemical and in situ hybridization analyses. J Comp Neurol. 2007;500:557–573. doi: 10.1002/cne.21193. [DOI] [PubMed] [Google Scholar]