

Abstract
Transcription and splicing must proceed over genomic distances of hundreds of kilobases in many human genes. However, the rates and mechanisms of these processes are poorly understood. We have used the compound 5,6-Dichlorobenzimidazole 1-b-D-ribofuranoside (DRB) that reversibly blocks gene transcription in vivo combined with quantitative RT-PCR to analyze the transcription and RNA processing of several long human genes. We found that the rate of RNA polymerase II transcription over long genomic distances is about 3.8 kb per minute and is nearly the same whether transcribing long introns or exon rich regions. We also determined that co-transcriptional pre-mRNA splicing of U2-dependent introns occurs within 5–10 minutes of synthesis irrespective of intron length between 1 kb and 240 kb. Similarly, U12-dependent introns were co-transcriptionally spliced within 10 minutes of synthesis confirming that these introns are spliced within the nuclear compartment. These results show that the expression of large genes is surprisingly rapid and efficient.
Transcription by RNA Polymerase II (RNAPII) is a multistep process consisting of promoter binding, transcription initiation, elongation and termination. In addition, RNAPII plays important roles in RNA processing events including 5′ cap formation, splicing, poly A tail formation, 3′ end processing and transport of the mRNA to the cytoplasm1-6. Much of the previous research on transcriptional regulation has focused on promoter regulation and transcription initiation. However, recent work has suggested that events subsequent to transcript initiation may be of equal or greater importance in regulating the output of genes7-10. Furthermore, the rate of elongation and pausing of RNAPII is emerging as a key contributory factor in the regulation of alternative splicing7,11-13. This in turn has generated considerable interest in the kinetics of transcriptional elongation by RNAPII.
Previous efforts to calculate the in situ elongation rate of RNAPII have employed a range of methods including RT-PCR14, nuclear run-on assays15, and fluorescent in situ hybridization16,17. These studies focused on specific genes and yielded apparent elongation rate estimates ranging from 1.1 to 4.3 kb per minute (kb min−1). More recent studies have utilized engineered gene constructs monitored by fluorescent imaging techniques in living cells17,18. Mathematical modeling was used to extract detailed kinetic information including rates of transcriptional elongation. The calculated elongation rates derived from these studies ranged from 1.9 to 4.3 kb min−1. Given the importance of post-initiation events in gene expression and the range of results produced by these methods, a simple technique capable of measuring the rates of transcription of many genes in their endogenous environments in many cell types would be a major advance.
The rate of splicing of introns in vivo has also been studied in a few cases. Analysis of introns of an endogenous gene19 or of small introns in artificial gene constructs with inducible promoters20 showed that splicing occurred on a time scale of 1 to 12 minutes. These studies examined relatively short introns. Since intron length can vary from less than 100 bases to hundreds of kilobases, it would be interesting to measure splicing rates over a range of gene and intron lengths.
There is strong evidence to conclude that RNA splicing of major class or U2-dependent introns frequently occurs co-transcriptionally and is aided by the elongating RNAPII3,21-23. In contrast, a recent report24 suggested that the majority of the minor class or U12-dependent spliceosome components may be localized predominantly in the cytoplasm and hence most if not all of the U12-dependent splicing would have to take place post-transcriptionally after the transcript is fully synthesized and transported to the cytoplasm. This study, which itself contradicted earlier results25, prompted other reports26,27 that demonstrated the preferential localization of U12-dependent spliceosomal RNA and protein components to the nucleus. However, in the absence of direct in vivo evidence on the temporal and spatial relationship of transcription and minor class intron splicing, the controversy has not been resolved28,29.
Here we report the development of a simple assay system with which to measure the rates of RNAPII transcription and pre-mRNA splicing in vivo on endogenous human genes. Using the reversible inhibitor DRB, we can switch off and rapidly switch on transcription by RNAPII of a large number of genes without apparent detrimental effect on the cellular machinery. We report on the use of this method in conjunction with real-time qRT-PCR to study the kinetics of transcription and splicing in a number of human genes in their natural chromatin environments. Our results demonstrate that RNAPII transcribes at a rate close to 3.8 kb min−1 over megabase distances and that the splicing of both U2-dependent and U12-dependent introns can occur rapidly and co-transcriptionally.
Results
DRB inhibits the P-TEFb dependent ser-2 phosphorylation of the C-terminal domain (CTD) of RNAPII resulting in its failure to progress from the initiation to the elongation phase of transcription30-33. Importantly, it does not block elongation of previously initiated transcripts already within the gene32. It should also be noted that, while most genes are blocked by DRB, there is a subset of promoters that do not respond to DRB34.
We used this property of DRB to develop a method to reversibly block gene transcription in cultured human cells. The cells were incubated for various times with DRB and total RNA was prepared from the cultures and assayed for the levels of unspliced pre-mRNA for several genes. During the time of DRB treatment, most of the unspliced pre-mRNA should be processed to mature mRNA by the splicing machinery or degraded (see Supplementary Fig. 1 online). Removal of DRB should then lead to release of RNAPII from promoter proximal regions starting fresh rounds of transcription. These newly started primary transcripts will be detectable due to the presence of introns.
Thus, human Tet-21 cells were treated for 3 hours with DRB and samples were taken at 5 minute intervals after removal of DRB. Quantitative RT-PCR was performed using primers spanning exon-intron junctions to detect pre-mRNA expression (Fig. 1a). As a test case we focused on the 561 kb Utrophin gene. The Utrophin gene contains 74 exons and 73 introns and its first intron is 111 kb long. The results show that the cells incubated with DRB were able to recover transcription of the exon 1 region of the Utrophin gene within minutes of DRB removal (Fig.1b). In contrast, recovery of expression of the exon 2 region shows a delay until 35 minutes after drug release consistent with a transcriptional lag due to the genomic distance between the first two exons. This suggests that RNAPII transcribed the 111 kb region of the Utrophin gene in roughly 30 minutes at a rate of ∼3.7 kb min−1.
Figure 1. DRB reversibly inhibits new transcription by RNA polymerase II.
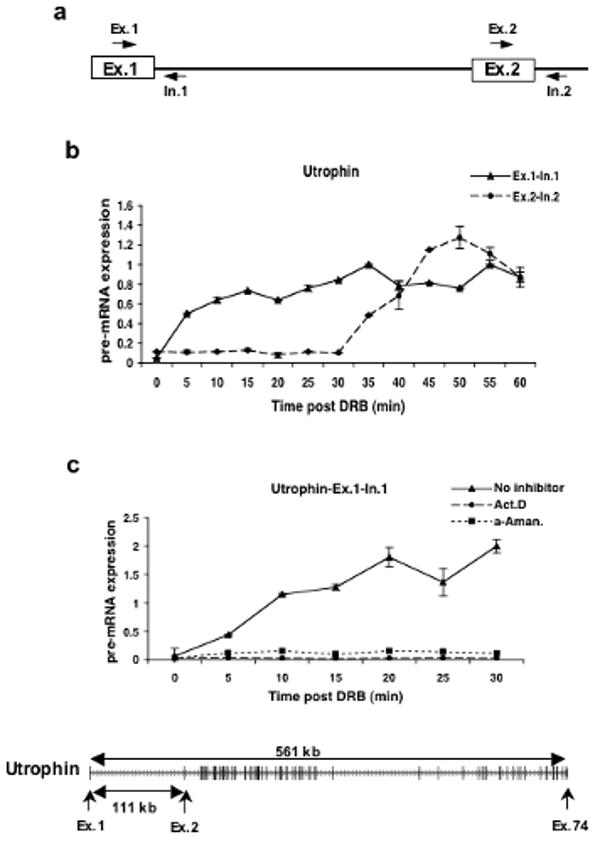
a) Schematic diagram showing the primer sets used to amplify the exon-intron junctions of exons 1 and 2 of the Utrophin gene.
b) Quantitative RT-PCR was used to measure the expression levels of Utrophin pre-mRNA in the exon 1 and exon 2 regions of the gene. Cells were first treated with 100μM of DRB for 3hrs and then fresh medium was added after DRB removal. The cells were harvested at 5 minute intervals for RNA isolation and qRT-PCR. The expression values are plotted relative to the expression level of the no treatment control which is set to 1 in all experiments.
c) Effect of actinomycin D (Act. D) and α-amanitin (α-Aman.) on transcription of the first exon-intron junction of the Utrophin gene after DRB treatment. Cells were treated with DRB for 3hrs and either actinomycin D or α-amanitin was added 30 minutes prior to the removal of DRB. Cells were released from DRB but maintained in the other drugs. Cells were harvested at 5 minute intervals and quantitative RT-PCR was performed as above to measure the levels of expression of the exon1-intron 1 region of the Utrophin gene. Shown at the bottom is the exon/intron structure of the Utrophin gene. Transcription is from left to right and the exons are represented as larger vertical bars.
It is possible that the 3 hour incubation in DRB might alter the chromatin state of the cells and thus affect the rate of transcription that we measure. To test this, we treated Tet-21 cells for only 30 minutes with DRB and isolated RNA at 5 minute intervals after removal of DRB. The results for the 178 kb long CTNNBL1 gene (Supplementary Figure 2 online) online show that transcription of the exon 1-intron 1 region recovers quickly after DRB removal while the pre-mRNA signal at the intron 15-exon 16 region decays slowly over 45 minutes and then recovers quickly. This suggests that RNAPII requires about 45 minutes to transcribe the 178 kb gene giving an elongation rate of ∼3.9 kb min−1 which is similar to the rate seen after a 3 hour DRB treatment.
Validation of the assay to study transcription elongation
In order to establish that the increase in pre-mRNA levels shown in Figure 1b for exon 1 and exon 2 of the Utrophin gene was due to new rounds of RNAPII dependent transcription, several control experiments were carried out. Treatment with either actinomycin D or α-amanitin blocked the recovery of the pre-mRNA signal following DRB release (Fig. 1c). This provides clear evidence that the signal we detected in Figure 1b was due to transcriptional activity of RNAPII.
We interpret the time lag between the reappearance of the signals for exons 1 and 2 of the Utrophin gene to be due to elongation through the long intron that separates them. If this is true, the time lag should be sensitive to changes in the elongation rate of RNAPII. The drug camptothecin (CPT), an inhibitor of topoisomerase I, has been shown to substantially reduce the rate of transcription elongation by RNAPII to about 1 kb min−1 or about 3 to 4 times slower than normal17,35-37. If we are measuring the rate of RNAPII elongation in our experiments, CPT treatment should dramatically increase the observed lag times.
To test this, DRB treated cells were incubated in the presence of CPT for 15 minutes before removal of DRB and were maintained in CPT after removal of DRB. Supplementary Figure 3 online shows that the recovery time of gene regions downstream of the promoters of two genes are substantially lengthened as expected. These results strongly support the view that these experiments are measuring new RNAPII transcription starting at or near the normal transcriptional start site and that the lag times observed in the expression of downstream sequences reflect the rate of elongation of RNAPII.
A concern with the above experiments is that DRB treatment might somehow alter the chromatin environment producing an incorrect elongation rate. We therefore used an independent method to induce transcription that did not rely on DRB. Treatment of responsive cells with cytokines such as interferon-β leads to rapid induction of many genes38. One example is the PKR gene (also known as EIF2AK2). We treated human HT1080 cells with interferon-β and monitored the pre-mRNA levels from the PKR gene at two positions that are 40 kb apart. Supplementary Figure 4 online shows that pre-mRNA levels rise at both sites after interferon treatment with a time difference of about 12 minutes suggesting an elongation rate of ∼3.3-kb min−1. Thus, an independent method of gene induction gives an apparent RNAPII elongation rate that is similar to the rate determined using DRB.
The experimental protocols described above measure the transcriptional output of RNAPII rather than the physical presence of the polymerase in different gene regions. To demonstrate a direct relationship between the transcriptional signal and the presence of polymerase, we performed chromatin immunoprecipitation (ChIP) using antibodies against RNAPII. Supplementary Figures 5a and b online show conventional gel analysis and real time PCR ChIP respectively of the promoter proximal regions of three genes and a distal region of one. In all cases, DRB treatment for three hours (0′ lanes) reduced the amount of RNAPII in these regions. The RNAPII signals recovered within 5 minutes of washing for the promoter proximal regions and within 10 minutes for downstream regions.
We also examined RNAPII occupancy at gene regions far downstream of the promoter region. Supplementary Figures 5c and d online show that the RNAPII ChIP signals for the ITPR1 gene show similar kinetics of appearance as the pre-mRNA signals (see below). These results confirm that the distribution of RNAPII reflects the RNA signals detected by RT-PCR analysis.
Elongation rates appear to be similar for most genes
Having shown that the DRB-release method allows us to follow the progress of RNAPII through chromatin, we investigated the rate of transcriptional elongation in several large human genes. The selected genes are listed in Table 1 and data for four of these is shown in Figure 2 along with their exon-intron structures.
Table 1.
Rates of elongation of RNAPII through various regions of different genes
| Gene | Region | Length (kb) | Time to transcribe (min) | Elongation Rate (kb/min) |
|---|---|---|---|---|
| Utrophin | Exon1-Exon2 | 111 | 30 | 3.70 |
| Utrophin | Ex2-Ex50 | 174 | 40 | 4.35 |
| Utrophin | Ex50-Ex51 | 101 | 25 | 4.04 |
| Utrophin | Ex51-Ex74 | 173 | 40 | 4.33 |
| Utrophin | Ex1-Ex74 | 561 | 140 | 4.01 |
| ITPR1 | Ex1-Ex5 | 133 | 40 | 3.33 |
| ITPR1 | Ex5-Ex40 | 105 | 25 | 4.20 |
| ITPR1 | Ex1-Ex40 | 238 | 65 | 3.66 |
| EfnA5 | Ex1-Ex2 | 243 | 70 | 3.47 |
| Bcl2 | Ex2-Ex3 | 189 | 50 | 3.78 |
| OPA1 | Ex1-Ex29 | 104 | 25 | 4.16 |
| IFT80 | Ex1-Ex20 | 142 | 35 | 4.06 |
| CTNNBL1 | Ex1-Ex16 | 178 | 45 | 3.96 |
| KIFAP3 | Ex1-Ex20 | 153 | 45 | 3.40 |
| SLC9A9 | Ex1-Ex16 | 583 | 160 | 3.64 |
| Average elongation rate of all genes | 3.79±0.26 | |||
The elongation rates for full length genes are depicted in bold and these values were used to compute the average and standard deviation of the elongation rate for all the genes.
Figure 2. Kinetics of RNAPII dependent transcription elongation.
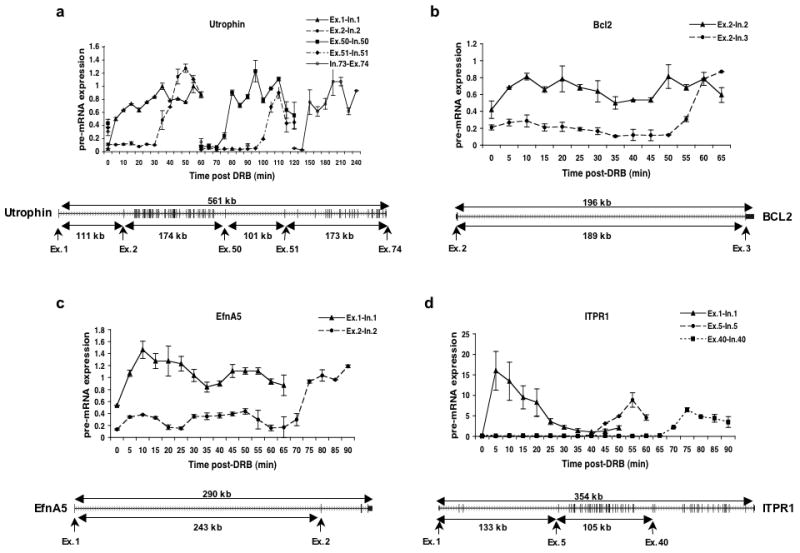
Cells were treated with DRB and transcription was analyzed as in Figure 1b. (a-d) Quantitative real time RT-PCR was performed using primer sets specific for different parts of the indicated genes to measure the levels of pre-mRNA expression. The gene structure is shown below each graph with arrows indicating the exon-intron junctions that were analyzed.
Several notable results can be seen in this data. First, as shown in Table 1, all gene regions appeared to be transcribed with a remarkably uniform rate of about 3.8 kb min−1. This value is quite close to the maximum elongation rate (4.3 kb min−1) determined by Darzacq et al.17. This suggests that, within genes in their natural chromosomal locations and chromatin states, transcription elongation can be very efficient.
A second interesting result is that there appears to be a fairly consistent progression of polymerase within each gene. As can be seen for the Utrophin gene shown in Figure 2a, there is relatively little dispersion of the signals as transcription proceeds through hundreds of kilobases of DNA. Even at the end of the 561 kb gene (exon 74), there is a sharp rise in the signal between 10 minute time points. This suggests that transcription elongation is highly uniform over individual genes in a cell population.
Third, the data suggests that there is little or no difference in elongation rate through gene regions that contain few or no exons (and thus long introns) compared to regions that contain multiple exons and short introns. Two of the genes shown in Figure 2, Utrophin and ITPR1, have structures that lend themselves well to this analysis. As shown in Figure 2a, the Utrophin gene can be divided into four regions with widely differing exon content. Intron 1 is 111 kb in length and intron 50 is 101 kb long. Between these two introns, a 174 kb region contains 49 exons while the 3′ 173 kb region contains the remaining 23 exons. The elongation rates within these individual regions are shown in Table 1 where it appears that the exon rich and exon poor gene regions are transcribed at similar rates. The ITPR1 gene shown in Figure 2d gave a similar result. In this case, a 133 kb region with only 5 exons is compared to a 105 kb region that contains 35 exons. Again, the apparent elongation rates in these two regions are quite similar (Table 1). Also similar are the transcription elongation rates of the very long introns of two other genes, Bcl2 and EfnA5, as shown in Table 1 and Figures 2b and c.
Splicing is rapid and independent of intron length
Previous results have shown that pre-mRNA splicing can occur both co-transcriptionally and post-transcriptionally. To measure the rate of pre-mRNA splicing, we determined the time between the new synthesis of an exon and the appearance of the splicing product of that exon and the immediately preceding exon. To avoid the signal due to pre-existing mRNA in the DRB treated cells, we used a primer in the downstream intron to amplify the partially spliced intermediate as diagramed in Figure 3a. Figure 3b-d shows the time course of splicing for three introns of the major or U2-dependent class. We also analyzed the splicing of six additional introns with the results summarized in Table 2 (see also Supplementary Fig. 6 online).
Figure 3. Kinetics of splicing of U2-dependent introns.
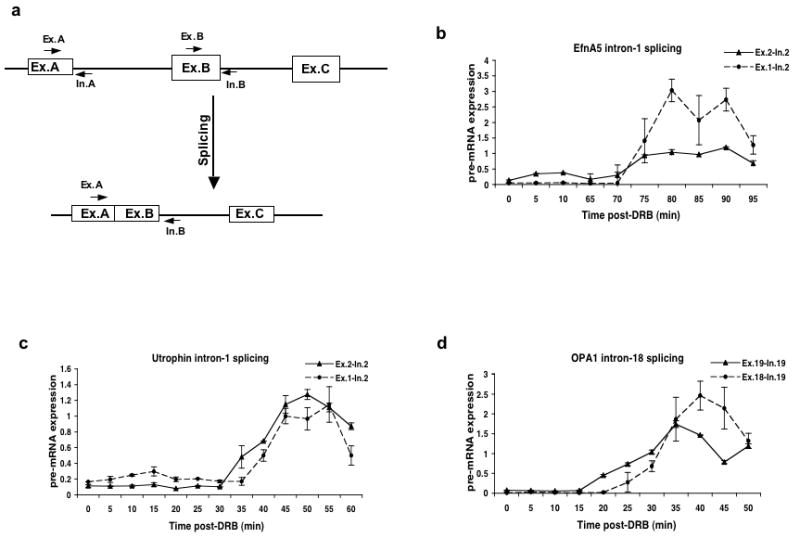
a) Schematic diagram showing the locations of primers used to measure the levels of partially spliced mRNA. Primer sets A and B detect the unspliced pre-mRNA containing the upstream and downstream exons flanking the intron respectively. The Ex-A and In-B primer pair detects partially spliced RNA in which intron A has been spliced out but intron B remains unspliced. RNA samples from DRB treated cells were analyzed by qRT-PCR using primer pairs that detect the transcription of the exon downstream of the indicated intron or the ligated exon product of splicing.
b) Transcription of exon 2 (Ex.2-In.2) and splicing of intron 1 (Ex.1-In.2) of the Ephrin A5 gene.
c) Transcription of exon 2 (Ex.2-In.2) and splicing of intron 1 (Ex.1-In.2) of the Utrophin gene.
d) Transcription of exon 19 (Ex.19-In.19) and splicing of intron 18 (Ex.18-In.19) of the OPA1 gene.
Table 2.
Time required to splice introns of different sizes and types.
| Gene | Intron type | Intron number | Kb from start | Intron size (kb) | Time to splice (min) | Total gene size (kb) |
|---|---|---|---|---|---|---|
| Utrophin | U2 | 1 | 0.15 | 111 | 5 | 561 |
| Utrophin | U2 | 13 | 155 | 2.8 | 10 | 561 |
| Utrophin | U2 | 43 | 245 | 1.5 | 5 | 561 |
| Utrophin | U2 | 50 | 285 | 101 | 5 | 561 |
| Ephrin A5 | U2 | 1 | 0.15 | 243 | 5 | 294 |
| OPA1 | U2 | 18 | 52 | 1.2 | 5 | 104 |
| ITPR1 | U2 | 44 | 284 | 2.1 | 5 | 354 |
| CTNNBL1 | U2 | 3 | 43 | 9 | 5 | 178 |
| CTNNBL1 | U2 | 5 | 63 | 7.5 | 5 | 178 |
| KiFAP3 | U12 | 7 | 40 | 2.7 | 5 | 153 |
| CTNNBL1 | U12 | 4 | 52 | 11 | 10 | 178 |
| SLC9A9 | U12 | 7 | 270 | 4.5 | 10 | 583 |
| IFT80 | U12 | 2 | 14.8 | 3 | 10 | 142 |
All of these introns are spliced within 5-10 min of transcription of the downstream exon (Figure 3 and Table 2). Furthermore, this splicing must occur co-transcriptionally since the splicing reaction is completed well before the gene is transcribed to the end. For example, introns 1 and 13 of the Utrophin gene were spliced within 40 and 60 minutes respectively after the start of transcription (Fig. 3c and supplementary Fig. 6a online) while the entire gene requires 140 minutes to transcribe (Table 1). As shown in Table 2, we analyzed splicing of introns of widely varying sizes ranging from 1.2 kb to 243 kb. The remarkable result is that there appears to be no relation between the length of an intron and the time required to splice it. In all the cases we analyzed, splicing of these introns occurred within 5 to 10 minutes of synthesis of the downstream exon and 3′ splice site.
U12-dependent splicing can occur co-transcriptionally
In addition to the U2-dependent introns, the human genome also contains a relatively small number of U12-dependent introns that are spliced by a distinct spliceosome. These introns are found in genes that also contain multiple U2-dependent introns. Previous reports have shown that some U12-dependent introns are spliced more slowly than U2-dependent introns39. This has been suggested to be a common property of U12-dependent introns perhaps due to the much lower abundance of the spliceosomal snRNAs specific to these introns or, controversially, to splicing in the cytoplasm24.
To examine the splicing of minor class introns, we chose several genes which were large in size and contained at least one U12-dependent intron in the proximal part of the gene so that we could determine whether or not U12-dependent splicing occurs co-transcriptionally. Genes were selected that were longer than 100 kb, contained a U12-dependent intron near the 5′ end of the gene and were widely expressed.
Figure 4 shows the results of quantitative RT-PCR of these genes to detect the partially spliced transcripts resulting from splicing of each U12-dependent intron but prior to the splicing of the downstream U2-dependent intron. In each case, the first and last exon-intron boundaries are assayed as well as the exon immediately downstream of the U12-dependent intron and the product of U12-dependent splicing. The results were similar to those shown above for the U2-dependent introns. In each case, splicing occurred within 5-10 min of transcription of the exon downstream of the U12-dependent intron. Also, our results clearly show that U12-dependent splicing takes place before RNAPII reaches the end of the gene in every instance. Therefore, splicing of this class of introns must be nuclear.
Figure 4. Kinetics of splicing of U12-dependent introns.
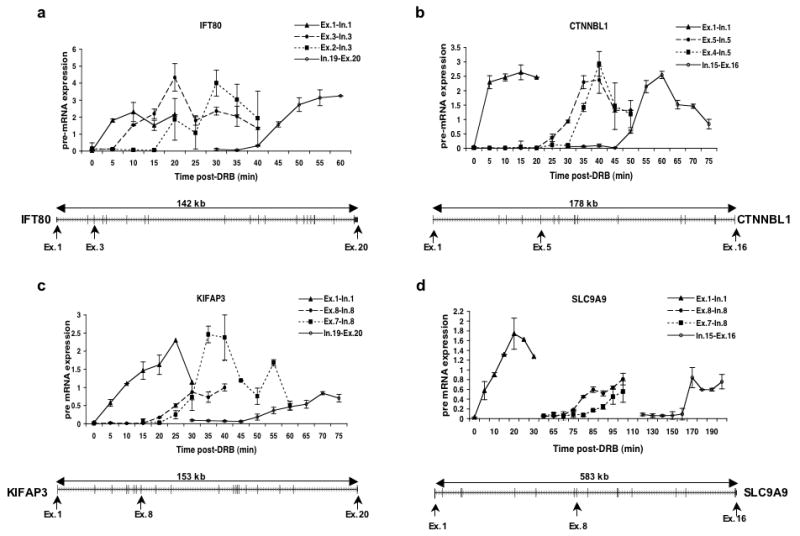
RNA samples from DRB treated cells were analyzed by qRT-PCR using primer pairs that detect the transcription of the exon downstream of the indicated intron or the ligated exon product of splicing.
a) Transcription of exons 1 (Ex.1-In.1), 3 (Ex.3-In.3) and 20 (In.19-Ex.20) as well as splicing of intron 2 (Ex.2-In.3) of the IFT80 gene.
b) Transcription of exons 1 (Ex.1-In.1), 5 (Ex.5-In.5) and 16 (In.15-Ex.16) as well as splicing of intron 4 (Ex.4-In.5) of the CTNNBL1 gene.
c) Transcription of exons 1 (Ex.1-In.1), 8 (Ex.8-In.8) and 20 (In.19-Ex.20) as well as splicing of intron 7 (Ex.7-In.8) of the KIFAP3 gene.
d) Transcription of exons 1 (Ex.1-In.1), 8 (Ex.8-In.8) and 16 (In.15-Ex.16) as well as splicing of intron 7 (Ex.7-In.8) of the SLC9A9 gene.
Discussion
The results presented here demonstrate a simple and efficient method to study the rates of transcriptional elongation and pre-mRNA splicing in endogenous human genes in their natural chromatin environment. Using this technique, we were able to measure the average rate of RNAPII transcription over genomic distances of hundreds of kilobases by examining transcription of large human genes. Our results show that, over large chromosomal distances, the average rate of RNAPII transcription is approximately 3.8 kb min−1. This rate is remarkably consistent among the genes analyzed (Table 1) and is similar in both the cell line used here (Tet-21) and in another human cell line, HEK293 (data not shown).
We also examined our results for a relationship between gene expression level and transcriptional elongation rate. To obtain an estimate of gene expression levels, we used the qRT-PCR levels of pre-mRNA spanning the first exon/intron junctions of the genes listed in Table 1. These are uncalibrated values but are likely more indicative of transcriptional activity than the relative levels of mature mRNAs. Within the set of genes in Table 1, Utrophin and OPA1 have the highest expression levels while CTNNBL1 has the lowest with a relative difference of 15-20 fold. Inspection of Table 1 shows that these three genes have very similar apparent elongation rates. Thus, within the limits of our measurements, there does not appear to be a strong relationship between elongation rate and transcriptional activity.
Our measured apparent elongation rate is considerably faster than several previous estimates that fell in the range of 1.1 to 2.5 kb per minute14,15,18,40. Many of these studies examined single genes that were transcriptionally silent prior to their activation. This may put them in a different chromatin environment than genes that are being continuously transcribed such as those analyzed here. Interestingly, our observed elongation rate is about 80% of the maximum rate of RNAPII transcription determined by Darzacq et al.17. These investigators derived a maximal rate of elongation of 4.3 kb min−1 and a residence time for paused polymerases of about 4 minutes. Comparing our average rate over long genes to the maximum rate found by Darzacq et al. suggests that at least the leading polymerases in our long genes pause very little or only for much briefer periods than measured by Darzacq et al.17. For example, over 100 kb, the time difference between an elongation rate of 3.8 kb min−1 and a rate of 4.3 kb min−1 would be about three minutes which, in turn, is less than the 4 minute pause time found by Darzacq et al.17. Thus, for the genes investigated here, the results suggest that RNAPII transcribes at close to its maximum rate with few or no pausing events over hundreds of kilobases of chromatin.
We also observed that the rate of transcription was very similar over long distances of pure intronic sequence or exon rich sequences. The Utrophin gene in particular provided a means to address this point since it contained two >100 kb introns as well as regions of >100 kb with more than thirty introns. Since there is evidence for the coupling of transcription and splicing (see below), these results show that, at least for these genes, this coupling does not appear to slow down the progress of the polymerase as it transcribes multiple exons and splice sites.
We were also able to measure the rate of pre-mRNA splicing in these large genes by following the synthesis of partially spliced RNA where the two upstream exons are spliced together but the downstream intron remains unspliced. These intermediates could be distinguished from mature mRNA due to their partially unspliced component. A surprising finding of our study is that splicing occurred at a similar rate for several U2-dependent introns spanning a wide range of sizes. We found lag times for splicing of 5-10 minutes for introns ranging from 1.2 kb to 240 kb (Table 2). These times represent the time, after synthesis of the downstream exon, for the appearance of the spliced exons. The splicing rates for small introns agree well with previous measurements19,20. However, this appears to be the first report for the rate of splicing of introns greater than 100 kb in length. The speed with which long introns are spliced suggests that there are mechanisms for keeping the 5′ splice site near the elongating polymerase to allow for efficient joining to the newly synthesized 3′ splice site. Possible mechanisms include recursive splicing41 and association of the 5′ exon with the CTD of RNAPII21,42-46.
We also investigated the splicing of four U12-dependent introns. We found that these introns were spliced slower than the U2-dependent introns by a factor of ∼2. This result is consistent with previous data showing that these introns are spliced slower than comparable U2-dependent introns39. However, this previous study reported larger rate differences than we see. The 10 minute splicing rates observed for U12-dependent introns suggests that slow splicing of these introns may not represent a significant impediment to expression of their host genes as has been suggested by previous work39.
Another significant finding from our study is that pre-mRNA splicing occurs co-transcriptionally irrespective of the class of introns. For U2-dependent introns, this is in close agreement with many other reports4,21,22. For U12-dependent introns, however, our results are in direct contradiction to a recent report which argued that the U12-dependent spliceosome machinery resides predominantly in the cytoplasm and therefore the majority of U12-dependent splicing must take place in the cytoplasm after the pre-mRNA is fully transcribed and transported to the cytoplasm24. Our results show that splicing of U12-dependent introns occurs long before the ends of the genes are transcribed. Thus, from a temporal perspective, these introns are spliced co-transcriptionally which directly counters the hypothesis of Konig et al.24 and lends unequivocal support to other reports that U12 spliceosomal complexes reside in the nucleus and function there26,27,29.
Methods
Cell culture
A derivative of the neuroblastoma cell line SH-EP Tet-2147 was provided by Dr. J. Shohet (Baylor College of Medicine, Houston, TX) with permission from Dr. M. Schwab (Deutsches Krebsforschungszentrum, Heidelberg, Germany) and was grown in RPMI medium (Mediatech) and 10% (v/v) fetal bovine serum (Atlanta Biologicals), supplemented with 0.8% L-glutamine and 100 units/ml penicillin and 0.1mg/ml streptomycin (Mediatech). HT1080 fibrosarcoma cells were obtained from ATCC and were grown in DMEM (Mediatech) with 10% fetal bovine serum supplemented with 0.8% L-glutamine and 100 units/ml penicillin and 0.1mg/ml streptomycin.
Inhibition and re-initiation of transcription
We grew cells overnight on 60mm plates to 70-80% confluency and then treated them with 100μM of 5,6-Dichlorobenzimidazole 1-β–D-ribofuranoside (DRB) (Sigma) in culture medium for 3 hours. The cells were washed twice with PBS to remove the DRB and incubated in fresh medium for various time periods. Following the incubation period, cells were directly lysed and total RNA was isolated using a High Pure RNA isolation kit (Roche) as per the manufacturer's instructions. Actinomycin D (Sigma) or α-amanitin (Sigma) whenever used, were added at a concentration of 1μg/ml and 2μg/ml respectively. Camptothecin (CPT) (Sigma) was used at 15μM concentration and was added 15 minutes before completion of the DRB treatment. The PBS that was used for subsequent washings also contained 15μM CPT and then fresh medium containing 15μM CPT was added to the cells for various time periods.
Induction of gene expression by interferon-β
We used HT1080 fibrosarcoma cells to study the rate of elongation of interferon-β inducible genes. We grew cells overnight to ∼80% confluency in 60mm cell culture dishes and then treated them with 1000u/ml of interferon-β (Calbiochem) for 5-40 minutes. Cells were harvested at various time points to isolate total RNA for quantitative RT-PCR.
Quantitative RT-PCR
We carried out reverse transcription using ImProm-II Reverse Transcriptase (Promega) as per the manufacturer's instructions using random hexamer primers (Promega) and 1μg RNA per 20μl reaction. The cDNA (1μl/well) was used for quantitative RT-PCR using iQ-SYBR Green Supermix and an iQ-iCycler (Bio-Rad). The real-time primers spanning the exon-intron junctions of various genes were designed using the IDT primer designing software PrimerQuest on the IDT website (www.idtdna.com). All primers were purchased from IDT and the sequence information for all of the primers used is available in Supplementary Table 1 online. All the primers were checked for their specificity first in silico (http://genome.ucsc.edu) and then by standard PCR before being used for q-PCR. A total of 40-50 PCR cycles were performed in a 2-step cycling procedure with an initial denaturation at 94°C for 3 minutes and subsequently at 94°C for 15 sec. and 60°C for 30 sec. The calculated threshold values were determined by the maximum curvature approach and ΔCt was calculated as Ct GAPDH-Ct sample. Final values for each sample were plotted relative to the value in control cells which was set to 1.0. Reactions were performed in triplicate. Results are shown as means with standard deviations from a single experiment.
Chromatin immunoprecipitation (ChIP)
We grew cells on 150mm plates to 80% confluency and treated them with DRB for 3hrs. They were then washed, fresh medium was added and the cells were harvested at various time points. Subsequently, we fixed the cells with 1% (v/v) formaldehyde and we performed ChIP assays using a ChIP-IT Express Kit (Active Motif) as per the manufacturer's instructions. We carried out immunoprecipitations using antibodies specific to RNA Polymerase II (N20, Santa Cruz Biotechnology) and with an isotype matched IgG control. We reserved 1/10 of each chromatin sample as the total input DNA. We then de-cross linked the immunoprecipitated chromatin fragments and used them as templates for either real time or standard PCR reactions using primers complementary to various exon-intron junctions of various genes. We carried out standard PCR using the Hot-start-IT PCR enzyme (USB) in a 3-step cycling procedure with an initial denaturation at 94°C for 2 minutes and subsequently 35-40 PCR cycles at 94°C for 30 sec., 60°C for 30 sec. and 72°C for 1min.
Gene structure and splice site identification
We obtained all of the sequence information used for designing the exon-intron junction primers for the genes used in this study from the UCSC genome browser (http://genome.ucsc.edu). We performed the splice site determination (U2- versus U12-dependent) using SpliceRack (http://katahdin.cshl.edu/SpliceRack/).
Supplementary Material
Acknowledgments
We wish to thank Rosemary Dietrich and Sakuntala Saikia for technical assistance, J. Shohet (Baylor College of Medicine, Houston, TX) and M. Schwab (Deutsches Krebsforschungszentrum, Heidelberg, Germany) for cell lines used in this study, D. Price for suggesting the use of DRB and D. Luse for careful reading of the manuscript. This work was supported by grants to R.A.P. from the National Institutes of Health and the Ralph Wilson Medical Research Foundation.
Footnotes
Author Contributions: R.P conceived and coordinated the project. J.S. performed all experimental work and compiled the data. Both authors analyzed the data and wrote the manuscript.
References
- 1.Rasmussen EB, Lis JT. In vivo transcriptional pausing and cap formation on three Drosophila heat shock genes. Proc Natl Acad Sci U S A. 1993;90:7923–7927. doi: 10.1073/pnas.90.17.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Proudfoot NJ, Furger A, Dye MJ. Integrating mRNA processing with transcription. Cell. 2002;108:501–512. doi: 10.1016/s0092-8674(02)00617-7. [DOI] [PubMed] [Google Scholar]
- 3.Neugebauer KM. On the importance of being co-transcriptional. J Cell Sci. 2002;115:3865–3871. doi: 10.1242/jcs.00073. [DOI] [PubMed] [Google Scholar]
- 4.Bentley DL. Rules of engagement: co-transcriptional recruitment of pre-mRNA processing factors. Curr Opin Cell Biol. 2005;17:251–256. doi: 10.1016/j.ceb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Hirose Y, Manley JL. RNA polymerase II is an essential mRNA polyadenylation factor. Nature. 1998;395:93–96. doi: 10.1038/25786. [DOI] [PubMed] [Google Scholar]
- 6.Hirose Y, Tacke R, Manley JL. Phosphorylated RNA polymerase II stimulates pre-mRNA splicing. Genes Dev. 1999;13:1234–1239. doi: 10.1101/gad.13.10.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de la Mata M, et al. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell. 2003;12:525–532. doi: 10.1016/j.molcel.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Caceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet. 2002;18:186–193. doi: 10.1016/s0168-9525(01)02626-9. [DOI] [PubMed] [Google Scholar]
- 9.Core LJ, Lis JT. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science. 2008;319:1791–1792. doi: 10.1126/science.1150843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batsche E, Yaniv M, Muchardt C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat Struct Mol Biol. 2006;13:22–29. doi: 10.1038/nsmb1030. [DOI] [PubMed] [Google Scholar]
- 11.Roberts GC, Gooding C, Mak HY, Proudfoot NJ, Smith CW. Co-transcriptional commitment to alternative splice site selection. Nucleic Acids Res. 1998;26:5568–5572. doi: 10.1093/nar/26.24.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howe KJ, Kane CM, Ares M., Jr Perturbation of transcription elongation influences the fidelity of internal exon inclusion in Saccharomyces cerevisiae. RNA. 2003;9:993–1006. doi: 10.1261/rna.5390803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kornblihtt AR. Chromatin, transcript elongation and alternative splicing. Nat Struct Mol Biol. 2006;13:5–7. doi: 10.1038/nsmb0106-5. [DOI] [PubMed] [Google Scholar]
- 14.Tennyson CN, Klamut HJ, Worton RG. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat Genet. 1995;9:184–190. doi: 10.1038/ng0295-184. [DOI] [PubMed] [Google Scholar]
- 15.O'Brien T, Lis JT. Rapid changes in Drosophila transcription after an instantaneous heat shock. Mol Cell Biol. 1993;13:3456–3463. doi: 10.1128/mcb.13.6.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Femino AM, Fogarty K, Lifshitz LM, Carrington W, Singer RH. Visualization of single molecules of mRNA in situ. Methods Enzymol. 2003;361:245–304. doi: 10.1016/s0076-6879(03)61015-3. [DOI] [PubMed] [Google Scholar]
- 17.Darzacq X, et al. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol. 2007;14:796–806. doi: 10.1038/nsmb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boireau S, et al. The transcriptional cycle of HIV-1 in real-time and live cells. J Cell Biol. 2007;179:291–304. doi: 10.1083/jcb.200706018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kessler O, Jiang Y, Chasin LA. Order of intron removal during splicing of endogenous adenine phosphoribosyltransferase and dihydrofolate reductase pre-mRNA. Mol Cell Biol. 1993;13:6211–6222. doi: 10.1128/mcb.13.10.6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Audibert A, Weil D, Dautry F. In vivo kinetics of mRNA splicing and transport in mammalian cells. Mol Cell Biol. 2002;22:6706–6718. doi: 10.1128/MCB.22.19.6706-6718.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das R, et al. Functional coupling of RNAP II transcription to spliceosome assembly. Genes Dev. 2006;20:1100–1109. doi: 10.1101/gad.1397406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Listerman I, Sapra AK, Neugebauer KM. Cotranscriptional coupling of splicing factor recruitment and precursor messenger RNA splicing in mammalian cells. Nat Struct Mol Biol. 2006;13:815–822. doi: 10.1038/nsmb1135. [DOI] [PubMed] [Google Scholar]
- 23.Kornblihtt AR, de la Mata M, Fededa JP, Munoz MJ, Nogues G. Multiple links between transcription and splicing. Rna. 2004;10:1489–1498. doi: 10.1261/rna.7100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Konig H, Matter N, Bader R, Thiele W, Muller F. Splicing segregation: the minor spliceosome acts outside the nucleus and controls cell proliferation. Cell. 2007;131:718–729. doi: 10.1016/j.cell.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 25.Matera AG, Ward DC. Nucleoplasmic organization of small nuclear ribonucleoproteins in cultured human cells. J Cell Biol. 1993;121:715–727. doi: 10.1083/jcb.121.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pessa HK, et al. Minor spliceosome components are predominantly localized in the nucleus. Proc Natl Acad Sci U S A. 2008;105:8655–8660. doi: 10.1073/pnas.0803646105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friend K, Kolev NG, Shu MD, Steitz JA. Minor-class splicing occurs in the nucleus of the Xenopus oocyte. RNA. 2008;14:1459–1462. doi: 10.1261/rna.1119708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konig H, Muller F. Minor splicing: nuclear dogma still in question. Proc Natl Acad Sci U S A. 2008;105:E37. doi: 10.1073/pnas.0804939105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steitz JA, et al. Where in the cell is the minor spliceosome? Proc Natl Acad Sci U S A. 2008;105:8485–8486. doi: 10.1073/pnas.0804024105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marshall NF, Price DH. Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol Cell Biol. 1992;12:2078–2090. doi: 10.1128/mcb.12.5.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price DH. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol. 2000;20:2629–2634. doi: 10.1128/mcb.20.8.2629-2634.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng B, Price DH. Properties of RNA polymerase II elongation complexes before and after the P-TEFb-mediated transition into productive elongation. J Biol Chem. 2007;282:21901–21912. doi: 10.1074/jbc.M702936200. [DOI] [PubMed] [Google Scholar]
- 33.Chodosh LA, Fire A, Samuels M, Sharp PA. 5,6-Dichloro-1-beta-D-ribofuranosylbenzimidazole inhibits transcription elongation by RNA polymerase II in vitro. J Biol Chem. 1989;264:2250–2257. [PubMed] [Google Scholar]
- 34.Gomes NP, et al. Gene-specific requirement for P-TEFb activity and RNA polymerase II phosphorylation within the p53 transcriptional program. Genes Dev. 2006;20:601–612. doi: 10.1101/gad.1398206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Capranico G, et al. The effects of camptothecin on RNA polymerase II transcription: roles of DNA topoisomerase I. Biochimie. 2007;89:482–489. doi: 10.1016/j.biochi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 36.Mondal N, Parvin JD. DNA topoisomerase IIalpha is required for RNA polymerase II transcription on chromatin templates. Nature. 2001;413:435–438. doi: 10.1038/35096590. [DOI] [PubMed] [Google Scholar]
- 37.Khobta A, et al. Early effects of topoisomerase I inhibition on RNA polymerase II along transcribed genes in human cells. J Mol Biol. 2006;357:127–138. doi: 10.1016/j.jmb.2005.12.069. [DOI] [PubMed] [Google Scholar]
- 38.Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel AA, McCarthy M, Steitz JA. The splicing of U12-type introns can be a rate-limiting step in gene expression. EMBO J. 2002;21:3804–3815. doi: 10.1093/emboj/cdf297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ucker DS, Yamamoto KR. Early events in the stimulation of mammary tumor virus RNA synthesis by glucocorticoids. Novel assays of transcription rates. J Biol Chem. 1984;259:7416–7420. [PubMed] [Google Scholar]
- 41.Burnette JM, Miyamoto-Sato E, Schaub MA, Conklin J, Lopez AJ. Subdivision of large introns in Drosophila by recursive splicing at nonexonic elements. Genetics. 2005;170:661–674. doi: 10.1534/genetics.104.039701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fong N, Bird G, Vigneron M, Bentley DL. A 10 residue motif at the C-terminus of the RNA pol II CTD is required for transcription, splicing and 3′ end processing. EMBO J. 2003;22:4274–4282. doi: 10.1093/emboj/cdg396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeng C, Berget SM. Participation of the C-terminal domain of RNA polymerase II in exon definition during pre-mRNA splicing. Mol Cell Biol. 2000;20:8290–8301. doi: 10.1128/mcb.20.21.8290-8301.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirose Y, Ohkuma Y. Phosphorylation of the C-terminal domain of RNA polymerase II plays central roles in the integrated events of eucaryotic gene expression. J Biochem. 2007;141:601–608. doi: 10.1093/jb/mvm090. [DOI] [PubMed] [Google Scholar]
- 45.Dye MJ, Gromak N, Proudfoot NJ. Exon tethering in transcription by RNA polymerase II. Mol Cell. 2006;21:849–859. doi: 10.1016/j.molcel.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 46.Rosonina E, Blencowe BJ. Analysis of the requirement for RNA polymerase II CTD heptapeptide repeats in pre-mRNA splicing and 3′-end cleavage. Rna. 2004;10:581–589. doi: 10.1261/rna.5207204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.West AB, et al. N-myc regulates parkin expression. J Biol Chem. 2004;279:28896–28902. doi: 10.1074/jbc.M400126200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.