

Abstract
(R)-Lacosamide ((R)-2, (R)-N-benzyl 2-acetamido-3-methoxypropionamide), has recently gained regulatory approval for the treatment of partial-onset seizures in adults. Whole animal pharmacological studies have documented that (R)-2 function is unique. A robust strategy is advanced for the discovery of interacting proteins associated with function and toxicity of (R)-2 through the use of (R)-2 analogs, 3, that contain “affinity bait (AB)” and “chemical reporter (CR)” functional groups. In 3, covalent modification of the interacting proteins proceeds at the AB moiety, and detection or isolation of the selectively captured protein occurs through the bioorthogonal CR group upon reaction with an appropriate probe. We report the synthesis, pharmacological evaluation, and interrogation of the mouse soluble brain proteome using 3 where the AB group is an isothiocyanate moiety. One compound, (R)-N-(4-isothiocyanato)benzyl 2-acetamido-3-(prop-2-ynyloxy)propionamide ((R)-9), exhibited excellent seizure protection in mice and, like (R)-2, anticonvulsant activity principally resided in the (R)-stereoisomer. Several proteins were preferentially labeled by (R)-9 compared with (S)-9, including collapsin response mediator protein 2.
Introduction
Epilepsy is a major neurological disorder that affects all populations.1 Epilepsy describes the types of recurrent seizures produced by paroxysmal excessive neuronal discharges in the brain.2,3 The mainstay of treatment has been the long-term and consistent administration of anticonvulsant drugs.4-6 Unfortunately, current medications are ineffective for approximately one-third of patients with epilepsy.7 Many continue to have seizures, while others experience disturbing side effects (e.g., drowsiness, dizziness, nausea, liver damage).8 The shortcomings of current regimens highlight the need for new agents with novel mechanisms of action. The ability to identify specific nervous system pathways and protein target sites responsible for seizures would accelerate the development of new and safer therapeutic agents.
In 1985, we discovered a novel class of anticonvulsant agents, termed functionalized amino acids (FAAa, 1).9-18 The lead compound, (R)-lacosamide ((R)-2, (R)-N-benzyl 2-acetamido-3-methoxypropionamide18), has recently gained regulatory approval for adjunctive therapy for partial-onset seizures in adults.19 Whole animal pharmacological studies have documented that (R)-2 function is unique and prevents seizure spread by mechanisms different from current clinical agents.18,20 Radioligand displacement assays using more than 100 potential binding sites, which include both ligand- and voltage-gated receptors, failed to identify high-affinity binding targets,21 suggesting that either the target site(s) are novel or the binding is weak or both. Only recently has (R)-2 function been reported to be correlated to the modulation of both the slow inactivation gate of sodium channels22 and collapsin response mediator protein 2 (CRMP2).21 The full spectrum of pharmacological pathways for (R)-2 remains elusive.

These findings have led us to suggest that (R)-2 may interact with several targets and that (R)-2 drug efficacy is a summation of multiple beneficial interactions, similar to other neurological agents.23 Using two antiepileptic agents as examples,24 carbamazepine interacts with sodium channels, reduces sodium and calcium influx through N-methyl D-aspartate (NMDA)-activated channels at glutamate receptor sites, blocks the reuptake of norepinephrine into presynaptic terminals, and has adenosine receptor antagonistic activity. Similarly, topiramate blocks sodium channels, enhances gamma-aminobutyric acid (GABA)A-mediated chloride influx, acts as an antagonist to α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) and kainate receptors, and inhibits carbonic anhydrase.
A major objective of our laboratories is to identify and characterize the target(s) and mechanism(s) of (R)-2 function and to advance molecular tools useful to elucidate the pathways leading to seizure control. Here, we report three modified (R)-2 agents each with an appended isothiocyanate moiety that have been designed to search the brain proteome for site(s) of drug binding. These three compounds were used to interrogate soluble mouse brain lysate. We show that CRMP2 along with other potential lacosamide target(s) are selectively captured.
Results
a. The Proposed Method
Central to our efforts is the expansion of traditional and contemporary methods of identifying small-molecular-weight ligand protein target sites where binding is modest and where moderate-to-extensive ligand structural change abolishes target binding. Accordingly, we have designed (R)-2 analogs that contain an “affinity bait (AB)” and a “chemical reporter (CR)”25 group. These compounds are defined as lacosamide AB&CR agents (R)-3. In Scheme 1, we diagram the proposed pathway for (R)-3 function. We anticipate that (R)-3 reversibly binds to the (R)-2 receptor(s) 4 to give 5, provided that the AB and CR moieties do not perturb binding. This interaction is followed by an irreversible, covalent modification wherein receptor capture occurs through the AB moiety to give 6. In most instances, the AB group is either an electrophilic or a photoaffinity agent. Both groups have been extensively utilized to capture biomolecular targets. Removal of the selectively captured protein 6 occurs through the bioorthogonal25a CR group upon reaction with a probe (P, 7) to provide 8. While similar target discovery approaches have been reported,26 few, if any, have been designed to identify receptors of small-molecular-weight ligands (drugs) where binding does not occur at the enzyme catalytic site. The captured (R)-2-bound target 8 in the reaction mixture (e.g., cell lysate) is either detected by in-gel fluorescence through the use of a reporter group tethered to 7 or is selectively removed from the mixture using affinity-based chromatographic methods (e.g., biotin-(strept)avidin).
Scheme 1.

“Affinity Bait” and “Chemical Reporter” (AB&CR) Strategy to Scan the Brain Proteome for (R)-Lacosamide Target(s)
b. Structural and Chemical Criteria for Lacosamide AB&CR Probes ((R)-3)
Six criteria have been established for our AB&CR ((R)-3) agents. First, the compound must conform to the structure-activity relationship (SAR) observed for 1.9-18 In this case, we must limit the size of the AB and the CR groups because of 1’s SAR.9-18 Second, the reactive AB group must have documented success in previous target adduction studies.27 In this report, we describe (R)-3 agents into which an electrophilic affinity bait has been installed. For the electrophilic AB, we expect that this group must be correctly aligned with the target-based nucleophile if covalent modification is to take place.27 Third, the CR group must be inert under the conditions necessary to bait the target protein yet must be prone to react with probe P. Fourth, syntheses of both enantiomeric forms of the lacosamide AB&CR agents 3 are necessary. A distinguishing feature of 1 is that its anticonvulsant activity resides principally in the D-configuration.11-13,15,18 Accordingly, preferential enantioselective irreversible inactivation with the AB&CR agents serves as an important measure of target site specificity.28,29 Fifth, the electrophilic (photoaffinity) labels should be incorporated at different sites within the (R)-2 structure to capture all its possible binding partners. Sixth, the (R)-3 agents used to identify target sites should ideally demonstrate significant anticonvulsant activity in in vivo assays (e.g., maximal electroshock (MES)-seizure test30). Documentation of similar pharmacological activity provides preliminary evidence that the mode of action of (R)-3 is comparable with (R)-2. We recognize that promising pharmacological data do not guarantee that these compounds will serve as site-selective agents since the ability of the AB unit to efficiently modify the protein targets is a composite of variables, some of which are unrelated to function. Correspondingly, compounds inactive in vivo may still function as in vitro affinity labels since chemical and cellular (e.g., transport, metabolic) processes may prevent ligand-target site binding. Nonetheless, demonstrating significant and stereospecific in vivo anticonvulsant activity provides an initial stringent test for our agents.
c. Compound Selection
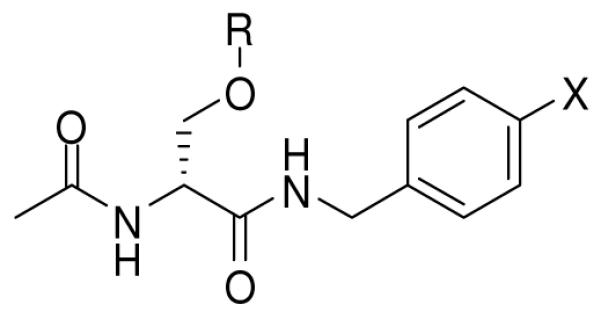
In this study, we selected (R)- and (S)-9–11 as our initial AB&CR agents where AB is the electrophilic isothiocyanate moiety.31-42 We have chosen small AB and CR groups to minimize adverse binding of our agents with possible receptor sites. In 9 and 11, we have incorporated the isothiocyanate moiety at the 4′ N-benzyl position. Earlier studies showed that N-benzyl 4′-substituted 1 compounds exhibited enhanced anticonvulsant activities compared with their corresponding 3′-isomers.43 In 9, the CR group is an alkyne, while in 11 it is an azide. In compound 10, the sites of AB (isothiocyanate) and CR (alkyne) incorporation have been interchanged from that in 9.

The isothiocyanate AB group displays an excellent balance between stability and reactivity; it does not readily react with hydroxylic solvents (permitting its dissolution in aqueous solutions) but does react with nucleophiles (e.g., thiols (cysteines), amines (lysines)).27 Significantly, isothiocyanate electrophilic units have proven effective in selectively modifying target proteins (e.g., GABA,37 opioid31,34,35) within brain membrane fractions and have been used for in vivo modification studies after intracerebroventricular administration.31c Nonetheless, the isothiocyanate AB unit has limitations. First, only lysine and cysteine residues are likely to be modified. This restriction may prevent covalent adduction of some receptors. Fortunately, the extended C(α) methylene chain within lysines gives this nucleophilic amino acid considerable conformational flexibility to reach a bound AB&CR agent. Another concern is that hydrophobic drug binding pockets may not contain nearby lysine residues. Considerable support exists for the selected CR moieties (i.e., alkyne, azide) groups and for the notion that protein capture with P occurs through a Cu(I)-mediated cycloaddition reaction (“click chemistry”).25
d. Synthesis of Agents
The synthetic pathways used to prepare 9–11 are provided in Scheme 2. Care was taken to install the AB unit in the last step to avoid possible isothiocyanate-related reactions during the synthesis and reaction workup. In general, commercially available D- or L-serine methyl ester hydrochloride (12) was converted to the corresponding (R)- and (S)-methyl 1-acetylaziridine-2-carboxylates (16), followed by stereospecific ring opening with the appropriate alcohol using catalytic amounts of BF3•Et2O to give the desired (R)- and (S)-methyl 2-acetamido-3-(alkoxy)propanoates (17, 18).44,45 The esters 17 and 18 were converted to the free acids 19 and 20, respectively, using stoichiometric amounts of LiOH at 0 °C. Under these conditions, we detected little or no racemization. The free acids were coupled with the appropriate substituted benzylamine using either the mixed anhydride coupling method46a (i.e., isobutyl chloroformate (IBCF), N-methyl morpholine (NMM)) or 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM)46b to give the lacosamide analogs 23–25. The syntheses of 9–11 concluded with AB isothiocyanate moiety installation. For 9 and 10, we used di(2-pyridyl) thionocarbonate (DPT)47a and for 11, 1,1′-thiocarbonyldiimidazole (DITC).47b The spectral and analytical data for 9–11 and all intermediates were in agreement with their proposed structures. We documented the enantiopurity of the final compounds using the chiral resolving agent (R)-mandelic acid and 1H NMR spectroscopy.48
Scheme 2.

General Synthetic Pathway to Prepare Lacosamide AB&CR Agents 9–11
In order to assess the individual effects of the AB (isothiocyanate) and CR (alkyne, azide) units on anticonvulsant activities, the corresponding lacosamide analogs that contained only the AB (27) or the CR (28–30) units have also been prepared (Supplementary Schemes S1-S3 in the Supporting Information). We have previously reported the synthesis and anticonvulsant activity for the lacosamide isothiocyanate (AB) probes (R)-31 and (S)-31.43a

e. Pharmacological Evaluation
Initial criteria we established for our lacosamide AB&CR agents are that they prove effective in preventing MES-induced seizures in animal models30 and that the principal activity resides in the (R)-stereoisomer (D-configuration). In the MES test, tonic hindlimb seizures are induced by corneal electrical stimulation. This assay is an established method to identify compounds effective against generalized tonic-clonic seizures30 and was the initial assay used to discover (R)-2. Table 1 lists the MES anticonvulsant activities obtained for the lacosamide AB&CR compounds (9, 11), the analogs into which only an AB (27, 31) or a CR (28–30) moiety was installed in the lacosamide framework, (R)-2,18 and other clinical antiepileptic agents. The compounds were also evaluated in the subcutaneous metrazol (scMet) seizure model but provided no protection (data not shown). The absence of seizure protection in this assay is a hallmark of 1 activity.9-18
Table 1.
Pharmacological Evaluation of Lacosamide AB&CR Agents
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mice (ip)a |
Rat (po)b |
||||||||
| Compd. No. | R | X | MESc, ED50 (mg/kg) | Toxd, TD50 (mg/kg) |
PIe | 6Hz, ED50 (mg/kg) (32 mA) |
MES, ED50 (mg/kg) |
Tox, TD50f (mg/kg) |
PI |
| (R)-9 | CH2C≡CH | NCS | 45 [1] (41 – 48) |
110 [0.25] (76 – 150) |
2.4 | 53 [0.5] (42 – 68) |
> 30 | > 30 | |
| (S)-9 | CH2C≡CH | NCS | > 300 | > 300 | NDg | ND | ND | ||
| (R)-28 | CH2C≡CH | H | 16 [0.25] (13 – 19) |
59 [0.25] (55 – 66) |
3.7 | 29 [0.5] (21 – 40) |
7.9 [0.5] (4.7 – 11) |
> 500 | >63 |
| (S)-28 | CH2C≡CH | H | > 300 | > 300 | > 30 | > 30 | > 30 | ||
| (R)-31h | CH3 | NCS | 24 [0.5] (21 – 27) |
47 [0.25] (43 – 50) |
2.0 | ND | 4.2 [4] (2.4 – 8.0) |
> 250 | > 59 |
| (S)-31 | CH3 | NCS | > 100, < 300 | > 30, < 100 | ND | > 180 | > 30 | ||
| (R)-10 | CH2CH2NCS | C≡CH | ND | ND | ND | ND | ND | ||
| (S)-10 | CH2CH2NCS | C≡CH | ND | ND | ND | ND | ND | ||
| (R)-29 | CH3 | C≡CH | > 3, < 10 [0.5] | >10, <30 [0.5] | ND | < 30 [0.5 –4.0] | < 30 [0.5 – 4.0] | ||
| (S)-29 | CH3 | C≡CH | > 300 [0.5] | > 300 [0.5] | ND | ND | ND | ||
| (R)-27 | CH2CH2NCS | H | >300 [0.5] | > 30, < 100 | ND | < 30 [4.0] | >30 [4.0] | ||
| (S)-27 | CH2CH2NCS | H | > 300 [0.5] | > 30, < 100 [0.5] | ND | < 30 [4.0] | ND | ||
| (R)-11 | CH2CH2N3 | NCS | > 100, < 300 [4.0] | > 100, < 300 [0.5] | ND | ND | ND | ||
| (S)-11 | CH2CH2N3 | NCS | > 100, < 300 [0.25] | > 300 | ND | ND | ND | ||
| (R)-30 | CH2CH2N3 | H | > 100, < 300 [4.0] | > 100, < 300 [4.0] | 44 [0.5] (32 – 65) |
>40 [0.25] | >30 | ||
| (S)-30 | CH2CH2N3 | H | > 300 | > 300 | ND | ND | ND | ||
| (R)-31 | CH3 | NCS | 24 [0.5] (21 – 27) |
47 [0.25] (43 – 50) |
2.0 | ND | 4.2 [4] (2.4 – 8.0) |
> 250 | > 59 |
| (S)-31 | CH3 | NCS | > 100, < 300 | > 30, < 100 | ND | > 180 | > 30 | ||
| (R)-2i | 4.5 [0.5] (3.7-5.5) |
27 [0.25] (38-47) |
6.0 | 10 | 3.9 [0.5] (2.9-5.5) |
>500 [0.5] | > 130 | ||
| phenytoinj | 9.5 [2] (8.1 – 10) |
66 [2] (53 – 72) |
6.9 | 30 [4] (22 – 39) |
k | > 100 | |||
| phenobarbital | 22 [1] (15 – 23) |
69 [0.5] (63 – 73) |
3.2 | 9.1 [5] (7.6 – 12) |
61 [0.5] (44 – 96) |
6.7 | |||
| valproate | 270 [0.25] (250 – 340) |
430 [0.25] (370 – 450) |
1.6 | 490 [0.5] (350 – 730) |
280 [0.5] (190 – 350) |
0.6 | |||
The compounds were administered intraperitoneally. Numbers in parentheses are 95% confidence intervals. The dose effect data was obtained at the “time of peak effect” (indicated in hours in the brackets).
The compounds were administered orally.
MES = maximal electroshock seizure test.
Tox = neurologic toxicity determined from rotorod test.
PI = protective index (TD50/ED50).
Tox = behavioral toxicity.
ND = not determined.
LeTiran, A.; Stables, J. P.; Kohn, H. J. Med. Chem. 2002, 45, 4762-4773.
Choi, D.; Stables, J. P.; Kohn, H. J. Med. Chem. 1996, 39, 1907-1916.
Porter, R. J.; Cereghino, J. J.; Gladding, G. D.; Hessie, B. J.; Kupferberg, H. J.; Scoville, B.; White, B. G. Cleveland Clin. Q. 1984, 51, 293-305.
No ataxia observed up to 3000 mg/kg.
We found that we could incorporate a small moiety in the 4′ position of the N-benzylamide unit of (R)-2 without appreciable loss of MES anticonvulsant activity compared with unmodified (R)-2 (MES ED50 = 4.5 mg/kg) in mice (ip). For example, excellent seizure protection was observed for CR (R)-29 (MES ED50 = >3, <10 mg/kg) and AB (R)-31 (MES ED50 = 24 mg/kg).43 We also found that some modifications of the C(2) side chain in (R)-2 led to excellent activity retention in the MES seizure test, while others did not. The MES ED50 values for the CR O-propargyl ((R)-28), the CR O-2-azidoethyl ((R)-30), and the AB O-2-isothiocyanatoethyl ((R)-27) lacosamide analogs were 16, 100-300, and >300 mg/kg, respectively. We do not know if transport, efflux or metabolic factors are responsible for the diminished activity for (R)-27 and (R)-30 in the MES test. Interestingly, (R)-30 showed appreciable activity in the psychomotor seizure test49 (6 Hz ED50 = 44 mg/kg). Consistent with the pharmacological pattern observed for 218 and other FAAs (1),11-13,15 we found that for 28, 29, and 31, the (R)-stereoisomer was consistently more potent than the (S)-stereoisomer.
The findings for the lacosamide AB and the lacosamide CR analogs guided our investigation of 9–11 where both an AB and a CR moiety were installed in the molecule. Here, we found that 9, containing a N-benzyl 4′-isothiocyanate moiety and an O-propargyl moiety, provided significant seizure protection (MES ED50 = 45 mg/kg) and that activity principally resided in the (R)-stereoisomer. For 11, we observed that both stereoisomers gave only minimal seizure protection, a finding that mirrored the anticonvulsant activity for the corresponding CR-only analogs (R)-30 and (S)-30. Finally, we did not evaluate (R)-10 and (S)-10 in animal models since the corresponding AB compounds (R)-27 and (S)-27 were inactive at 300 mg/kg.
f. Chemical Evaluation of the AB and CR Units in 3
The AB and CR moieties have been installed within 9–11 for specific chemical functions. The isothiocyanate AB unit serves as an electrophilic agent to trap either lysine or cysteine residue(s) situated near the (R)-2 binding site, and either the alkyne or azide CR group acts as a bioorthogonal unit that can be selectively captured by the corresponding azide- or alkyne-containing probe via a Cu(I)-mediated cycloaddition reaction.50,51 Prior to conducting our studies we evaluated the chemical reactivity of these groups using model reactions (Figure 1). Treating AB (R)-31 with EtNH2 gave the expected thiourea (R)-32 in high yield (94%).43 Substituting the aliphatic isothiocyanate 33 for (R)-31 gave the expected thiourea 34 (77% yield).43a Correspondingly, when (R)-9 was reacted with 1.0 equiv of the biotin-containing azide probe 35,52 CuSO4 and sodium ascorbate,50 we isolated the desired product, (R)-36, in 51% yield. No modification of the isothiocyanate moiety was observed under these Cu(I)-mediated cycloaddition conditions. Finally, we treated aliphatic azide (R)-30 with alkyne 37 under similar conditions to obtain (R)-38 (96% yield).
Figure 1.

Chemical evaluation of the AB and the CR units in 3
g. Using Mouse Brain Soluble Lysates to Identify Lacosamide Targets
The utility of the lacosamide AB&CR agents 9–11 to identify drug binding targets was first assessed using mouse brain soluble lysates prepared from male (2 months) ICR mice (Rockland Immunochemicals, Gilbertsville, PA). The lysates were treated with (R)- and (S)-9–11 (5 μM) for 0.5 h at room temperature, and the lysate mixture reacted with an appropriate rhodamine-containing probe (39,53 40) under Cu(I)-mediated cycloaddition conditions (CuSO4, tris(2-carboxyethyl)phosphine (TCEP)). The reaction samples were separated by electrophoresis using a 10% SDS-PAGE gel and then analyzed for proteins labeled by the AB&CR agents by in-gel fluorescence using a typhoon scanning laser (emission 532 nm, absorption 580 nm) (Figure 2a). Gels were Coomassie-stained to visualize the proteins (Figure 2c).
Figure 2.

Proteome reactivity profiles of three different isothiocyanate (NCS) agents 9–11.
Figure 2A; AB&CR agent-labeled proteins were detected by in-gel fluorescence scanning after Cu(I)-mediated cycloaddition to either a rhodamine-azide reporter probe 39 or a rodamine-alkyne reporter probe 40. Figure 2B; CRMP2 protein was detected by western blot using anti-CRMP2. Figure 2C; All proteins in lysate were visualized by Coomassie blue staining after in-gel fluorescence scanning. (All images shown in gray scale).

Figure 2a showed that AB&CR 9 efficiently labeled many proteins in the lysate while 10 did not. The in-gel fluorescence pattern for 11 suggested the labeling of many of the proteins seen with 9, along with additional background proteins. Significantly, the rhodamine-based probe used for 9- and 10-modified reactions was different from that used for 11-modified reaction. The alkyne CR moieties in 9 and 10 required the use of the azide probe 39 for the Cu(I)-mediated cycloaddition step while the azide CR group in 11 required the alkyne-based probe 40. Cravatt and co-workers have previously reported higher levels of background protein adduction with alkyne probes compared with their azide counterparts.53 This observation was consistent with the gel patterns observed for 11 versus 9 and 10 (Figure 2a) that showed more proteins modified by the probe 40 compared with probe 39 under Cu(I)-mediated cycloaddition conditions. Similarly, we observed more labeled proteins with 40 compared with 39 in the absence of the AB&CR agent (Figure 2a, no drug control lanes).
Analysis of the (R)-9- and (S)-9-modified lysate gel patterns showed that some proteins were more extensively modified by (R)-9 than (S)-9 (Figure 2a, see bands labeled by arrow and asterisk). One of these bands appeared to be ~62-kDa (arrow), the approximate molecular weight of CRMP2.54 To confirm that the ~62-kDa band contained CRMP2, we performed a western blot analysis using an anti-CRMP2 antibody and observed prominent bands consistent with full-length and truncated forms of CRMP254 (Figure 2b). In addition to the ~62-kDa band, we observed another protein band (Figure 2a) with stronger intensity for the (R)-9 lane than for (S)-9 (Figure 2a, see band labeled by asterisk). The identity of this protein(s) is currently under investigation.
We recognized that both the increased fluorescence of the ~62 kDa band for the (R)-9 treated lysate compared with the (S)-9 reaction, and the identification of CRMP2 by western blot analysis do not demonstrate, by themselves, the preferential modification of CRMP2 by (R)-9. We suspect that the ~62 kDa band consists of several proteins, and thus the observed fluorescence pattern may be due to a protein(s) other than CRMP2. Accordingly, additional studies were conducted to verify the selective modification of CRMP2 by (R)-9. First, the (R)-9 lysate adduction was repeated. The modified lysate was treated with biotinyl CR probe 35 using Cu(I)-mediated cycloaddition conditions. Substitution of probe 35 for 39 permits the selective isolation of the (R)-9 captured proteins from the lysate. The biotinylated proteins were captured with streptavidin beads, the beads stringently washed to remove background proteins adsorbed on the resin, and then the protein adducts released with SDS treatment. SDS-PAGE electrophoresis showed bands at ~62 kDa upon Coomassie blue staining. The 62 kDa band was excised, trypsinized, and analyzed by mass spectrometry (MS). Analysis of the tryptic fragments indicated the presence of several proteins (e.g., CRMP2, CRMP1, serum albumin) whose molecular weights were consistent with their electrophoretic mobility. The protein with the most number of identified fragments in the (R)-9 treated reaction was CRMP2 (17 peptides including 11 peptides unique to CRMP2 and 6 peptides common to other CRMP proteins, 45% coverage), followed by CRMP1 (6 peptides including 4 peptides unique to CRMP1 and 2 peptides common to other CRMP proteins, 17% coverage), and serum albumin (4 peptides, 10% coverage), indicating that CRMP2 was a likely major constituent in the ~62 kDa band. Correspondingly, CRMP2 and other proteins were not observed in ~62 kDa band in the no drug control reaction. We made a quantitative comparison of the 62 kDa trypsin digests using differential stable isotope labeling (isobaric tag for relative and absolute quantitation (iTRAQ)).55 The trypsin digests from the (R)-9, (S)-9, the no drug control reaction, and iTRAQ buffer alone were each separately treated with distinct iTRAQ isotope labels, the samples combined, and then analyzed by LC-MS/MS (Supplementary Table S1 in the Supporting Information). There was no difference in observed iTRAQ ratio between fragments unique to CRMP2 and those common to other CRMP proteins. The peptide fragments showed, on average, an ~1.3:1 increase in the relative amounts of each tryptic fragment from the (R)-9 treated sample compared with (S)-9, after normalization to the tubulin fragments, based on the assumption of no stereochemical preference for lacosamide binding to tubulin. Minor amounts (<6% compared with the (R)-9 sample) of CRMP2 fragments were observed in the no drug sample. Analysis of the CRMP1 fragments also showed a 1.3:1 increase in the (R)-9 reaction compared with the (S)-9 reaction. This differential amount in tryptic fragment abundances was in agreement with the observed ratio for the fluorescence intensities of the ~62 kDa band in the (R)-9 reaction versus the (S)-9 experiment (Figure 2a), substantiating the identification of CRMP1 and CRMP2 as the relevant components of the gel bands from which they were derived. Finally, we overexpressed CRMP2 as a glutathione-S-transferase (GST)-fusion protein.56 Attachment of the GST-tag permitted protein purification and increased the size of the CRMP2 protein by ~25 kDa. The purified recombinant GST-CRMP2 protein (10 μg) was included in the lysate (50 μL, 2 mg/mL) and the AB&CR adduction reaction repeated with (R)-9, (S)-9, and no drug. The reaction products were treated with rhodamine azide 39 under Cu(I)-mediated cycloaddition conditions and then separated by 10% SDS-PAGE (Figure 3). The electrophoretic mobility of the GST-CRMP2 placed it in a region (~82 kDa) that was relatively free of other soluble lysate proteins. We observed an approximate 1.5-fold increase in fluorescent intensity for the (R)-9 modified GST-CRMP2 protein compared with the corresponding (S)-9 modified recombinant protein (double arrowhead, Figure 3a). By comparison, the (R)-9-modified endogenous 62-kDa band was approximately 1.8-fold more intense than the corresponding (S)-9-modified band (arrow, Figure 3a). Western blot analysis of the ~82 kDa band was in agreement with the presence of CRMP2 and GST (Figure 3b). Collectively, these studies show that CRMP2 is preferentially modified in the lysate by lacosamide AB&CR agent (R)-9, and that (R)-9 leads to higher CRMP2 adduction levels than the corresponding (S)-9 derivative.
Figure 3.

In vitro labeling of externally added GST-CRMP2 and endogenous CRMP2 in mixture of mouse lysate and overexpressed GST-CRMP2.
Figure 3A; AB&CR agent-labeled proteins were detected by in-gel fluorescence scanning after Cu(I)-mediated cycloaddition to rhodamine-azide reporter probe 39. Figure 3B; CRMP2 and GST proteins were detected by western blot using anti-CRMP2 and anti-GST antibodies. Figure 3C; All proteins in lysate were visualized by Coomassie blue staining after in-gel fluorescence scanning. (All images shown in gray scale).
Discussion
A robust strategy is advanced for the discovery of proteins that interact (bind) with small-molecular-weight ligands (drugs) that explicate function. Our target discovery approach could be general. As a test case, we have searched for the central nervous system (CNS) binding partners for (R)-2, a therapeutic agent recently approved for the treatment of partial-onset seizures.19 Whole animal pharmacological studies indicated that (R)-2 function is unique.20
Our strategy requires the installation of AB and CR units within the drug molecule (Scheme 1). The AB group in 3 covalently modifies the drug target to give 6 rendering permanently a reversible binding interaction. The CR moiety in 3 is a biologically inert group that specifically reacts with the bioorthogonal probe 7 designed to either detect or isolate the modified protein adduct. Specific criteria have been established for the designed AB&CR units. These requirements are stringent and have been adopted to minimize false positive results. In this study, we sought lacosamide AB&CR agents that exhibited potent anticonvulsant activity in rodent models for epilepsy and where the pharmacological activity principally resided in the enantiomer corresponding to D-serine, similar to that seen for (R)-2. This stereochemical selectivity for (R)-2 function and related 1s is a distinguishing feature for this class of compounds,11-13,15,18 and we now show that this enantiospecificity may extend to receptor tagging as well.
An efficient synthesis was devised to permit the preparation of enantiomerically pure samples of the AB&CR agents 9–11 and the corresponding AB 27 and 31, and CR 28–30 compounds (Scheme 1, Schemes S1-S3 in the Supporting Information). Key to our route was the BF3•Et2O catalyzed ring opening of aziridine 16.44 We found this method versatile, and it reliably gave optically pure 2-alkoxypropionamide derivatives. The isothiocyanate moiety was chosen as the AB moiety in 9–11 because of its relative stability in aqueous solutions and its reactivity with amines and thiols.27 We installed this group from the corresponding amine in the final step of the synthesis to minimize product loss. For amines 23 and 26, we used di(2-pyridyl) thionocarbonate,47a and for amine 25 we employed 1,1′-thiocarbonyldiimidazole 47b for this step. We further tested the reactivity of the different AB and CR groups to validate their use in our proteomic target search (Figure 1).
An important design component for our AB&CR agents is the use of small AB and CR units in order to minimize possible adverse interactions that could either prevent or alter target binding. In the case of the C(2) site in (R)-2, for example, we demonstrated that progressive size increases of the oxygen substituent led to a steady loss in seizure protection in the MES test (mice, ip).57 A similar structure-activity relationship has been found for the 4′ benzyl position in (R)-2 derivatives.58 Thus, in (R)-9, we incorporated an isothiocyanate group at the 4′ N-benzyl site for the AB moiety and a propargyl unit at C(2) alkoxy site for the CR moiety. We found that (R)-9 exhibited notable anticonvulsant activity in mice (ip) (MES ED50 = 45 mg/kg) while the corresponding (S)-9 did not (MES ED50 = >300 mg/kg). The anticonvulsant activities of the corresponding AB (R)-31 (MES ED50 = 24 mg/kg)43a and CR (R)-28 (MES ED50 = 16 mg/kg) derivatives exceeded that of the AB&CR agent (R)-9 (MES ED50 = 45 mg/kg). The loss of activity observed for (R)-9 is consistent with the cumulative adverse effects that may have occurred when two groups were appended to the lacosamide framework. We observed only modest MES seizure protection for AB&CR agent (R)-11 (MES ED50 = 100-300 mg/kg). Moreover, the animal behavioral studies did not differentiate the activities of (R)-11 and its stereoisomer (S)-11. The 6 Hz psychomotor seizure test49 for (R)-30 (mice, ip; 32 mA) provided evidence to support the use of the 2-azidoethoxy group in this lacosamide AB&CR agent. The absence of detectable anticonvulsant activity of (R)-27 in mice (>300 mg/kg) in the MES-seizure test (Table 1) discouraged our preparation and evaluation of the structural isomer of (R)-11 where the positions of the isothiocyanate and azide moieties are reversed.
Collectively, the whole animal pharmacological data showed that incorporating small AB and CR units at either the 4′ N-benzyl site or the C(2)-alkoxy position did not markedly perturb anticonvulsant function and likely did not impede binding to the drug’s cognate receptors. Our data demonstrated that the seizure protection provided by (R)-9 and (S)-9 mirrored that of (R)-2 and (S)-2, respectively. Correspondingly, the whole animal pharmacological data for 10 and 11 provided neither support nor evidence against their use for in vitro proteomic searches. Placement of either an isothiocyanate or azido group at the C(2) aliphatic site may diminish their activity, in part, presumably by preventing these compounds from reaching the CNS due to chemical, transport, efflux, and metabolic processes. We chose to interrogate the mouse brain lysates with all three AB&CR agents (9–11) to gain further information about the suitability of different AB and CR units for receptor identification, with the understanding that only animal pharmacological data supported the use of 9.
We first chose to examine the soluble proteome from Charles River male ICR mouse brains. Comparison of AB&CR agents (R)- and (S)-9–11 showed that (R)-9 and (S)-9 provided the cleanest in-gel fluorescent patterns while still showing appreciable protein labeling (Figure 2a). This is particularly apparent when the lanes for 9 are compared with that for 11. These sets of agents contain the same AB unit (4′-isothiocyanate N-benzyl moiety) but have different CR moieties. Consistent with results reported in the literature, we observed that the number of proteins modified by using alkyne-based probe 40 were considerably higher than with azide-based probe 39. We have attributed this, in part, to non-specific lysate modification by the rhodamine alkyne 40 probe.53 This unwanted reaction complicates receptor identification. When the protein band pattern for lacosamide AB&CR agent 10 was compared with that for 9, we observed a marked decrease in the extent of protein modification. Compounds 9 and 10 can be considered functional isomers where the structural positions of the AB and CR units have been reversed. Together, our findings indicate that AB&CR agent 9 may be best suited for lacosamide target identification.
This study showed that CRMP2 was preferentially modified in the lysate by lacosamide AB&CR agent (R)-9, and that (R)-9 leads to higher CRMP2 adduction levels than the corresponding reaction with the (S)-9 derivative. Interrogation of the mouse brain soluble lysate is complicated by the complexity of the proteome. Thus, several findings were used to validate CRMP2 labeling. First, western blot analysis verified the presence of CRMP2 in the ~62 kDa band after lysate modification by (R)-9 and treatment with 39 (Figure 2b). Second, MS analysis of the trypsin digest of the modified proteins in the ~62 kDa band indicated that CRMP2 was among the most abundant proteins present in this gel slice. Third, the enhanced adduction by (R)-9 compared with (S)-9 was confirmed using iTRAQ analysis of the tryptic fragments from the ~62 kDa band. A modest increase (1.3-fold) in the abundance of CRMP2 fragments was observed in the (R)-9 adduction experiment compared to (S)-9 reaction when compared to an internal protein (tubulin alpha chain) (Table S1 in the Supporting Information). The increased (R)-9 adduction levels are important since whole animal pharmacological studies have demonstrated that the principal anticonvulsant activity for 2 resided in the (R)-stereoisomer. Finally, we overexpressed GST-CRMP2 and then added a small amount of the fusion protein to the mouse lysate. Repetition of the 9 adduction experiments followed by Cu(I)-mediated cycloaddition with probe 39 and SDS-PAGE showed increased fluorescent levels (1.5–1.8-fold) for both the exogenous ~82 kDa and the endogenous ~62 kDa bands in the (R)-9 reaction compared with the (S)-9 experiment (Figure 3a).
The relative ratios of (R)-9 versus (S)-9 modification of CRMP2 proteins (1.3–1.8-fold) were modest compared with the observed MES ED50 values for (R)-2 and (S)-2 in this seizure model in mice (>22-fold).18 Several factors likely contributed to this finding. First, the MES ED50 values for (R)-9 and (S)-9 in rodents represent a composite value that encompasses numerous pharmacological factors that include binding to target(s), metabolism, transport, efflux, and nonspecific binding to abundant cellular proteins. Therefore, the in vitro modification data is not likely to fully recapitulate the in vivo pharmacological results. Second, different (R)-2 targets likely display different stereochemical preferences for the 9 enantiomers, and this stereochemical preference may be further influenced by the AB and CR groups that are incorporated within the lacosamide framework to facilitate in vitro analysis. Finally, we expect that the conditions of adduction such as drug and target concentration, temperature, and time will affect the levels of (R)-9 and (S)-9 CRMP2 adduction. For our experiments, we used relatively high amounts of 9 and moderate reaction conditions to provide substantial protein adduction levels.
Our results support a report by Beyreuther and coworkers that CRMP2 may interact with (R)-2.21 In their study, (R)-2-like derivatives (precise structures not disclosed) that contained an appended isothiocyanate group and a biotin moiety were prepared and then reacted with mouse brain lysates. Subsequent treatment of the modified lysate with streptavidin removed the biotinylated tagged proteins. Differentiating Beyreuther and colleagues’ study from ours was their use of significantly larger (R)-2-type agents and the absence of whole animal pharmacological data to support their use. Knowing these collective findings, detailed studies are warranted to determine the site of (R)-3 CRMP2 adduction and to validate this protein as an (R)-2 target.
We observed other possible (R)-2 binding target(s) (Figure 2a, asterisk band). The identity of this band is under investigation. Only mouse soluble brain proteome was used in this study. Future studies will examine the membrane-bound fraction. In this regard, (R)-2 has been reported to selectively enhance the slow inactivation of voltage-gated sodium channels.22 The combined use of our AB&CR (R)-3 agents, enzymatic digestion, and MS of the protein fragments may help reveal the site(s) of drug binding.
Conclusions
We advance a strategy to identify molecular targets for ligands (e.g., drugs) whose binding to their cognate receptors is modest and where moderate-to-extensive structural change abolishes target binding. Our method requires the installation of AB and CR units within the ligand where the AB group covalently modifies the target and the CR group is used to either identify or isolate the ligand-modified receptor. Strict guidelines for the use of the AB&CR strategy have been established to increase the success of this method and to minimize the isolation of false positive sites. Among these guidelines are that the AB and CR groups must conform to the SAR for the ligand and that the pharmacological profile for the AB&CR agent mirror that of the ligand. Using this approach we interrogated the mice brain soluble proteome for (R)-2 binding sites. We identified several potential drug targets including CRMP2, a protein previously suggested to interact with (R)-2. Further studies will be needed to validate these targets and to identify the sites of drug interaction.
Experimental Section
General Methods
Melting points were determined in open capillary tubes using a Thomas-Hoover melting point apparatus and are uncorrected. Infrared spectra (IR) were run on an ATI Mattson Genesis FT-IR spectrometer. Absorption values are expressed in wavenumbers (cm−1). Optical rotations were obtained on a Jasco P-1030 polarimeter at the sodium D line (589 nm) using a 1 dm path length cell. NMR spectra were obtained at 300 MHz (1H) and 75 MHz (13C) using TMS as an internal standard. Chemical shifts (δ) are reported in parts per million (ppm) from tetramethylsilane. Low-resolution mass spectra were obtained with a BioToF-II-Bruker Daltonics spectrometer by Drs. Matt Crowe and S. Habibi at the University of North Carolina Department of Chemistry. The high-resolution mass spectrum was performed on a Bruker Apex-Q 12 Telsa FTICR spectrometer by Drs. Matt Crowe and S. Habibi. Microanalyses were performed by Atlantic Microlab, Inc. (Norcross, GA). Reactions were monitored by analytical thin-layer chromatography (TLC) plates (Aldrich, Cat # Z12272-6) and analyzed with 254 nm light. The reaction mixtures were purified by MPLC (CombiFlash Rf) with self-packed columns (silica gel from Dynamic Adsorbents Inc., Cat # 02826-25) or by flash column chromatography using silica gel (Dynamic Adsorbents Inc., Cat # 02826-25). All chemicals and solvents were reagent grade and used as obtained from commercial sources without further purification. THF was distilled from blue sodium benzophenone ketyl. Yields reported are for purified products and were not optimized. All compounds were checked by TLC, 1H and 13C NMR, MS, and elemental analyses. The analytical results are within +0.40% of the theoretical value. The TLC, NMR and the analytical data confirmed the purity of the products was ≥95%.
General procedure for the aziridine ring opening of (R)/(S)-16 with alcohols. Method A
To a cooled CH2Cl2 solution (ice bath) containing the N-acetylaziridine methyl ester (R)/(S)-16 ([C] ~ 0.5–1 M) and the appropriate alcohol (1–3 eq.) was added BF3•Et2O (0.1–1 equiv) dropwise while stirring. After addition, the mixture was warmed to room temperature and stirred (90 min—2 h), and then an equal volume of saturated aqueous NaHCO3 was added. The reaction was vigorously stirred (15 min) and the organic layer separated. The aqueous layer was extracted with CH2Cl2 until no product could be detected (TLC analysis) and then all the organic layers were combined, dried (Na2SO4), and concentrated in vacuo to yield a residue that was purified by flash column chromatography.
General procedure for the ester hydrolysis of (R)/(S)-17 or (R)/(S)-18 with LiOH. Method B
To a THF solution of methyl ester (2 volumes, [C] ~ 0.1 M) was added an aqueous LiOH (1 equiv) solution (1 volume). The reaction was stirred at room temperature (90 min—12 h), after which time the aqueous layer was washed with Et2O (2 volumes). The aqueous layer was acidified (pH ~ 1) by the dropwise addition of aqueous concentrated HCl at 0 °C, saturated with NaCl, and extracted with EtOAc until no further product was detected (TLC analysis). The combined organic layers were combined, dried (Na2SO4), and evaporated to an oily residue that was used directly for the next step, or recrystallized when needed from EtOAc and hexanes to provide an analytical sample.
General procedure for the mixed anhydride coupling reaction. Method C
To a cooled THF solution (−78 °C, dry ice acetone bath) of acid (R)/(S)-19 ([C] ~ 0.1 M) were successively added NMM (1.0–1.1 equiv), stirred for 2 min, IBCF (1.0–1.2 equiv), stirred for 5 min, and then the desired benzylamine (1.0–1.2 equiv). Upon addition the reaction mixture was allowed to warm to room temperature and further stirred (2–3 h). The salts were filtered and rinsed with THF and the filtrate was concentrated in vacuo. The residue obtained was purified by flash chromatography, followed by recrystallization from EtOAc and hexanes when necessary.
General procedure for the DMTMM amide coupling reaction. Method D
To a THF solution of acid (R)/(S)-20 ([C] ~ 0.1 M) at room temperature was added the desired benzylamine (1.2 equiv). The solution was stirred (5–10 min) until the benzylammonium carboxylate precipitated. While stirring, DMTMM (1.2 equiv) was added all at once, and the resulting suspension was stirred at room temperature (3–12 h). In those cases where a salt did not precipitate, DMTMM was added after 15 min to the solution. The salts were removed by filtration, washed with THF, and the solvent was removed in vacuo. The residue obtained was purified by flash column chromatography to afford the benzylamide, and then recrystallized from EtOAc and hexanes.
(R)-Methyl 2-Acetamido-3-(prop-2-ynyloxy)propionate ((R)-17)
Utilizing Method A, (R)-methyl 1-acetylaziridine-2-carboxylate44 ((R)-16) (1.50 g, 10.49 mmol), propargyl alcohol (0.92 mL, 15.74 mmol) and BF3•Et2O (1.39 mL, 11.01 mmol) gave 1.43 g (68%) of (R)-17 as a white solid after recrystallization with cold hexanes: mp 58.5–59.5 °C; [α]25D −64.6° (c 1.0, CHCl3); Rf = 0.35 (3/1 EtOAc/hexanes); IR (nujol mull) 3301, 2919, 2114, 1750, 1639, 1542, 1457 cm−1; 1H NMR (CDCl3) δ 2.06 (s, CH3C(O)), 2.46 (t, J = 2.3 Hz, CH2CCH), 3.78 (s, OCH3), 3.79 (dd, J = 3.3, 9.6 Hz, CHH’OCH2), 3.98 (dd, J = 2.9, 9.6 Hz, CHH’OCH2), 4.09–4.21 (m, OCH2C), 4.77–4.82 (m, CH), 6.33–6.35 (br d, J = 7.2 Hz, NHCH); 13C NMR (CDCl3) δ 23.3 (CH3C(O)), 52.6 (CH), 52.8 (OCH3), 58.7 (CH2CCH), 69.6 (CH2OCH2), 75.3 (CH2CCH), 79.0 (CH2CCH), 170.1, 170.8 (2 C(O)); Mr (+ESI) 200.0916 [M+H]+ (calcd for C9H13NO4H+ 200.0923 [M+H]+). Anal. (C9H13NO4): C, H, N.
(S)-Methyl 2-Acetamido-3-(prop-2-ynyloxy)propionate ((S)-17)
Utilizing the preceding procedure and using (S)-1644 (1.50 g, 10.49 mmol), propargyl alcohol (0.92 mL, 15.74 mmol) and BF3•Et2O (1.39 mL, 11.01 mmol) gave 1.32 g (63%) of (S)-17 as a white solid: mp 59.0–59.5 °C; [α]25D +65.3° (c 1.0, CHCl3); Rf = 0.35 (3/1 EtOAc/hexanes); IR (nujol mull) 3296, 2924, 2116, 1740, 1641, 1543, 1457 cm−1; 1H NMR (CDCl3) δ 2.06 (s, CH3C(O)), 2.46 (t, J = 2.4 Hz, CH2CCH), 3.78 (s, OCH3), 3.79 (dd, J = 3.2, 9.4 Hz, CHH’OCH2), 3.98 (dd, J = 3.0, 9.4 Hz, CHH’OCH2), 4.12 (1/2HH’q, J = 2.4, 16.2 Hz, OCHH’C), 4.18 (1/2HH’q, J = 2.4, 16.2 Hz, OCHH’C), 4.77–4.82 (m, CH), 6.32–6.34 (br d, J = 7.2, NHCH); 13C NMR (CDCl3) δ 23.3 (CH3C(O)), 52.6 (CH), 52.8 (OCH3), 58.7 (CH2CCH), 69.6 (CH2OCH2), 75.3 (CH2CCH), 79.0 (CH2CCH), 170.1, 170.7 (2 C(O)); Mr (+ESI) 200.0917 [M+H]+ (calcd for C9H13NO4H+ 200.0923 [M+H]+)). Anal. (C9H13NO4) C, H, N.
(R)-2-Acetamido-3-(prop-2-ynyloxy)propionic Acid ((R)-19)
Utilizing Method B, (R)-17 (1.00 g, 5.03 mmol), LiOH (0.6 M, 8.4 mL, 5.03 mmol) gave 0.87 g of (R)-19 (94%) as a yellow oil: Rf = 0.35 (15/5/1 EtOAc/hexanes/AcOH); IR (nujol mull) 3264, 2924, 2120, 1726, 1642, 1548, 1459 cm−1; 1H NMR (CD3OD) δ 2.01 (s, CH3C(O)), 2.88 (t, J = 2.6 Hz, CH2CCH), 3.77 (dd, J = 3.6, 9.7 Hz, CHH’OCH2), 3.92 (dd, J = 5.0, 9.7 Hz, CHH’OCH2), 4.18 (d, J = 2.6 Hz, OCH2C), 4.61–4.64 (m, CH); 13C NMR (CD3OD) δ 22.5 (CH3C(O)), 54.1 (CH), 59.3 (CH2CCH), 70.4 (CH2OCH2), 76.5 (CH2CCH), 80.2 (CH2CCH), 173.1, 173.5 (2 C(O)); Mr (+ESI) 186.0761 [M+H]+ (calcd for C8H11NO4H+ 186.0766 [M+H]+). Anal. (C8H11NO4•0.14H2O): C, H, N.
(S)-2-Acetamido-3-(prop-2-ynyloxy)propionic Acid ((S)-19)
Utilizing the preceding procedure and using (S)-17 (1.00 g, 5.03 mmol) and LiOH (0.6 M, 8.4 mL, 5.03 mmol) gave 0.86 g (93%) of (S)-19 as a yellow oil: Rf = 0.35 (15/5/1 EtOAc/hexanes/AcOH); IR (nujol mull) 3270, 2924, 2120, 1729, 1641, 1547, 1459 cm−1; 1H NMR (DMSO-d6) δ 1.87 (s, CH3C(O)), 3.48 (t, J = 2.4 Hz, CH2CCH), 3.62 (dd, J = 4.1, 9.6 Hz, CHH’OCH2), 3.74 (dd, J = 2.4, 9.6 Hz, CHH’OCH2), 4.16 (d, J = 2.4 Hz, OCH2C), 4.41–4.47 (m, CH), 8.21–8.23 (d, J = 8.4 Hz, NHCH); 13C NMR (DMSO-d6) δ 22.4 (CH3C(O)), 52.1 (CH), 57.8 (CH2CCH), 69.2 (CH2OCH2), 77.6 (CH2CCH), 79.9 (CH2CCH), 169.6, 171.5 (2 C(O)); Mr (+ESI) 186.0760 [M+H]+ (calcd for C8H11NO4H+ 186.0766 [M+H]+). Anal. (C8H11NO4•0.25H2O) C, H, N.
(R)-Methyl 2-Acetamido-3-(2-azidoethoxy)propionate ((R)-18)
Using Method A, compound (R)-1644 (2.35 g, 16.4 mmol), 2-azidoethanol (4.5 mL, 65.6 mmol) and BF3•Et2O (1 mL, 8.2 mmol) gave 1.56 g (41%) of (R)-18 after purification: [α]25D +49.1° (c 1.0; EtOAc); Rf = 0.47 (EtOAc); IR (neat) 3302, 2948, 2107, 1746, 1668, 1534, 1443 cm−1; 1H NMR (CDCl3) δ 2.06 (s, CH3C(O)NH), 3.23–3.42 (m, CH2N3), 3.31–3.64 (m, OCH2CH2N3, CHCHH’OCH2), 3.78 (s, OCH3), 3.96 (dd, J = 3.0, 9.1 Hz CHCHH’OCH2), 4.76–4.81 (m, CHCH2O), 6.30–6.41 (br d, CH C(O)NH); 13 3 C NMR (CDCl3) δ 23.2 (CH3C(O)), 50.0 (OCH2CH2N3), 52.6 (OCH3 or CHCH2O), 52.8 (CHCH2O or OCH3), 70.9 (CHCH2O or OCH2CH2N3), 71.1 (OCH2CH2N3 or CHCH2O), 170.1, 170.6 (C(O)OCH3, CH3C(O)NH); Mr (+ESI) 253.0906 [M+Na]+ (calcd for C8H14N4O4Na+ 253.0913 [M+Na]+).
(S)-Methyl 2-Acetamido-3-(2-azidoethoxy)propionate ((S)-18)
Utilizing the preceding procedure, and using (S)-1644 (3.30 g, 23 mmol), 2-azidoethanol (6.3 mL, 92 mmol), and BF3•Et2O (1.4 mL, 11.5 mmol) in CH2Cl2 (115 mL) gave 1.67 g (31%) of a colorless residue: [α]25D −48.0° (c 1.0; EtOAc); Rf = 0.47 (EtOAc); IR (neat) 3302, 2948, 2107, 1746, 1667, 1534, 1443 cm−1; 1H NMR (CDCl3) δ 2.06 (s, CH3C(O)NH), 3.23–3.42 (m, CH2N3), 3.31–3.64 (m, OCH2CH2N3, CHCHH’OCH2), 3.78 (s, OCH3), 3.96 (dd, J = 3.0, 9.1 Hz CHCHH’OCH2), 4.76–4.81 (m, CHCH2O), 6.30–6.41 (br d, CH3C(O)NH); 13C NMR (CDCl3) δ 23.3 (CH3C(O)), 50.1 (OCH2CH2N3), 52.7 (OCH3 or CHCH2O), 52.9 (CHCH2O or OCH3), 71.0 (CHCH2O or OCH2CH2N3), 71.2 (OCH2CH2N3 or CHCH2O), 170.2, 170.7 (CH3C(O)NH and C(O)OCH3); Mr (+ESI) 253.0906 [M+Na]+ (calcd for C8H14N4O4Na+ 253.0913 [M+Na]+).
(R)-2-Acetamido-3-(2-azidoethoxy)propionic Acid ((R)-20)
Utilizing Method B, and using compound (R)-18 (1.56 g, 6.78 mmol) in THF (60 mL) and LiOH (195 mg, 8.14 mmol) in H2O (30 mL) gave 750 mg (51%) of (R)-20 as a white solid. An analytical sample was obtained by recrystallization from EtOAc and hexanes: mp 99–100 °C; [α]25D −12.6° (c 1.8; MeOH); Rf = 0.21 (1/9 MeOH/CHCl3); IR (nujol mull) 3356, 2119, 1735, 1624, 1547 cm−1; 1H NMR (CDCl3) δ 2.07 (s, CH3C(O)NH), 3.23– 3.42 (m, CH2N3), 3.60–3.80 (m, OCH2CH2N3, CHCHH’O), 4.01 (dd, J = 3.0, 9.1 Hz, CHCHH’O), 4.76–4.80 (m, CHCH2O), 6.00–7.00 (br, CO2H), 6.48 (d, J = 7.5 Hz, CH3C(O)NH), no signal could be detected for the COOH proton; 13C NMR (DMSO-d6) 22.3 (CH3C(O)), 49.9 (OCH2CH2N3), 52.2 (CHCH2O), 69.4 (CHCH2O or OCH2CH2N3), 70.1 (OCH2CH2N3 or CHCH2O), 169.4 (CH3C(O)NH or C(O)OH), 171.5 (C(O)OH or CH3C(O)NH); Mr (+ESI) 239.0750 [M+Na]+ (calcd for C7H12N4O4Na+ 239.0756, [M+Na]+). Anal. (C7H12N4O4•0.07EtOAc): C, H, N.
(S)-2-Acetamido-3-(2-azidoethoxy)propionic Acid ((S)-20)
Utilizing the preceding procedure and using (S)-18 (1.67 g, 7.26 mmol) in THF (60 mL), and LiOH (209 mg, 8.71 mmol) in H2O (30 mL) gave 862 mg (55%) of (S)-20 as a white solid upon work-up and evaporation. An analytical sample was obtained by recrystallization from EtOAc and hexanes: mp 99–100 °C; [α]25D +12.4° (c 1.3; MeOH); Rf = 0.21 (1/9 MeOH/CHCl3); IR (nujol mull) 3356, 2119, 1735, 1624, 1547 cm−1; 1H NMR (DMSO-d6) δ 1.86 (s, CH3C(O)NH), 3.30–3.45 (m, CH2N3), 3.50–3.78 (m, OCH2CH2N3, CHCH2OCH2), 4.39–4.48 (m, CHCH2O), 8.13 (d, J = 8.1 Hz, CH3C(O)NH), no signal could be detected for the COOH proton; 13C NMR (DMSO-d6) 22.3 (CH3C(O)), 49.9 (OCH2CH2N3), 52.2 (CHCH2O), 69.4 (CHCH2O or OCH2CH2N3), 70.1 (OCH2CH2N3 or CHCH2O), 169.4 (CH3C(O)NH or C(O)OH), 171.5 (C(O)OH or CH3C(O)NH); Mr (+ESI) 239.0750 [M+Na]+ (calcd for C7H12N4O4Na+ 239.0756 [M+Na]+). Anal. (C7H12N4O4•0.07EtOAc): C, H, N.
(R)-N-(4-Amino)benzyl 2-Acetamido-3-(prop-2-ynyloxy)propionamide ((R)-23)
Utilizing Method C, (R)-19 (0.25 g, 1.37 mmol), NMM (0.23 mL, 2.06 mmol), IBCF (0.23 mL, 1.73 mmol), and 4-aminobenzylamine (22) (0.21 mL, 1.85 mmol) gave 0.24 g (61%) of (R)-23 as a light yellow solid: mp 97.5–98.5 °C; [α]25D +4.2° (c 1.0, MeOH); Rf = 0.40 (1/9 MeOH/CHCl3); IR (nujol mull) 3279, 2930, 2111, 1628, 1536, 1459 cm−1; 1H NMR (CDCl3) δ 2.00 (s, CH3C(O)), 2.44 (t, J = 2.4 Hz, CH2CCH), 3.62 (dd, J = 6.9, 9.3 Hz, CHH’OCH2), 3.56–3.72 (br m, NH2), 3.88 (dd, J = 4.1, 9.3 Hz, CHH’OCH2), 4.12 (1/2HH’q, J = 2.4, 15.9 Hz, OCHH’C), 4.19 (1/2HH’q, J = 2.4, 15.9 Hz, OCHH’C), 4.32 (d, J = 5.7 Hz, CH2Ar), 4.54–4.60 (m, CH), 6.55–6.58 (br d, J = 7.2 Hz, NHCH), 6.60–6.65 (m, 2 ArH), 6.65-6.71 (br m, NHCH2), 7.03-7.06 (m, 2 ArH); 13C NMR (CDCl3) δ 23.4 (CH3C(O)), 43.5 (CH2Ph), 52.7 (CH), 58.8 (CH2CCH), 69.4 (CH2OCH2), 75.5 (CH2CCH), 79.1 (CH2CCH), 115.4, 127.7, 129.1, 146.1 (ArC), 169.6, 170.5 (2 C(O)); Mr (+ESI) 290.1500 [M+H]+ (calcd for C15H19N3O3H+ 290.1505 [M+H]+). Anal. (C15H19N3O3): C, H, N.
(S)-N-(4-Amino)benzyl 2-Acetamido-3-(prop-2-ynyloxy)propionamide ((S)-23)
Utilizing the preceding procedure and using (S)-19 (0.25 g, 1.37 mmol), NMM (0.20 mL, 1.78 mmol), IBCF (0.19 mL, 1.44 mmol), and 22 (0.16 mL, 1.44 mmol) gave 0.30 g (77%) of (S)-23 as a light yellow solid: mp 97.0–98.0 °C; [α]25D −4.1° (c 1.0, MeOH); Rf = 0.40 (1/9 MeOH/CHCl3); IR (nujol mull) 3282, 2924, 2112, 1630, 15372, 1458 cm−1; 1H NMR (CDCl3) δ 2.03 (s, CH3C(O)), 2.45 (t, J = 2.4 Hz, CH2CCH), 3.62 (dd, J = 7.1, 9.1 Hz, CHH’OCH2), 3.62–3.74 (br m, NH2), 3.92 (dd, J = 4.2, 9.1 Hz, CHH’OCH2), 4.14 (1/2HH’q, J = 2.4, 15.9 Hz, OCHH’C), 4.22 (1/2HH’q, J = 2.4, 15.9 Hz, OCHH’C), 4.35 (d, J = 6.0, CH2Ar), 4.53–4.59 (m, CH), 6.42–6.44 (br d, J = 6.3 Hz, NHCH), 6.48–6.56 (br m, NHCH2), 6.62–6.67 (m, 2 ArH), 7.03–7.08 (m, 2 ArH); 13C NMR (CDCl3) δ 23.6 (CH3C(O)), 43.8 (CH2Ph), 52.9 (CH), 59.0 (CH2CCH), 69.7 (CH2OCH2), 75.7 (CH2CCH), 79.4 (CH2CCH), 115.6, 128.0, 129.4, 146.3 (C6H4), 169.9, 170.7 (2 C(O)); Mr (+ESI) 290.1499 [M+H]+ (calcd for C15H19N3O3H+ 290.1505 [M+H]+). Anal. (C15H19N3O3): C, H, N.
(R)-N-(4-Isothiocyanato)benzyl 2-Acetamido-3-(prop-2-ynyloxy)propionamide ((R)-9)
To a dry THF solution (20 mL) of (R)-23 (0.17 g, 0.60 mmol) was added dropwise a solution of DPT (0.21 mL, 0.89 mmol) in THF (8 mL). The reaction solution was stirred under Ar at room temperature (2 h). The solvent was removed in vacuo, and the product was purified by silica gel column chromatography (1/9 acetone/EtOAc) to give 0.15 g (77%) of (R)-9 as a white solid: mp 157–159 °C; [α]25D −10.9° (c 1.5, CHCl3); Rf = 0.40 (1/9 acetone/EtOAc); IR (nujol mull) 3270, 2924, 2084, 1634, 1553, 1459 cm−1; 1H NMR (CDCl3) δ 2.03 (s, CH3C(O)), 2.48 (t, J = 2.5 Hz, CH2CCH), 3.65 (dd, J = 7.1, 9.3 Hz, CHH’OCH2), 3.92 (dd, J = 4.2, 9.3 Hz, CHH’OCH2), 4.15 (1/2HH’q, J = 2.5, 15.9 Hz, OCHH’C), 4.22 (1/2HH’q, J = 2.5, 15.9 Hz, OCHH’C), 4.40 (1/2HH’q, J = 5.9, 15.5 Hz, CHH’Ar), 4.49 (1/2HH’q, J = 5.9, 15.5 Hz, CHH’Ar), 4.60–4.66 (m, CH), 6.52 (br d, J = 6.9 Hz, NHCH), 6.96 (br t, J = 5.9 Hz, NHCH2), 7.16–7.19 (m, 2 ArH), 7.24–7.27 (m, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-9 gave only a single signal for the acetyl methyl protons and the alkyne proton, addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-9 and (S)-9 (1:2 ratio) gave two signals for the acetyl methyl protons (δ 2.019 (R) and 2.033 (S) (Δppm = 0.014)), and two signals for the alkyne proton (δ 2.424 (S) and 2.462 (R) (Δppm = 0.038)); 13C NMR (CDCl3) δ 23.4 (CH3C(O)), 43.1 (CH2Ph), 52.7 (CH), 58.9 (CH2CCH), 69.3 (CH2OCH2), 75.6 (CH2CCH), 79.0 (CH2CCH), 126.1, 128.8, 130.6 (3 ArC), 135.7 (NCS), 137.5 (1 ArC), 170.0, 170.6 (2 C(O)); Mr (+ESI) 332.1065 [M+H]+ (calcd for C16H17N3O3SH+ 332.1069 [M+H]+). Anal. (C16H17N3O3S): C, H, N, S.
(S)-N-(4-Isothiocyanato)benzyl 2-Acetamido-3-(prop-2-ynyloxy)propionamide ((S)-9)
Utilizing the preceding procedure, and using (S)-23 (0.30 g, 1.02 mmol), and DPT (0.32 g, 1.38 mmol) gave 0.25 g (75%) of pure (S)-9 as a white solid: mp 157–158 °C; [α]25D +10.6° (c 1.5, CHCl3); Rf = 0.40 (1/9 MeOH/CHCl3); IR (nujol mull) 3272, 2924, 2084, 1633, 1553, 1459 cm−1; 1H NMR (CDCl3) δ 2.04 (s, CH3C(O)), 2.48 (t, J = 2.5 Hz, CH2CCH), 3.65 (dd, J = 7.1, 9.2 Hz, CHH’OCH2), 3.94 (dd, J = 4.1, 9.2 Hz, CHH’OCH2), 4.16 (1/2HH’q, J = 2.5, 16.0 Hz, OCHH’C), 4.24 (1/2HH’q, J = 2.5, 16.0 Hz, OCHH’C), 4.42 (1/2HH’q, J = 6.3, 15.5 Hz, CHH’Ar), 4.50 (1/2HH’q, J = 6.3, 15.5 Hz, CHH’Ar), 4.57–4.63 (m, CH), 6.41–6.43 (br d, J = 6.6 Hz, NHCH), 6.81 (br t, J = 6.3 Hz, NHCH2), 7.17–7.20 (m, 2 ArH), 7.25-7.27 (m, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (S)-9 gave only a single signal for the acetyl methyl protons and the alkyne proton, addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-9 and (S)-9 (1:2 ratio) gave two signals for the acetyl methyl protons (δ 2.019 (R) and 2.033 (S) (Δppm = 0.014)), and two signals for the alkyne proton (δ 2.424 (S) and 2.462 (R) (Δppm = 0.038)); 13C NMR (CDCl3) δ 23.3 (CH3C(O)), 43.1 (CH2Ph), 52.7 (CH), 58.8 (CH2CCH), 69.3 (CH2OCH2), 75.6 (CH2CCH), 79.0 (CH2CCH), 126.1, 128.7, 130.5 (3 ArC), 135.7 (NCS), 137.5 (1 ArC), 170.0, 170.7 (2 C(O)); Mr (+ESI) 332.1063 [M+H]+ (calcd for C16H17N3O3SH+ 332.1069 [M+H]+). Anal. (C16H17N3O3S): C, H, N, S.
(R)-N-(4-Isothiocyanato)benzyl 2-Acetamido-3-(2-azidoethoxy)propionamide ((R)-11)
Utilizing Method D, and using acid (R)-20 (1130 mg, 5.2 mmol), 4-aminobenzylamine (22) (760 mg, 6.3 mmol), and DMTMM (1730 mg, 6.3 mmol) in THF (100 mL) gave 715 mg (42%) of a dark brown residue. The residue was directly dissolved in dry acetonitrile (40 mL) and DITC (90%, 510 mg, 2.57 mmol) was added all at once. The reaction solution was stirred at room temperature (1 h), the solvent was removed in vacuo, and the residue purified using flash chromatography (5/95 MeOH/CHCl3) and then recrystallized from EtOAc to yield 502 mg (27%, 2 steps) of (R)-11 as an off-white solid: mp 142–143 °C; [α]25D −28.6° (c 0.81; CHCl3); Rf = 0.45 (1/9 acetone/EtOAc); IR (nujol mull) 3276, 2181, 2114, 1631, 1547, 1459, 1374, 1285 cm−1; 1H NMR (CDCl3) δ 2.05 (s, CH3C(O)NH), 3.30–3.50 (m, CH2N3), 3.55 (dd, J = 7.5, 9.3 Hz, CHCHH’OCH2), 3.62–3.80 (m, OCH2CH2N3), 3.95 (dd, J = 4.0, 9.3 Hz, CHCHH’OCH2), 4.46 (d, J = 6.0 Hz, C(O)NHCH2Ph), 4.54–4.60 (m, CHCH2O), 6.45 (br d, J = 6.2 Hz, CH3C(O)NH), 6.84–6.92 (br t, C(O)NHCH2Ph), 7.18–7.22 (m, 2 ArH), 7.23–7.29 (m, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-11 gave only one signal for the acetyl protons, addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (S)-11 and (R)-11 (1:2 ratio) gave two signals for the acetyl methyl protons (δ 2.033 (S) and 2.020 (R) (Δppm = 0.013); 13C NMR (CDCl3) 23.2 (CH3C(O)), 41.0 (NHCH2Ph), 50.7 (CH2N3), 52.4 (CHCH2O), 70.0 (OCH2CH2N3 or CHCH2O), 70.3 (CHCH2O or OCH2CH2N3), 125.9, 128.6, 130.5 (3 ArC), 135.6 (NCS), 137.4 (1 ArC), 169.8, 169.2 (CH3C(O)NH, C(O)NHCH2); Mr (+ESI) 363.1236 [M+H]+ (calcd for C15H18N6O3SH+ 363.1239 [M+H]+). Anal. (C15H18N6O3S): C, H, N, S.
(S)-N-(4-Isothiocyanato)benzyl 2-Acetamido-3-(2-azidoethoxy)propionamide ((S)-11)
Utilizing the preceding procedure, and using acid (S)-20 (1.61 g, 7.52 mmol), 22 (1.02 mL, 9.03 mmol) and DMTMM (2.50 g, 9.03 mmol) in anhydrous THF (200 mL) followed by DITC (90%, 1.58 g, 7.90 mmol) in acetonitrile (20 mL) gave 706 mg (26%, two steps) of (S)-11 as an off-white solid after flash chromatography and recrystallization: mp 142–143 °C; [α]25D +28.5° (c 0.95; CHCl3); Rf = 0.45 (1/9 acetone/EtOAc); IR (nujol mull) 3280, 2177, 2108, 1637, 1548, 1452, 1374, 1293 cm−1; 1H NMR (CDCl3) δ 2.01 (s, CH3C(O)NH), 3.28–3.48 (m, CH2N3), 3.55 (dd, J = 7.2, 9.3 Hz, CHCHH’OCH2), 3.62-3.78 (m, OCH2CH2N3), 3.91 (dd, J = 4.0, 9.3 Hz, CHCHH’OCH2), 4.43 (d, J = 6.0 Hz, C(O)NHCH2Ph), 4.56–4.62 (m, CHCH2O), 6.56 (br d, J = 6.2 Hz, CH3C(O)NH), 6.84–6.92 (br t, J = 6.0 Hz, C(O)NHCH2Ph), 7.14–7.20 (m, 2 ArH), 7.22–7.28 (m, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (S)-11 gave only one signal for the acetyl protons, addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (S)-11 and (R)-11 (1:2 ratio) gave two signals for the acetyl methyl protons (δ 2.033 (S) and 2.020 (R) (Δppm = 0.013); 13C NMR (CDCl3) 23.3 (CH3C(O)), 43.2 (NHCH2Ph), 50.9 (CH2N3), 52.7 (CHCH2O), 70.3 (OCH2CH2N3 or CHCH2O), 70.4 (CHCH2O or OCH2CH2N3), 126.1, 128.8, 130.6 (3 ArC), 135.8 (NCS), 137.6 (1 ArC), 170.0, 170.7 (CH3C(O)NH, C(O)NHCH2); Mr (+ESI) 363.1241 [M+H]+ (calcd for C15H18N6O3SH+ 363.1239 [M+H]+). Anal. (C15H18N6O3S): C, H, N, S.
(R)-N-(4-Ethynyl)benzyl 2-Acetamido-3-(2-azidoethoxy)propionamide ((R)-24)
Using Method C, (R)-2-acetamido-3-(2-azidoethoxy)propionic acid ((R)-20) (0.31 g, 1.4 mmol), NMM (189 μL, 1.7 mmol), IBCF (228 μL, 1.7 mmol), 4-ethynylbenzylamine (21) (225 mg, 1.7 mmol), and THF (20 mL) gave 0.26 g (55%) of (R)-24 as a white solid: Rf = 0.18 (EtOAc); mp 80–87 °C (dec); [α]25D +4.8° (c 0.5, DMSO); IR (nujol mull) 3282, 2103, 1636, 1548, 1458, 1375, 1303, 1258, 1124, 982, 821, 721 cm−1; 1H NMR (CDCl3) δ 2.03 (s, CH3C(O)), 3.08 (s, HC≡C), 3.27-3.46 (m, CH2N3), 3.56 (dd, J = 7.2, 9.6 Hz, OCHH’), 3.62–3.77 (m, CH2CH2O), 3.92 (dd, J = 3.6, 9.6 Hz, OCHH’), 4.46 (d, J = 5.7 Hz, CH2N), 4.56–4.62 (m, CH), 6.53 (d, J = 6.9 Hz, NH), 6.88–7.01 (br t, CH2NH), 7.22 (d, J = 8.1 Hz, 2 ArH), 7.45 (d, J = 8.1 Hz, 2 ArH); 13C NMR (CDCl3) δ 23.1 (CH3CO), 43.3 (CH2N), 50.7 (CH2N3), 52.5 (CH), 70.1 (CH2O), 70.3 (CH2O), 77.4 (C≡C), 83.3 (C≡C), 121.3, 127.4, 132.4, 138.7 (4 ArC), 169.8, 170.5 (2 C(O)); Mr (+ESI) 330.1569 [M+H]+ (calcd for C16H19N5O3H+ 330.1566 [M+H]+).
(S)-N-(4-Ethynyl)benzyl 2-Acetamido-3-(2-azidoethoxy)propionamide ((S)-24)
Utilizing the preceding procedure, and using THF (20 mL), (S)-20 (0.31 g, 1.4 mmol), NMM (189 μL, 1.7 mmol), IBCF (225 μL, 1.7 mmol) and 21 (225 mg, 1.7 mmol) gave 165 mg of a white solid (35%): Rf = 0.18 (EtOAc); mp 107–109 °C; [α]25D −6.0° (c 0.5, DMSO); IR (nujol mull) 3274, 2100, 1635, 1548, 1457, 1375, 1301, 1257, 1121, 980, 820, 719 cm−1; 1H NMR (CDCl3) δ 2.02 (s, CH3C(O)), 3.08 (s, HC≡C), 3.27–3.46 (m, CH2N3), 3.56 (dd, J = 7.5, 9.6 Hz, OCHH’), 3.61–3.76 (m, CH2CH2O), 3.91 (dd, J = 3.6, 9.6 Hz, OCHH’), 4.45 (d, J = 6.0 Hz, CH2N), 4.56–4.63 (m, CH), 6.54 (d, J = 6.0 Hz, NH), 6.95–7.05 (br t, CH2NH), 7.22 (d, J = 7.9 Hz, 2 ArH), 7.45 (d, J = 7.9 Hz, 2 ArH); 13C NMR (CDCl3) δ 23.1 (CH3C(O)), 43.2 (CH2N), 50.6 (CH2N3), 52.4 (CH), 70.1, 70.2 (2 CH2O), 77.3 (C≡C), 83.2 (C≡C), 121.2, 127.4, 132.3, 138.7 (4 ArC), 169.8, 170.4 (2 C(O)); Mr (+ESI) 352.1387 [M+Na]+ (calcd for C16H19N5O3Na+ 352.1386 [M+Na]+).
(R)-N-(4-Ethynyl)benzyl) 2-Acetamido-3-(2-isothiocyanato-ethoxy)propionamide ((R)-10)
Triphenylphosphine polymer bound (Fluka, cat # 93094) (1.6 mol/g, 3.0 mmol) was added to an aqueous (180 μL, 10.0 mmol)/THF (10 mL) solution of (R)-24 (329 mg, 1.0 mmol). The mixture was shaken until the starting material was no longer evident by TLC analysis (18 h). The triphenylphosphine-based support was filtered, washed with CH2Cl2, and the filtrate was concentrated in vacuum to obtain the free amine (R)-26: 1H NMR (CDCl3) δ 2.03 (s, CH3C(O)), 2.76-2.92 (m, CH2NH2), 3.07 (s, HC≡C), 3.48–3.62 (m, OCHH’, CH2O), 3.84 (dd, J = 4.2, 10.2 Hz, OCHH’), 4.39–4.52 (m, CH2N), 4.54–4.60 (m, CH), 6.71–6.79 (br d, CH3C(O)NH), 7.23 (d, J = 7.9 Hz, 2 ArH), 7.45 (d, J = 7.9 Hz, 2 ArH), 7.82–7.91 (br m, NHCH2); 13C NMR (CD3CN) δ 23.1 (CH3CO), 42.5, 43.2 (2 CH2N), 54.7 (CH), 71.2, 74.2 (2 CH2O), 78.7 (C≡C), 84.2 (C≡C), 121.5, 128.3, 133.0, 141.7 (ArC), 171.2, 171.4 (2 C(O)).
Anhydrous CH2Cl2 (5 mL) was added to the free amine (R)-26 and DPT (232 mg, 1.0 mmol) was added. The yellow solution was stirred at room temperature (18 h). The solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography (EtOAc to 90/10 EtOAc/MeOH) to obtain (R)-10 as a white solid (80 mg, 23%); Rf = 0.45 (EtOAc); mp 124–126 °C; [α]25D +8.0° (c 0.5, DMSO); IR (nujol mull) 3266, 2571, 2201, 2103, 1631, 1538, 1458, 1375, 1304, 1120, 820, 725 cm−1; 1H NMR (CDCl3) δ 2.08 (s, CH3C(O)), 3.07 (s, HC≡C), 3.55–3.77 (m, CH2NCS, OCHH’, CH2CH2O), 4.01 (dd, J = 3.3, 9.3 Hz, OCHH’), 4.43–4.57 (m, CH2N), 4.58–4.64 (m, CH), 6.54 (d, J = 6.3 Hz, CH3C(O)NH), 6.79–6.88 (br t, NHCH2), 7.25 (d, J = 8.6 Hz, 2 ArH), 7.46 (d, J = 8.6 Hz, 2 ArH); 13C NMR (CDCl3) δ 23.3 (CH3CO), 43.3 (CH2N), 45.4 (CH2NCS), 52.7 (CH), 69.3, 70.0 (2 CH2O), 77.4 (C≡C), 83.3 (C≡C), 121.3, 127.5, 132.4, 138.8 (4 ArC), 169.7, 170.6 (2 C(O)), the signal for the isothiocyanate carbon resonance was not detected under the conditions used for the acquisition of the NMR spectrum; Mr (+ESI) 368.1048 [M+Na]+ (calcd for C17H19N3O3SNa+ 368.1045 [M+Na]+). Anal. (C17H19N3O3S•0.2H2O): C, H, N, S.
(S)-N-(4-Ethynyl)benzyl) 2-Acetamido-3-(2-isothiocyanato-ethoxy)propionamide ((S)-10)
Utilizing the preceding procedure, polymer bound (Fluka, cat # 93094) (1.6 mol/g, 3.3 mmol), H2O (205 μL, 11.4 mmol)/THF (10 mL) solution, and (S)-24 (375 mg, 1.1 mmol) gave the free amine (S)-26; 1H NMR (CDCl3) δ 2.01 (s, CH3C(O)), 2.77–2.91 (m, CH2NH2), 3.07 (s, HC≡C), 3.49–3.57 (m, OCHH’, CH2O), 3.80 (dd, J = 4.5, 9.9 Hz, OCHH’), 4.38–4.51 (m, CH2N), 4.56–4.62 (m, CH), 6.88 (d, J = 6.9 Hz, CH3C(O)NH), 7.23 (d, J = 8.7 Hz, 2 ArH), 7.45 (d, J = 8.7 Hz, 2 ArH), 7.88–8.01 (br t, NHCH2); Mr (+ESI) 304.1662 [M+H]+ (calcd for C16H21N3O3H+ 304.1661 [M+H]+).
Utilizing the preceding procedure, the free amine (S)-26 was coupled with DPT (125 mg, 32%) to give (S)-10: Rf = 0.45 (EtOAc); mp 127 °C; [α]25D −9.6° (c 0.5, DMSO); IR (nujol mull) 3277, 2730, 2208, 2107, 1635, 1549, 1458, 1375, 1307, 1130, 819, 726 cm−1; 1H NMR (CDCl3) δ 2.07 (s, CH3C(O)), 3.07 (s, HC≡C), 3.53–3.77 (m, CH2NCS, OCHH’, CH2CH2O), 4.00 (dd, J = 3.3, 9.3 Hz, OCHH’), 4.42–4.56 (m, CH2N), 4.59–4.64 (m, CH), 6.55 (d, J = 6.9 Hz, CH3C(O)NH), 6.81-6.85 (br t, NHCH2), 7.24 (d, J = 8.4 Hz, 2 ArH), 7.46 (d, J = 8.4 Hz, 2 ArH); 13C NMR (CDCl3) δ 23.2 (CH3CO), 43.3 (CH2N), 45.4 (CH2NCS), 52.7 (CH), 69.3, 70.0 (2 CH2O), 77.3 (C≡C), 83.3 (C≡C), 121.3, 127.5, 132.4, 138.7 (4 ArC), 169.7, 170.6 (2 C(O)), the signal for the isothiocyanate carbon resonance was not detected under the conditions used for the acquisition of the NMR spectrum; Mr (+ESI) 368.1046 [M+Na]+ (calcd for C17H19N3O3SNa+ 368.1045 [M+Na]+). Anal. (C17H19N3O3S•0.16CH3OH): C, H, N, S.
Preparative Reaction of (R)-N-(4-Isothiocyanato)benzyl) 2-Acetamido-3-(prop-2-ynyloxy)propionamide ((R)-9) and N-2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethyl Biotin Amide (35) to Give (R)-36
(R)-9 (50 mg, 0.15 mmol) and 35 (81 mg, 0.18 mmol) were dissolved in t-BuOH/H2O (1:2, 3 mL) and then a freshly prepared aqueous 1 M sodium ascorbate solution (15 μL, 15.0 mol) was added, followed by a freshly prepared aqueous 0.1 M CuSO4·5H2O solution (15 μL, 1.5 μmol). The reaction mixture was vigorously stirred at room temperature (15 h), evaporated in vacuo, and then purified by column chromatography (SiO2; 1/9 MeOH/CHCl3) to yield 60 mg (51%) of (R)-36 as a white sticky foam: Rf = 0.30 (1/9 MeOH/CHCl3); IR (nujol) 3284, 2925, 2108, 1657, 1544, 1459 cm−1; 1H NMR (CDCl3) δ 1.37-1.44 (m, C(6)H2), 1.55-1.67 (m, CH2CH2C(6)), 2.02 (s, CH3C(O)), 2.14 (t, J = 7.5 Hz, C(10)CH2), 2.70 (d, J = 12.6 Hz, C(5)HH’), 2.89 (dd, J = 4.8, 12.6 Hz, C(5)HH’), 3.09-3.15 (m, C(2)H), 3.38-3.41 (m, OCH2CH2NHC(O)), 3.52-3.62 (m, 2 OCH2CH2O, triazole-CH2CH2O), 3.67 (dd, J = 6.5, 9.8 Hz, CHCHH’OCH2), 3.86-3.94 (m, OCH2CH2NHC(O), CHCHH’OCH2), 4.25-4.30 (m, C(3)H), 4.40-4.49 (m, CH2Ar, C(4)H), 4.55 (t, J = 5.0 Hz, triazole-CH2CH2O), 4.62-4.68 (m, CHCH2OCH2-triazole, CHCH2OCH2-triazole), 5.57, 6.52 (s, N(1′)H, N(3′)H), 6.84 (t, J = 5.4 Hz, OCH2CH2NHC(O)), 7.10-7.15 (m, 2 ArH), 7.24 (d, J = 8.7 Hz, 2 ArH), 7.40 (d, J = 7.5 Hz, CH3C(O)NHCH), 7.80 (s, CH triazole), 7.83 (t, J = 6.0 Hz, NHCH2Ar); 13C NMR (CD3OD) δ 23.3 (CH3C(O)), 25.7 (C(6)), 28.3 (C(7)), 28.4 (C(8)), 35.9 (C(9)), 39.3 (OCH2CH2NHC(O)), 40.8 (C(5)), 43.0 (CH2Ar), 50.4 (triazole-CH2CH2O), 53.2 (CHCH2OCH2-triazole), 55.8 (C(2)), 60.3 (C(3)), 62.1 (C(4)), 64.6 (CHCH2OCH2-triazole), 69.5 (CHCH2OCH2-triazole), 70.0, 70.1, 70.2, 70.4, 70.5, 70.6 (3 CH2OCH2 in PEG linker), 124.3 (triazole CH), 126.0, 128.7, 130.1, 138.1 (C6H4), 135.4 (NCS), 144.3 (triazole C), 164.1, 170.6, 171.2, 173.6 (4 C(O)); HRMS (ESI) 776.3215 [M + H+] (calcd. for C34H50N9O8S2 776.3224).
(R)- N-Benzyl 2-Acetamido-3-(2-(4-(methoxymethyl)-1H-1,2,3-triazol-1-yl)ethoxy)propionamide ((R)-38)
Compound (R)-30 (400 mg, 1.3 mmol) was dissolved in a THF:H2O mixture (1:1, 50 mL) and while stirring, propargyl methyl ether (1 mL, 11.8 mmol), sodium ascorbate (25 mg, 0.13 mmol), and CuSO4 (3 mg, 0.01 mmol), were successively added. The reaction was stirred at room temperature (24 h), and saturated aqueous NaHCO3 (100 mL) was added. The aqueous layer was extracted with CH2Cl2 (2 × 100 mL), the combined organic layers were washed with brine (100 mL) and filtered through a Celite® bed. Removal of solvents in vacuo gave an off-white solid that was purified by flash chromatography (10/90 MeOH/CH2Cl2) to yield (R)-38 (470 mg, 96%) as a crystalline solid: mp 127-129 °C; [α]25D +1.8° (c 0.34; MeOH); Rf = 0.49 (10/90 MeOH/CH2Cl2); IR (nujol mull) 3296, 2861, 1641, 1545, 1457, 1377, 1143, 1095 cm−1; 1H NMR (CDCl3) δ 2.00 (s, CH3C(O)), 3.38 (s, CH2OCH3), 3.50 (dd, J = 6.5, 9.6 Hz, CHH’OCH2CH2), 3.76–3.85 (m, OCH2CH2N(N)CH), 3.92 (dd, J = 4.2, 9.6 Hz, CHH’OCH2CH2), 4.34–4.51 (m, NHCH2Ph, OCH2CH2N(N)CH, CH2OCH3), 6.63 (d, J = 6.6 Hz, NHCHC(O)), 7.00–7.10 (br t, NHCH2Ph), 7.18–7.35 (m, C6H5), 7.52 (s, NCHC(N)), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-38 gave only one signal for the acetyl protons; 13C NMR (CDCl3) 23.2 (CH3C(O)), 43.7 (NHCH2Ph), 50.0 (CH2N(N)CH), 52.9 (CHC(O)NH), 58.6 (CH2OCH3), 66.0 (OCH2CH2N), 69.4, 70.5 (CHCH2OCH2, CH2OCH3), 123.8 (NCHC(N)), 127.6, 127.7, 128.8, 138.3 (C6H5), 145.3 (NCHC(N)), 169.7, 170.8 (CH3C(O), CHC(O)NH); Mr (+ESI) 398.1806 [M+Na]+ (calcd for C18H25N5O4Na+ 398.1804 [M+Na]+). Anal. (C18H25N5O4) C, H, N.
Preparation of Mouse Soluble Brain Proteosomes
Mouse brains (male, 2 months, ICR mice [Rockland Immunochemicals, Gilbertsville, PA]) were Dounce-homogenized in 50 mM HEPES buffer (pH 7.4). The lysate was centrifuged at slow speed (1200 × g for 12 min at 4 °C) to remove debris. The supernatant was then centrifuged at high speed (100,000 × g for 1 h at 4 °C). The resulting supernatant was collected and stored at −80 °C until use. The total protein concentration was determined by using the Bradford assay.
Three NCS Probe Labeling, Cycloaddition Reaction, and In-Gel Fluorescence Scanning
Mouse brain lysate (1 mL, 50 mM HEPES buffer (pH 7.4)) was passed through a NAP-10 column (GE Healthcare) to exchange buffer to an aqueous 50 mM HEPES buffer (pH 8.0). Lysate aliquots (50 μL of 2.0 mg/mL protein in 50 mM HEPES buffer (pH 8.0)) were treated with lacosamide AB (NCS) & CR (alkyne, N3) compounds (5 μM) at room temperature (30 min). The modified lysates were sequentially treated with rhodamine reporter tag (50 μM) (either rhodamine-azide (Rho-N3, 39) or rhodamine-alkyne (Rho-PEG-≡, 40)), TCEP (1 mM), TBTA (200 μM) and CuSO4 (1 mM). Samples were shaken and allowed to rotate using Roto-shake (8 rpm, Scientific Industries Inc., Model No. SI-1100, Bohemia, NY) at room temperature (1 h). Proteins were separated by SDS-PAGE after addition of 4× SDS-PAGE loading buffer and visualized by in-gel fluorescence using a typhoon 9400 scanner (GE Healthcare/Amersham Bioscience) with excitation at 555 nm and detection at 580 nm.
Probe Labeling of GST-CRMP2 in Mouse Lysate, Cycloaddition, and In-Gel Fluorescence Scanning
Utilizing the previous method, lysate aliquots (150 μL, pH 8.0) were prepared after passage through a NAP-10 column. Overexpressed GST-CRMP2 (30 μg) was added to the lysate (150 μL) and divided into three equal aliquots. The aliquots (50 μL) were treated with (R)- and (S)-9 (5 μM) at room temperature (30 min). Using the previous method, the modified lysates were then treated with 39 under Cu(I)-mediated cycloaddition conditions, followed by SDS-PAGE separation, and in-gel fluorescence.
Pharmacology
Compounds were screened under the auspices of the National Institutes of Health’s Anticonvulsant Screening Program. Experiments were performed in male rodents [albino Carworth Farms No. 1 mice (intraperitoneal route, ip), albino Spague-Dawley rats (oral route, po)]. Housing, handling, and feeding were in accordance with recommendations contained in the ‘Guide for the Care and Use of laboratory Animals’. Anticonvulsant activity was established using the MES test59,60 and the scMet test,59 using previously reported methods.43a
Supplementary Material
Acknowledgments
The authors thank the NINDS and the Anticonvulsant Screening Program (ASP) at the National Institutes of Health, for kindly performing the pharmacological studies via the ASP’s contract site at the University of Utah with Drs. H. Wolfe, H.S. White, and K. Wilcox. Funds for this study was generously provided by grants R01NS054112 (H.K., R.L.) and F31NS060358 (P.M.) from the National Institute of Neurological Disorders and Stroke, and by the Korean Research Foundation Grant funded by the Korean Government (MOEHRD) grant KRF-2006-352-C00042 (KDP). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke or the National Institutes of Health. Harold Kohn has a royalty-stake position in (R)-2.
Footnotes
- FAA
- functionalized amino acids
- AB
- affinity bait
- CR
- chemical reporter
- CRMP2
- collapsin response mediator protein 2
- NMDA
- N-methyl D-aspartate
- GABA
- gamma-aminobutyric acid
- AMPA
- α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate
- SAR
- structure-activity relationship
- MES
- maximal electroshock
- IBCF
- isobutyl chloroformate
- NMM
- N-methyl morpholine
- DMTMM
- 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride
- DPT
- di(2-pyridyl) thionocarbonate
- DITC
- 1,1′-thiocarbonyldiimidazole
- scMet
- subcutaneous metrazol
- TCEP
- tris(2-carboxyethyl)phosphine
- iTRAQ
- isobaric tag for relative and absolute quantitation
- GST
- glutathione-S-transferase
- CNS
- central nervous system
Supporting Information Available: Synthetic procedures for the preparation of (R)- and (S)-13, 15, 16, 27–30, 42, 44–46, 52-55, and 21, 39, 40, 49, 50, 56–59, 61, 63–65, elemental analyses, 1H and 13C NMR spectra of compounds reported in this study, and MS table. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Hauser WA, Annegers JF, Kurland LT. The prevalence of epilepsy in Rochester, Minnesota, 1940-80. Epilepsia. 1991;32:429–445. doi: 10.1111/j.1528-1157.1991.tb04675.x. [DOI] [PubMed] [Google Scholar]
- 2.Evans JH. Post-traumatic epilepsy. Neurology. 1962;12:665–674. doi: 10.1212/wnl.12.10.665. [DOI] [PubMed] [Google Scholar]
- 3.Lindsay JM. Genetics and epilepsy. Epilepsia. 1971;12:47–54. doi: 10.1111/j.1528-1157.1971.tb03914.x. [DOI] [PubMed] [Google Scholar]
- 4.(a) Rogawski MA, Porter RJ. Antiepileptic drugs: Pharmacological mechanisms and clinical efficacy with consideration of promising development stage compounds. Pharmacol. Reviews. 1997;42:223–286. [PubMed] [Google Scholar]; (b) McNamara JO. Ch. 21. In: Hardman JG, Limbird LE, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 10th Ed McGraw-Hill; New York: 2001. pp. 521–547. [Google Scholar]; (c) Aiken SP, Brown WM. Treatment of epilepsy: Existing therapies and future developments. Frontiers in Bioscience. 2000;5:124–152. doi: 10.2741/aiken. [DOI] [PubMed] [Google Scholar]
- 5.Brodie MJ, Dichter MA. Antiepileptic drugs. New England J. Med. 1996;334:168–175. doi: 10.1056/NEJM199601183340308. [DOI] [PubMed] [Google Scholar]
- 6.Dichter MA, Brodie MJ. New antiepileptic drugs. New England J. Med. 1996;334:1583–1590. doi: 10.1056/NEJM199606133342407. [DOI] [PubMed] [Google Scholar]
- 7.(a) McCorry D, Chadwick D, Marson A. Current drug treatment of epilepsy in adults. Lancet Neurol. 2004;3:729–735. doi: 10.1016/S1474-4422(04)00935-4. [DOI] [PubMed] [Google Scholar]; (b) Duncan JS. The promise of new antiepileptic drugs. Brit. J. Clin. Pharmacol. 2002;53:123–131. doi: 10.1046/j.0306-5251.2001.01540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bauer J, Reuber M. Medical treatment of epilepsy. Expert Opinion on Emerging Drugs. 2003;8:457–467. doi: 10.1517/14728214.8.2.457. [DOI] [PubMed] [Google Scholar]; (d) Mattson RH, Cramer JA, Collins JF, Smith DB. Comparison of carbamazepine, phenobarbital, phenytoin, and primidone in partial and secondarily generalized tonic-clonic seizures. New Eng. J. Med. 1985;313:145–151. doi: 10.1056/NEJM198507183130303. [DOI] [PubMed] [Google Scholar]
- 8.Pellock JM, Willmore LJ. A rational guide to monitoring in patients receiving anticonvulsants. Neurology. 1991;41:961–964. doi: 10.1212/wnl.41.7.961. [DOI] [PubMed] [Google Scholar]
- 9.Cortes S, Liao Z-K, Watson D, Kohn H. Effect of structural modification of the hydantoin ring on anticonvulsant activity. J. Med. Chem. 1985;28:601–606. doi: 10.1021/jm50001a012. [DOI] [PubMed] [Google Scholar]
- 10.Conley JD, Kohn H. Functionalized D, L-amino acid derivatives. Potent new agents for the treatment to epilepsy. J. Med. Chem. 1987;30:567–574. doi: 10.1021/jm00386a021. [DOI] [PubMed] [Google Scholar]
- 11.Kohn H, Conley JD. New antiepileptic agents. Chem. Br. 1988;24:231–234. [Google Scholar]
- 12.Kohn H, Conley JD, Leander JD. Marked stereospecificity in a new class of anticonvulsants. Brain Res. 1988;457:371–375. doi: 10.1016/0006-8993(88)90709-3. [DOI] [PubMed] [Google Scholar]
- 13.Kohn H, Sawhney KN, LeGall P, Conley JD, Robertson DW, Leander JD. Preparation and anticonvulsant activity of a series of functionalized α-aromatic and α-heteroaromatic amino acids. J. Med. Chem. 1990;33:919–926. doi: 10.1021/jm00165a006. [DOI] [PubMed] [Google Scholar]
- 14.Kohn H, Sawhney KN, LeGall P, Robertson DW, Leander JD. Preparation and anticonvulsant activity of a series of functionalized α-heteroatom-substituted amino acids. J. Med. Chem. 1991;34:2444–2452. doi: 10.1021/jm00112a020. [DOI] [PubMed] [Google Scholar]
- 15.Kohn H, Sawhney KN, Bardel P, Robertson DW, Leander JD. Synthesis and anticonvulsant activities of α-heterocyclic α-acetamido-N-benzylacetamide derivatives. J. Med. Chem. 1993;36:3350–3360. doi: 10.1021/jm00074a016. [DOI] [PubMed] [Google Scholar]
- 16.Bardel P, Bolanos A, Kohn H. Synthesis and anticonvulsant activities of α-acetamido-N-benzylacetamide derivatives containing an electron-deficient α-heteroaromatic substituent. J. Med. Chem. 1994;37:4567–4571. doi: 10.1021/jm00052a017. [DOI] [PubMed] [Google Scholar]
- 17.Kohn H, Sawhney KN, Robertson DW, Leander JD. Anticonvulsant properties of N-substituted α,α-diamino acid derivatives. J. Pharm. Sci. 1994;83:689–691. doi: 10.1002/jps.2600830519. [DOI] [PubMed] [Google Scholar]
- 18.Choi D, Stables JP, Kohn H. Synthesis and anticonvulsant activities of N-benzyl-2-acetamidopropionamide derivatives. J. Med. Chem. 1996;39:1907–1916. doi: 10.1021/jm9508705. [DOI] [PubMed] [Google Scholar]
- 19.Perucca E, Yasothan U, Clincke G, Kirkpatrick P. Lacosamide. Nature Rev. Drug Disc. 2008;7:973–974. doi: 10.1038/nrd2764. [DOI] [PubMed] [Google Scholar]
- 20.Stoehr T, Kupferberg HJ, Stables JP, Choi D, Harris RH, Kohn H, Walton N, White HS. Lacosamide, a novel anticonvulsant drug, shows efficacy with a wide safety margin in rodent models for epilepsy. Epil. Res. 2007;74:147–154. doi: 10.1016/j.eplepsyres.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 21.Beyreuther B, Freitag J, Heers C, Krebsfaenger N, Scharfenecker U, Stoehr T. Lacosamide: a review of preclinical properties. CNS Drug Rev. 2007;13:21–42. doi: 10.1111/j.1527-3458.2007.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Errington AC, Stohr T, Heers C, Lees G. The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol. Pharmacol. 2008;73:157–169. doi: 10.1124/mol.107.039867. [DOI] [PubMed] [Google Scholar]; (b) Sheets PL, Heers C, Stoehr T, Cummins TR. Differential block of sensory neuronal voltage-gated sodium channels by lacosamide, lidocaine and carbamazepine. J. Pharmacol. Exp. Ther. 2008;326:89–99. doi: 10.1124/jpet.107.133413. [DOI] [PubMed] [Google Scholar]
- 23.(a) Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Dis. 2004;3:353–359. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]; (b) Cavalli A, Bolognesi ML, Minarini A, Rosini M, Tumiatti V, Recanatini M, Melchiorre C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008;51:347–372. doi: 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- 24.(a) Brunton LL, Lazo JS, Parker KL. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 11th Ed McGraw-Hill; New York: 2006. [Google Scholar]; (b) Errington AC, Stoehr T, Lees G. Voltage-gated ion channels: targets for anticonvulsant drugs. Curr. Top. Med. Chem. 2005;5:15–30. doi: 10.2174/1568026053386872. [DOI] [PubMed] [Google Scholar]
- 25.(a) Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]; (b) Speers AE, Cravatt BF. Chemical strategies for activity-based proteomics. ChemBioChem. 2004;5:41–47. doi: 10.1002/cbic.200300721. [DOI] [PubMed] [Google Scholar]; (c) Ovaa H, van Swieten PF, Kessler BM, Leeuwenburgh MA, Fiebiger E, van den Nieuwendijk AMCH, Galardy PJ, van der Marel GA, Ploegh HL, Overkleeft HS. Chemistry in living cells: Detection of active proteasomes by a two-step labeling strategy. Angew. Chem. Int. Ed. 2003;42:3626–3629. doi: 10.1002/anie.200351314. [DOI] [PubMed] [Google Scholar]; (d) Kho Y, Kim SC, Jiang C, Barma D, Kwon SW, Cheng J, Jaunbergs J, Weinbaum C, Tamanoi F, Falck J, Zhao Y. A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12479–12484. doi: 10.1073/pnas.0403413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Evans MJ, Saghatelian A, Sorensen EJ, Cravatt BF. Target discovery in small-molecule cell-based screens by in situ proteome reactivity profiling. Nat. Biotech. 2005;23:1303–1307. doi: 10.1038/nbt1149. [DOI] [PubMed] [Google Scholar]; (b) Hosoya T, Hiramatsu T, Ikemoto T, Nakanishi M, Aoyama H, Hosoya A, Iwata T, Maruyama K, Endo M, Suzuki M. Novel bifunctional probe for radioisotope-free photoaffinity labeling: compact structure comprised of photospecific ligand ligation and detectable tag anchoring units. Org. Biomol. Chem. 2004;2:637–641. doi: 10.1039/b316221d. [DOI] [PubMed] [Google Scholar]; (c) Ballell L, van Scherpenzeel M, Buchalova K, Liskamp RMJ, Pieters RJ. A new chemical probe for the detection of the cancer-linked galectin-3. Org. Biomol. Chem. 2006;4:4387–4394. doi: 10.1039/b611050a. [DOI] [PubMed] [Google Scholar]; (d) Gubbens J, Ruijter E, De Fays LEV, Damen JMA, de Kruijff B, Slijper M, Rijkers DTS, Liskamp RMJ, de Kroon AIPM. Photocrosslinking and click chemistry enable the specific detection of proteins interacting with phospholipids at the membrane interface. Chem. Biol. 2009;16:3–14. doi: 10.1016/j.chembiol.2008.11.009. [DOI] [PubMed] [Google Scholar]; (e) Staub I, Sieber SA. β-Lactam probes as selective chemical-proteomic tools for the identification and functional characterization of resistance associated enzymes in MRSA. J. Am. Chem. Soc. 2009;131:6271–6276. doi: 10.1021/ja901304n. [DOI] [PubMed] [Google Scholar]
- 27.(a) Takemori AE, Portoghese PS. Affinity labels for opioid receptors. Ann. Rev. Pharmacol. Toxicol. 1985;25:193–223. doi: 10.1146/annurev.pa.25.040185.001205. [DOI] [PubMed] [Google Scholar]; (b) Robertson JG. Mechanistic basis of enzyme-targeted drugs. Biochemistry. 2005;44:5561–5571. doi: 10.1021/bi050247e. [DOI] [PubMed] [Google Scholar]; (c) Drahl C, Cravatt BF, Sorensen EJ. Protein-reactive natural products. Angew. Chem. Intl. Ed. 2005;44:5788–5809. doi: 10.1002/anie.200500900. [DOI] [PubMed] [Google Scholar]
- 28.Walle T, Walle UK. Pharmacokinetic parameters obtained with racemates. TIPS. 1986:155–158. [Google Scholar]
- 29.Ariëns EJ. Stereochemistry: A source of problems in medicinal chemistry. Medicinal Res. Rev. 1986;6:451–466. doi: 10.1002/med.2610060404. [DOI] [PubMed] [Google Scholar]
- 30.Levy RH, Mattson R, Meldrum B. Antiepileptic Drugs. 4th Ed Raven Press; New York: 1995. pp. 99–110. [Google Scholar]
- 31.(a) de Costa BR, Rothman RB, Bykov V, Jacobson AE, Rice KC. Selective and enantiospecific acylation of κ opioid receptors by (1S,2S)-trans-2-isothiocyanato-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]- benzeneacetamide. Demonstration of κ receptor heterogeneity. J. Med. Chem. 1989;32:281–283. doi: 10.1021/jm00122a001. [DOI] [PubMed] [Google Scholar]; (b) Burke TR, Jr., Jacobson AE, Rice KC, Silverton JV, Simonds WF, Streaty RA, Klee WA. Probes for narcotic receptor mediated phenomena. 12. cis-(+)-3-Methylfentanyl isothiocyanate, a potent site-directed acylating agent for δ opioid receptors. Synthesis, absolute configuration, and receptor enantioselectivity. J. Med. Chem. 1986;29:1087–1093. doi: 10.1021/jm00156a030. [DOI] [PubMed] [Google Scholar]; (c) de Costa BD, Rothman RB, Bykov V, Band L, Pert A, Jacobson AE, Rice KC. Probes for narcotic receptor mediated phenomena. 17.1 Synthesis and evulation of a series of trans-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzeneacetamide (U50,488 related isothiocyanate derivatives as opioid receptor affinity ligands. J. Med. Chem. 1990;33:1171–1176. doi: 10.1021/jm00166a014. [DOI] [PubMed] [Google Scholar]; (d) de Costa BR, Rothman RB, Bykov V, Jacobson AE, Rice KC. Selective and enantiospecific acylation of κ opioid receptors by (1S,2S)-trans-2-isothiocyanato-N-methyl-N-[2,(1-pyrrolidinyl)cyclohexyl]benzeneacetamide. Demonstration of κ receptor heterogeneity. J. Med. Chem. 1989;32:281–283. doi: 10.1021/jm00122a001. [DOI] [PubMed] [Google Scholar]; (e) Burke TR, Jr., Bajwa BS, Jacobson AE, Rice KC, Streaty RA, Klee WA. Probes for narcotic receptor mediated phenomena. 7.1 Synthesis and pharmacological properties of irreversible ligands specific for μ or δ opiate receptors. J. Med. Chem. 1984;27:1570–1574. doi: 10.1021/jm00378a008. [DOI] [PubMed] [Google Scholar]
- 32.de Costa BR, Lewin AH, Rice KC, Skolnick P, Schoenheimer JA. Novel site-directed affinity ligands for GABA-gated chloride channels: Synthesis, characterization, and molecular modeling of 1-(Isothiocyanatophenyl)-4-tert-butyl-2,6,7-trioxabicyclo[2.2.2]octanes. J. Med. Chem. 1991;34:1531–1538. doi: 10.1021/jm00109a002. [DOI] [PubMed] [Google Scholar]
- 33.Williams EF, Rice KC, Paul SM, Skolnick P. Heterogeneity of benzodiazepine receptors in the central nervous system demonstrated with kenazepine, an alkylating benzodiazepine. J. Neurochem. 1980;35:591–597. doi: 10.1111/j.1471-4159.1980.tb03695.x. [DOI] [PubMed] [Google Scholar]
- 34.(a) Rice KC, Jacobson AE, Burke TR, Jr., Bajwa BS. Irreversible ligands with high selectivity toward μ or δ opiate receptors. Science. 1983;220:314–316. doi: 10.1126/science.6132444. [DOI] [PubMed] [Google Scholar]; (b) Lessor RA, Rice KC, Streaty RA, Klee WA, Jacobson AE. Probes for narcotic receptor mediated phenomena. 10. Irreversibly ligands to opioid receptors based on biologically potent endoethenooripavines. Reversible binding of fit to mu and delta opioid receptors. Neuropeptides. 1984;5:229–232. doi: 10.1016/0143-4179(84)90069-6. [DOI] [PubMed] [Google Scholar]
- 35.(a) Burke TR, Jr., Bajwa BS, Jacobson AE, Rice KC, Streaty RA, Klee WA. Probes for narcotic receptor mediated phenomena. 7. Synthesis and pharmacological properties of irreversible ligands specific for μ or δ opiate receptors. J. Med. Chem. 1984;27:1570–1574. doi: 10.1021/jm00378a008. [DOI] [PubMed] [Google Scholar]; (b) Burke TR, Jr., Jacobson AE, Rice KC, Silverton JV, Simonds WF, Streaty RA, Klee WA. Probes for narcotic receptor mediated phenomena. 12.1 cis-(+)-3-Methylfentanyl isothiocyanate, a potent site-directed acylating agent for δ opioid receptors. Synthesis, absolute configuration, and receptor enantioselectivity. J. Med. Chem. 1986;29:1087–1093. doi: 10.1021/jm00156a030. [DOI] [PubMed] [Google Scholar]
- 36.Rice KC, Brossi A, Tallman J, Paul SM, Skolnick P. Irazepine, and noncompetitive, irreversible inhibitor of 3H-diazepam binding to benzodiazepine receptors. Nature (London) 1979;278:854–855. doi: 10.1038/278854a0. [DOI] [PubMed] [Google Scholar]
- 37.Perret P, Sarda X, Wolff M, Wu T-T, Bushey Goeldner, M. Interaction of non-competitive blockers within the γ-aminobutyric acid type A chloride channel using chemical reactive probes as chemical sensors for cysteine mutants. J. Biol. Chem. 1999;274:25350–25354. doi: 10.1074/jbc.274.36.25350. [DOI] [PubMed] [Google Scholar]
- 38.Shelton RL, Langdon RG. Location of the maltose isothiocyanate binding site on the human erythrocyte glucose transporter. Biochemistry. 1985;24:2397–2400. doi: 10.1021/bi00331a001. [DOI] [PubMed] [Google Scholar]
- 39.Morse KL, Fournier DJ, Li X, Grzybowska J, Makriyannis A. A novel electrophilic high affinity irreversible probe for the cannabinoid receptor. Life Sci. 1995;56:1957–1962. doi: 10.1016/0024-3205(95)00176-7. [DOI] [PubMed] [Google Scholar]
- 40.Daphne A, Plotek Y, Miskin I. An affinity label for α2-adrenergic receptors in rat brain. Eur. J. Biochem. 1982;126:537–541. doi: 10.1111/j.1432-1033.1982.tb06814.x. [DOI] [PubMed] [Google Scholar]
- 41.Soskic V, Maelicke A, Petrovic G, Ristic B, Petrovic J. Synthesis of some phenothiazine derivatives as potential affinity ligands for the central dopamine receptors. J. Pharm. Pharmacol. 1991;43:27–31. doi: 10.1111/j.2042-7158.1991.tb05442.x. [DOI] [PubMed] [Google Scholar]
- 42.Casalotti SO, Kozikowski AP, Fauq A, Tuckmantel W, Krueger KE. Design of an irreversible affinity ligand for the phencyclidine recognition site on N-methyl-D-aspartate-type glutamate receptors. J. Pharmacol. Exper. Therap. 1992;260:21–28. [PubMed] [Google Scholar]
- 43.(a) LeTiran A, Stables JP, Kohn H. Design and evaluation of affinity labels of functionalized amino acid anticonvulsants. J. Med. Chem. 2002;45:4762–4773. doi: 10.1021/jm020225f. [DOI] [PubMed] [Google Scholar]; (b) Kohn H, Robertson DW, Leander JD. unpublished results.
- 44.Bucciarelli M, Forni A, Moretti I, Prati F, Torre G. Regioselectivity of ring-opening reactions of optically active N-acety-2-methoxycarbonylaziridine. Tetrahedron Asym. 1995;6:2073–2080. [Google Scholar]
- 45.(a) Larsson U, Carlson R. Synthesis of amino acids with modified principal properties 2: amino acids with polar side chains. Acta Chem. Scand. 1994;48:511–516. [Google Scholar]; (b) Belohradsky M, Ridvan L, Zavada J. Synthesis of homochiral acyclic mono- and bis(alpha-amino acids) with oligo(oxyethylene) chains. Collect. Czech. Chem. Commun. 2003;68:1319–1328. [Google Scholar]
- 46.(a) Anderson GW, Zimmerman JE, Callahan FM. A reinvestigation of the mixed carbonic anhydride method of peptide synthesis. J. Am. Chem. Soc. 1967;87:5012–5017. doi: 10.1021/ja00995a032. [DOI] [PubMed] [Google Scholar]; (b) Kunishima M, Kawachi C, Monta J, Terao K, Iwasaki F, Tani S. 2-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride: an efficient condensing agent leading to the formation of amides and esters. Tetrahedron. 1999;55:13159–13170. [Google Scholar]
- 47.(a) Kim S, Yi KY. Di-2-pyridyl thioncarbonate. A new reagent for the preparation of isothiocyantes and carbodiimides. Tetrahedron Lett. 1985;26:1661–1664. [Google Scholar]; (b) Potashman MH, Bready J, Coxon A, DeMelfi TM, DiPietro L, Doerr N, Elbaum D, Estrada J, Gallant P, Germain J, Gu Y, Harmage J, Kaufman SA, Kendall R, Kim JL, Kumar GN, Long AM, Neervannan S, Patel VF, Polverino A, Rose P, van der Plas S, Whittington D, Zanon R, Zhao H. Design, synthesis, and evaluation of orally active benzimidazoles and benxoxazoles as vascular endothelial growth factor-2 receptor tyrosine kinase inhibitors. J. Med. Chem. 2007;50:4351–4373. doi: 10.1021/jm070034i. [DOI] [PubMed] [Google Scholar]
- 48.For comparable procedures for resolving stereoisomers, see the following: Weisman GR. In: Asymmetric Synthesis-Analytical Methods. Morrison JD, editor. Vol. 1. Academic Press; New York: 1983. pp. 153–171.Parker D, Taylor RJ. Direct 1H NMR assay of the enantiomeric composition of amines and β-amino alcohols using O-acetyl mandelic acid as a chiral solvating agent. Tetrahedron. 1987;43:5431–5456.
- 49.Barton ME, Klein BD, Wolff HH, White HS. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epil. Res. 2001;47:217–227. doi: 10.1016/s0920-1211(01)00302-3. [DOI] [PubMed] [Google Scholar]
- 50.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 51.Tornoe CW, Christensen C, Meldal M. Peptidotriazoles on solid phases [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 52.(a) Park KD, Liu R, Kohn H. Useful tool for biomolecule isolation, detection, and identification: acylhydrazone-based cleavable linker. Chem. Biol. 2009;16:763–772. doi: 10.1016/j.chembiol.2009.06.005. [DOI] [PubMed] [Google Scholar]; (b) Sun X-L, Stabler CL, Cazalis CS, Chaikof EL. Carbohydrate and protein immobilization onto solid surfaces by sequential Diels-Alder and azide-alkyne cycloadditions. Bioconj. Chem. 2006;17:52–57. doi: 10.1021/bc0502311. [DOI] [PubMed] [Google Scholar]
- 53.Speers AE, Cravatt BF. Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Z, Ottens AK, Sadasivan S, Kobeissy FH, Fang T, Hayes RL, Wang KKW. Calpain-mediated collapsing response mediator protein-1, -2, and -4 proteolysis after neurotoxic and traumatic brain injury. J. Neurotrauma. 2007;24:460–472. doi: 10.1089/neu.2006.0078. [DOI] [PubMed] [Google Scholar]
- 55.Ross PL, Huang UN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 56.Cotten S, Liu R. unpublished data.
- 57.Morieux P, Stables JP, Kohn H. Synthesis and anticonvulsant activities of N-benzyl (2R)-2-acetamido-3-oxysubstituted propionamide derivatives. Bioorg. Med. Chem. 2008;16:8968–8975. doi: 10.1016/j.bmc.2008.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Salome C, Salome E, Park KD, Morieux P, Swendiman R, DeMarco E, Stables JP, Kohn H. unpublished data.
- 59.Stables JP, Kupferberg HJ. In: Molecular and Cellular Targets for Antiepileptic Drugs. Avanzini G, Tanganelli P, Avoli M, editors. John Libbey; London: 1977. pp. 191–198. [Google Scholar]
- 60.Krall RL, Penry JK, Kupferberg HJ, Swinyard EA. Antiepileptic drug development: I. History and a program for progress. Epilepsia. 1978;19:393–408. doi: 10.1111/j.1528-1157.1978.tb04506.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.