

Abstract
Fetal alcohol exposure is known to induce cell death through apoptosis. We found that colivelin (CLN), a novel peptide with the sequence SALLRSIPAPAGASRLLLLTGEIDLP, prevents this apoptosis. Our initial experiment revealed that CLN enhanced the viability of primary cortical neurons exposed to alcohol. We then used a mouse model of fetal alcohol exposure to identify the intracellular mechanisms underlying these neuroprotective effects. On embryonic day 7 (E7), weight-matched pregnant females were assigned to the following groups: (1) ethanol liquid diet (ALC) 25% (4.49%, v/v) ethanol derived calories; (2) pair-fed control; (3) normal chow; (4) ALC combined with administration (i.p.) of CLN (20 μg/20 g body weight); and (5) pair-fed combined with administration (i.p.) of CLN (20 μg/20 g body weight). On E13, fetal brains were collected and assayed for TUNEL staining, caspase-3 colorimetric assay, ELISA, and MSD electrochemiluminescence. CLN blocked the alcohol-induced decline in brain weight and prevented alcohol-induced: apoptosis, activation of caspase-3 and increases of cytosolic cytochrome c, and decreases of mitochondrial cytochrome c. Analysis of proteins in the upstream signaling pathway revealed that CLN down-regulated the phosphorylation of the c-Jun N-terminal kinase. Moreover, CLN prevented alcohol-induced reduction in phosphorylation of BAD protein. Thus, CLN appears to act directly on upstream signaling proteins to prevent alcohol-induced apoptosis. Further assessment of these proteins and their signaling mechanisms is likely to enhance development of neuroprotective therapies.
Keywords: colivelin, ADNF-9, neuroprotection, fetal alcohol exposure, cytochrome c, mitochondria, apoptosis, caspase-3, TUNEL
INTRODUCTION
Prenatal alcohol exposure impedes cerebral and cerebellar growth (Bauer-Moffett and Altman, 1975, 1977, Kornguth et al., 1979, Samson and Diaz, 1981, Sulik et al., 1981, Barron et al., 1988, Bonthius and West, 1990), and induces brain growth deficits (Sari and Gozes, 2006) through an apoptotic mechanism (Ikonomidou et al., 2000). Little is known, however, about the underlying signaling pathways of the apoptotic mechanism. Alcohol exposure may accelerate apoptosis in the developing brain through the direct activation of intrinsic mitochondrial apoptotic signaling pathways or by the indirect activation of extrinsic pathways such as receptor-mediated systems (Cheema et al., 2000, de la Monte et al., 2000). The extrinsic signaling pathways may involve c-Jun N-terminal kinase (JNK) and extracellular signal regulated kinase (ERK1/2) (Ashkenazi and Dixit, 1998). The intrinsic signaling pathways involve the Bax protein, which appears to be translocated from the cytosol to the outer mitochondrial membrane. Translocation increases mitochondrial membrane permeability and the subsequent release of cytochrome c, which binds to apoptotic protease activating factor-1 (Apaf1) and activates caspase-9, which then cleaves caspase-3 (Green and Reed, 1998). Prenatal alcohol exposure also induces mitochondrial dysfunction, including decreased mitochondrial glutathione concentration, decreased activities of respiratory chain complex IV and ATP synthase, and increased mitochondrial permeability transition (Ramachandran et al., 2001, Spong et al., 2001, Xu et al., 2005, Green et al., 2006). The outer mitochondrial membrane proteins Bcl-2, Bcl-xl, Bax, Bcl-xs, and Bad have been implicated in ethanol-induced apoptosis in the neonatal brain (Moore et al., 1999, Cory and Adams, 2002, Heaton et al., 2003, Young et al., 2003, Ge et al., 2004). These proteins may regulate apoptosis during fetal brain development. Studies investigating the action of prenatal alcohol exposure in the signaling pathways that involve extrinsic and intrinsic mitochondrial factors may provide important information for the identification of mechanisms for neuroprotection and allow for the development of intervention procedures.
Possible prevention strategies may focus on the treatment of pregnant mice with peptides shown to be neuroprotective in vitro (Brenneman et al., 1998, Bassan et al., 1999) and in vivo (Spong et al., 2001, Poggi et al., 2003, Sari and Gozes, 2006, Sari, 2009). These peptides include: SALLRSIPA (SAL or ADNF-9), derived from activity-dependent neurotrophic factor (ADNF) (Brenneman and Gozes, 1996, Brenneman et al., 1998), and NAPVSIPQ ( NAP), derived from activity-dependent neuroprotective protein (ADNP) (Bassan et al., 1999, Zamostiano et al., 2001). Both peptides protect against oxidative stress associated with alcohol exposure [for review see ref. (Sari and Gozes, 2006)].
A novel synthetic hybrid peptide, colivelin (CLN, SALLRSIPAPAGASRLLLLTGEIDLP, patent # 61/098013), which is composed of ADNF-9 (SALLRSIPA) and a humanin derivative (C8A-HN, MAPRGFSALLLLTSEIDLPVKRRA), has been found to prevent cell death induced by Alzheimer’s disease-causative genes and the β-amyloid peptide (Chiba et al., 2005, Yamada et al., 2008). Unlike ADNF-9, however, CLN is neuroprotective over a wide range of concentrations. Here, we investigated if CLN protects against alcohol-induced apoptosis. In addition, we determined the downstream and upstream signaling pathways that are implicated in neuroprotection against alcohol-induced cell death in the fetal mouse brain exposed to alcohol between embryonic days 7 and 13 (E7 and E13, respectively).
EXPERIMENTAL PROCEDURES
1. In vitro studies
1.1. Peptides, antibodies, and drugs
Colivelin, humanin and ADNF-9 were synthesized as described previously (Hashimoto et al., 2001, Chiba et al., 2005). Rabbit antibodies against phospho-JNK (Thr183/Tyr185) were purchased from Cell Signaling Technology (Beverly, MA). Rabbit polyclonal antibodies against total-JNK (sc-571) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). PD98059, Wortmannin, AG490 and SP600125 were purchased from Calbiochem (San Diego, CA). Other reagents described here were commercially available.
1.2. Cell culture and phosphorylation assays
Primary cortical neurons (PCNs) (5.0 × 104 cells per well in poly-L-lysine-coated 96-well plate) were prepared from E14 mouse embryos as described previously (Chiba et al., 2005). On day 3 in vitro (DIV3), PCNs were similarly pre-treated with the CLN peptide (100 pM) or indicated concentrations of inhibitors (5 μM PD98059 for MEK, 10 nM Wortmannin for PI3-kinase, 1 μM AG490 for JAK2, 10 nM SP600125 for JNK; all inhibitors contain 0.5% DMSO) for 30 minutes and then further cultured for 72 hours with or without alcohol (ALC, 1000 mg/dl). Cultured media containing inhibitors and ethanol were refreshed on DIV6 and DIV7. Cell viability was measured with WST-8 assay. WST-8 assay was performed with 2-(2-methoxy-4-nitrophenyl)-3-(4nitrophenyl)- 5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt, using Cell Counting kit-8 (Wako Pure Chemicals Industries, Tokyo, Japan), as described previously (Chiba et al., 2004).
The xCELLigence system (Roche Applied Science in partnership with ACEA Biosciences Inc.) monitors cellular events in real time without the incorporation of labels. The system measures electrical impedance across interdigitated microelectrodes integrated on the bottom of tissue culture E-Plates (96 well). The impedance measurement provides quantitative information about the biological status of the cells, including cell number, viability, and morphology. Using the xCELLLigence system, we continuously monitored the “Cell Index”, which is derived from the measured impedances and reflects the cell viability under ethanol exposure together with peptide treatment (CLN at 1 pM, 100 pM, or 10 nM; HNA at 10 nM; ADNF-9 at 100 pM, n= 6).
1.3. Immunoblot analysis
PCNs (at 2 × 106 per well in a poly-L-lysine-coated 6-well plate on DIV4) were incubated with or without 1000 mg/dl ALC for the indicated time. Cells were harvested in a lysis buffer (50 mM Tris HCl pH7.4, 150 mM NaCl, 1% Triton-X 100, protease inhibitors, 1 mM EDTA, phosphatase inhibitor cocktails 1 and 2 [Sigma-Aldrich]). Samples (35μg/lane) were subjected to normal SDS-PAGE and were then blotted onto PVDF membranes. After washing the membranes in PBST at 37°C for 10 minutes, they were submerged in stripping buffer (100mM 2-Mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl pH 6.7) and incubated at 50°C for 30 minutes with occasional agitation. The membranes were then blocked with 10% nonfat dried milk in PBST for 1 hour at room temperature prior to immunodetection. The membranes were soaked with the appropriate primary antibodies (phopsho-JNK, at 1:1000) and then with HRP-labeled secondary antibodies (BioRad Laboratories, Hercules, CA). Immunoreactive bands were detected with the Western Blotting Detection Reagents (Amersham Bioscience, Uppsala, Sweden). Total protein levels were detected on the identical membranes using anti-total protein antibodies (t-JNK, at 1:3000) after stripping the anti-phospho-protein antibody. We performed three independent experiments (n=3).
2. In vivo studies
2.1. Animals
Both male and female C57BL/6 mice were obtained at 6–7 weeks of age (20–23g) from Harlan Laboratory at Indianapolis, Indiana, USA. All mice were housed in the departmental animal colony in a vivarium with a controlled climate (temperature 22°C, 30% humidity), on a reverse 12 hours light/dark cycle (off at 700 hour). Pregnant mice had free access to liquid diet or food for 24 hours during the treatment period. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the Indiana University Bloomington and were in accordance with the guidelines of the Institutional Animal Care and Use Committee of the NIH, and the Guide for the Care and Use of Laboratory Animals.
2.2. Breeding and treatment procedure
Female mice were placed into male home cages for 2 hours. The females were then checked for a sperm plug by vaginal smear immediately afterwards. If the plugs were positive, we designated this time point as embryonic day 0 (E0). Weight-matched pregnant females were assigned pseudo randomly (matched for weight) on E7 to the following groups: (1) Ethanol liquid diet group (ALC, n=8) had free access to chocolate sustacal (supplemented with vitamins and minerals) liquid diet 25% (4.49%, v/v) ethanol derived calories (EDC), (2) Pair-fed control group (PF, pair-fed to ethanol-fed group, n=9), fed with a maltose-dextrin solution which was made isocaloric to the dose of ethanol used, (3) Chow group (Chow, n=5) fed with mouse chow pellets and water, (4) treatment group, CLN i.p. injection alongside alcohol exposure in liquid diet (ALC/CLN, 20 μg/20 g, n=6), and (5) Pair-fed alongside i.p. injection of CLN group (PF/CLN, 20 μg/20 g, n=5). The PF, PF/CLN, and Chow served as control groups. The PF group dam yoked individually to an ALC dam and was given daily amounts of matched isocaloric liquid diet, with maltose-dextrin substituted for ethanol. Pregnant mice had continuous, 24-hour access, to the alcohol diet or PF liquid diet for 7 days.
As published by Middaugh and colleagues, the fortified liquid diet contained 237 ml of chocolate-flavored sustacal (Mead Johnson), 1.44 g Vitamin Diet Fortification Mixture, and 1.2 g Salt Mixture XIV. For the ethanol diet, 15.3 ml (4.49% v/v; 25% EDC) of 95% ethanol was added to the fortified sustacal formula with water to make 320 ml of diet with 1 cal/ml (ethanol). For the isocaloric control diet, 20.2 g Maltose Dextrin was added to the fortified sustacal formula with water to bring it to 1 cal/ml (Middaugh et al., 1988, Middaugh and Boggan, 1995). One day before treatment, the ALC, PF, PF/CLN, and ALC/CLN dams were adapted to the liquid diet. The dams were weighed. The volume of liquid diet consumed during the previous 24 hours was recorded from 30-ml graduated screw-cap tubes, and a freshly prepared diet was provided. The ALC group had free-access to an ethanol liquid diet delivering 25% EDCs as the sole source of nutrients; the PF control yoked individually to an ALC dam and were given matched daily amounts of free-access isocaloric liquid diet, with maltose-dextrin substituted for ethanol; or ad libitum chow and water (Chow) at all times during gestation (E7–E13).
2.3. Maternal Blood Alcohol Levels
Maternal blood alcohol levels were tested in a separate group of C57Bl/6 dams with the 25% (4.49%, v/v) ethanol derived calorie (EDC) diet as described previously (Sari, 2009). In brief, pregnant mice were given the same feeding protocol as the other experimental dams (EDC diet provided on E7 at 700 hour), and two 50-μl tail blood samples were obtained (at 900 and 1100 hour) on E8 and E11. The blood samples were collected in heparinized capillary tubes and centrifuged. The 5-μl plasma samples were analyzed for alcohol concentrations using the Analox Alcohol Analyzer, calibrated with a 100 mg/dl ethanol standard. Blood alcohol concentrations (BACs) were evaluated at 2 and 4 hours after onset of the dark cycle on E8 and E11. The BACs were consistently higher at 2 hours. The average peaks obtained in the 25% EDC group at 2 hours were ~40 mg/dl on E8 and ~55 mg/dl on E11. Although the present study focuses on the E13 stage, we have previously reported that E14 plasma BAC was about 142.7 ± 39.5 mg/dl (Sari and Zhou, 2004, Zhou et al., 2004, Zhou et al., 2005). Thus, it is possible that the BAC might be increased at the E13 stage as well.
2.4. Fetal brains
Pregnants mice were euthanized by CO2 followed by cervical dislocation on E13 and the fetuses were removed. This method is consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association. In addition, the number of animals used was minimized similar to our previous studies. The fetal brains were further dissected from the base of the primordium olfactory bulb to the base of the metencephalon. From the same dam, the group of fetal brains was frozen and stored at −70°C until used for chemical assays unlike the other group of fetal brains, which were fixed in 4% paraformaldehyde (PAF) for TUNEL-positive cell detection and for fetal brain weight analyses. Note that only one fetal brain from each dam was randomly assigned chemical assay.
2.5. In vivo optical imaging
A pregnant female CD-1 mouse (E14) administered i.p. with Alexa Fluor 680 (Invitrogen, Carlsbad, CA)-labeled CLN (CLN-Alexa680), was imaged with the Optix MX2 system (ART, Inc., Montreal, Canada). This system enables real time-scanning of fluorophores in vivo. To avoid strong auto-fluorescence from the hair, we shaved the abdominal side before imaging. We used a 679-nm pulse laser for excitation and a 700-nm long-pass filter on the emission side. Regions of interest (ROI) were manually set for each mouse, and each ROI was scanned with the resolution of a 1.5-mm grid. The laser setting (power of the laser and integration time) for each ROI was automatically optimized by the Optix system and collected data were analyzed by OptiView software (Ver.2.0.1, ART, Inc.). The experiment was repeated and the same results were obtained.
2.6. Analysis of the concentrations of active caspase-3
The concentrations of active caspase-3 were determined by using the caspase-3 colorimetric assay kit in frozen forebrain from E13 Chow, PF, PF/CLN, PF/ALC, ALC, and ALC/CLN (Assay Designs, Inc., Ann Arbor, MI). Protein extractions and colorimetric assay were performed as recently reported (Sari, 2009). For tissue extracts, frozen E13 fetal brains of different groups were ground to a powder with a pestle. The powdered tissue was mixed with TNE buffer (10 mM Tris, pH 7.4; 0.15 M NaCl; 1 mM ethylenediaminetetraacetic acid) supplemented with protease inhibitor cocktail (Sigma) under continuous grinding until the suspension was homogeneous. The suspension was centrifuged at 16,100 rcf for 10 minutes at 4°C. The supernatant was collected and frozen at −70°C until a later date for detection of protein and/or active caspase-3 concentration. The total protein estimation in each sample was evaluated with the Bio-Rad (Hercules, CA) protein assay. Extracted proteins in TNE buffer from different sample groups were plated in 96 microplates in variant dilutions, standards, p-nitroaniline calibrator (pNA), and blank controls were plated in duplicate in 96 microplates. The conversion of substrate into the colored product was measured after 3 hours of incubation at 37°C, and the reaction was stopped by a 1 N solution of hydrochloric acid. The multiple samples, standards, pNA, and blank controls were read rapidly by an absorbance reader (SUNRISE, Phoenix Research Products, Phoenix, AZ). The average net optical density (OD) for each standard and sample was calculated by subtracting the average blank OD from the average OD for each standard and sample. The activity measurements were quantitated by comparisons of the optical densities obtained with standards or with the pNA. By using graphing software (Prizm, HalloGram Publishing, Aurora, CO), the concentration of active caspase-3 in the samples was determined by interpolation of the average net OD for each standard versus the actual concentration of active caspase-3 substrate for the standards. The concentrations of active caspase-3 in the samples for all groups were expressed as units per milligram of total protein tissue (Units/mg of total protein). The colorimetric assay was repeated twice for confirmation of the quantitative analyses.
2.7. Analysis of cytosolic and mitochondrial fractions of cytochrome c
The detections of both cytosolic and mitochondrial fractions of cytochrome c were performed as described recently (Sari, 2009). Frozen fetal brains were ground to a powder with a pestle. The powdered tissue was mixed with digitonin (0.05%) in a lysis buffer (250 mM Sucrose, 20 mM HEPES pH 7.4, 10 mM KCl, 5 mM MgCl2, 1 mM EGTA, 1mM EDTA, 1:100 Protease cocktail inhibitor). The homogenates were then centrifuged at 13,400 rfc for 12 minutes at 4°C. The supernatant (cytosolic fraction) was removed and stored at −80°C. The pellet was resuspended in a second lysis buffer for 30 minutes (133 mM NaCl, 50 mM Tris pH 8.0, SDS 0.1% [w/v], sodium deoxycholate 0.5% [w/v], Igepal CA630 1.0% [v/v] and protease cocktail inhibitor 1:100). The mixture was then centrifuged at 13,400 rfc for 15 minutes at 4°C, and the supernatant (mitochondrial fraction) was collected and stored at −80°C until Enzyme-Linked ImmunoSorbent Assay (ELISA) (Chemicon International) testing for cytochrome c. Cytosolic and mitochondrial fractions were assayed with ELISA for the determination of cytochrome c as detailed in the manufacture protocol. In brief, an anti-cytochrome c monoclonal coating antibody was absorbed onto a 96 microtiter plate. Cytochrome c from samples or standards was incubated with the absorbed antibody for 2 hours at room temperature. After washing with the wash buffer, samples and standards were incubated with a biotin-conjugated monoclonal anti-cytochrome c diluted with assay diluent (provided by the manufacturer) for 2 hours at room temperature. Following incubation, unbound biotin-conjugated anti-cytochrome c was removed by several steps of washes with wash buffer. Streptavidin-HRP diluted with assay diluent was incubated for 1 hour at room temperature. After washes, a substrate solution reactive with HRP was added to each well for 5 minutes at room temperature. The enzymatic reaction was stopped with stop solution and the absorbance was read immediately on a spectrophotometer at 450 nm. To standardize the differences in concentration due to the differences in tissue mass, the total protein concentration in the cytosolic or mitochondrial fraction was evaluated with the Bio-Rad (Hercules, CA) protein assay. This allowed us to calculate the average nanograms of cytochrome c in cytosolic and/or mitochondrial fractions per milligram of protein tissue.
2.8. MSD Electrochemiluminescence assay
To detect the intracellular phosphorylation signaling pathways, the Meso Scale Discovery (MSD) electrochemiluminescence (ECL) assay, which is equivalent to the sandwich ELISA (MAPK Panel p-p38 multiplex assay [MULTI-SPOT 4, 96-well plate; Meso Scale Discovery, Gaithersburg, MD] and MSD Phospho (Ser112)/Total BAD assay [Duplex assay]) was performed using MSD Sector Imager 2400 according to the manufacturer’s protocol. The MSD assay is based on the sandwich immunoassay which utilizes ECL to measure protein levels. Frozen fetal brains were ground to a powder with a pestle. The powdered tissue was mixed with Tris lysis buffer (20 mM Tris pH7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100) containing protease and phosphatase inhibitors (included in the kit) and were then centrifuged at 17,360 rcf for 15 minutes at 4°C to obtain a soluble fraction. BCA assays were performed to determine the protein concentrations in the lysates. After incubating for 1 hour with blocking buffer containing 3% bovine serum albumin (BSA), 100 μg-protein lysates in 25 μl-lysis buffer were added to the multi-spotted ELISA plates. After incubation at 4°C overnight, the plates were washed four times with 150 μl of the provided wash buffer (50 mM Tris pH7.5, 1.5 M NaCl, 0.2% Tween-20). Then, 25 μl of the appropriate SULFO-TAG-labeled detection antibody solution was added to the wells and the plates were incubated at room temperature for 2 hours. The SULFO-TAG (Ruthenium (II) trisbiphyridine N-hydroxysuccinimide ester) emits light following electrical stimulation of the plates when it is in the bottom of the well. The plates were again washed four times with the wash buffer and 150 μl of the provided buffer T while surfactant was added to the wells, which prevented the introduction of any bubbles. The plates were analyzed with the SECTOR Imager 2400, in which a voltage applied to the plate electrodes causes the SULFO-TAG bound to the electrode surface to emit light. ECL intensities (arbitrary units, AU) obtained from p-p38 were normalized by the non-specific binding of the lysates to BSA-coated spot. BAD phosphorylation levels (% Phosphoprotein) were calculated by using “(2*(Phospho signal))/(Phospho signal + Total Signal)*100”.
2.9. TUNEL immunostaining reaction for cell death detection and cell counts
Cell death was determined using TUNEL reaction (TdT-mediated dUTP Nick End Labeling). To keep consistent conditions of immunostaining for both the experimental and the control groups, we embedded the fetal brains from ALC, ALC/CLN, and PF groups in 10% gelatin grade A. All fetal brains were aligned at the same level in gelatin, and serial 50-μm coronal sections were cut using a vibratome apparatus. Fetal brain sections were treated with Proteinase K (10–20 μg/ml) for 5 minutes at 37°C, rinsed with PBS 3 times for 5 minutes and then incubated with 3% H2O2 in methanol for 10 minutes at room temperature. The sections were again rinsed with PBS 3 times for 5 minutes and then incubated in a permeabilisation solution (0.1% TX-100 in 0.1% sodium citrate) for 2 minutes at 4°C. After the sections were rinsed twice in PBS for 5 minutes, they were incubated with a TUNEL reaction mixture (50 μl from bottle 1 and 450 μl from bottle 2, Roche Pharmaceuticals, Inc.) for 1 hour at 37°C. The control was prepared by incubating the solution from bottle 2. The sections were rinsed 3 times for 5 minutes with PBS and incubated in converter-POD for 30 minutes at 37°C. After the sections were rinsed with TBS, they were incubated in 0.05% 3′-3′-diaminobenzidine tetrahydrochloride and 0.003% H2O2 in TBS to reveal peroxidase activity. Sections were Nissl-counterstained with 0.5 % cresyl violet to observe the cellular profile of the sections.
TUNEL-positive cells were counted by a blind experimenter in the primordium frontal cortex of E13 fetal brains from PF, ALC, and ALC/CLN groups. The penetration of TUNEL-staining through a thickness of 50 μm was verified at 100X magnification. The expected shrinkage of a 50 μm-thick section in the z plane was averaged to approximately 14 μm. The number of sections in the selected brain region for TUNEL-positive cell counting was also considered and controlled in this study to avoid the bias of any missing sections from PF, ALC, and ALC/CLN groups. The entire population of TUNEL-positive cells was counted manually in every other section of the primordium frontal cortex, and this measure overcomes the bias of over-counting the number of TUNEL-positive cells in adjacent sections. The number of TUNEL-positive cells was averaged per section and analyzed statistically.
2.10. Immunohistochemistry for detection of phosphorylated form of JNK
Histological analysis was performed as described recently (Chiba et al., 2009). Fetal brains fixed in 4% PFA were frozen and embedded in Tissue-Tek O.C.T. compound (Sakura Fine-technical Co., Ltd., Tokyo, Japan) and then sliced into 10-μm thick sections using a cryostat microtome (CM3050, Leica Microsystems, Nussloch, Germany). After blocking with normal goat serum in TBST (Tris-buffered saline with 0.1% Tween20), slices were soaked with the primary antibody against the phosphorylated form of JNK (anti-p-JNK antibody, 1:100) at 4°C for 72 hours and then with biotinylated goat anti-rabbit IgG antibody. Sections were further soaked with HRP-labeled streptoavidin-biotin complex (Vectastain Elite ABC Kit; Vector, Burlingame, CA) and were visualized by Tyramide-FITC (TSA kit; NEN-Perkin-Elmer). The specimens were mounted in VECTASHIELD (Vector, Burlingame, CA) and were observed with a fluorescence microscope (BIOREVO BZ-9000, KEYENCE, Osaka, Japan).
2.11. Statistical analyses
Values in the figures are shown as means ± SEM (means ± SD for Fig. 2). Statistical analysis was performed with either one-way analysis of variance (ANOVA) or two-way ANOVA in which the P value was set at 0.05 as indicated in the figure legends. Newman-Keuls’s Test or Dunnett’s Test post-hoc multiple comparisons were made between individual groups.
Figure 2.

Effect of colivelin (CLN), C8A-humanin, and ADNF-9 against alcohol exposure in primary cortical neurons (PCNs). PCNs were treated with or without the indicated concentrations of peptides along with ALC. Cell index, which reflects cell viability, measured by xCELLigence system is shown as mean ± SD (n=6). Cell index of PCNs without alcohol exposure is equal to 1.0 [the baseline]. Control (water), red; 1 pM CLN, light green; 100 pM CLN, navy; 10 nM CLN, purple; 10 nM C8A-humanin (HNA), light blue; and 100 pM ADNF-9, thin purple. The one-way ANOVA at 8 hours revealed a significant difference between groups (F(5,35)=7.940 [p<0.0001]). * p<0.05, ** p<0.01, *** p<0.001, n.s. means no significant difference (Dunnett’s post hoc test).
RESULTS
1. In vitro data analyses
1.1. Colivelin protected primary cortical neurons from alcohol-induced neurotoxicity
Alcohol treatment for 72 hours significantly decreased the viability of PCNs (p<0.05) as shown in Fig. 1. We have performed dose-dependent toxicity analysis of alcohol on primary neurons and determined that only higher doses of alcohol 1000–1500 mg/dl were effective in inducing toxicity. This is probably due to the fact that the use of 10% FBS might be neuroprotective against lower doses of alcohol exposure. In addition, simultaneous treatment with CLN recovered the viability of alcohol-treated PCNs to the normal levels. However, CLN alone did not increase the viability of PCNs (Fig. 1).
Figure 1.

Effect of colivelin (CLN) against the effects of alcohol exposure in primary cortical neurons (PCNs). PCNs were treated with or without CLN together with or without alcohol. Relative viability measured with WST-8 is shown as means ± SEM (n=3). The two-way ANOVA (p<0.05) demonstrated a significant difference between groups (for alcohol exposure, ALC, F(1,16)=37.26 [p<0.0001]; for CLN treatment, F(1,16)=3.626 [p=0.0750]; for interaction, F(1,16)=6.060 [p=0.256]). Values are shown as mean ± SEM. * p<0.05, n.s. means no significant difference (Newman-Keul’s post hoc test). mQ: autoclaved milli-Q grade water.
Real time measurement of cell viability in PCNs demonstrates that alcohol exposure caused rapid decrease in Cell Index, which reflects impedance or cell viability (Fig. 2). CLN dose-dependently suppressed a decrease in Cell Index induced by alcohol exposure, suggesting that CLN inhibited alcohol-induced neurotoxicity. Both C8A-HN at 10 nM and ADNF-9 at 100 pM (both peptides were treated at effective concentrations) only showed marginal effects on alcohol-induced toxicity in PCNs (Fig. 2).
1.2. Pharmacological characterization of alcohol-induced neurotoxicity
To characterize the neurotoxicity induced by alcohol exposure, we examined the effect of various protein kinase inhibitors on alcohol-induced decreases in the viability of neuronal cells. Alcohol treatment for 72 hours at dose of 1000 mg/dl significantly decreased the viability of PCNs (Fig. 3). Treatment with a MEK inhibitor, PD98059, augmented the toxicity of alcohol, while treatment with a JNK inhibitor, SP600125, prevented alcohol induced decreases in cell viability. Treatment with inhibitors for the PI3-kinase/Akt pathway (Wortmannin) and the JAK2/STAT3 pathway (AG490) did not affect the neurotoxicity induced by alcohol treatment on PCNs. Consequently, it is assumed that the MEK-JNK pathways may play a critical role in neuroprotection against alcohol-induced apoptosis.
Figure 3.

Effect of protein kinase inhibitors on alcohol-mediated neurotoxicity. PCNs were pre-treated with protein kinase inhibitors (vehicle, 0.5% dmso; PD98059 for MEK, Wortmannin for PI3-kinase, AG490 for JAK2, and SP600125 for JNK) and then with ALC (1000 mg/dl as indicated) to examine the effect of alcohol exposure, n=3. The two-way ANOVA demonstrated a significant difference between groups (for alcohol exposure, F(4,20)=44.10 [p<0.0001]; for inhibitors, F(4,20)=2.048 [p=0.1261]; for interaction, F(4,20)=2.048 [p=0.1261]). Values are shown as mean ± sem. * p<0.05, ** p<0.01, n.s. means no significant difference (Newman-Keul’s post hoc test). mQ: autoclaved milli-Q grade water.
1.3. Time-course analysis of phosphorylation of the MAP kinase pathway in PCNs treated with alcohol and the role of CLN in neuroprotection
Next, we performed a time-course analysis of phosphorylation levels of the MAP kinase pathways in PCNs treated with ALC at the dose of 1000 mg/dl. Alcohol exposure did not affect p-JNK levels at an acute phase (1–24 hour) (Fig. 4A, B). At chronic phase (48 hours), we observed a substantial increase in p-JNK levels by alcohol exposure. Considering the fact that chronic up-regulation of p-JNK (Hashimoto et al., 2003) is involved in the neuronal death, the modulation of the MAP kinase pathways by alcohol exposure is likely to be directly involved in the alcohol neurotoxicity or alcohol-induced neuronal death.
Figure 4.
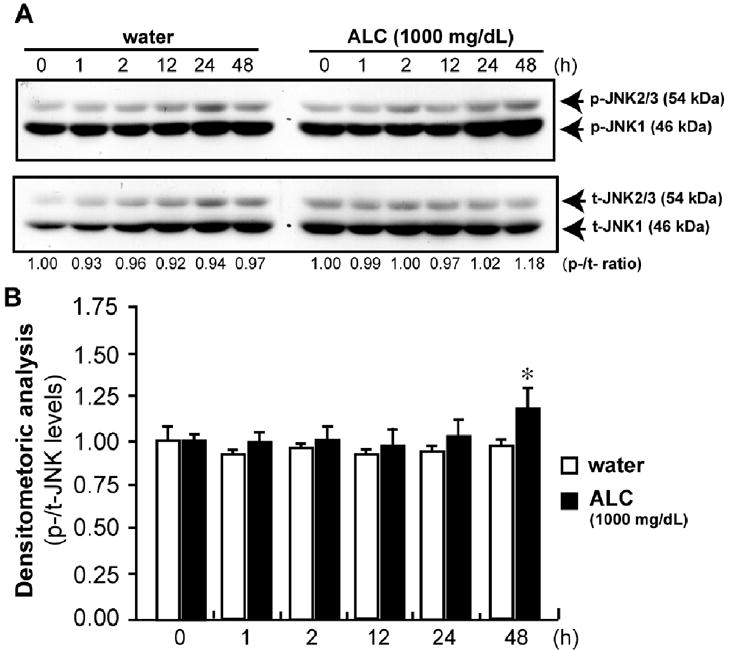
Immunoblots of p-JNK (A). PCNs were treated with or without ALC at the dose of 1000 mg/dl for the indicated duration (h), n=3. Immunoblot analyses for p-JNK are shown in B. Values are shown as mean ± SEM. * p<0.05
2. In Vivo data analyses
2.1. Protective effects of CLN administration on fetal brain weight against alcohol exposure
Statistical analyses of fetal brains weights show significant reduction in individual fetal brain weight of the ALC group as compared to the control Chow (p<0.01), PF (p<0.001) and PF/CLN (p<0.001) groups (Fig. 5). CLN administration alongside alcohol exposure prevented such weight reduction and stabilized brain weights comparable to the control groups (Chow, PF, and PF/CLN); however, there were significant differences between the CLN treatment group (ALC/CLN) and ALC group (p<0.01). Also, there were no significant differences between the Chow, PF, PF/CLN and ALC/CLN groups. These results demonstrate the role of CLN in neuroprotection against alcohol-induced growth restriction. Note that there were no significant differences in the number of fetuses between control and treatment groups: Chow (8.00 ± 0.40), PF (7.66 ± 0.33), PF/CLN (8.25 ± 0.25), ALC (7.9 ± 0.31), and ALC/CLN (7.87 ± 0.52). These data indicate that the average of the number of fetuses is about 8 fetuses per dam for each control or treatment group.
Figure 5.

Protective effect of colivelin (CLN) on fetal brain weight in mice exposed prenatally to alcohol. The one-way ANOVA demonstrated a significant difference between groups (p=0.0001). Prenatal alcohol exposure induced significant fetal brain reduction as compared to the Chow, PF, and PF/CLN control groups (p<0.01, p<0.001, and p<0.001 respectively). CLN treatment prevented the reduction in fetal brain weight found in the ALC group (p<0.05). No significant differences were found between the Chow, PF, PF/CLN, and ALC/CLN groups. Values are shown as mean ± SEM. (Chow, n=5; PF, n=9; PF/CLN, n=5; ALC, n=8; ALC/CLN, n=6). * p<0.01, ** p<0.001 (Newman-Keul’s post hoc test).
2.2. Biodistribution of CLN into the fetal brain by crossing the placenta
We found significant uterus-like fluorescence intensity in the abdominal region of CLN-Alexa680-injected mice as compared to control pregnant mice injected with saline (Fig. 6). Fluorescence intensity was also detected in the uterus of CLN-Alexa680-injected mice. Furthermore, we found that CLN-Alexa680 was distributed in the fetal brains of the embryos (Fig. 6).
Figure 6.
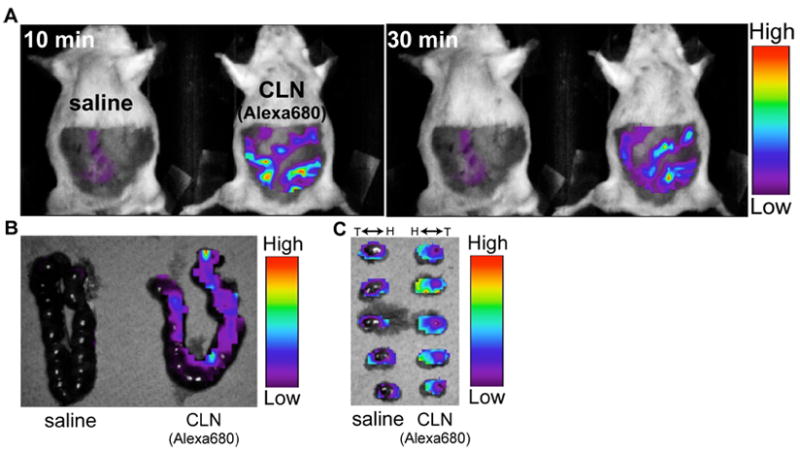
In vivo optical imaging of colivelin (CLN) labeled with Alexa680 (CLN-Alexa680). Pregnant female mice (E14) were i.p. administered with saline or saline containing 7 nmol of CLN-Alexa680. Thirty minutes after the injection, mice were optically imaged with Optix MX2 (A). The uterus was then dissected from the mice and imaged (B). The embryos were further dissected from the uterus and imaged (C). H, head; T, tail.
2.3. Neuroprotective effect of CLN administration against prenatal alcohol-induced apoptosis
To determine if CLN plays a role in the prevention of alcohol-induced apoptosis, we used TUNEL-staining assay to evaluate cell death. Immunohistochemical staining for detection of TUNEL-positive cells in the primordial frontal cortex revealed that alcohol exposure increased cell death (Fig. 7B) relative to the PF group (Fig. 7A). Importantly, CLN administration along with alcohol exposure prevented the increases in TUNEL-positive cells (Fig. 7C). Statistical analyses of cell counts show a significant increase in TUNEL-positive cells in primordial frontal cortex of the ALC group compared to the PF group (p<0.01) (Fig. 7D). Additionally, there was also a significant difference between the ALC/CLN and ALC groups (p<0.01), but not between the PF and ALC/CLN groups (p>0.05).
Figure 7.
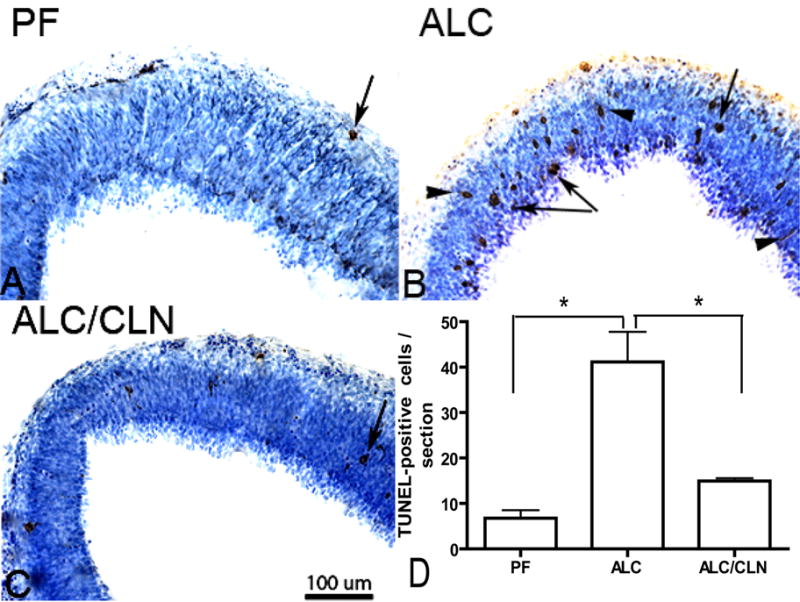
Prenatal alcohol exposure increases the number of TUNEL-positive cells in primordium frontal cortex (A, B, C) of E13 fetal brains. Administration of colivelin (CLN) alongside alcohol exposure prevented alcohol-induced increases in TUNEL-positive cells. Arrowheads indicate cell undergoing apoptosis as indicated by cell processes, and arrows indicate the final stage of cell death. Scale bars: 100 μm. The one-way ANOVA demonstrated a significant difference between groups (p=0.0032). An increase in TUNEL-positive cells was found in the ALC group as compared to the PF group (p<0.01) (D). Importantly, CLN administration alongside alcohol exposure prevented a significant alcohol-induced increase in TUNEL-positive cells (p<0.01) (D). Values are shown as mean ± SEM. (PF, n=5; ALC, n=6; ALC/CLN, n=5). * p<0.01 (Newman-Keul’s post hoc test).
2.4. Neuroprotective effect of CLN administration against prenatal alcohol-induced caspase-3 activation
Caspase-3 colorimetric assay was used to evaluate the concentration of active caspase-3 in the E13 fetal brain to determine if the neuroprotective effect of CLN is mediated through the caspase-3 downstream apoptotic pathway. The activity of the caspase-3 enzyme indicated that cells entered the apoptotic pathway. The concentration of active caspase-3 in the fetal brain was increased in the ALC group compared to the Chow (p<0.05), PF (p<0.01), and PF/CLN (p<0.05) groups (Fig. 8). CLN administration with alcohol exposure, however, prevented the alcohol-induced increase in the concentration of active caspase-3 (p<0.05). No significant difference was found between the ALC/CLN and control (Chow and PF) groups (Fig. 8).
Figure 8.

Neuroprotective effect of colivelin (CLN) against the insult of prenatal alcohol exposure is mediated through caspase-3 activation as tested by caspase-3 colorimetric assay in E13 fetal brains. The one-way ANOVA demonstrated a significant difference between groups (p=0.0039). Prenatal alcohol exposure induced a significant increase in the concentrations of active caspase-3 as compared to the Chow and PF control groups (p<0.05 and p<0.01, respectively). CLN administration prevented significantly the effect of alcohol-induced increases in the concentrations of active caspase-3 (p<0.05). There were no significant differences between the control and CLN treatment groups. Values are shown as mean ± SEM. (Chow, n=5; PF, n=9; ALC, n=8; ALC/CLN, n=6). * p<0.05, ** p<0.01 (Newman-Keul’s post hoc test).
2.5. Neuroprotective effect of CLN administration against prenatal alcohol-induced increase in cytosolic cytochrome c and mitochondrial cytochrome c
Next we investigated whether cytosolic cytochrome c release is correlated with a change in concentrations of active caspase-3 and whether the neuroprotective effect of CLN are mediated through the downstream signaling pathways that involve cytosolic cytochrome c. Statistical analyses show that prenatal alcohol exposure induced a significant increase in cytosolic cytochrome c level compared to the Chow and PF groups (p<0.01; Fig. 9A). CLN administration alongside alcohol exposure prevented the increase in cytosolic cytochrome c (p<0.01; Fig. 9A). However, no significant difference was found in the concentration of cytosolic cytochrome c in the fetal brains between the CLN treated group and the control groups (Chow and PF).
Figure 9.

Neuroprotective effect of colivelin (CLN) against the insult of prenatal alcohol exposure is mediated through cytosolic cytochrome c as tested by ELISA in E13 fetal brains. The data generated with ELISA demonstrated a significant difference in the concentration of cytosolic and mitochondrial cytochrome c between groups (p<0.05) as shown by one-way ANOVA. (A) Prenatal alcohol exposure induced significant increase in the concentrations of cytosolic cytochrome c as compared to the Chow (p<0.01) and PF (p<0.01) control groups. CLN administration prevented significantly the increases in the concentrations of cytosolic cytochrome c as compared to the ALC group (p<0.01). (B) Prenatal alcohol exposure induced significant decrease in the concentration of mitochondrial cytochrome c as compared to the Chow and PF control groups (p<0.01 and p<0.001, respectively). CLN administration prevented the reduction in mitochondrial cytochrome c as compared to ALC (p<0.01). There were no significant differences between the control and CLN treatment groups in the level of mitochondrial and cytosolic cytochrome c. Values are shown as mean ± SEM. (Chow, n=5; PF, n=5; ALC, n=6; ALC/CLN, n=6). * p<0.01, ** p<0.001 (Newman-Keul’s post hoc test).
We also examined if the change in the cytosolic cytochrome c is correlated with mitochondrial cytochrome c. Statistical analyses revealed that prenatal alcohol exposure induced a significant decrease in mitochondrial cytochrome c compared to the Chow (p<0.01) and PF (p<0.001) controls (Fig. 9B). Administration of CLN alongside alcohol exposure prevented the alcohol-induced reduction in mitochondrial cytochrome c (p<0.01; Fig. 9B). However, no significant differences were found between the ALC/CLN, Chow, and PF groups.
2.6 Neuroprotective effect of CLN administration against prenatal alcohol exposure is mediated through the MAP kinase pathway
We next examined whether MAP kinase pathway is involved in the neuroprotective effect of CLN in vivo. Immunohistochemical staining for detection of the active form (or phosphorylated form) of JNK between groups showed that prenatal alcohol exposure induced increases in the phoshporylated form of JNK (p-JNK) as compared to the PF group (Fig. 10). Interestingly, CLN prevented alcohol-induced increases in p-JNK in both fetal brain regions: primordium frontal cortex (Fig. 10c) and basal ganglia eminence (Fig. 10f).
Figure 10.
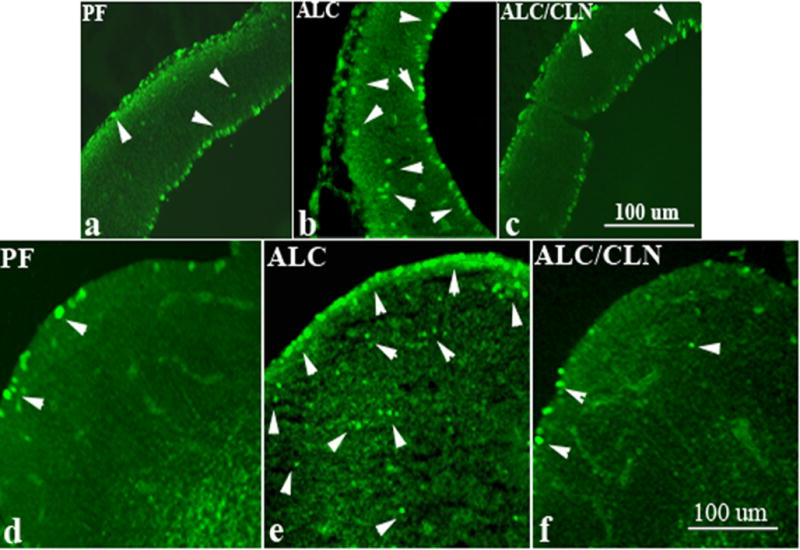
Prenatal alcohol exposure increases the number of p-JNK-positive cells in primordium frontal cortex (b) and basal ganglia eminence (e) of E13 fetal brains in the ALC group as compared to the control group (a, d). Administration of colivelin (CLN) alongside alcohol exposure prevented alcohol-induced increases in TUNEL-positive cells in both regions (c, f). Arrowheads indicate cells expressing p-JNK positive staining. Scale bars: 100 μm.
We also performed MSD assays for determination of the involvement of p-38 in fetal brain lysates from alcohol exposed groups. There were no significant differences in p-p38 levels between control and treatment groups (data not shown). To further examine the role of the MAP kinase pathways in alcohol-induced neurotoxicity, we also quantified p-Bad levels in fetal brain. Prenatal alcohol exposure decreased significantly p-Bad levels in the fetal brains as compared to the PF group (data not shown). CLN treatment restored p-Bad levels to control levels.
DISCUSSION
Our results reveal some of the signaling pathways underlying CLN-mediated neuroprotection against alcohol-induced apoptosis both in vitro and in vivo. The in vivo experiments were tested in a specific window of pregnancy (E7–E13). The rationale for the choice of period of exposure from E7 to E13 in this study was based on previous findings demonstrating that the effects of prenatal alcohol exposure depend on the timing of alcohol treatment with different brain areas exhibiting differential susceptibility (Webster et al., 1983). This study, performed by Webster et al. 1983, reported that treatment with alcohol on day 7 or 8 caused a variety of facial abnormalities, some of which were comparable to those seen in children with fetal alcohol syndrome (FAS) or fetal alcohol effect (FAE). Webster and colleagues suggested that during the period between E7 to E13 and also later ages, the developing brain exhibited the highest susceptibility to alcohol exposure. Moreover, using similar drinking paradigm (25% EDC), we have shown that prenatal alcohol exposure from E7 to E13, E15 and/or E18 caused brain growth deficits, reduction in the size of fetal brain regions and reduction in the number of serotonin neurons (Sari et al., 2001, Sari and Zhou, 2004, Sari and Gozes, 2006, Sari, 2009).
In this study, we examined the neuroprotective effect of a novel hybrid peptide CLN on alcohol toxicity. Our in vitro model of alcohol exposure showed that CLN recovered the viability of alcohol-treated PCNs to the normal levels. CLN by itself, however, did not increase the viability of PCNs, which suggests the specific role of CLN in preventing alcohol-induced apoptosis. Importantly, our in vivo data revealed that CLN prevented the alcohol-induced reduction of the fetal brain weight, increased cytosolic cytochrome c and caspase-3 activity, and decreased mitochondrial cytochrome c in E13 fetal brain exposed prenatally to alcohol. In consistence, TUNEL- staining of apoptotic neurons revealed that CLN suppressed alcohol-induced apoptosis in the primordial frontal cortex. These findings indicate that CLN significantly suppressed neurotoxicity caused by prenatal alcohol exposure and that CLN-mediated neuroprotection may involve mitochondrial control. We have recently observed similar effects involving these downstream signaling pathways with NAP administration (Sari, 2009). But the upstream signaling pathway of NAP might be different from that of CLN and this is warranted to be investigated in the future.
Regarding the molecular mechanisms underlying CLN-mediated neuroprotection against alcohol-induced neurotoxicity, the analyses of the effects of several protein kinase inhibitors on PCNs exposed to alcohol demonstrated that JNK is a potential candidate in the mechanism involving alcohol-induced neurotoxicity. Importantly, our vivo data revealed that CLN administration prevented alcohol-induced increases in p-JNK, which leads to neuroprotection. This is in accordance with previous work demonstrating that glial-derived neurotrophic factor prevented alcohol-induced apoptosis through decreases in the level of p-JNK (McAlhany et al., 2000). JNK-mediated 14-3-3 phosphorylation is reported to induce dissociation of Bad, a pro-apoptotic member of the Bcl-2 family that can bind to and antagonizes anti-apoptotic proteins such as Bcl-2 and Bcl-xL (Yang et al., 1995), from 14-3-3 and consequential dephosphorylation of Bad (Sunayama et al., 2005).
Meanwhile, suppression of p-JNK levels may antagonize 14-3-3 phosphorylation and Bad translocation to the mitochondria (Yang et al., 2008). Importantly, it is suggested that CLN up-regulates p-Bad through suppression of p-JNK levels, which is leading to the relocation of Bad to the cytoplasm and prevents its interaction with mitochondrial Bcl-2 (Datta et al., 1997). Interestingly, proteomics quantitative analyses have demonstrated that prenatal alcohol exposure down-regulated the expression of Bcl-2 and 14-3-3 proteins in E13 fetal brains (Sari and Mechref, 2008). Direct phosphorylation of Bad by p90 ribosomal S6 kinase, which is one of the downstream targets of MEK/ERK pathway, is also reported to suppress neuronal apoptosis (Jin et al., 2002, Koh, 2007). This may support our in vitro data that a MEK inhibitor, PD98059, augmented neuronal death caused by alcohol exposure. In addition, we demonstrate here that prenatal alcohol exposure significantly decreased p-Bad levels in E13 fetal brains and that CLN treatment significantly restored p-Bad levels to the control levels. Given that the decrease in p-Bad levels results in neuronal apoptosis (Wang et al., 1999), it is likely that the CLN-induced neuroprotection against alcohol neurotoxicity involves both direct phosphorylation of Bad and indirect inactivation of JNK.
As to the potential involvement of the Bcl-2 family in the mechanism of neuroprotection by CLN, it has been reported that a humanin (HN) fragment of CLN has an endogenous role in protecting cells from apoptosis (Hashimoto et al., 2001, Sponne et al., 2004) by interfering with pro-apoptotic Bcl-2 family proteins such as Bax (Guo et al., 2003), Bid (Zhai et al., 2005) and BimEL (Luciano et al., 2005). Binding of HN to these pro- apoptotic Bcl-2 family proteins prevents their translocation from the cytosol to the mitochondria and suppresses cytochrome c release. Collectively, these findings suggest that CLN may exert neuroprotective effect on alcohol toxicity through direct binding to and inhibition of the pro- and anti-apoptotic Bcl-2 family proteins. Although we do not have evidence that extracellular CLN peptide is transported into the cytosol and binds to the Bcl-2 family proteins in our experimental systems.
There are other potential mechanisms of action of CLN based on previous works on the ADNF-9 fragment of CLN, which is known to play a critical role in neuroprotection against alcohol-induced brain deficit in models of FAE and FAS (Spong et al., 2001, Sari and Gozes, 2006). In a model of FAS, research has shown that ADNF–9 may interfere with the decrease in oxidized glutathione levels that may be caused by alcohol exposure (Spong et al., 2001). In vitro evidence suggests that among the characteristics of neuroprotection of ADNF-9 are maintenance of mitochondrial function and a reduction in the accumulation of intracellular reactive oxygen species (Glazner et al., 1999). Moreover, using a model of Familial Amyotrophic lateral Sclerosis-linked mutant Cu/Zn-superoxide dismutase-1 (SOD1), we (Chiba et al., 2004) have demonstrated that ADNF-9-mediated neuroprotection involves the Ca++/calmodulin-dependent protein kinase IV (CaMKIV) signaling pathway (Chiba et al., 2004). Together, these findings suggest that ADNF-9 mediates neuroprotection directly and indirectly through the mitochondria. Interestingly, we also revealed that CLN-induced neuroprotection is mediated through two pathways: CaMKIV (triggered by ADNF-9) and STAT3 (triggered by HN) (Chiba et al., 2005). In other models that induce oxidative stress, studies have revealed that ADNF-9 is associated with stimulation of the PKA and PKC pathways (Gressens et al., 1999). Thus, CLN may involve the PKA and PKC pathways which indirectly interact with cytosolic and/or mitochondrial pro-apoptotic proteins. Further studies are warranted to determine the precise signaling pathways involving CLN in neuroprotection against alcohol-induced apoptosis.
In conclusion, our results show for the first time that a novel compound, CLN, protects against alcohol-induced cell death in E13 fetal brains. Administration of CLN alongside prenatal alcohol exposure significantly prevented caspase-3 activation caused by mitochondrial release of cytochrome c to the cytosol, which comprises the downstream components of the alcohol-mediated neurotoxicity. Importantly, neuroprotective effect of CLN is suggested to involve the regulation of MAPK pathways such as up-regulation of the phosphorylated form of the Bad protein, which in turn down-regulates the phosphorylated form of JNK caused by alcohol exposure. This mechanism may lead to deceases in cytosolic cytochrome c and consequently prevention of alcohol-induced apoptosis. Thus, CLN may be considered as a potential compound for the prevention of alcohol-induced intoxication during pregnancy.
Acknowledgments
We would like to thank NIH-NIAAA for their full support in this project, grant number R21AA016115. This work was also supported in part by grants from the Japan Society for the Promotion of Science (JSPS, #19790615) and NOEVIR CO., LTD. We also would like to thank METACyt funded by a Lilly endowment for providing the resources and equipment to accomplish this work. The author would like to thank Eta Isiorho for her technical assistance in TUNEL detection assay and ELISA. We finally would like to thank Faye Caylor for her administrative assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Barron S, Gagnon WA, Mattson SN, Kotch LE, Meyer LS, Riley EP. The effects of prenatal alcohol exposure on odor associative learning in rats. Neurotoxicol Teratol. 1988;10:333–339. doi: 10.1016/0892-0362(88)90036-0. [DOI] [PubMed] [Google Scholar]
- Bassan M, Zamostiano R, Davidson A, Pinhasov A, Giladi E, Perl O, Bassan H, Blat C, Gibney G, Glazner G, Brenneman DE, Gozes I. Complete sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J Neurochem. 1999;72:1283–1293. doi: 10.1046/j.1471-4159.1999.0721283.x. [DOI] [PubMed] [Google Scholar]
- Bauer-Moffett C, Altman J. Ethanol-induced reductions in cerebellar growth of infant rats. Exp Neurol. 1975;48:378–382. doi: 10.1016/0014-4886(75)90164-8. [DOI] [PubMed] [Google Scholar]
- Bauer-Moffett C, Altman J. The effect of ethanol chronically administered to preweanling rats on cerebellar development: a morphological study. Brain Res. 1977;119:249–268. doi: 10.1016/0006-8993(77)90310-9. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, West JR. Alcohol-induced neuronal loss in developing rats: increased brain damage with binge exposure. Alcohol Clin Exp Res. 1990;14:107–118. doi: 10.1111/j.1530-0277.1990.tb00455.x. [DOI] [PubMed] [Google Scholar]
- Brenneman DE, Gozes I. A femtomolar-acting neuroprotective peptide. J Clin Invest. 1996;97:2299–2307. doi: 10.1172/JCI118672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman DE, Hauser J, Neale E, Rubinraut S, Fridkin M, Davidson A, Gozes I. Activity-dependent neurotrophic factor: structure-activity relationships of femtomolar-acting peptides. J Pharmacol Exp Ther. 1998;285:619–627. [PubMed] [Google Scholar]
- Cheema ZF, West JR, Miranda RC. Ethanol induces Fas/Apo [apoptosis]-1 mRNA and cell suicide in the developing cerebral cortex. Alcohol Clin Exp Res. 2000;24:535–543. [PubMed] [Google Scholar]
- Chiba T, Hashimoto Y, Tajima H, Yamada M, Kato R, Niikura T, Terashita K, Schulman H, Aiso S, Kita Y, Matsuoka M, Nishimoto I. Neuroprotective effect of activity-dependent neurotrophic factor against toxicity from familial amyotrophic lateral sclerosis-linked mutant SOD1 in vitro and in vivo. J Neurosci Res. 2004;78:542–552. doi: 10.1002/jnr.20305. [DOI] [PubMed] [Google Scholar]
- Chiba T, Yamada M, Hashimoto Y, Sato M, Sasabe J, Kita Y, Terashita K, Aiso S, Nishimoto I, Matsuoka M. Development of a femtomolar-acting humanin derivative named colivelin by attaching activity-dependent neurotrophic factor to its N terminus: characterization of colivelin-mediated neuroprotection against Alzheimer’s disease-relevant insults in vitro and in vivo. J Neurosci. 2005;25:10252–10261. doi: 10.1523/JNEUROSCI.3348-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba T, Yamada M, Sasabe J, Terashita K, Shimoda M, Matsuoka M, Aiso S. Amyloid-beta causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol Psychiatry. 2009;14:206–222. doi: 10.1038/mp.2008.105. [DOI] [PubMed] [Google Scholar]
- Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Ganju N, Banerjee K, Brown NV, Luong T, Wands JR. Partial rescue of ethanol-induced neuronal apoptosis by growth factor activation of phosphoinositol-3-kinase. Alcohol Clin Exp Res. 2000;24:716–726. [PubMed] [Google Scholar]
- Ge Y, Belcher SM, Pierce DR, Light KE. Altered expression of Bcl2, Bad and Bax mRNA occurs in the rat cerebellum within hours after ethanol exposure on postnatal day 4 but not on postnatal day 9. Brain Res Mol Brain Res. 2004;129:124–134. doi: 10.1016/j.molbrainres.2004.06.034. [DOI] [PubMed] [Google Scholar]
- Glazner GW, Boland A, Dresse AE, Brenneman DE, Gozes I, Mattson MP. Activity-dependent neurotrophic factor peptide (ADNF9) protects neurons against oxidative stress-induced death. J Neurochem. 1999;73:2341–2347. doi: 10.1046/j.1471-4159.1999.0732341.x. [DOI] [PubMed] [Google Scholar]
- Green CR, Watts LT, Kobus SM, Henderson GI, Reynolds JN, Brien JF. Effects of chronic prenatal ethanol exposure on mitochondrial glutathione and 8-iso-prostaglandin F2alpha concentrations in the hippocampus of the perinatal guinea pig. Reprod Fertil Dev. 2006;18:517–524. doi: 10.1071/rd05128. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Gressens P, Marret S, Bodenant C, Schwendimann L, Evrard P. Activity-dependent neurotrophic factor-14 requires protein kinase C and mitogen-associated protein kinase kinase activation to protect the developing mouse brain against excitotoxicity. J Mol Neurosci. 1999;13:199–210. doi: 10.1385/JMN:13:1-2:199. [DOI] [PubMed] [Google Scholar]
- Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423:456–461. doi: 10.1038/nature01627. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Ito Y, Sudo H, Hata M, Arakawa E, Abe Y, Kita Y, Nishimoto I. Detailed characterization of neuroprotection by a rescue factor humanin against various Alzheimer’s disease-relevant insults. J Neurosci. 2001;21:9235–9245. doi: 10.1523/JNEUROSCI.21-23-09235.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Tsuji O, Niikura T, Yamagishi Y, Ishizaka M, Kawasumi M, Chiba T, Kanekura K, Yamada M, Tsukamoto E, Kouyama K, Terashita K, Aiso S, Lin A, Nishimoto I. Involvement of c-Jun N-terminal kinase in amyloid precursor protein-mediated neuronal cell death. J Neurochem. 2003;84:864–877. doi: 10.1046/j.1471-4159.2003.01585.x. [DOI] [PubMed] [Google Scholar]
- Heaton MB, Moore DB, Paiva M, Madorsky I, Mayer J, Shaw G. The role of neurotrophic factors, apoptosis-related proteins, and endogenous antioxidants in the differential temporal vulnerability of neonatal cerebellum to ethanol. Alcohol Clin Exp Res. 2003;27:657–669. doi: 10.1097/01.ALC.0000060527.55252.71. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Jin K, Mao XO, Zhu Y, Greenberg DA. MEK and ERK protect hypoxic cortical neurons via phosphorylation of Bad. J Neurochem. 2002;80:119–125. doi: 10.1046/j.0022-3042.2001.00678.x. [DOI] [PubMed] [Google Scholar]
- Koh PO. Estradiol prevents the injury-induced decrease of 90 ribosomal S6 kinase (p90RSK) and Bad phosphorylation. Neurosci Lett. 2007;412:68–72. doi: 10.1016/j.neulet.2006.10.060. [DOI] [PubMed] [Google Scholar]
- Kornguth SE, Rutledge JJ, Sunderland E, Siegel F, Carlson I, Smollens J, Juhl U, Young B. Impeded cerebellar development and reduced serum thyroxine levels associated with fetal alcohol intoxication. Brain Res. 1979;177:347–360. doi: 10.1016/0006-8993(79)90785-6. [DOI] [PubMed] [Google Scholar]
- Luciano F, Zhai D, Zhu X, Bailly-Maitre B, Ricci JE, Satterthwait AC, Reed JC. Cytoprotective peptide humanin binds and inhibits proapoptotic Bcl-2/Bax family protein BimEL. J Biol Chem. 2005;280:15825–15835. doi: 10.1074/jbc.M413062200. [DOI] [PubMed] [Google Scholar]
- McAlhany RE, Jr, West JR, Miranda RC. Glial-derived neurotrophic factor (GDNF) prevents ethanol-induced apoptosis and JUN kinase phosphorylation. Brain Res Dev Brain Res. 2000;119:209–216. doi: 10.1016/s0165-3806(99)00171-6. [DOI] [PubMed] [Google Scholar]
- Middaugh LD, Boggan WO. Perinatal maternal ethanol effects on pregnant mice and on offspring viability and growth: influences of exposure time and weaning diet. Alcohol Clin Exp Res. 1995;19:1351–1358. doi: 10.1111/j.1530-0277.1995.tb01624.x. [DOI] [PubMed] [Google Scholar]
- Middaugh LD, Randall CL, Favara JP. Prenatal ethanol exposure in C57 mice: effects on pregnancy and offspring development. Neurotoxicol Teratol. 1988;10:175–180. doi: 10.1016/0892-0362(88)90082-7. [DOI] [PubMed] [Google Scholar]
- Moore DB, Walker DW, Heaton MB. Neonatal ethanol exposure alters bcl-2 family mRNA levels in the rat cerebellar vermis. Alcohol Clin Exp Res. 1999;23:1251–1261. doi: 10.1111/j.1530-0277.1999.tb04286.x. [DOI] [PubMed] [Google Scholar]
- Poggi SH, Goodwin K, Hill JM, Brenneman DE, Tendi E, Schinelli S, Abebe D, Spong CY. The role of activity-dependent neuroprotective protein in a mouse model of fetal alcohol syndrome. Am J Obstet Gynecol. 2003;189:790–793. doi: 10.1067/s0002-9378(03)00834-2. [DOI] [PubMed] [Google Scholar]
- Ramachandran V, Perez A, Chen J, Senthil D, Schenker S, Henderson GI. In utero ethanol exposure causes mitochondrial dysfunction, which can result in apoptotic cell death in fetal brain: a potential role for 4-hydroxynonenal. Alcohol Clin Exp Res. 2001;25:862–871. [PubMed] [Google Scholar]
- Samson HH, Diaz J. Altered development of brain by neonatal ethanol exposure: zinc levels during and after exposure. Alcohol Clin Exp Res. 1981;5:563–569. doi: 10.1111/j.1530-0277.1981.tb05362.x. [DOI] [PubMed] [Google Scholar]
- Sari Y. Activity-dependent neuroprotective protein-derived peptide, NAP, preventing alcohol-induced apoptosis in fetal brain of C57BL/6 mouse. Neuroscience. 2009;158:1426–1435. doi: 10.1016/j.neuroscience.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari Y, Gozes I. Brain deficits associated with fetal alcohol exposure may be protected, in part, by peptides derived from activity-dependent neurotrophic factor and activity-dependent neuroprotective protein. Brain Res Brain Res Rev. 2006;52:107–118. doi: 10.1016/j.brainresrev.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Sari Y, Mechref Y. Differential expression of proteins in fetal brains of alcohol-treated prenatally C57BL/6 mice: a proteomic investigation. Soc Neurosc. 2008 doi: 10.1002/elps.200900385. 663.1/II26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari Y, Powrozek T, Zhou FC. Alcohol deters the outgrowth of serotonergic neurons at midgestation. J Biomed Sci. 2001;8:119–125. doi: 10.1007/BF02255980. [DOI] [PubMed] [Google Scholar]
- Sari Y, Zhou FC. Prenatal alcohol exposure causes long-term serotonin neuron deficit in mice. Alcohol Clin Exp Res. 2004;28:941–948. doi: 10.1097/01.alc.0000128228.08472.39. [DOI] [PubMed] [Google Scholar]
- Spong CY, Abebe DT, Gozes I, Brenneman DE, Hill JM. Prevention of fetal demise and growth restriction in a mouse model of fetal alcohol syndrome. J Pharmacol Exp Ther. 2001;297:774–779. [PubMed] [Google Scholar]
- Sponne I, Fifre A, Koziel V, Kriem B, Oster T, Pillot T. Humanin rescues cortical neurons from prion-peptide-induced apoptosis. Mol Cell Neurosci. 2004;25:95–102. doi: 10.1016/j.mcn.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Sulik KK, Johnston MC, Webb MA. Fetal alcohol syndrome: embryogenesis in a mouse model. Science. 1981;214:936–938. doi: 10.1126/science.6795717. [DOI] [PubMed] [Google Scholar]
- Sunayama J, Tsuruta F, Masuyama N, Gotoh Y. JNK antagonizes Akt-mediated survival signals by phosphorylating 14–3-3. J Cell Biol. 2005;170:295–304. doi: 10.1083/jcb.200409117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Webster WS, Walsh DA, McEwen SE, Lipson AH. Some teratogenic properties of ethanol and acetaldehyde in C57BL/6J mice: implications for the study of the fetal alcohol syndrome. Teratology. 1983;27:231–243. doi: 10.1002/tera.1420270211. [DOI] [PubMed] [Google Scholar]
- Xu Y, Liu P, Li Y. Impaired development of mitochondria plays a role in the central nervous system defects of fetal alcohol syndrome. Birth Defects Res A Clin Mol Teratol. 2005;73:83–91. doi: 10.1002/bdra.20110. [DOI] [PubMed] [Google Scholar]
- Yamada M, Chiba T, Sasabe J, Terashita K, Aiso S, Matsuoka M. Nasal Colivelin treatment ameliorates memory impairment related to Alzheimer’s disease. Neuropsychopharmacology. 2008;33:2020–2032. doi: 10.1038/sj.npp.1301591. [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Yang X, Luo C, Cai J, Pierce WM, Tezel G. Phosphorylation-dependent interaction with 14-3-3 in the regulation of bad trafficking in retinal ganglion cells. Invest Ophthalmol Vis Sci. 2008;49:2483–2494. doi: 10.1167/iovs.07-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young C, Klocke BJ, Tenkova T, Choi J, Labruyere J, Qin YQ, Holtzman DM, Roth KA, Olney JW. Ethanol-induced neuronal apoptosis in vivo requires BAX in the developing mouse brain. Cell Death Differ. 2003;10:1148–1155. doi: 10.1038/sj.cdd.4401277. [DOI] [PubMed] [Google Scholar]
- Zamostiano R, Pinhasov A, Gelber E, Steingart RA, Seroussi E, Giladi E, Bassan M, Wollman Y, Eyre HJ, Mulley JC, Brenneman DE, Gozes I. Cloning and characterization of the human activity-dependent neuroprotective protein. J Biol Chem. 2001;276:708–714. doi: 10.1074/jbc.M007416200. [DOI] [PubMed] [Google Scholar]
- Zhai D, Luciano F, Zhu X, Guo B, Satterthwait AC, Reed JC. Humanin binds and nullifies Bid activity by blocking its activation of Bax and Bak. J Biol Chem. 2005;280:15815–15824. doi: 10.1074/jbc.M411902200. [DOI] [PubMed] [Google Scholar]
- Zhou FC, Sari Y, Powrozek TA. Fetal alcohol exposure reduces serotonin innervation and compromises development of the forebrain along the serotonergic pathway. Alcohol Clin Exp Res. 2005;29:141–149. doi: 10.1097/01.alc.0000150636.19677.6f. [DOI] [PubMed] [Google Scholar]
- Zhou FC, Sari Y, Powrozek TA, Spong CY. A neuroprotective peptide antagonizes fetal alcohol exposure-compromised brain growth. J Mol Neurosci. 2004;24:189–199. doi: 10.1385/JMN:24:2:189. [DOI] [PubMed] [Google Scholar]