

Abstract
Islet amyloid polypeptide (amylin) is the main component in amyloid deposits formed in type II diabetes. We used triplet quenching to probe the dynamics of contact formation between the N-terminal disulfide loop and a C-terminal tryptophan in monomeric amylins from human and rat. Quenching rates measured in the absence of denaturant are four times larger than those in 6 M guanidinium chloride, indicating a decrease in the average end-to-end distance (collapse) at low denaturant concentrations. We were surprised to find an even greater (sevenfold) increase in quenching rates on removal of denaturant for a hydrophilic control peptide containing the disulfide loop compared to the same peptide without the loop (twofold change). These results suggest that collapse is driven by backbone-backbone and backbone-side chain interactions involving the disulfide loop portion of the chain rather than by the formation of side-chain hydrophobic contacts. Molecular dynamics simulations of the control peptide show that the collapse results from hydrogen-bonding interactions between the central residues of the chain and the disulfide loop. The quenching experiments also indicate that the monomer of the human, amyloidogenic form of amylin is more compact than the rat form, which does not form amyloid. We discuss these newly observed differences between human and rat amylin in solution and their possible relation to aggregation and to the physiological function of amylin binding to the calcitonin receptor.
Introduction
Islet amyloid polypeptide (IAPP or amylin) is a 37-residue peptide that is cosecreted with insulin in the β-cells of the pancreas. Human IAPP (hIAPP) is the main component found in amyloid deposits of type II diabetes and forms amyloid fibrils in vitro (1,2). There is no known folded structure of the hIAPP monomer, and circular dichroism suggests that it is largely unstructured in aqueous solution (3,4). As for other amyloid peptides, the link between the states of this peptide observed in vitro and disease is not well understood (5–9).
A feature of all amylin variants is the disulfide loop between residues 2 and 7, which appears to be a functional requirement for amylin to stimulate the calcitonin receptor (10–12). The sequences of hIAPP, rat IAPP (rIAPP, which does not form fibrils), and two control peptides, referred to as cc-(AGQ)9 and c(AGQ)9, are shown in Fig. 1. There is extensive structural information on the monomeric form of rIAPP, because it does not aggregate even at high concentrations. NMR chemical shift data show significant helix propensity for residues 5–19, as well as structure in the disulfide loop (residues 2–7), whereas the C-terminal residues appear to be less structured (13,14). It has been suggested that an α-helical segment extending from the disulfide at residue 7 to residue 22 is formed when IAPP is dissolved in lipids (3,13,15) and when it is bound to the calcitonin-RAMP complex, and α-helical structures of the N-terminal 19 residues of both hIAPP and rIAPP incorporated in dodecylphosphocholine vesicles have recently been reported (16). The possible roles of the membrane-bound form in both initiation of fibril formation (3,17,18) and cell death, have been discussed (15,19,20). Recently, a structure of hIAPP in amyloid fibrils has been proposed by Tycko and co-workers, based on solid-state NMR restraints (21).
Figure 1.

Sequences of hIAPP (Human), rIAPP (Rat), and the control peptides cc-AGQ9 and c-AGQ9. The disulfide bond between residues 2 and 7, forming a loop at the N-terminus of the sequences, is indicated in blue. Note that the tyrosine at the C-terminus of native IAPP sequences (Y37) has been modified to a tryptophan (W37).
Unlike rIAPP, the conformational distribution of the monomeric state of hIAPP has remained elusive because of its fibril-forming propensity at the high concentrations required for many experiments. However, the properties of the monomer are expected to be important in the control of its assembly and in determination of receptor binding affinity. We have recently shown that monomeric hIAPP can be prepared in aqueous buffer by sedimentation before the assembly of fibrils is complete, yielding concentrations high enough for spectroscopic analysis, without the need for chemical denaturants or organic solvents. The supernatant contains only hIAPP monomers (22), which, although metastable relative to the fibrillar aggregate, persist long enough to be experimentally characterized.
To increase our understanding of the structure and dynamics of both hIAPP and rIAPP monomers in aqueous solution, we investigated the rates at which contacts are formed between chain termini within a monomer, using the rate of quenching of the triplet state of a C-terminal tryptophan by the N-terminal disulfide. These measurements can be readily carried out on short polypeptides at low concentrations and provide information on both the volume accessible to the chain ends, which depends on the end-to-end distance distribution, and the rate at which these conformations are sampled by end-to-end diffusion. The technique has been developed to obtain dynamical and structural information about a range of problems including disordered peptides, denatured proteins, the helix-coil transition, and ultrafast protein folding (23–26). Here, we have used tryptophan triplet quenching to measure the rate of contact formation between Trp37 and the Cys2-Cys7 disulfide in human and rat amylin both in high denaturant conditions and in aqueous buffer in the absence of denaturant. As a control for these studies, using the triplet quenching technique we investigated the properties of a well studied peptide sequence (25,27,28) to which the disulfide loop of hIAPP was added, cc-(AGQ)9 (Fig. 1). To provide a detailed structural interpretation of the results, we carried out all-atom molecular dynamics studies of all three peptides in water.
Theory of experiment
Quenching of the tryptophan triplet state by cysteine or disulfides is a short-range process, requiring that these side chains be roughly within 0.3 nm of contact (29). The quenching is described by a two-step kinetic model in which the ends of the peptide diffuse together with rate kD+ to form an encounter complex; the cystine then either quenches the tryptophan triplet state at a rate q or diffuses away at a rate kD−. The observed quenching rate (kobs) is given by
| (1) |
where the quenching yield, ϕ, is the probability of quenching during the lifetime of the encounter complex. Equation 1 can be rearranged to give
| (2) |
where the reaction-limited rate, kR, is given by kR = qKeq, and Keq (=kD+/kD−) is the equilibrium constant for forming the encounter complex. The reaction-limited and diffusion-limited (kD+) rates can be obtained experimentally by measuring the dependence of the observed quenching rates on solution viscosity (see the Supporting Material). By considering the contact formation as a one-dimensional diffusion along the end-to-end distance coordinate, the reaction- and diffusion-limited rates can be related to the end-to-end distance distribution, p(r), and the diffusion coefficient, D. In the case where the quenching rate is given by a δ-function, qδ(r − a), where a is the distance of the van der Waals contact, the Szabo, Schulten, Schulten theory gives the reaction-limited rates as .
For a Gaussian chain and small a, this reduces to
| (3) |
where . The diffusion-limited rate is given by
| (4) |
This simple case illustrates that both rates are inversely proportional to Vchain; in addition, kR depends on the quenching rate, q, whereas kD+ depends on the diffusion coefficient, D. Note that if both q and D are independent of solution conditions that alter Vchain, then both rates would scale identically. For example, increasing the average end-to-end distance by increasing chain length or by swelling the chain via repulsive interactions will increase Vchain and decrease both kR and kD+.
Methods
Sample preparation
Highly purified, lyophilized hIAPP, rIAPP, and cc-AGQ9 peptides, with amidated C-terminus and a disulfide bond between Cys2 and Cys7, were supplied by R. Tycko's group. For each experiment, solutions of monomeric IAPP (rat or human) and cc-AGQ9 were freshly prepared as described by Vaiana et al. (22). Buffer was 50 mM NaOAc at pH 4.9 with or without 6 M GdmHCl.
Tryptophan triplet quenching experiments
The dynamics of end-to-end contact formation were measured by monitoring the lifetime of the triplet state of tryptophan (at the N-terminus of the peptide), which is quenched by close contact with cystine and disulfides (at the C-terminus of the peptide) (25,28). The tryptophan is excited to its triplet state by a nanosecond ultraviolet laser pulse and the population of the triplet state as a function of time is monitored by time-resolved triplet-triplet absorption. For each sample, measurements were performed as a function of temperature at values ranging from 0° to 50°C. Viscosity dependence of the quenching rates was measured in sucrose solutions at concentrations ranging from 0 to 40% w/w in buffer and from 0 to 32% w/w in GdmCl.
Independent fits to determine relaxation rates, kobs, at each temperature
Data were initially independently fit to an exponential relaxation, together with a time-independent offset and a second offset function that was constant for times <10−4 s and varied linearly with log(t) for times >10−4 s (empirically accounting for the observed slower decay due to a radical photoproduct (25)). This yielded the exponential relaxation rates (kobs), which were plotted as a function of temperature for each peptide and solution condition (see Fig. 3).
Figure 3.
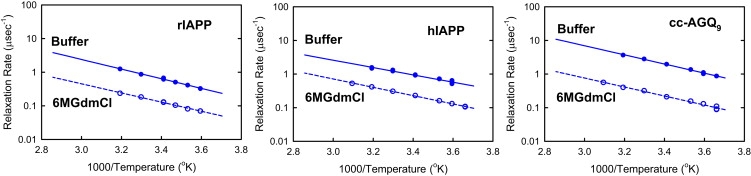
Arrhenius plots of the exponential relaxation rates (quenching rates) obtained from fits of Fig. 2 (and Fig. 4 and Fig. S2). Comparison between the three peptides, rIAPP, hIAPP, and cc-AGQ9, in 6 M GdmCl and in aqueous buffer.
Molecular dynamics simulations
Molecular dynamics simulations of c-(AGQ)9, cc-(AGQ)9, hIAPP, and rIAPP were run in explicit solvent using the GROMACS (30) molecular simulation package, with the Amber ff03 protein force field (31) and TIP3P water (32). Simulations of the AGQ-derived peptides were run for 94 ns, initiated from configurations chosen at 50-ps intervals from a constant volume simulation at 800 K, for a total of ∼0.75 μs of data. Several 20-ns trajectories of hIAPP and rIAPP, initiated from configurations having the same contacts as observed in the “kinked” cc-(AGQ)9 population, were run at 300 K. The temperature was maintained at 300 K using a Nose-Hoover (33,34) thermostat, and the pressure at 1 atm with a Parinello-Rahman (35) barostat.
Further details are provided in the Supporting Material.
Results
Quenching rates for rIAPP and hIAPP
To probe the end-to-end contact dynamics of IAPP, we mutated the C-terminal tyrosine to tryptophan and utilized the disulfide bridge between residues 2 and 7 as the quencher of tryptophan phosphorescence (Fig. 1). It is important to note that the tryptophan (W37) mutation introduces no observable differences in the fibril-forming properties of hIAPP (see Fig. S7 in the Supporting Material, as well as related text). The disulfide bridge produces a cystine, the quenching properties of which have been characterized previously in a cystine dipeptide and a cyclic disulfide lipoate (25,36). We find that the bimolecular rate at which the disulfide loop of amylin, KCNTATCA (ccA), quenches n-acetyl tryptophan amide (2.1 (108) M−1 s−1; see Fig. S1) is comparable to those measured for both cysteine and cystine (25), where we have previously established that quenching is almost completely reaction-limited. Consequently, we can assume that changes in rates produced by changing solution conditions are primarily due to changes in the disulfide-tryptophan distance distribution. A more sophisticated analysis, described in the Supporting Material, shows that this is indeed the case.
The decay of tryptophan triplet-triplet absorption measured after nanosecond excitation in 6 M GdmCl and in buffer is shown in Fig. 2 for hIAPP and in Fig. S2 for rIAPP. The measured absorbance traces show a fast exponential decay (corresponding to the decay of the triplet state) followed by a slowly decaying tail of much smaller amplitude (corresponding to the decay of a radical photoproduct) (25). Arrhenius plots of the exponential relaxation rates obtained by independently fitting the data for each temperature are shown in Fig. 3 (see the Methods section of the Supporting Material). Two results are immediately apparent: 1), the relaxation rates are ∼2-fold faster for hIAPP than for rIAPP under all conditions, indicating that rIAPP is always more expanded than hIAPP; and 2), the rates for both rIAPP and hIAPP increase by approximately a factor of 4 in aqueous buffer relative to 6 M GdmCl (Fig. 3), indicating a significant compaction of both peptides in buffer. This increase of the quenching rate is ∼2 times larger than that observed in earlier studies of c(AGQ)9 (27), a peptide of the same length as IAPP (Fig. 1). This result suggested that these peptides, which have larger, more hydrophobic residues, are experiencing larger attractive interactions than c(AGQ)9 in aqueous buffer, presumably due to attractive interactions between hydrophobic side chains.
Figure 2.
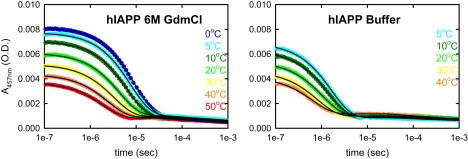
Decay of tryptophan triplet-triplet absorption measured after nanosecond excitation in solutions of monomeric hIAPP at different temperatures (50 mM NaOAc, pH 4.9, c∼70 μM) in 6 M GdmCl (left) and aqueous buffer (right). Solid lines represent independent fits using an exponential decay followed by a sloping baseline (see Methods section in the Supporting Material). Exponential decay corresponds to the quenching of the tryptophan triplet state by cystine (disulfide bridge between residues 2 and 7) due to end-to-end contact formation.
Quenching rates for cc-(AGQ)9
To test this hypothesis, we synthesized a control peptide, cc-(AGQ)9, in which the C-terminal cysteine of c(AGQ)n (25,27–29) was substituted with the disulfide terminal loop of IAPP (Fig. 1). The decay of tryptophan triplet-triplet absorption observed for this peptide is shown in Fig. 4 with the fitted exponential relaxation rates in Fig. 3. The striking result is that the rates for cc-(AGQ)9 increase even more in buffer relative to 6 M GdmCl than those for the IAPP sequences. An ∼9-fold increase is observed for cc-(AGQ)9, compared with the ∼4-fold increase for rIAPP and hIAPP (Fig. 3) and a <2-fold increase for c(AGQ)9 in the absence of the disulfide loop (27). These results indicate that the end-to-loop distribution of cc-(AGQ)9 becomes much more compact as the denaturant concentration is lowered, whereas a much smaller increase in size is inferred for c-(AGQ)9, strongly suggesting that the collapse results from attractive interactions between the repeating AGQ sequence and the disulfide loop. The most reasonable hypothesis is that the high local concentration of backbone residues and hydrogen-bonding side chains in the disulfide loop increases the probability of forming favorable interactions with the more C-terminal residues of cc-(AGQ)9. A control experiment in which the disulfide bond of cc-(AGQ)9 was reduced by adding tris(2-carboxyethyl)phosphine (a reducing agent) showed that the quenching rate for the reduced peptide increases only by a factor of 2 in going from buffer to 6 M GdmCl, similar to that for c-(AGQ)9. This result rules out the possibility that the collapse is due to the addition of the particular C-terminal sequence rather than the disulfide loop. The fact that the increase in rate for cc-(AGQ)9 is larger than that observed for either of the more hydrophobic hIAPP and rIAPP sequences further suggests that compaction of the amylin sequences does not result primarily from interactions between hydrophobic side chains, but instead must also be driven by interactions between the chain backbone and the disulfide loop.
Figure 4.
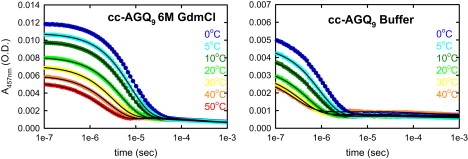
Decay of tryptophan triplet-triplet absorption for solutions of monomeric cc-AGQ9 (as in Fig. 2).
Molecular dynamics simulations of c-(AGQ)9 and cc-(AGQ)9
To test this hypothesis, we carried out molecular dynamics simulations of c-(AGQ)9 and cc-(AGQ)9 in water; for each peptide, eight independent trajectories were run, starting from randomized conformations drawn from a run at high temperature. It can be seen from the sample trajectories in Fig. 5, A and B, that both peptides sample a comparable range of end-to-end distances (3–35 Å) (remaining trajectories are given in Fig. S5). A striking difference, however, is that the c-(AGQ)9 trajectories often sample the entire distribution within the ∼90-ns timescale of a single MD run, whereas the cc-(AGQ)9 trajectories sample only small subsets of the distribution on this timescale. This requires that there be relatively long-lived interactions in the ensemble of states of cc-(AGQ)9. In the limited set of trajectories that have been calculated, the distances from the disulfide to the tryptophan (Fig. 6, A and B) for cc-(AGQ)9 are significantly smaller and have more defined structure than the cysteine-tryptophan distances in c-(AGQ)9. The major portion of the population is observed in a relatively long-lived set of states in which this distance is 12–5 Å.
Figure 5.

Sample trajectories of end-to-end distances from explicit solvent MD simulations of the control sequence AGQ9 with and without the addition of the N-terminal disulfide loop of amylin. (A) End-to-end distance without disulfide loop c-AGQ9. (B) End-to-end distance with disulfide loop cc-AGQ9. Red lines indicate the boundaries between the three subpopulations, selected on the basis of the distance between the tryptophan and a cysteine sulfur atom: “contact” (rw-c < 5 Å), “kinked” (5 Å < rw-c < 12 Å), and “open” (12 Å < rw-c). Insets in B show representative structures from the kinked population taken from each trajectory. (C) Average secondary structure formation in the kinked population calculated using STRIDE; red, α-helix; black, β-sheet.
Figure 6.
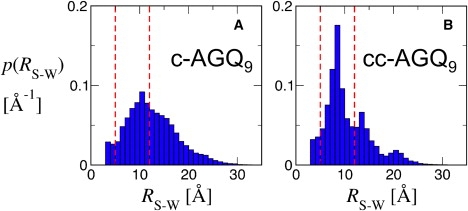
Distribution of end-to-end distances calculated from trajectories of c-AGQ9 (A) and cc-AGQ9 (B). Red lines indicate boundaries between “contact”, “kinked”, and “open” subsets (as in Fig. 5).
To determine the structural features responsible for this behavior, we examined three subpopulations from the trajectories selected on the basis of the distance between the tryptophan and a cysteine sulfur atom, rw-c: “contact”, with rw-c < 5 Å; “kinked”, with 5 Å < rw-c < 12 Å; and “open”, with 12 Å < rw-c. We calculated contact maps for these subsets from the c-(AGQ)9 and cc-(AGQ)9 trajectories. The borders of these categories are indicated by red dashed lines in Figs. 5 and 6 for reference. The maps for the contact and kinked subsets are shown in Fig. S6. In the kinked population of c-(AGQ)9, there is evidence for a β-hairpin-like structure with a turn that includes Gly9, which is present in about one-third of the population. A set of antiparallel hydrogen bonds between these residues would decrease the length of the free chain from 31 to ∼16 residues and contribute to the peak in the distribution at rw-c = 10–11 Å. In the contact subset, these interactions are even more probable, with occupancies as high as 0.5. In cc-(AGQ)9, a completely different set of interactions shortens the length of the free (AGQ)n sequence to <15 residues. In the kinked population, interactions between residues 2–4 of the loop and residues 10–20 in the repeating (AGQ)n sequence are present with probabilities as high as 0.5. These interactions become even more probable in the contact subset. Schematic illustrations of these populations are depicted in Fig. 7. The high probability of these interactions in the complete ensemble is therefore responsible for the decrease in end-to-end distance observed in the simulations of cc-(AGQ)9.
Figure 7.
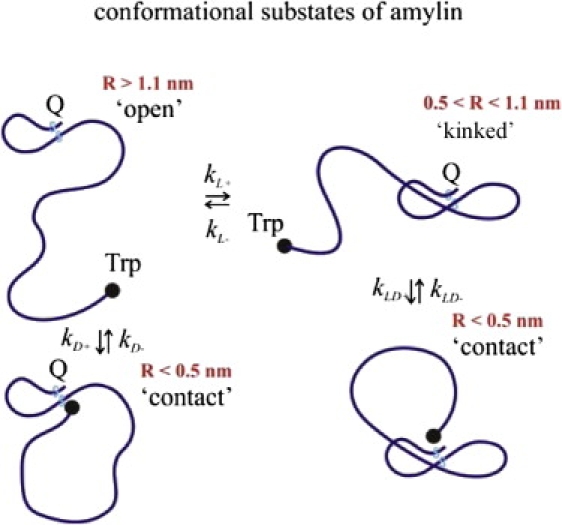
Schematic depiction of representative members of the “contact”, “kinked”, and “open” populations for peptides with the N-terminal disulfide loop.
When examined more closely, we find that a number of potential hydrogen bonds stabilize kinked conformations in which residues 10–20 in the (AGQ)n sequence interact with the disulfide loop. These interactions include backbone-backbone hydrogen bonds between residues 2 and 5 of the disulfide loop and residues 10 and 20 in the (AGQ)n sequence and side chain-backbone interactions between residues N3 and T4 and the same set of residues. The presence of these interactions effectively shortens the free (AGQ)n chain from ∼30 to ∼16 residues, dramatically increasing the rate of end-to-end contact formation. An important feature of the disulfide loop that favors the formation of stable hydrogen-bonded complexes is its very low flexibility. The backbone root mean-square fluctuations of the disulphide loop residues (2–7) over all 0.75 μs of simulation time is only ∼0.33 Å. The loop therefore provides a rigid template, reducing the entropy cost for the formation of intrapeptide hydrogen bonds. The residues from the (AGQ)n sequence that are most likely to be involved in these interactions with the loop are presumably selected by minimizing the loss in chain entropy required to immobilize this portion of the chain, and are therefore those closest to the disulfide (residues 10–20). However, we do also observe a significant population of long-range contacts between the disulfide loop and the C-terminus of the chain, whose formation may be facilitated by the intervening short-range contacts. In the “contact” population, the long-range contacts are replaced by a direct interaction of the terminal W37 side chain with the disulfide. Note that the most frequently formed contacts in the map do not arise from a single specific structure and that the precise interactions in the kinked population are quite heterogeneous (see structures in Fig. 5 B, inset). There is no persistent secondary structure in the kinked peptides, with only a small helical population near the C-terminus (Fig. 5 C); however, the force field used is known to be slightly biased in favor of helix formation. The effects we observe in sequences containing the amylin disulfide loop are much more pronounced than those observed for linear (gly-ser) peptides, for which the formation of random hydrogen bonds between backbone residues was invoked to explain the dependence of the loop formation dynamics and end-to-end distances on denaturant concentrations (37,38).
Molecular dynamics simulations of hIAPP and rIAPP
A series of molecular dynamics simulations of hIAPP and rIAPP was carried out starting from open states prepared at high temperature, as for cc-(AGQ)9. In these trajectories, the peptides often appear to have become “stuck” in configurations with lifetimes comparable to the length of the trajectories, so it was not possible to sample the conformational space occupied by these peptides even approximately. From the limited data, the duration of these events appears to be longer than in the cc-(AGQ)9 trajectories, suggesting that the addition of bulky, hydrophobic residues results in somewhat more stable intramolecular interaction in these peptides. Since these trajectories tended to become stuck in minima that could be reached rapidly from the starting conformations, they did not frequently sample conformations that contained the contacts found in the cc-(AGQ)9 simulations. In an effort to determine whether such kinked species might also be stable for the amylin sequences, a second series of short trajectories was calculated from starting conformations chosen at regular intervals from high-temperature simulations biased to favor the formation of contacts found in the contact maps obtained from the cc-(AGQ)9 simulations. These typically formed ∼60% of all the possible contacts observed in cc-(AGQ)9 and included species classified as “open”, “kinked”, and “contact” conformations. The rw-c distances from these trajectories, particularly those for which rw-c was <10 Å, varied less than those in Fig. 5, strongly suggesting that these contacts, or similar hydrogen-bonded interactions, may also be stable for the amylin sequences.
Determination of reaction- and diffusion-limited rates
Although we have assumed that the increase in observed tryptophan triplet quenching rates entirely reflects a decrease in average end-to-end distance (i.e., a change in the equilibrium end-to-end distance distribution), the observed quenching rates in principle also depend on the diffusional dynamics of the chain. To verify that the faster rates are not caused by faster dynamics, we measured the quenching rates as a function of solvent viscosity (cf. Lapidus et al. (28)) and analyzed the temperature and viscosity dependence as described in the Methods section of the Supporting Material (Fig. S3). This allows the reaction-limited rate kR (which entirely reflects changes in the equilibrium end-to-end distance distribution) to be separately determined from the effects of diffusion. The results from this analysis lead to conclusions qualitatively identical to those discussed above.
Arrhenius plots of the reaction-limited (kR) and diffusion-limited (ηkD+) rates obtained by this analysis are shown in Fig. S4. Since the temperature dependence of the rates in 6 M GdmCl is very similar for all three peptides, and analogous results were obtained in buffer, we confine our discussion to the rates at 20°C, the median temperature of our experiments. The values of kR and ηkD+ for cc-AGQ9, hIAPP, and rIAPP at this temperature are summarized in Table 1. As was observed for the rates shown in Fig. 3, in 6 M GdmCl, the values of kR and ηkD+ for hIAPP are very similar to those for cc-AGQ9, whereas for rIAPP, both rates are ∼2-fold slower. When the rates in buffer are compared to those in 6 M GdmCl, an ∼4-fold increase is observed in both kR and ηkD+ for both rIAPP and hIAPP, whereas a nearly sevenfold increase is observed for cc-AGQ9. As noted above, these increases are significantly larger than the almost twofold increase observed for c-AGQ9 in previous studies (27). The finding that dependence of kR on solution conditions is nearly identical to that of ηkD+ is consistent with our interpretation that the differences result primarily from interactions between the chain and the disulfide loop that alter Vchain by affecting the end-to-end distribution, peq(r) (cf. Eqs. 3 and 4).
Table 1.
Comparison of rates in aqueous buffer and 6 M GdmCl
|
kobs (μs−1) |
kR (μs−1) |
ηkD (cP μs−1) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peptide | Buffer | 6 M GdmCl | Buffer | 6 M GdmCl | Buffer | 6 M GdmCl | ||||||
| cc-AGQ9 | 2.0 ± 0.2 | 0.21 ± 0.02 | 9.5 ± 1.7 | 2.4 ± 0.2 | 0.35 ± 0.03 | 6.9 ± 1.2 | 6.4 ± 1.2 | 0.94 ± 0.17 | 6.8 ± 2.6 | |||
| hIAPP | 0.93 ± 0.07 | 0.23 ± 0.02 | 4.0 ± 0.7 | 1.2 ± 0.1 | 0.32 ± 0.03 | 3.8 ± 0.7 | 3.6 ± 0.6 | 1.3 ± 0.2 | 2.8 ± 1.1 | |||
| rIAPP | 0.64 ± 0.05 | 0.13 ± 0.01 | 4.9 ± 0.9 | 0.89 ± 0.07 | 0.19 ± 0.02 | 4.7 ± 0.8 | 2.0 ± 0.4 | 0.62 ± 0.11 | 3.2 ± 1.2 | |||
Uncertainty in kobs (∼9%) was estimated by calculating the standard deviation of log kobs values obtained from independent fits of the relaxation rates relative to the best-fit estimates obtained from the global fits of viscosity dependence at all temperatures. The number of degrees of freedom was (N – 2) × (number of temperature values – 1), where N is the total number of points. The errors for kR were assumed to be the same as for kobs (∼9%) and the error on ηkD was estimated from the error on the slope of a straight line passing through two values of kobs (by simple error propagation: δkD/kD = 2 × δ kobs/Δ kobs = 18%). The errors on the ratios obtained by error propagation are ∼13% for kR/kR and ∼26% for ηkD/ηkD.
Discussion
Difference between hIAPP and rIAPP
Comparing the three peptides in 6 M GdmCl, it is evident from both the raw data (Fig. 3) and the reaction- and diffusion-limited rates kR and ηkD+ (Fig. S4 and Table 1) that the chain volume for hIAPP is comparable to that for the control sequence cc-AGQ9, but that the chain volume for rIAPP is significantly larger than that of the control. We interpret these results as reflecting changes in the stiffness of the chain between the disulfide and the C-terminal tryptophan. The ordering between the three peptides is reasonable, given their differences in sequence. The additional bulky side chains in the hIAPP chain, which has 17, would be expected to result in somewhat larger excluded-volume interactions compared to the nine bulky side chains in cc-AGQ9, leading to increased occupancy of expanded configurations. In rIAPP, three additional residues from the hIAPP sequence with small side chains (Ala25, Ser28, and Ser29) are substituted with proline, which, because of both side-chain volume and restriction in accessible (ϕ–ψ) space, is expected to increase the stiffness of rIAPP. These results are consistent with a recent NMR study that appeared after submission of this article (39).
Chain volume and dynamics: role of the disulfide loop
Taken together, our results for hIAPP, rIAPP, and cc-AGQ9 in buffer and 6 M GdmCl, when compared with those for c-AGQ9, show that there is significant collapse of all three peptides in buffer relative to their structure in 6 M GdmCl and suggest that, instead of side-chain hydrophobic interactions, hydrogen bonding between the N-terminal disulfide loop and the rest of the backbone plays an important role in the collapse of these peptides (40). The simulations support this interpretation and indicate that the collapse of cc-AGQ9 is driven primarily by hydrogen-bonding backbone-backbone and backbone-sidechain interactions between the chain and the disulfide loop. The simulations also support the contention that these interactions play a significant role in the collapse of the IAPP sequences. The smaller increases observed for hIAPP and rIAPP, however, suggest that the bulky side chains in the IAPP sequences in some way moderate the extent of collapse of these peptides relative to cc-AGQ9.
Our finding that the collapse of cc-AGQ9 is significantly enhanced by the introduction of the disulfide loop can be rationalized in terms of the effective contact order (ECO), proposed by Fiebig and Dill (41,42). ECO is defined as the length of the shortest path (following covalent or noncovalent bonds between adjacent residues) between two residues, i and j, that form a contact. Fiebig and Dill pointed out that the entropic cost of forming a contact is determined not by the positions of two residues in sequence, but by the ECO. The stable disulfide loop in IAPP and in our model peptide cc-AGQ9 serves to decrease the ECO for interactions between residues within the loop. As shown by our simulations, this increases the occupancy of an ensemble of conformations that has clusters of weak interactions between residues within the loop and residues in the free chain that are closest to the loop, resulting in shortened distances between the disulfide and the C-terminal tryptophan. By clustering several potential interaction sites, formation of the disulfide loop increases the probability that multiple interactions will occur within the kinked subensemble, lowering its free energy. From a kinetic perspective, this additional constraint accelerates the conformational search for interacting partners by >100-fold relative to the free chain. The effective decrease in free energy required to produce a kinked state that has significant occupancy can be estimated from the diffusional lifetime of the pair state, kD− ≈ 1–2 × 108 s−1 (25), and the diffusion-limited rate of loop formation, kD+ ≈ 2–4 × 106 s−1, to be ΔG ∼ –RTln(kD/kD+) = –2 kcal/mol, which would require that the ensemble contain, on average, only a couple of hydrogen bonds.
Possible functional significance of the disulfide loop
The presence of the disulfide loop could modulate interactions between amylin and other proteins in a number of ways. First, the formation of intramolecular hydrogen bonds between residues in the chain and the loop will disfavor the formation of structural features in which these residues participate in other intramolecular hydrogen bonds. In particular, the formation of both solvated and transmembrane helices would be expected to be less probable in the presence of the loop. Second, the loop could serve as a template for the formation of intermolecular interactions with hydrogen bonding partners from other proteins. The interactions between the disulfide loop and the AGQ backbone are quite weak: the dissociation constant, estimated as the reciprocal of the chain volume for a 7- to 10-residue chain, is 40–100 mM, consistent with the observation in sedimentation studies of rIAPP and hIAPP (22) that the population of dimers is negligible at loading concentrations of ∼50 μM. The probability of forming such interactions will be determined, to a large degree, by the entropy cost that must be paid to form them. For a free chain, the entropy loss should be comparable to the ΔSconf of ∼4 eu for folding (43), so the affinity of the loop for an immobilized chain having an optimal conformation could increase by as much as a factor of 105. Interactions with a region of a chain that is already constrained by other interactions to be close in space to the loop would be particularly likely and could compete with intramolecular interactions.
The N-terminal disulfide loop between Cys2 and Cys7 is highly conserved among IAPP sequences, a structural feature also shared with the larger family of genetically, structurally, and functionally related hormone peptides: calcitonin (Ct), calcitonin gene-related peptide, and adrenomedullin, referred to as the Ct peptide family. The loop has been shown to be necessary for these peptides to activate cell response when binding to their receptors, although its absence does not prevent binding with high affinity. The capacity of the loop to form multiple H-bond interactions could play a role in modulating binding and receptor activation. H-bonding of residues in the transmembrane sequence to the loop could, for example, reduce affinity of this portion of the sequence for the receptor. Conversely, the transmembrane binding of the peptide (thought to involve some of residues 8–37) to its receptor (12), would be expected to favor attractive intermolecular interactions between the loop and other nearby extracellular portions of the receptor. It has been suggested that this is a necessary step for biological activation (12)).
Although the available structural data argue that the disulfide loop does not participate in the core structure of the amyloid fiber (21), it has been shown to alter the aggregation kinetics (44). Our observations suggest that fibrillization might be modulated by the loop in several ways. First, the formation of intramolecular interactions could compete with fibril formation by decreasing the population of chains that can adopt the fibril-forming structure. This is consistent with the finding that removing the disulfide bond eliminates the “activation phase” in seeded kinetics of hIAPP (44). Such a scenario, in which assembly occurs via an “assembly-competent” structure (N∗) (45), has been observed in several simulation studies (46,47), where it has also been found that altering the kinetic barrier separating the natively unfolded state U from N∗, can have a large effect on the aggregation kinetics (47). Second, the finding that under all solution conditions studied, rIAPP preferentially occupies states with larger end-to-end distances compared to hIAPP is expected to affect both stability and kinetics of fibril formation, though it is unlikely that this alone causes the dramatic differences observed between rIAPP and hIAPP. Third, the presence of an array of potential hydrogen-bonding groups at the ends of the growing fibril could provide a template to which the disulfide loop could bind. An intriguing possibility is that the disulfide loops within the fibril may act as secondary nucleation sites, as has been recently suggested (44).
Summary and Conclusions
We find that the presence of the N-terminal disulphide loop of amylin (residues 2–7) induces a significant collapse of the three peptides studied, hIAPP, rIAPP, and the model peptide cc-AGQ9 in aqueous buffer. We were surprised to find that the collapse is greatest for the least hydrophobic sequence, cc-AGQ9, and is dramatically enhanced by the introduction of the disulfide loop, suggesting that the collapse is driven by backbone-backbone and backbone-side chain interactions rather than by interactions between hydrophobic side chains. Molecular dynamics simulations on this control sequence show that hydrogen-bonding interactions between the central residues of the chain and the disulfide loop reduce the length of the free chain by ∼2-fold. We interpret this in terms of ECO, which is dramatically reduced in the presence of the disulphide bond and favors the formation of clusters of individually short-lived H-bonds, stabilizing the population of a kinked state. We propose that a similar mechanism is responsible for the observed collapsed state of rIAPP and hIAPP in buffer, that is, that these peptides collapse by forming a significantly occupied, kinked subensemble of states stabilized primarily by the formation of backbone-backbone and side chain-backbone hydrogen bonds between residues in the N-terminal disulfide loop and residues nearby in the linear portion of the polypeptide chain. In this case, though, the presence of large hydrophobic residues and the increased stiffness of the IAPP sequences relative to cc-AGQ9 act to screen the attractive interactions, causing a reduced collapse relative to that of the more flexible and minimally hydrophobic sequence cc-AGQ9.
Finally, we note that the experimental results described here represent the first, to our knowledge, direct comparison between monomeric rIAPP and hIAPP in aqueous solution to show a clear difference in the conformational states sampled by these two peptides (these results are consistent with a recent NMR study that appeared after we submitted this article (39)). The differences in the chain properties point out the importance of both topology and sequence in determining the extent of collapse and suggests that collapse is likely to occur in other disordered peptides featuring tight N-terminal disulphide loops, with the sequence modulating the collapse through excluded volume and specific side chain-side chain interactions. The differences in rIAPP and hIAPP that we observe, however, do not appear to be capable of rationalizing the large differences in fibrillogenesis and pathogenesis of these peptides, so interpeptide interactions clearly need to be considered. Further studies are needed to understand how conformational states sampled by the monomer in solution are linked to its aggregation propensity. Understanding the interplay between intra- and interprotein interactions remains the main challenge for understanding amyloid aggregation and disease.
Supporting Material
Methods, eight figures, and references are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01432-5.
Supporting Material
Acknowledgments
We thank Rob Tycko's group for supplying the peptides.
S.M.V., W.M.Y., W.A.E., and J.H. were supported by the intramural research program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, and made use of the Biowulf cluster at the National Institutes of Health. R.B.B. is supported by a Royal Society University Research Fellowship.
Footnotes
Sara M. Vaiana's present address is Dept. of Physics, Arizona State University, Tempe AZ 85287-1504.
Contributor Information
Sara M. Vaiana, Email: sara.vaiana@asu.edu.
James Hofrichter, Email: jameshof@niddk.nih.gov.
References
- 1.Westermark P., Wernstedt C., Wilander E., Hayden D.W., Obrien T.D. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc. Natl. Acad. Sci. USA. 1987;84:3881–3885. doi: 10.1073/pnas.84.11.3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cooper G.J.S., Willis A.C., Clark A., Turner R.C., Sim R.B. Purification and characterization of a peptide from amyloid-rich pancreases of type-2 diabetic patients. Proc. Natl. Acad. Sci. USA. 1987;84:8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jayasinghe S.A., Langen R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry. 2005;44:12113–12119. doi: 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- 4.Kayed R., Bernhagen J., Greenfield N., Sweimeh K., Brunner H. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J. Mol. Biol. 1999;287:781–796. doi: 10.1006/jmbi.1999.2646. [DOI] [PubMed] [Google Scholar]
- 5.Hoppener J.W.M., Lips C.J.M. Role of islet amyloid in type 2 diabetes mellitus. Int. J. Biochem. Cell Biol. 2006;38:726–736. doi: 10.1016/j.biocel.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 6.Lansbury P.T., Lashuel H.A. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- 7.Lorenzo A., Razzaboni B., Weir G.C., Yankner B.A. Pancreatic-islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature. 1994;368:756–760. doi: 10.1038/368756a0. [DOI] [PubMed] [Google Scholar]
- 8.Kapurniotu A. Arnyloidogenicity and cytotoxicity of islet amyloid polypeptide. Biopolymers. 2001;60:438–459. doi: 10.1002/1097-0282(2001)60:6<438::AID-BIP10182>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 9.Jaikaran E.T.A.S., Clark A. Islet amyloid and type 2 diabetes: from molecular misfolding to islet pathophysiology. Biochim. Biophys. Acta. 2001;1537:179–203. doi: 10.1016/s0925-4439(01)00078-3. [DOI] [PubMed] [Google Scholar]
- 10.Hay D.L., Christopoulos G., Christopoulos A., Poyner D.R., Sexton P.M. Pharmacological discrimination of calcitonin receptor: receptor activity-modifying protein complexes. Mol. Pharmacol. 2005;67:1655–1665. doi: 10.1124/mol.104.008615. [DOI] [PubMed] [Google Scholar]
- 11.Hay D.L., Christopoulos G., Christopoulos A., Sexton P.M. Amylin receptors: molecular composition and pharmacology. Biochem. Soc. Trans. 2004;32:865–867. doi: 10.1042/BST0320865. [DOI] [PubMed] [Google Scholar]
- 12.Conner A.C., Simms J., Barwell J., Wheatley M., Poyner D.R. Ligand binding and activation of the CGRP receptor. Biochem. Soc. Trans. 2007;35:729–732. doi: 10.1042/BST0350729. [DOI] [PubMed] [Google Scholar]
- 13.Williamson J.A., Miranker A.D. Direct detection of transient α-helical states in islet amyloid polypeptide. Protein Sci. 2007;16:110–117. doi: 10.1110/ps.062486907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yonemoto I.T., Kroon G.J.A., Dyson H.J., Balch W.E., Kelly J.W. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry. 2008;47:9900–9910. doi: 10.1021/bi800828u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jayasinghe S.A., Langen R. Membrane interaction of islet amyloid polypeptide. Biochim. Biophys. Acta. 2007;1768:2002–2009. doi: 10.1016/j.bbamem.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 16.Nanga R.P.R., Brender J.R., Xu J., Veglia G., Ramamoorthy A. Structures of rat and human islet amyloid polypeptide IAPP(1–19) in micelles by NMR spectroscopy. Biochemistry. 2008;47:12689–12697. doi: 10.1021/bi8014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knight J.D., Miranker A.D. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 2004;341:1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 18.Knight J.D., Hebda J.A., Miranker A.D. Conserved and cooperative assembly of membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry. 2006;45:9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 19.Engel M.F.M., Khemtemourian L., Kleijer C.C., Meeldijk H.J.D., Jacobs J. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA. 2008;105:6033–6038. doi: 10.1073/pnas.0708354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brender J.R., Hartman K., Reid K.R., Kennedy R.T., Ramamoorthy A. A single mutation in the nonamyloidogenic region of islet amyloid polypeptide greatly reduces toxicity. Biochemistry. 2008;47:12680–12688. doi: 10.1021/bi801427c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luca S., Yau W.M., Leapman R., Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaiana S.M., Ghirlando R., Yau W.M., Eaton W.A., Hofrichter J. Sedimentation studies on human amylin fail to detect low-molecular-weight oligomers. Biophys. J. 2008;94:L45–L47. doi: 10.1529/biophysj.107.125146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buscaglia M., Kubelka J., Eaton W.A., Hofrichter J. Determination of ultrafast protein folding rates from loop formation dynamics. J. Mol. Biol. 2005;347:657–664. doi: 10.1016/j.jmb.2005.01.057. [DOI] [PubMed] [Google Scholar]
- 24.Buscaglia M., Schuler B., Lapidus L.J., Eaton W.A., Hofrichter J. Kinetics of intramolecular contact formation in a denatured protein. J. Mol. Biol. 2003;332:9–12. doi: 10.1016/s0022-2836(03)00891-x. [DOI] [PubMed] [Google Scholar]
- 25.Lapidus L.J., Eaton W.A., Hofrichter J. Measuring the rate of intramolecular contact formation in polypeptides. Proc. Natl. Acad. Sci. USA. 2000;97:7220–7225. doi: 10.1073/pnas.97.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lapidus L.J., Eaton W.A., Hofrichter J. Measuring dynamic flexibility of the coil state of a helix-forming peptide. J. Mol. Biol. 2002;319:19–25. doi: 10.1016/S0022-2836(02)00193-6. [DOI] [PubMed] [Google Scholar]
- 27.Buscaglia M., Lapidus L.J., Eaton W.A., Hofrichter J. Effects of denaturants on the dynamics of loop formation in polypeptides. Biophys. J. 2006;91:276–288. doi: 10.1529/biophysj.105.071167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lapidus L.J., Steinbach P.J., Eaton W.A., Szabo A., Hofrichter J. Effects of chain stiffness on the dynamics of loop formation in polypeptides. Appendix: testing a 1-dimensional diffusion model for peptide dynamics. J. Phys. Chem. B. 2002;106:11628–11640. [Google Scholar]
- 29.Lapidus L.J., Eaton W.A., Hofrichter J. Dynamics of intramolecular contact formation in polypeptides: distance dependence of quenching rates in a room-temperature glass. Phys. Rev. Lett. 2001;87:258101. doi: 10.1103/PhysRevLett.87.258101. [DOI] [PubMed] [Google Scholar]
- 30.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 31.Sorin E.J., Pande V.S. Exploring the helix-coil transition via all-atom equilibrium ensemble simulations. Biophys. J. 2005;88:2472–2493. doi: 10.1529/biophysj.104.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jorgensen W.L., Chandrasekhar J., Madura J.D. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 33.Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 34.Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984;52:255–268. [Google Scholar]
- 35.Parinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- 36.Gonnelli M., Strambini G.B. Phosphorescence lifetime of tryptophan in proteins. Biochemistry. 1995;34:13847–13857. doi: 10.1021/bi00042a017. [DOI] [PubMed] [Google Scholar]
- 37.Moglich A., Joder K., Kiefhaber T. End-to-end distance distributions and intrachain diffusion constants in unfolded polypeptide chains indicate intramolecular hydrogen bond formation. Proc. Natl. Acad. Sci. USA. 2006;103:12394–12399. doi: 10.1073/pnas.0604748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moglich A., Krieger F., Kiefhaber T. Molecular basis for the effect of urea and guanidinium chloride on the dynamics of unfolded polypeptide chains. J. Mol. Biol. 2005;345:153–162. doi: 10.1016/j.jmb.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 39.Soong R., Brender J.R., Macdonald P.M., Ramamoorthy A. Association of highly compact type II diabetes related islet amyloid polypeptide intermediate species at physiological temperature revealed by diffusion NMR spectroscopy. J. Am. Chem. Soc. 2009;131:7079–7085. doi: 10.1021/ja900285z. [DOI] [PubMed] [Google Scholar]
- 40.Bolen D.W., Rose G.D. Structure and energetics of the hydrogen-bonded backbone in protein folding. Annu. Rev. Biochem. 2008;77:339–362. doi: 10.1146/annurev.biochem.77.061306.131357. [DOI] [PubMed] [Google Scholar]
- 41.Fiebig K.M., Dill K.A. Protein core assembly processes. J. Chem. Phys. 1993;98:3475–3487. [Google Scholar]
- 42.Dill K.A., Fiebig K.M., Chan H.S. Cooperativity in protein-folding kinetics. Proc. Natl. Acad. Sci. USA. 1993;90:1942–1946. doi: 10.1073/pnas.90.5.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henry E.R., Eaton W.A. Combinatorial modeling of protein folding kinetics: free energy profiles and rates. Chem. Phys. 2004;307:163–185. [Google Scholar]
- 44.Koo B.W., Miranker A.D. Contribution of the intrinsic disulfide to the assembly mechanism of islet amyloid. Protein Sci. 2005;14:231–239. doi: 10.1110/ps.041051205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thirumalai D., Klimov D.K., Dima R.I. Emerging ideas on the molecular basis of protein and peptide aggregation. Curr. Opin. Struct. Biol. 2003;13:146–159. doi: 10.1016/s0959-440x(03)00032-0. [DOI] [PubMed] [Google Scholar]
- 46.Dima R.I., Thirumalai D. Exploring protein aggregation and self-propagation using lattice models: phase diagram and kinetics. Protein Sci. 2002;11:1036–1049. doi: 10.1110/ps.4220102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reddy G., Straub J.E., Thirumalai D. Influence of preformed Asp23-Lys28 salt bridge on the conformational fluctuations of monomers and dimers of Aβ peptides with implications for rates of fibril formation. J. Phys. Chem. B. 2009;113:1162–1172. doi: 10.1021/jp808914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.