

Abstract
The Dynameomics project aims to simulate a representative sample of all globular protein metafolds under both native and unfolding conditions. We have identified protein unfolding transition state (TS) ensembles from multiple molecular dynamics simulations of high-temperature unfolding in 183 structurally distinct proteins. These data can be used to study individual proteins and individual protein metafolds and to mine for TS structural features common across all proteins. Separating the TS structures into four different fold classes (all proteins, all-α, all-β, and mixed α/β and α + β) resulted in no significant difference in the overall protein properties. The residues with the most contacts in the native state lost the most contacts in the TS ensemble. On average, residues beginning in an α-helix maintained more structure in the TS ensemble than did residues starting in β-strands or any other conformation. The metafolds studied here represent 67% of all known protein structures, and this is, to our knowledge, the largest, most comprehensive study of the protein folding/unfolding TS ensemble to date. One might have expected broad distributions in the average global properties of the TS relative to the native state, indicating variability in the amount of structure present in the TS. Instead, the average global properties converged with low standard deviations across metafolds, suggesting that there are general rules governing the structure and properties of the TS.
Introduction
The major rate-limiting step is a critical juncture along the protein folding pathway. The corresponding transition state (TS) ensemble contains contacts that are important for folding, including nonnative interactions that cannot be predicted solely from the study of native state structures. Characterizing the general properties of the TS ensemble can help us to deduce which residues play important roles in the protein folding/unfolding pathway.
The TS is inherently unstable and difficult to characterize experimentally. High-temperature, all-atom molecular dynamics (MD) unfolding simulations have been used to obtain atomic-level detail about putative TS structures. For example, a combination of MD simulations and experimental Ф-value analysis revealed a diffuse TS for chymotrypsin inhibitor 2 (CI2), with a small cluster of hydrophobic residues that act as the nucleus for folding (1–5). Experimental folding studies of SH3 domains have revealed a more polarized TS structure, with a nucleus for folding but little structure elsewhere along the sequence (6–8). MD simulations of SH3 domains have revealed atomic-level detail of the TS structures and emphasized the importance of the native topology in determining the folding/unfolding pathway (9–11). Experimental data are available for only a small number of proteins, thus far limiting our ability to study the general features of the TS ensemble across all proteins.
We have used MD simulations of both high- and moderate-temperature unfolding, as well as simulations including chemical denaturants (2,12), to characterize TS ensembles for nearly 20 years (1,2,13–18). We have extensively characterized the unfolding behavior for CI2, as well as other systems, including the engrailed homeodomain (EnHD) (15), c-myb (19), E3BD (20), WW domains (16,21,22), protein A (23,24), the FF domain (25,26), α-spectrin (27), FKBP12 (14), and barnase (13). In each case, we compared the unfolding pathways from simulation to all available experimental data.
To illustrate in particular what our approach can provide with respect to prediction, the TS of EnHD contains nativelike secondary structure and a partially packed hydrophobic core, which is consistent with a framework mechanism of folding. The calculated and experimental Φ-values for the TS are in good agreement (R = 0.85) (15,28). The simulated unfolding process is independent of temperature, and essentially the same transition states are obtained at 348, 373, and 498 K (17,29). The TS was done as a prediction: the TS structures and their description were first published in 2000 (29). The experimental results became available 3 years later, and we published the MD and experimental Φ-value analysis together in 2003 (15). From the transition state, reorientation of the helices, expansion, and disruption of the helix docking lead to the intermediate state. This intermediate state has a high helical content and few tertiary contacts. We first reported this structure in 2000, and expanded the description in 2003 and 2004 (17,28,29). In 2005, the structure of the intermediate was determined by NMR, and it is very similar to our prediction (30).
In addition, we have directly demonstrated the principle of microscopic reversibility, showing that the unfolding pathway is the same as the folding pathway in continuous simulations at the Tm for EnHD (31) and CI2 (32). The TSs identified from unfolding simulations at a range of temperatures show similar amounts of structure for EnHD (15,17), CI2 (2,33), and FBP28 WW domain (16); that is, raising the temperature affects the rate of the process but not the pathway. Furthermore, we have found that the urea-induced TS is similar to the thermally derived TS for CI2, in agreement with experiment (2). However, neither single-molecule pulling nor force-induced unfolding yielded the same TS as either bulk experiments or thermal unfolding by MD for barnase (it came as no surprise that pulling altered the pathway) (34). Given our success with previous systems, and our desire to more systematically and thoroughly sample fold space, as part of the Dynameomics project (35), we have extended our approach to the identification and comprehensive characterization of TS properties from MD simulations in a high-throughput manner across a diverse set of protein metafolds.
The first step in the Dynameomics project was the identification of a set of 1129 protein metafolds that represent essentially all known globular protein structures in the Protein Data Bank (36). Our initial goal is to simulate a single representative from each fold where possible, as there is evidence that the properties of the folding pathway are shared across members of a fold family (15,37,38). To date, we have studied the native state behavior of a set of 188 structurally diverse proteins (35), which represent ∼67% of all known protein structures. The native simulations are stable and agree well with NMR chemical shifts, NMR nuclear Overhauser effect crosspeaks, and crystallographic B-factors for those proteins with readily accessible experimental data for comparison (35). These native simulations now serve as a reference for our study of protein folding/unfolding pathways.
Here, we present the properties of the TS ensemble identified from the high-temperature unfolding simulations of this 188-protein set. These results represent the largest, most comprehensive description of protein folding/unfolding TS ensemble to date. These data can now be used to study individual proteins and fold families and to mine for TS structural features across protein folds.
Methods
From 181 Dynameomics protein metafolds, 188 proteins were taken to be considered here (35). A single native state 298 K simulation was performed for each protein, and the analyses have been described (35,39). Each protein was also simulated multiple times at high temperature (498 K) to study unfolding pathways, resulting in over 1300 high-temperature unfolding simulations. All simulations were performed using in lucem molecular mechanics (40) and the potential function of Levitt et al. (41) with the microcanonical ensemble (NVE, or constant number of particles, volume, and energy).
Simulation protocol
Each starting structure was obtained from the Protein Data Bank (42), and any missing atoms were added. All Cys residues were reduced to allow unfolding in the absence of disulfide bonds. Each target protein of interest was minimized for 1000 steps of steepest-descent minimization in vacuo. The structure was then solvated in a periodic box of flexible F3C water molecules (43) with a density of 0.829 g/ml (44) for 498 K. The solvent box extended at least 10 Å from the protein in all dimensions. The water molecules alone were then minimized for 500 steps, followed by 1 ps of MD. The energy of the water molecules was then minimized for another 500 steps, followed by 500 steps of minimization of the protein. Each simulation used a 2-fs time step for integration and an 8-Å force-shifted cutoff for nonbonded interactions (41,45). We ran two simulations for at least 31 ns each at 498 K for each protein. To further sample the TS and early unfolding events, we ran three to five additional shorter simulations (for at least 2 ns) at 498 K. Specifics of the methods have been presented previously (46).
TS identification
We identified TS ensembles from unfolding simulations using the conformational clustering method of Li and Daggett (5,47). In short, the Cα root mean-square deviation (RMSD) was calculated between each pair of structures from the unfolding simulation. We used classic multidimensional scaling on the resulting Cα RMSD matrix to produce a three-dimensional representation of the data. Structures with similar Cα RMSDs cluster together in the 3D projection. The 3D representation was then examined visually to identify cluster exits. The TS ensemble was defined as the 5-ps window of structures immediately preceding the exit from the first, nativelike cluster. To identify the TS ensemble, we typically zoom in on the early events in the unfolding pathway by clustering separately over the first 500 ps, 1 ns, and 2 ns. We independently confirmed the TS ensembles using a newly developed one-dimensional reaction coordinate based on 15 physical properties (R. D. Toofanny, A. L. Jonsson, and V. Daggett, unpublished results), in this case the sparsely populated region between the native and the denatured states.
Simulation analysis
All analyses were performed with in lucem molecular mechanics (40). The standard analyses have been described previously (35). We accessed the Dynameomics database (49,50) to average properties over the TS structures from the unfolding simulations. Pairwise residue contacts were also computed for the aggregate TS ensemble and compared with the native-state simulations. The number of times each pairwise contact was present in the TS ensemble for one protein was calculated and then divided by the number of structures used in the calculation. To determine the average properties over all proteins, the average pairwise contacts for each protein were combined and divided by the total number of proteins. The same calculation was then carried out over 1–21 ns of all 298-K simulations. The native value was then subtracted from the value for the TS ensemble. Neighboring (i → i and i → i + 1) contacts were excluded in all cases.
Results
We identified putative TS ensembles from unfolding simulations for 183 of the 188 proteins in our data set. The remaining five proteins (Protein Data Bank codes 1du5, 1f8d, 1vmo, 2sil, and 2trc) unfolded too rapidly for the TS ensemble to be identified. All 498 K simulations were subjected to our standard analyses (35). Structure indices, or S values (1), were calculated over the TS ensemble for each simulation and averaged for each protein. S values are a product of two terms, S2° and S3°. S3° is the ratio of number of contacts made in the TS structures to the number of contacts in the reference structure. Contacts were calculated on a per-atom basis, excluding hydrogen atoms and atoms in adjacent residues. S2° is the ratio of native secondary structure present in the TS ensemble. Those residues residing in α-helix or β-strand in the native state were required to maintain their native secondary structure (for helix, −100° < ϕ < −30°, −80° < ψ < −5°; for β-strand, −170° < ϕ < −50°, 80° < ψ < −170°). All other residues were considered to maintain their native structure if their (ϕ,ψ) angles were within 35° of the corresponding angles in the reference structure (the simulation starting structure).
The resulting data have been analyzed at three different levels. First, the global TS properties were calculated over the whole data set and over particular fold classes. Second, we looked for trends on a per-residue basis, using S3° and S as measures of structure in the TS ensemble. Finally, we examined the distributions of pairwise contacts in the TS ensemble relative to the native state. Regarding fold classes, we examined four categories: all proteins, all-α, all-β, and mixed α/β proteins. The classes were defined using a combination of SCOP (51) and CATH (52) fold-type classifications. The all-α fold class contains 46 proteins from our data set, 44 are in the all-β class, and 84 are mixed α/β. Nine proteins in our set did not fall into these categories according to SCOP and CATH.
Global TS ensemble properties
The mean value over the TS ensemble for each protein was calculated for 27 physical properties. To compare among proteins, two different approaches were taken. First, the mean TS ensemble value for each protein was divided by the mean over the native simulation (35). The resulting values are displayed as a histogram (Fig. 1) and as the mean and SD calculated over the four fold classes (Table 1 and Table S1 in the Supporting Material). These results show the average change in the TS ensemble relative to the native state. There was a loss of total and native contacts, as well as hydrogen bonds, to ∼75% of the value from the native state simulations (Fig. 1, A–C). At the same time, the total solvent accessible surface area (SASA) of the TS ensemble was ∼30% greater (Fig. 1 D) and there was a 10% increase in radius of gyration (Table 1). The SDs overlapped for all four fold classes for all properties, indicating no significant difference between the fold classes in the average fractional change when compared to the native state.
Figure 1.
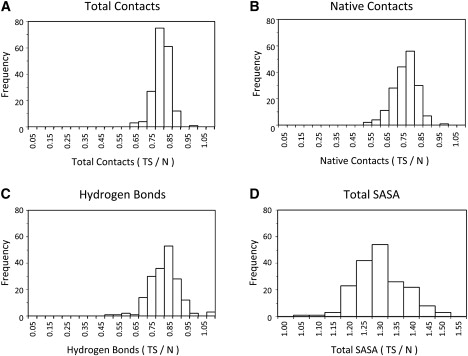
Typical distributions for the ratio of TS properties to native-state properties over all proteins. For each property, the mean TS value is calculated over all structures in the TS ensemble and the mean native state value. The ratio of the mean TS value over the mean native state value is plotted as a histogram.
Table 1.
Properties of the aggregate TS ensemble of 183 proteins relative to the aggregate native-state ensemble
| Property | Fold class |
|||
|---|---|---|---|---|
| All | All-α | All-β | Mixed α/β | |
| Total contacts | 0.79 (0.05) | 0.82 (0.05) | 0.77 (0.07) | 0.79 (0.04) |
| Native contacts | 0.75 (0.07) | 0.80 (0.08) | 0.70 (0.09) | 0.75 (0.06) |
| Nonnative contacts | 1.16 (0.35) | 1.14 (0.40) | 1.25 (0.43) | 1.13 (0.27) |
| Hydrogen bonds | 0.80 (0.09) | 0.86 (0.07) | 0.74 (0.10) | 0.80 (0.07) |
| Hydrophobic contacts | 0.73 (0.04) | 0.74 (0.05) | 0.73 (0.06) | 0.73 (0.04) |
| Other contacts | 0.82 (0.04) | 0.84 (0.04) | 0.80 (0.05) | 0.82 (0.04) |
| % α-structure (ϕ,ψ) | 0.79 (0.10) | 0.80 (0.10) | 0.77 (0.15) | 0.80 (0.08) |
| % β-structure (ϕ,ψ) | 1.06 (0.21) | 1.29 (0.31) | 0.96 (0.12) | 0.99 (0.07) |
| MC SASA | 1.35 (0.11) | 1.31 (0.10) | 1.38 (0.16) | 1.37 (0.10) |
| SC SASA | 1.26 (0.08) | 1.24 (0.08) | 1.26 (0.10) | 1.28 (0.07) |
| Total SASA | 1.28 (0.08) | 1.25 (0.07) | 1.28 (0.10) | 1.30 (0.08) |
| Cα RMSD | 1.85 (0.57) | 1.81 (0.63) | 1.93 (0.67) | 1.79 (0.47) |
| Cα radius of gyration | 1.09 (0.05) | 1.09 (0.06) | 1.10 (0.06) | 1.09 (0.05) |
| CONGENEAL Score | 1.90 (0.44) | 1.81 (0.45) | 2.03 (0.56) | 1.88 (0.37) |
For each property, the ratio of the mean value over the TS ensemble to the mean over the 298 K simulation (<xTS>/<xN>) was calculated. The values given in this table are the mean of <xTS>/<xN> over all proteins, with the SD in parentheses. MC, main chain; SC, side chain.
Next, the mean TS ensemble value for appropriate properties was divided by the number of residues in the protein and the mean ± SD was calculated over sets of proteins (Table 2 and Table S2). This approach allowed us to compare proteins of different sizes. Once again, there was no significant difference in the values across protein fold classes. Six properties—the fraction of residues with helical (ϕ,ψ) angles, the fraction of residues with β-(ϕ,ψ) angles, Cα RMSD, Cα RMSD100 (53), Cα radius of gyration, and CONGENEAL score (54)—were directly comparable across proteins and were not divided by the number of residues (Table 3).
Table 2.
Per-residue properties of the TS ensemble
| Property | Fold class |
|||
|---|---|---|---|---|
| All | All-α | All-β | Mixed α/β | |
| Number of total contacts∗ | 2.73 (0.31) | 2.80 (0.27) | 2.57 (0.31) | 2.81 (0.25) |
| Number of native contacts∗ | 2.23 (0.35) | 2.36 (0.31) | 2.02 (0.35) | 2.31 (0.27) |
| Number of nonnative contacts∗ | 0.50 (0.12) | 0.44 (0.12) | 0.55 (0.13) | 0.49 (0.12) |
| Number of hydrogen bonds | 0.55 (0.12) | 0.66 (0.11) | 0.44 (0.11) | 0.56 (0.09) |
| Number of hydrophobic contacts† | 10.78 (1.30) | 10.76 (1.28) | 10.46 (1.37) | 11.10 (1.10) |
| Number of other contacts† | 17.45 (2.01) | 19.25 (1.77) | 15.82 (1.71) | 17.57 (1.22) |
| Mc SASA (Å2) | 17.30 (3.11) | 16.12 (2.73) | 18.77 (3.00) | 16.70 (2.31) |
| Sc SASA (Å2) | 65.13 (8.25) | 70.46 (7.34) | 63.84 (7.39) | 62.60 (7.53) |
| Total SASA (Å2) | 82.43 (9.84) | 86.58 (8.43) | 82.61 (9.31) | 79.30 (8.92) |
For each property, the mean value over the TS ensemble was calculated and divided by the number of residues in the protein. The values given in this table are the mean over all proteins; the standard deviation is in parentheses. Mc, main chain; Sc, side chain.
Contacts counted on a residue-residue basis.
Contacts counted on an atom-atom basis.
Table 3.
Average properties of the aggregate TS ensemble
| Property | Fold class |
|||
|---|---|---|---|---|
| All | All-α | All-β | Mixed α/β | |
| Fraction lp α-structure (ϕ,ψ) | 0.32 (0.16) | 0.53 (0.12) | 0.14 (0.09) | 0.31 (0.09) |
| Fraction β-structure (ϕ,ψ) | 0.37 (0.14) | 0.18 (0.08) | 0.51 (0.09) | 0.38 (0.08) |
| Cα RMSD (Å) | 5.03 (0.88) | 4.82 (0.96) | 5.29 (0.91) | 4.94 (0.77) |
| Cα RMSD100 (Å) | 5.23 (1.84) | 5.53 (2.01) | 5.58 (1.52) | 4.16 (1.38) |
| Cα radius of gyration (Å) | 15.13 (2.81) | 14.37 (2.51) | 14.66 (2.67) | 15.83 (2.70) |
| CONGENEAL Score | 0.35 (0.06) | 0.32 (0.07) | 0.38 (0.08) | 0.33 (0.05) |
Mean value for each property was calculated for each protein. The values given in this table are the means over all proteins in the set, with the standard deviation given in parentheses.
Properties across fold classes were similar, with the exception of the fraction of residues with helical and β-(ϕ,ψ) angles. As expected, the all-α-proteins had more residues in the α-helical region of (ϕ,ψ) space than all-β proteins, and vice versa. The SDs of the average fraction of residues with α- and β-structure overlapped for all proteins, and the same is true for the mixed α/β-class. The mixed α/β class maintained a smaller fraction of residues with helical (ϕ,ψ) angles than all-α proteins, whereas the SD for the average fraction of β-residues overlapped between mixed α/β and all-β classes (Table 3). At the same time, the fractions α and β, when divided by the native state, were the same across fold classes and were similar to each other (Table 1).
TS ensemble structural properties by residue type
S3° and S values were used to probe the nature of the TS at the level of residue type. The mean values of S and S3° at each position were calculated for each protein. Residues were categorized by amino acid type, and then by the secondary structure in the native state. Residues were assigned as helix, β, or “other” in the native structure based on hydrogen bonding using DSSP (55). Residues were further classified by the SASA in the native simulation. The peak SASA from 100-ns simulations of GGXGG pentapeptides at 298 K (taken from our SLIRP database, www.dynameomics.org/SLIRP) was used as a reference maximum value for each amino acid (56). The data were separated into two classes, those residues in the native state with <10% of the maximum SASA were considered buried, and all other residues were considered exposed. Table 4 shows S and S3° values for residues categorized by secondary structure, and Table 5 shows S and S3° for a subset of residues classified by type, secondary structure, and SASA. In both tables, the mean value, with the SD in parentheses, and the value of the most highly populated bin are given. Figs. 2 and 3 provide a graphical representation of the data for Ala.
Table 4.
S and S3° values for amino acids categorized by type and secondary structure in starting native structure
| Residue | Helix |
β |
Other |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S3° |
S |
S3° |
S |
S3° |
S |
|||||||
| Mean | Mode | Mean | Mode | Mean | Mode | Mean | Mode | Mean | Mode | Mean | Mode | |
| Ala | 0.73 (0.16) | 0.64 | 0.58 (0.21) | 0.64 | 0.61 (0.21) | 0.64 | 0.43 (0.18) | 0.34 | 0.90 (1.18) | 0.49 | 0.22 (0.18) | 0.04 |
| Cys | 0.65 (0.14) | 0.64 | 0.53 (0.18) | 0.56 | 0.54 (0.19) | 0.56 | 0.37 (0.18) | 0.34 | 0.54 (0.25) | 0.49 | 0.17 (0.14) | 0.04 |
| Asp | 0.74 (0.23) | 0.64 | 0.55 (0.24) | 0.56 | 0.72 (0.37) | 0.64 | 0.46 (0.26) | 0.49 | 0.81 (0.85) | 0.64 | 0.25 (0.20) | 0.19 |
| Glu | 0.75 (0.22) | 0.79 | 0.59 (0.23) | 0.64 | 0.72 (0.26) | 0.56 | 0.54 (0.23) | 0.64 | 0.94 (1.53) | 0.64 | 0.27 (0.25) | 0.04 |
| Phe | 0.58 (0.16) | 0.56 | 0.48 (0.17) | 0.49 | 0.49 (0.14) | 0.41 | 0.38 (0.15) | 0.34 | 0.64 (1.43) | 0.41 | 0.17 (0.14) | 0.11 |
| Gly | 0.77 (0.19) | 0.79 | 0.55 (0.24) | 0.64 | 0.64 (0.31) | 0.56 | 0.35 (0.17) | 0.34 | 0.83 (0.58) | 0.64 | 0.22 (0.18) | 0.04 |
| His | 0.73 (0.25) | 0.56 | 0.57 (0.23) | 0.49 | 0.59 (0.16) | 0.49 | 0.42 (0.15) | 0.34 | 0.83 (0.93) | 0.49 | 0.24 (0.22) | 0.11 |
| Ile | 0.63 (0.13) | 0.64 | 0.52 (0.16) | 0.56 | 0.58 (0.16) | 0.49 | 0.45 (0.17) | 0.34 | 0.69 (1.08) | 0.41 | 0.23 (0.32) | 0.11 |
| Lys | 0.83 (0.23) | 0.79 | 0.64 (0.24) | 0.71 | 0.77 (0.30) | 0.64 | 0.56 (0.27) | 0.41 | 0.93 (1.18) | 0.71 | 0.29 (0.31) | 0.19 |
| Leu | 0.65 (0.15) | 0.64 | 0.54 (0.18) | 0.64 | 0.59 (0.17) | 0.49 | 0.45 (0.18) | 0.41 | 0.68 (0.66) | 0.49 | 0.23 (0.19) | 0.19 |
| Met | 0.67 (0.18) | 0.64 | 0.54 (0.18) | 0.56 | 0.57 (0.16) | 0.64 | 0.45 (0.15) | 0.34 | 0.76 (1.28) | 0.49 | 0.17 (0.18) | 0.04 |
| Asn | 0.70 (0.18) | 0.71 | 0.55 (0.21) | 0.64 | 0.63 (0.22) | 0.64 | 0.45 (0.21) | 0.49 | 0.77 (0.54) | 0.49 | 0.25 (0.20) | 0.11 |
| Pro | 0.70 (0.20) | 0.56 | 0.54 (0.21) | 0.49 | 0.70 (0.24) | 0.64 | 0.53 (0.21) | 0.49 | 0.82 (0.69) | 0.49 | 0.35 (0.29) | 0.19 |
| Gln | 0.73 (0.19) | 0.71 | 0.59 (0.22) | 0.64 | 0.62 (0.21) | 0.56 | 0.44 (0.19) | 0.56 | 0.89 (0.98) | 0.49 | 0.27 (0.24) | 0.19 |
| Arg | 0.77 (0.20) | 0.64 | 0.61 (0.22) | 0.64 | 0.72 (0.30) | 0.64 | 0.52 (0.24) | 0.49 | 1.02 (1.26) | 0.64 | 0.29 (0.22) | 0.19 |
| Ser | 0.80 (0.23) | 0.79 | 0.62 (0.22) | 0.56 | 0.69 (0.27) | 0.64 | 0.45 (0.22) | 0.34 | 0.91 (1.37) | 0.49 | 0.27 (0.22) | 0.19 |
| Thr | 0.74 (0.23) | 0.71 | 0.60 (0.24) | 0.64 | 0.66 (0.21) | 0.64 | 0.48 (0.21) | 0.34 | 0.76 (0.54) | 0.64 | 0.28 (0.24) | 0.19 |
| Val | 0.67 (0.16) | 0.64 | 0.54 (0.19) | 0.49 | 0.59 (0.16) | 0.64 | 0.46 (0.17) | 0.49 | 0.76 (1.18) | 0.49 | 0.24 (0.25) | 0.19 |
| Trp | 0.56 (0.16) | 0.56 | 0.45 (0.15) | 0.49 | 0.51 (0.17) | 0.49 | 0.37 (0.16) | 0.34 | 0.52 (0.35) | 0.34 | 0.18 (0.12) | 0.11 |
| Tyr | 0.60 (0.16) | 0.49 | 0.48 (0.19) | 0.49 | 0.55 (0.16) | 0.49 | 0.41 (0.16) | 0.34 | 0.62 (0.78) | 0.49 | 0.21 (0.19) | 0.11 |
Values were separated into 20 bins of 0.075 each. The mode is the most populated bin.
Table 5.
S and S3° values for selected amino acids (Ala, Asp, and Leu) categorized by type, native secondary structure, and SASA
| Amino acid environment | S3° |
S |
||
|---|---|---|---|---|
| Mean (SD) | Modal bin | Mean (SD) | Modal bin | |
| Ala other buried | 0.49 (0.15) | 0.49 | 0.18 (0.13) | 0.19 |
| Ala other exposed | 1.00 (1.31) | 0.64 | 0.23 (0.19) | 0.04 |
| Ala other | 0.90 (1.18) | 0.49 | 0.22 (0.18) | 0.04 |
| Ala β-buried | 0.58 (0.14) | 0.64 | 0.45 (0.17) | 0.34 |
| Ala β-exposed | 0.68 (0.31) | 0.64 | 0.40 (0.22) | 0.26 |
| Ala β | 0.61 (0.21) | 0.64 | 0.43 (0.18) | 0.34 |
| Ala helix buried | 0.67 (0.12) | 0.64 | 0.56 (0.18) | 0.64 |
| Ala helix exposed | 0.78 (0.18) | 0.79 | 0.59 (0.24) | 0.79 |
| Ala helix | 0.73 (0.16) | 0.64 | 0.58 (0.21) | 0.64 |
| Asp other buried | 0.58 (0.17) | 0.49 | 0.25 (0.17) | 0.19 |
| Asp other exposed | 0.83 (0.88) | 0.64 | 0.25 (0.20) | 0.19 |
| Asp other | 0.81 (0.85) | 0.64 | 0.25 (0.20) | 0.19 |
| Asp β-buried | 0.63 (0.17) | 0.64 | 0.45 (0.15) | 0.49 |
| Asp β-exposed | 0.75 (0.42) | 0.64 | 0.47 (0.28) | 0.49 |
| Asp β | 0.72 (0.37) | 0.64 | 0.46 (0.26) | 0.49 |
| Asp helix buried | 0.66 (0.14) | 0.64 | 0.50 (0.18) | 0.41 |
| Asp helix exposed | 0.76 (0.24) | 0.79 | 0.55 (0.24) | 0.56 |
| Asp helix | 0.74 (0.23) | 0.64 | 0.55 (0.24) | 0.56 |
| Leu other buried | 0.47 (0.13) | 0.49 | 0.21 (0.13) | 0.11 |
| Leu other exposed | 0.79 (0.78) | 0.49 | 0.23 (0.21) | 0.19 |
| Leu other | 0.68 (0.66) | 0.49 | 0.23 (0.19) | 0.19 |
| Leu β-buried | 0.55 (0.12) | 0.49 | 0.45 (0.15) | 0.56 |
| Leu β-exposed | 0.66 (0.21) | 0.49 | 0.46 (0.22) | 0.41 |
| Leu β | 0.59 (0.17) | 0.49 | 0.45 (0.18) | 0.41 |
| Leu helix buried | 0.61 (0.10) | 0.64 | 0.52 (0.15) | 0.64 |
| Leu helix exposed | 0.72 (0.17) | 0.71 | 0.56 (0.21) | 0.64 |
| Leu helix | 0.65 (0.15) | 0.64 | 0.54 (0.18) | 0.64 |
Figure 2.
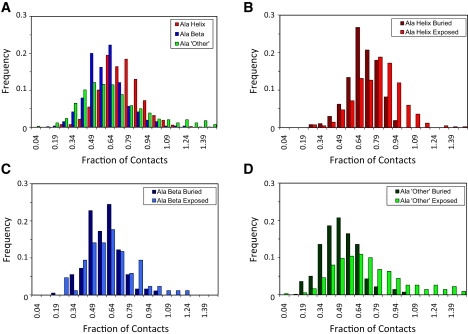
Histograms of S3° values for alanine residues in different types of secondary structure in the native state. (A) Histograms for alanine in “other”, β, and helical conformations. (B–D) Residues with each type of secondary structure in the starting state were further subdivided by the SASA in the native state. Residues >10% buried over the native simulation were separated from those whose mean SASA was >10%.
Figure 3.
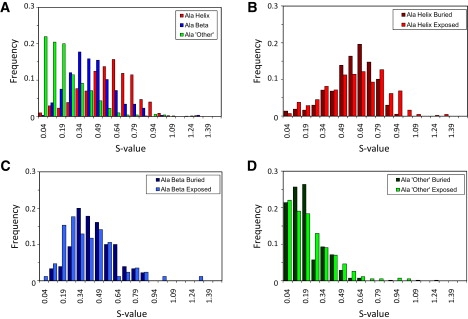
Histograms of S values for alanine residues that are in different types of secondary structure in the native state. (A) Histograms for Ala in “other”, β, and helical conformations. (B–D) Residues with each type of secondary structure in the starting state were further subdivided by the SASA in the native state. Residues >10% buried over the native simulation were separated from those whose mean SASA was >10%.
The distributions of the extent of tertiary contacts (S3°) in the TS ensembles were broad (Fig. 2). Although the distributions were centered on different values, the mean values for each secondary type fall within 1 SD of each other. When S3° values were further divided by SASA, the distributions for β-residues were almost identical. For “other” and helical Ala residues, there was a shift to lower tertiary contacts for buried residues compared to exposed residues (Fig. 2). The residues with the most contacts in the native state lost the most contacts in the TS ensemble (Table 5).
The addition of the secondary structural term in the S-value calculation led to less overlap between the distributions for residues with each secondary structure type. For example, the mean and modal values for Ala moved farther apart, with helical residues having a mean S value of 0.6, β-residues 0.4, and “other” residues 0.2 (Fig. 3). The pattern was repeated across the data set, though the magnitude of the difference in S between helical and β-residues depended on the residue type. When S values were further categorized by SASA, little difference was seen between the two distributions, and the secondary-structure component was dominant over the extent of burial.
Pairwise contacts in the TS ensemble
To determine whether certain types of contacts were lost or gained in the TS ensemble compared with the native state, pairwise contact maps were calculated. Analysis of the data for all proteins showed a loss of contacts between Ile, Val, Leu, and Ala residues in the TS, compared with the native state. In contrast, there was some gain in contacts between Asp-Lys charged residues (Fig. 4 A). When the data were classified by sequence separation, the greatest loss was long-range contacts (greater than i → i + 15), followed by medium- (between i → i + 6 and i → i + 15) and then short-range contacts (less than i → i + 6) (Fig. 4), as expected. There was no increase in short-range contacts between charged residues (Fig. 4 B); however, the number of medium-range charged contacts increased (Fig. 4 C).
Figure 4.
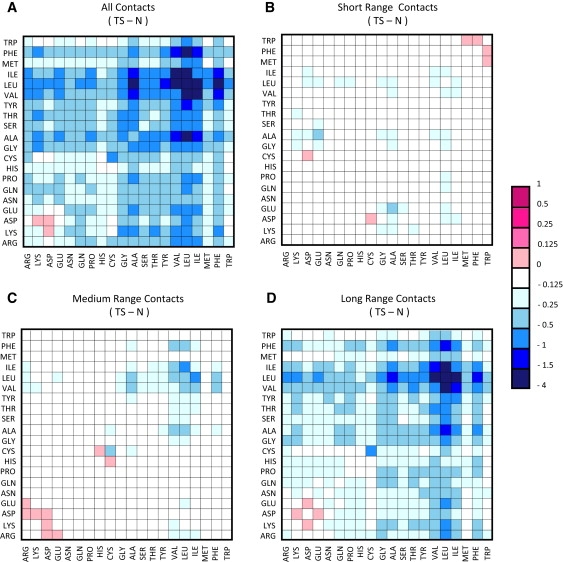
Pairwise contact difference maps between the TS and the native state for all proteins. The number of times each pairwise contact is present in the TS ensemble over all proteins is calculated. The same calculation is then carried out for the native simulations. The native value is subtracted from the TS. Shades of blue represent a decrease in the TS, shades of pink an increase. Neighboring (i → i and i → i + 1) contacts are excluded in all plots. (A) All contacts. (B) Short-range contacts. Only residue pairs separated by no more than five amino acids were considered. (C) Medium-range contacts. Only residue pairs separated by 6–15 amino acids were considered. (D) Long-range contacts. Only residue pairs separated by >15 amino acids were considered. Full color figure available online.
Across the fold categories, the number of short-range contacts in the TS was similar to that in the native state, with small increases. The increase in contacts was more uniform for the all-β-fold category (Fig. S2). All categories gained some medium-range charged contacts but lost hydrophobic contacts (Fig. S1, Fig. S2, and Fig. S3). The all-α-fold class had the smallest decrease in medium-range hydrophobic contacts. The all-α-, all-β-, and mixed α/β-fold categories all showed a slight increase in long-range charged contacts and a larger decrease in long-range hydrophobic contacts (Fig. S1, Fig. S2, and Fig. S3).
Discussion
For the past 15 years, our lab has compared predicted TS structures to experimental data, with much success. The comparison has been carried out using all helical proteins (EnHD (15,17,31), α-spectrin (27), the FF domain (25), EBD (20), c-myb (15,19), and protein A (23,24)), mixed α/β-proteins (CI2 (1–5), barnase (13), and FKBP12 (14)), and all-β-proteins (FBP28 WW domain (16)). We have now extended our efforts to studying the folding/unfolding pathways of a structurally diverse set of proteins as part of our Dynameomics project.
As a result of Dynameomics, we now have a database of over 6000 simulations, and we describe here the TS ensembles of 1303 simulations of 183 different proteins. These data can be used in a traditional manner by investigating the properties of a single protein in detail. We have compared our predicted TS structures in this data set with the Ф-values available for five of the proteins (EnHD, FKBP12, Fyn and α-spectrin SH3 domain, and Im7) for which good experimental data are available (R. D. Toofanny, A. L. Jonsson, and V. Daggett, unpublished results). The linear correlation coefficient between the experimental Ф-values and MD-derived S values ranges from 0.65 for Im7 to 0.91 for Fyn SH3 domain. The other TS ensembles identified here are predictions that await experimental confirmation.
The Dynameomics database is also useful for predicting the TS properties of proteins related to those that have been simulated. There is evidence that fold family members share similar TS structures. For example, Im7 and Im9 are four-helix bundle proteins in which three of the four helices are formed in the TS. There is significant similarity among the TS structures despite the fact that Im7 populates an on-pathway intermediate, whereas Im9 does not (58). Similarly, it has been shown, many members of the SH3 domain fold family share a common polarized TS (59). Using our Dynameomics database, we can make predictions across the 183 proteins. In addition, we hope that these data can aid in protein design and engineering. For example, MD-derived TS structures have been used to design faster folding variants of a protein (60).
In this study, we have focused on identifying the general properties of the TS ensemble by combining the TS structures of all 183 structurally diverse proteins in the largest description of TS ensembles to date. Over all proteins, there was an average 30% increase in the total SASA of the TS relative to the native state and a 10% increase in the radius of gyration (Table 1). The largest increases occurred in main-chain polar and side-chain nonpolar SASA values, indicating that the structures expanded to expose the hydrophobic residues and the main chain to solvent, as expected during unfolding. Over all fold classes, the TS structures maintained 74% of the native contacts, at the same time gaining on average 16% nonnative contacts relative to the native-state simulations. These data confirm that the TS structures are expanded versions of the native state.
Experimentally, the average degree of exposure of the TS ensemble can be measured using the Tanford β-value (βT). Three- and four-helix bundle proteins typically have high βT values: 0.83 for EnHD (15), 0.89 for Im7 (58), and 0.90 for hTRF1 (15), for example. Mixed α/β-proteins, such as CI2 and FKB12, have smaller values, 0.61 (61) and 0.67 (62), respectively. All-β-proteins also tend to have smaller βT values: 0.65 for FBP28 WW domain (16), 0.68 for Fyn SH3 domain (63), and 0.69 for Src SH3 domain (64). There are outliers in each case, for example, all-β cold shock protein A (CspA) and the all-α R17 domain from α-spectrin, with βT values of 0.90 (65) and 0.60 (27), respectively. Over the 183 proteins studied here, the average increase in solvent exposure in the TS ensemble relative to the native state was 28% (Table 1), resulting in a βT value of ∼0.72. This value was not significantly different over the different fold classes. However, we do obtain a range of possible βT values over all our proteins of 0.51–0.98, showing that we are capturing the range of values seen experimentally.
Properties categorized by native secondary structure revealed a preference for maintaining secondary structure when the residue was part of a helix in the native structure compared to residues starting in β and “other” secondary structure types. For each residue type, with the exception of Pro, the distribution of S values was skewed toward higher values if the residue began in a helical conformation than if the native secondary-structure conformation was β or “other”. Although the SDs of the average S value overlapped for each starting conformation, in each case, the most populated bin in the distribution of S values was highest for residues starting in a helical conformation (Table 4). In addition, residues that were buried in the starting structure had a higher proportion of residues with lower fractions of contacts compared to the native state. Therefore, residues that started with the most contacts lost the largest fraction of contacts by the TS.
We have described the general properties of the TS ensemble from a diverse set of protein structures, representing ∼67% of all known protein structures. We can imagine a scenario in which proteins from different folds pass through TS ensembles with vastly different properties, such as a large expansion in the TS when compared to the native state, as measured by SASA, or varied numbers of native contacts in the TS. This scenario would lead to broad distributions and large SDs in the average properties. Instead, across the diverse set of folds studied here, the average global properties have relatively small SDs (Tables 1–3). Even though the per-residue properties depend on the starting secondary-structure conformation and extent of burial, we have identified unifying features of the TS ensemble across folds, suggesting that there are very generic rules governing the structure and global properties of the TS ensemble.
Supporting Material
Two tables and three figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01458-1.
Supporting Material
Acknowledgments
We thank Dr. David Beck for running the simulations.
We are grateful for support from the Microsoft External Research Programs through Microsoft Research, www.microsoft.com/science (to V. D.) and from the National Institutes of Health (GM 50789). The MD trajectories were run using resources of the National Energy Resource Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy (contract No. DE-AC02-05CH11231).
Footnotes
Kathryn A. Scott's present address is Oxford Centre for Integrative Systems Biology, Department of Biochemistry, University of Oxford, Oxford, OX1 3QU, UK.
References
- 1.Daggett V., Li A.J., Itzhaki L.S., Otzen D.E., Fersht A.R. Structure of the transition state for folding of a protein derived from experiment and simulation. J. Mol. Biol. 1996;257:430–440. doi: 10.1006/jmbi.1996.0173. [DOI] [PubMed] [Google Scholar]
- 2.Day R., Daggett V. Sensitivity of the folding/unfolding transition state ensemble of chymotrypsin inhibitor 2 to changes in temperature and solvent. Protein Sci. 2005;14:1242–1252. doi: 10.1110/ps.041226005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itzhaki L.S., Otzen D.E., Fersht A.R. The structure of the transition state for folding of chymotrypsin inhibitor 2 analyzed by protein engineering methods: evidence for a nucleation-condensation mechanism for protein folding. J. Mol. Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 4.Otzen D.E., Itzhaki L.S., Elmasry N.F., Jackson S.E., Fersht A.R. Structure of the transition state for the folding/unfolding of the barley chymotrypsin inhibitor 2 and its implications for mechanisms of protein folding. Proc. Natl. Acad. Sci. USA. 1994;91:10422–10425. doi: 10.1073/pnas.91.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li A.J., Daggett V. Characterization of the transition state of protein unfolding by use of molecular dynamics: chymotrypsin inhibitor 2. Proc. Natl. Acad. Sci. USA. 1994;91:10430–10434. doi: 10.1073/pnas.91.22.10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grantcharova V.P., Riddle D.S., Santiago J.V., Baker D. Important role of hydrogen bonds in the structurally polarized transition state for folding of the src SH3 domain. Nat. Struct. Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
- 7.Martinez J.C., Serrano L. The folding transition state between SH3 domains is conformationally restricted and evolutionarily conserved. Nat. Struct. Biol. 1999;6:1010–1016. doi: 10.1038/14896. [DOI] [PubMed] [Google Scholar]
- 8.Northey J.G.B., Di Nardo A.A., Davidson A.R. Hydrophobic core packing in the SH3 domain folding transition state. Nat. Struct. Biol. 2002;9:126–130. doi: 10.1038/nsb748. [DOI] [PubMed] [Google Scholar]
- 9.Gsponer J., Caflisch A. Role of native topology investigated by multiple unfolding simulations of four SH3 domains. J. Mol. Biol. 2001;309:285–298. doi: 10.1006/jmbi.2001.4552. [DOI] [PubMed] [Google Scholar]
- 10.Gsponer J., Caflisch A. Molecular dynamics simulations of protein folding from the transition state. Proc. Natl. Acad. Sci. USA. 2002;99:6719–6724. doi: 10.1073/pnas.092686399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai J., Levitt M., Baker D. Hierarchy of structure loss in MD simulations of src SH3 domain unfolding. J. Mol. Biol. 1999;291:215–225. doi: 10.1006/jmbi.1999.2949. [DOI] [PubMed] [Google Scholar]
- 12.Bennion B.J., Daggett V. The molecular basis for the chemical denaturation of proteins by urea. Proc. Natl. Acad. Sci. USA. 2003;100:5142–5147. doi: 10.1073/pnas.0930122100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daggett V., Li A.J., Fersht A.R. Combined molecular dynamics and Φ-value analysis of structure-reactivity relationships in the transition state and unfolding pathway of barnase: structural basis of Hammond and anti-Hammond effects. J. Am. Chem. Soc. 1998;120:12740–12754. [Google Scholar]
- 14.Fulton K.F., Main E.R.G., Daggett V., Jackson S.E. Mapping the interactions present in the transition state for unfolding/folding of FKBP12. J. Mol. Biol. 1999;291:445–461. doi: 10.1006/jmbi.1999.2942. [DOI] [PubMed] [Google Scholar]
- 15.Gianni S., Guydosh N.R., Khan F., Caldas T.D., Mayor U. Unifying features in protein-folding mechanisms. Proc. Natl. Acad. Sci. USA. 2003;100:13286–13291. doi: 10.1073/pnas.1835776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petrovich M., Jonsson A.L., Ferguson N., Daggett V., Fersht A.R. Φ Analysis at the experimental limits: mechanism of β-hairpin formation. J. Mol. Biol. 2006;360:865–881. doi: 10.1016/j.jmb.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 17.Mayor U., Guydosh N.R., Johnson C.M., Grossmann J.G., Sato S. The complete folding pathway of a protein from nanoseconds to microseconds. Nature. 2003;421:863–867. doi: 10.1038/nature01428. [DOI] [PubMed] [Google Scholar]
- 18.Daggett V., Fersht A.R. Is there a unifying mechanism for protein folding? Trends Biochem. Sci. 2003;28:18–25. doi: 10.1016/s0968-0004(02)00012-9. [DOI] [PubMed] [Google Scholar]
- 19.White G.W.N., Gianni S., Grossmann J.G., Jemth P., Fersht A.R. Simulation and experiment conspire to reveal cryptic intermediates and a slide from the nucleation-condensation to framework mechanism of folding. J. Mol. Biol. 2005;350:757–775. doi: 10.1016/j.jmb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Ferguson N., Day R., Johnson C.M., Allen M.D., Daggett V. Simulation and experiment at high temperatures: ultrafast folding of a thermophilic protein by nucleation-condensation. J. Mol. Biol. 2005;347:855–870. doi: 10.1016/j.jmb.2004.12.061. [DOI] [PubMed] [Google Scholar]
- 21.Sharpe T., Jonsson A.L., Rutherford T.J., Daggett V., Fersht A.R. The role of the turn in β-hairpin formation during WW domain folding. Protein Sci. 2007;16:2233–2239. doi: 10.1110/ps.073004907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferguson N., Pires J.R., Toepert F., Johnson C.M., Pan Y.P. Using flexible loop mimetics to extend Φ-value analysis to secondary structure interactions. Proc. Natl. Acad. Sci. USA. 2001;98:13008–13013. doi: 10.1073/pnas.221467398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato S., Religa T.L., Daggett V., Fersht A.R. Testing protein-folding simulations by experiment: B domain of protein A. Proc. Natl. Acad. Sci. USA. 2004;101:6952–6956. doi: 10.1073/pnas.0401396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alonso D.O., Daggett V. Staphylococcal protein A: unfolding pathways, unfolded states, and differences between the B and E domains. Proc. Natl. Acad. Sci. USA. 2000;97:133–138. doi: 10.1073/pnas.97.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jemth P., Day R., Gianni S., Khan F., Allen M. The structure of the major transition state for folding of an FF domain from experiment and simulation. J. Mol. Biol. 2005;350:363–378. doi: 10.1016/j.jmb.2005.04.067. [DOI] [PubMed] [Google Scholar]
- 26.Jemth P., Gianni S., Day R., Li B., Johnson C.M. Demonstration of a low-energy on-pathway intermediate in a fast-folding protein by kinetics, protein engineering, and simulation. Proc. Natl. Acad. Sci. USA. 2004;101:6450–6455. doi: 10.1073/pnas.0401732101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scott K.A., Randles L.G., Moran S.J., Daggett V., Clarke J. The folding pathway of spectrin R17 from experiment and simulation: using experimentally validated MD simulations to characterize states hinted at by experiment. J. Mol. Biol. 2006;359:159–173. doi: 10.1016/j.jmb.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 28.DeMarco M.L., Alonso D.O., Daggett V. Diffusing and colliding: the atomic level folding/unfolding pathway of a small helical protein. J. Mol. Biol. 2004;341:1109–1124. doi: 10.1016/j.jmb.2004.06.074. [DOI] [PubMed] [Google Scholar]
- 29.Mayor U., Johnson C.M., Daggett V., Fersht A.R. Protein folding and unfolding in microseconds to nanoseconds by experiment and simulation. Proc. Natl. Acad. Sci. USA. 2000;97:13518–13522. doi: 10.1073/pnas.250473497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Religa T.L., Markson J.S., Mayor U., Freund S.M., Fersht A.R. Solution structure of a protein denatured state and folding intermediate. Nature. 2005;437:1053–1056. doi: 10.1038/nature04054. [DOI] [PubMed] [Google Scholar]
- 31.McCully M.E., Beck D.A.C., Daggett V. Microscopic reversibility of protein folding in molecular dynamics simulations of the engrailed homeodomain. Biochemistry. 2008;47:7079–7089. doi: 10.1021/bi800118b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Day R., Daggett V. Direct observation of microscopic reversibility in single-molecule protein folding. J. Mol. Biol. 2007;366:677–686. doi: 10.1016/j.jmb.2006.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Day R., Bennion B.J., Ham S., Daggett V. Increasing temperature accelerates protein unfolding without changing the pathway of unfolding. J. Mol. Biol. 2002;322:189–203. doi: 10.1016/s0022-2836(02)00672-1. [DOI] [PubMed] [Google Scholar]
- 34.Best R.B., Li B., Steward A., Daggett V., Clarke J. Can non-mechanical proteins withstand force? Stretching barnase by atomic force microscopy and molecular dynamics simulation. Biophys. J. 2001;81:2344–2356. doi: 10.1016/S0006-3495(01)75881-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beck D.A., Jonsson A.L., Schaeffer R.D., Scott K.A., Day R. Dynameomics: mass annotation of protein dynamics and unfolding in water by high-throughput atomistic molecular dynamics simulations. Protein Eng. Des. Sel. 2008;21:353–368. doi: 10.1093/protein/gzn011. [DOI] [PubMed] [Google Scholar]
- 36.Day R., Beck D.A., Armen R.S., Daggett V. A consensus view of fold space: combining SCOP, CATH, and the Dali Domain Dictionary. Protein Sci. 2003;12:2150–2160. doi: 10.1110/ps.0306803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gunasekaran K., Eyles S.J., Hagler A.T., Gierasch L.M. Keeping it in the family: folding studies of related proteins. Curr. Opin. Struct. Biol. 2001;11:83–93. doi: 10.1016/s0959-440x(00)00173-1. [DOI] [PubMed] [Google Scholar]
- 38.Clarke J., Cota E., Fowler S.B., Hamill S.J. Folding studies of immunoglobulin-like β-sandwich proteins suggest that they share a common folding pathway. Structure. 1999;7:1145–1153. doi: 10.1016/s0969-2126(99)80181-6. [DOI] [PubMed] [Google Scholar]
- 39.Benson N.C., Daggett V. Dynameomics: large-scale assessment of native protein flexibility. Protein Sci. 2008;17:2038–2050. doi: 10.1110/ps.037473.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beck D.A.C., Alonso D.O.V., Daggett V. University of Washington; Seattle: 2000–2009. In lucem Molecular Mechanics (ilmm) [Google Scholar]
- 41.Levitt M., Hirshberg M., Sharon R., Daggett V. Potential energy function and parameters for simulations of the molecular dynamics of proteins and nucleic acids in solution. Comput. Phys. Commun. 1995;91:215–231. [Google Scholar]
- 42.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levitt M., Hirshberg M., Sharon R., Laidig K.E., Daggett V. Calibration and testing of a water model for simulation of the molecular dynamics of proteins and nucleic acids in solution. J. Phys. Chem. B. 1997;101:5051–5061. [Google Scholar]
- 44.Haar L., Gallagher J.S., Kell G.S. Hemisphere; Washington, DC: 1984. NBS/NRC Steam Tables: Thermodynamic and Transport Properties and Computer Programs for Vapor and Liquid States of Water in SI Units. [Google Scholar]
- 45.Beck D.A.C., Armen R.S., Daggett V. Cutoff size need not strongly influence molecular dynamics results for solvated polypeptides. Biochemistry. 2005;44:609–616. doi: 10.1021/bi0486381. [DOI] [PubMed] [Google Scholar]
- 46.Beck D.A.C., Daggett V. Methods for molecular dynamics simulations of protein folding/unfolding in solution. Methods Enzymol. 2004;34:112–120. doi: 10.1016/j.ymeth.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 47.Li A.J., Daggett V. Identification and characterization of the unfolding transition state of chymotrypsin inhibitor 2 by molecular dynamics simulations. J. Mol. Biol. 1996;257:412–429. doi: 10.1006/jmbi.1996.0172. [DOI] [PubMed] [Google Scholar]
- 48.Reference deleted in proof.
- 49.Kehl C., Simms A.M., Toofanny R.D., Daggett V. Dynameomics: a multi-dimensional analysis-optimized database for dynamic protein data. Protein Eng. Des. Sel. 2008;21:379–386. doi: 10.1093/protein/gzn015. [DOI] [PubMed] [Google Scholar]
- 50.Simms A.M., Toofanny R.D., Kehl C., Benson N.C., Daggett V. Dynameomics: design of a computational lab workflow and scientific data repository for protein simulations. Protein Eng. Des. Sel. 2008;21:369–377. doi: 10.1093/protein/gzn012. [DOI] [PubMed] [Google Scholar]
- 51.Murzin A.G., Brenner S.E., Hubbard T., Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 52.Orengo C.A., Michie A.D., Jones S., Jones D.T., Swindells M.B. CATH: a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 53.Carugo O., Pongor S. A normalized root-mean-square distance for comparing protein three-dimensional structures. Protein Sci. 2001;10:1470–1473. doi: 10.1110/ps.690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yee D.P., Dill K.A. Families and the structural relatedness among globular proteins. Protein Sci. 1993;2:884–899. doi: 10.1002/pro.5560020603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 56.Beck D.A.C., Alonso D.O.V., Inoyama D., Daggett V. The intrinsic conformational propensities of the 20 naturally occurring amino acids and reflection of these propensities in proteins. Proc. Natl. Acad. Sci. USA. 2008;105:12259–12264. doi: 10.1073/pnas.0706527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reference deleted in proof.
- 58.Friel C.T., Capaldi A.P., Radford S.E. Structural analysis of the rate-limiting transition states in the folding of lm7 and lm9: similarities and differences in the folding of homologous proteins. J. Mol. Biol. 2003;326:293–305. doi: 10.1016/s0022-2836(02)01249-4. [DOI] [PubMed] [Google Scholar]
- 59.Riddle D.S., Grantcharova V.P., Santiago J.V., Alm E., Ruczinski I. Experiment and theory highlight role of native state topology in SH3 folding. Nat. Struct. Biol. 1999;6:1016–1024. doi: 10.1038/14901. [DOI] [PubMed] [Google Scholar]
- 60.Ladurner A.G., Itzhaki L.S., Daggett V., Fersht A.R. Synergy between simulation and experiment in describing the energy landscape of protein folding. Proc. Natl. Acad. Sci. USA. 1998;95:8473–8478. doi: 10.1073/pnas.95.15.8473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jackson S.E., Fersht A.R. Folding of chymotrypsin inhibitor-2. 1. Evidence for a 2-state transition. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 62.Jackson S.E. How do small single-domain proteins fold? Fold. Des. 1998;3:R81–R91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 63.Plaxco K.W., Guijarro J.I., Morton C.J., Pitkeathly M., Campbell I.D. The folding kinetics and thermodynamics of the Fyn-SH3 domain. Biochemistry. 1998;37:2529–2537. doi: 10.1021/bi972075u. [DOI] [PubMed] [Google Scholar]
- 64.Grantcharova V.P., Baker D. Folding dynamics of the src SH3 domain. Biochemistry. 1997;36:15685–15692. doi: 10.1021/bi971786p. [DOI] [PubMed] [Google Scholar]
- 65.Reid K.L., Rodriguez H.M., Hillier B.J., Gregoret L.M. Stability and folding properties of a model β-sheet protein, Escherichia coli CspA. Protein Sci. 1998;7:470–479. doi: 10.1002/pro.5560070228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.